

Abstract
Major histocompatibility complex (MHC) class I antigens are constitutively expressed yet highly induced by interferon (IFN) during inflammation. We found that not only IFN-induced but also normal basal expression of MHC I required IFN receptors and signal transducer and activator of transcription (STAT)1, providing genetic evidence for continuous IFN signaling. Surprisingly, an IFN-independent requirement for STAT1 was also found, specifically in T lymphocytes, where MHC class I expression was not fully accounted for by IFN signaling. This IFN-independent pathway maintained tyrosine phosphorylation of STAT1 in T but not B lymphocytes even in the absence of IFN receptors. Interestingly, interleukin (IL)-7 selectively activated STAT1 and induced MHC class I in mature T but not B cells. These loss of function studies demonstrate an essential role of endogenous IFN and activated STAT1 for constitutive MHC class I expression in normal mice and define IL-7–dependent but IFN-independent regulation of STAT1 restricted to T lymphocytes.
Keywords: interferon, STAT1, interleukin-7, gene regulation, tyrosine phosphorylation
MHC class I antigens, which are cell surface glycoproteins composed of a heavy chain and a light chain (β2-microglobulin [β2m]), are ubiquitously expressed on most nucleated cells. The levels of MHC class I can be regulated by various cytokines, including IFNs and TNF-α, and by mitogens such as LPS, phorbol esters, and Con A 1. Expression of MHC class I is tightly regulated during development 2 3 4 and differentiation 5 and is required for immune recognition of virus infection and for T cell development. Antigen presentation by MHC class I is essential for both the development and the effector function of CD8+ CTLs. Differential expression of MHC class I on thymic epithelial and dendritic cells influences both positive and negative selection of immature thymocytes 6.
Expression of MHC class I genes is controlled by both transcriptional and posttranslational mechanisms. Transcriptional regulation of MHC class I heavy chain is controlled through transcription factor binding sites in the promoters of the MHC class I genes 7 8. Conserved cis-acting elements include enhancer A, IFN consensus sequence (ICS),1 and site α. Enhancer A contains regions I and II recognized by members of the retinoic acid receptor family 5 and the nuclear factor (NF)-κB/Rel family 9, respectively. The binding of NF-κB is essential for constitutive class I expression and for induction by mitogens and by cytokines such as TNF 10. ICS contains an IFN-stimulated response element that is the target site for transcription factors of the IFN regulatory factor (IRF) family, such as IRF-1, IRF-2, IRF-9 (ISGF3γ p48), and IRF-8 (ICS-binding protein), and it mediates the induction of MHC class I expression by IFNs. The interactions of different transcription factors at enhancer A and ICS sites can achieve synergistic effects on the regulation of MHC class I genes 11. MHC class II transactivator (CIITA), a global regulator for MHC class II molecules, is also involved in the constitutive and IFN-γ–induced expression of MHC class I genes through site α 12 13.
Two sets of molecules involved in antigen processing and presentation are also essential for cell surface expression of MHC class I molecules 14 15. LMP2 and LMP7, two subunits associated with the 20S proteasome, are required for enhanced processing of cytosolic proteins into small peptides 16. Mice lacking LMP2 and LMP7 are deficient in presenting antigens and in MHC class I surface expression 17 18. Transporter associated with antigen processing (TAP)1 and TAP2 are transporter proteins that form heterodimers and facilitate movement of peptides from the cytosol into the lumen of the endoplasmic reticulum, where they are loaded into the groove of the MHC class I heavy chain, and these transporters are also required for MHC class I expression 19. For instance, RMA-S, a mutant cell line lacking TAP2, is defective in the expression of MHC class I on the cell surface owing to its rapid degradation in the absence of peptide 20. MHC class I structural genes as well as the processing machinery and some of the regulatory transcription factors are all induced by IFN during inflammation.
Induction of the MHC complex by type I and type II IFN is abrogated in the absence of signal transducer and activator of transcription (STAT)1 21 22, demonstrating the absolute requirement of STAT1 for IFN-stimulated transcriptional responses. As no STAT1 binding site was found in the promoter of the MHC class I gene, the role of STAT1 in IFN-γ–mediated MHC class I induction is likely to be indirect. Several transcription factors implicated in MHC class I expression are themselves targets for STAT1, such as IRF-1 and CIITA. However, during IFN-α/β responses, STAT1 may directly induce class I gene expression by forming the ISGF3 complex in conjunction with STAT2 and IRF-9, which in turn binds to the ICS of MHC class I promoters 23. The fact that IFN-β was capable of inducing MHC class I expression in IRF-1−/− fibroblasts suggests that the binding of ISGF3 to the ICS is sufficient for MHC class I induction 24.
JAK–STAT signaling, first characterized in the IFN system, has been shown to be essential for many cytokine responses 25 26. One of the surprises revealed by genetic studies of STAT deficiency is the remarkable specificity of individual STAT molecules for distinct cytokine signaling systems 27 in spite of the more promiscuous activation observed in cell culture. However, we have also explored possible roles for STAT1 outside of inflammatory responses to IFN. For instance, a role for STAT1 in response to fibroblast growth factor was detected during chondrocyte development in organ cultures 28, and in this paper we provide evidence that STAT1 is required for basal expression of MHC class I. Part of this requirement for STAT1 is due to a constitutive rather than an inflammatory role for IFN and demonstrates a steady-state requirement for IFN in the absence of infection. Unexpectedly, we also observed an IFN-independent and cell type–restricted role for STAT1 specifically in T but not B lymphocytes.
The cell type specificity of the IFN-independent STAT1 requirement corresponds to the responsiveness of cells to IL-7. IL-7 is a cytokine that is essential for lymphoid development 29 30 31, with its specific receptor IL-7Rα expressed on T and B lineage progenitors. In the thymus, double-negative (DN) and single-positive (SP) cells but not double-positive (DP) cells express IL-7Rα, and the receptor is expressed on peripheral T but not B lymphocytes. IL-7 signals through JAK1 and JAK3 and has been shown to activate STAT5a and STAT5b 32 and possibly STAT1 33, and it promotes the survival of lymphoid cells, which is particularly critical for thymic precursors 34. Recently, IL-7 was also shown to induce immune recognition molecules, including MHC class I, in acute myeloid leukemia and lymphoid leukemia patients 35. Here, we show that IL-7 also activates STAT1 in mature T lymphocytes, leading to increased expression of MHC class I.
Materials and Methods
Animals.
Generation of STAT1−/− 36, IFN-α receptor (IFNAR)1−/− 37, and IFN-γ receptor (IFNGR)1−/− mice 38 has been described. IFNAR and IFNGR double-knockout mice (AR+GR)−/− were derived by interbreeding IFNAR−/− and IFNGR−/− mice and screening for compound homozygous mutant offspring. Strain backgrounds were either 129 or C57BL/6 (eighth backcross generation) and were compared with wild-type mice of the same strain. Mice were housed under specific pathogen–free conditions, and all work with animals conformed to guidelines approved by the Institutional Animal Care and Use Committee of New York University School of Medicine.
Flow Cytometry.
Lymphocytes of thymi, spleens, and lymph nodes were prepared from 6–8-wk-old mice. Triple staining for surface markers was performed by incubating ∼106 cells first with M1/42.3 (TIB-126; American Type Culture Collection), a rat mAb that recognizes a framework epitope common to mouse MHC class I, followed by staining cells with FITC-conjugated anti–rat IgG (Caltag Labs.). Cells were subsequently incubated with anti–CD4-TC (tricolor) and anti–CD8-PE or anti–B220-PE antibodies (Caltag Labs.) in the presence of normal rat serum to block cross-reactions. Stained cells were washed two times with cold staining buffer (0.2% BSA and 0.1% sodium azide in PBS) and fixed with 1% paraformaldehyde in staining buffer, followed by FACS™ analysis. Additional surface markers were analyzed using anti–H-2Kb–PE (PharMingen) and anti–I-Ab–biotin, anti–IgM-biotin, and streptavidin–allophycocyanin (Caltag Labs.). For comparing MHC class I induction in response to IFN-γ and IL-7, selected lymphocyte populations were incubated in the presence or absence of cytokine for 48 h, followed by FACS™ analysis. Mean channel shift was the average increase in mean fluorescence intensity 39 and varied <10% for three separate experiments.
Northern Blot Analysis.
20 μg of total RNA prepared from splenocytes or thymocytes using Trizol reagent (Life Technologies) was resolved on 1.5% denaturing agarose gels and transferred to nitrocellulose membranes. cDNA probes were derived from splenic RNA by reverse transcriptase–PCR. The primer sets for amplifying different genes were as follows: LMP2, forward primer, 5′-ATGCTGCGGGCAGGAGCACCTACCGC-3′ and reverse primer, 5′-TCACTCATCGTAGAATTTTGGCAGCTC-3′; LMP7, forward primer, 5′-ATGGCGTTACTGGATCTGTGCGGTGC-3′ and reverse primer, 5′-TCACAGAGCGGCCTCTCCGTACTTGTA-3′; TAP1, forward primer, 5′-AGTGTCTCGGGAATGCTGCTGAAGGTG-3′ and reverse primer, 5′-AGT-GTGCAGTCCAGAGGCCTTGTCGTCTG-3′; β-actin, forward primer, 5′-GTGGGGCGCCCCAGGCACCA-3′ and reverse primer, 5′-CTCCTTATTGTCACGCACGATTTC-3′; MHC I, forward primer, 5′-GCACAGATTCCCCAAAGG-3′ and reverse primer, 5′-ATCTCAGGGTGAGGGGCTCA-3′; β2m, forward primer, 5′-GTGACCCTAGTCTTTCTGGTG-3′ and reverse primer, 5′-TGAATCTTCAGAGCATCATG-3′; IRF-1, forward primer, 5′-TCCATGGAAGCACGCTGCTA-3′ and reverse primer, 5′-AGACTGCTGCTGACGACACA-3′.
The amplified DNA fragments were purified and labeled using RadPrime DNA labeling kits (Life Technologies). Hybridization, washing, and stripping of the nitrocellulose membrane followed standard procedures 40. To quantify specific signals, membranes were exposed to PhosphorImager screens (Molecular Dynamics) and analyzed according to the manufacturer's instructions. Quantitative measurements were normalized to values for actin.
Retroviral Transduction of Bone Marrow Cells and Bone Marrow Transplantation.
Production of retrovirus was as described 41. A murine stem cell virus vector expressing humanized green fluorescent protein (GFP; a gift of Dr. W. Pear, University of Pennsylvania, Philadelphia, PA) was transfected into the Phoenix packaging cell line (a gift of Dr. G.P. Nolan, Stanford, CA), and culture supernatant was used as viral stock. Transduction of bone marrow cells with retrovirus was as described 42. In brief, C57BL/6 wild-type or STAT1−/− mice were treated with 150 mg/kg 5-fluorouracil (Sigma Chemical Co.) 6 d before harvest. Bone marrow cells were harvested and incubated on RetronectinTM (Takara-Shuzo) coated dishes containing bone marrow transfer medium (IL-3, 6 ng/ml; stem cell factor, 10 ng/ml; and IL-6, 10 ng/ml, all from PeproTech, Inc., and 20% FBS in RPMI 1640) and 1 ml of recombinant virus for 48 h. Cultures were supplemented with 1 ml of fresh virus after the first 24 h. 1.6 million bone marrow cells were injected intravenously into lethally irradiated (900 rads) wild-type or STAT1−/− recipients. Mononuclear cells recovered from peripheral blood of recipient mice were analyzed by double staining with anti–H-2Kb–PE and anti–TCR-allophycocyanin or IgM-allophycocyanin after gating for GFP fluorescence to score donor-derived lymphocytes.
Separation of T and B Lymphocytes and Analysis of Nuclear Extracts.
Freshly isolated splenocytes were incubated with rat anti-B220 at 0.2 μg/106 cells for 30 min at 4°C. Antibody-bound cells were washed twice with 1× PBS and incubated with anti–rat antibody–coated M450 Dynabeads (Dynal) for 30 min at 4°C on a mixer. B cells were collected using a magnet after three washes with 1× PBS containing 0.2% BSA. Nuclear extracts prepared from positively selected B cells and negatively selected T cells were resolved in 7% SDS-PAGE, followed by Western blot analysis as described 43. The antibodies used for immunoblots were anti–phosphotyrosyl-STAT1 (Zymed Labs.) and anti–STAT1 COOH terminus (STAT1C; a gift from Dr. C. Schindler, Columbia University, New York, NY). Purity of selected cell populations was verified by flow cytometry. B cell fractions were typically >95% pure; T cell fractions were >80% pure.
Electrophoretic Mobility Gel Shift.
Nuclear extracts prepared from Dynabead-selected T or B cells, which were left untreated or treated with human recombinant IL-7 (10 ng/ml; StemCell) or IFN-γ (100 U/ml; Boehringer Mannheim) for 30 min, were analyzed using a 32P-labeled probe containing a high-affinity GAS sequence 44 in the presence or absence of anti-STAT1C antibody as described 43.
Results
Decreased Surface Expression of MHC Class I on STAT1−/− Lymphocytes.
Type I and type II IFNs are known modulators of MHC antigen expression during inflammation and viral infection 45. Because STAT1 is a signal transducer shared by both types of IFNs, we examined the expression of MHC antigens on freshly isolated lymphocytes from wild-type or STAT1−/− mice with antibody to either MHC class I (M1/42.3, an mAb recognizing a framework epitope of MHC class I) or class II (I-Ab). Cells from STAT1−/− mice housed under pathogen-free conditions showed a two- to threefold reduction of surface MHC class I compared with lymphocytes from wild-type mice (Fig. 1 A). The expression of MHC class II, however, was comparable on lymphocytes of both genotypes (Fig. 1 B), even though both class I and class II expression can be induced by IFN, and both show an absolute requirement for STAT1 for induction 21. Reduced expression and absence of induction of MHC class I was not due to a global defect, however, because class I expression was induced in response to mitogenic stimulation of both B and T splenocytes devoid of STAT1 (data not shown), reflecting an intact NF-κB pathway 46 47 48.
Figure 1.


Reduced surface expression of MHC class I, but not class II, on STAT1−/− lymphocytes. Freshly isolated splenocytes prepared from wild-type (thin lines) or STAT1−/− mice (bold lines) were stained with antibodies against MHC class I (A) or class II (anti–I-Ab) (B), followed by FACS™ analysis. Dotted/dashed lines represent staining with secondary antibody alone.
Reduced Expression of mRNA for MHC Class I Genes and Antigen Processing and Presentation Molecules in STAT1−/− Lymphocytes.
To explore the mechanisms underlying the reduction of cell surface expression of MHC class I in the absence of STAT1, we examined the levels of MHC class I heavy chain and light chain (β2m) mRNA by Northern hybridization. To assess the potential role of constitutive IFN in this regulation, we included lymphocytes lacking receptors for either IFN-α or IFN-γ derived from IFNAR−/− or IFNGR−/− mice. As shown in Fig. 2 A, MHC class I heavy chain and β2m mRNA levels were reduced significantly in both thymi and spleens of STAT1−/− mice. Quantitation of the mRNA levels of MHC class I heavy and light chains after normalization with the values for actin showed ∼50–70% reduction compared with wild-type levels. The mRNA levels of MHC class I heavy chain and, to a lesser degree, light chain were also decreased in the thymi of IFNAR−/− and IFNGR−/− mice, showing that at least part of the regulation of basal class I expression requires an intact IFN signaling pathway. Similar reductions were also observed in splenocytes. To study the possible role of other transcription factors in reduced MHC class I expression, we examined the level of IRF-1 mRNA, which is a STAT1 target and has been shown to modulate the levels of both MHC class I heavy and light chain in response to IFN-γ 49 50. IRF-1 mRNA levels were decreased substantially (30–50%) in spleens and thymi of STAT1−/− mice (Fig. 2 A) and to a lesser extent in lymphocytes from IFNAR−/− and IFNGR−/− mice. These results suggest that IFNs, operating through STAT1 and possibly IRF-1, are required to maintain full basal mRNA levels of MHC class I heavy and light chains. Interestingly, class I heavy and light chain mRNA expression was much higher in spleen than thymus (Fig. 2 A). Similarly increased cell surface class I protein levels were observed in peripheral relative to immature lymphocytes, suggesting a developmental upregulation of class I molecules during maturation.
Figure 2.

Genes for MHC class I antigen processing and presentation are regulated by STAT1. (A) Reduced expression of heavy and light chain mRNA in the absence of STAT1. 20 μg of total RNA prepared from lymphocytes of thymi or spleens of wild-type, STAT1−/−, IFNAR−/−, or IFNGR−/− mice were hybridized with 32P-labeled probes specific for MHC class I heavy chain, light chain (β2m), and IRF-1. (B) Reduced constitutive mRNA for antigen processing and presentation–related molecules. Abundance of TAP1, LMP2, LMP7, and actin was measured as described for A. (C) PML is regulated by STAT1 and is differentially expressed in thymus and spleen. Abundance of PML and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was analyzed by Northern hybridization.
As antigen processing and presentation molecules are also critical for surface expression of the MHC class I complex, we measured the mRNA levels of these accessory molecules. As shown in Fig. 2 B, TAP1, LMP2, and LMP7 expression was reduced to ∼35% of wild-type levels in splenocytes and thymocytes from STAT1−/− mice. Smaller reductions of TAP1, LMP2, and LMP7 mRNA levels were seen in thymus and spleen of IFNAR−/− and IFNGR−/− mice. These results showed that all of the MHC class I complex and related genes examined were regulated by STAT1 and that the regulation was at least partially due to constitutive IFN signaling. In all cases, loss of STAT1 produced a more severe reduction in MHC class I and accessory molecules than did loss of a single IFN receptor, suggesting that the phenotype of STAT1−/− mice was due to either the additive effect of loss of both type I and type II IFN receptors or to an IFN-independent mechanism.
A nuclear protein recently implicated in the control of MHC class I gene expression and antigen processing is the promyelocytic leukemia (PML) protooncogene 51. PML mRNA expression was reduced approximately twofold in spleen cells from STAT1−/− mice (Fig. 2 C). Because this protein may act as a master regulator of MHC class I cell surface expression that functions downstream of STAT1, its reduction in STAT1−/− cells may contribute to reduced MHC class I cell surface levels. Interestingly, like class I heavy and light chain genes, PML was also expressed at higher levels in spleen relative to thymus, indicating its potential involvement in the developmental switch in class I expression in lymphocytes.
STAT1 Regulates Constitutive MHC Class I Expression on Lymphocytes by a Cell-intrinsic Mechanism.
Reduced expression of MHC class I molecules observed in the absence of STAT1 could be due to either a cell-intrinsic role for STAT1 in lymphocytes, e.g., downstream of a cytokine receptor, or to a more global role elsewhere in the animal, e.g., regulating production of a secreted cytokine that ultimately targets lymphocytes. To determine if the reduction of MHC class I was a primary, cell-autonomous defect, we performed bone marrow transplantation (BMT). Bone marrow cells from either wild-type or STAT1−/− mice were used to reconstitute either STAT1+/+ or STAT−/− animals that had been lethally irradiated. To distinguish donor-derived lymphocytes from residual endogenous cells of the recipient mice, donor cells were infected with a retrovirus expressing GFP. The levels of MHC class I on mature peripheral blood lymphocytes were monitored 6 wk after BMT by staining with antibodies against MHC class I plus TCR for mature T cells or plus IgM for mature B cells and gating on GFP-positive lymphocytes.
Four types of transplants were performed: STAT1+/+ cells transferred into STAT1+/+ or STAT1−/− mice and STAT1−/− cells transferred into STAT1+/+ or STAT1−/− mice. Wild-type and STAT1−/− bone marrow was capable of reconstituting both mature T and B lymphocytes in mice of both genotypes (Fig. 3). STAT1−/− bone marrow was somewhat less efficient at reconstituting mature T lymphocytes than wild-type donors, although the cause of this deficiency is unclear. Nonetheless, on both subsets of lymphocytes, MHC class I expression levels reflected the genotype of the donor rather than that of the recipient. STAT1+/+ cells transferred into STAT1−/− hosts acquired an MHC class I phenotype similar to that of wild-type cells (Fig. 3A and Fig. B, second rows from top), whereas STAT1−/− cells transferred to STAT1+/+ hosts maintained their reduced levels of MHC class I (Fig. 3, third rows from top). Autologous transplants also maintained the level of MHC class I appropriate for their genotype (Fig. 3, first and fourth rows from top). These results demonstrated that basal expression of MHC class I on T and B lymphocytes is regulated by a cell-intrinsic process that depends on the STAT1 status of the cell itself, independent of the environment in which it matures.
Figure 3.
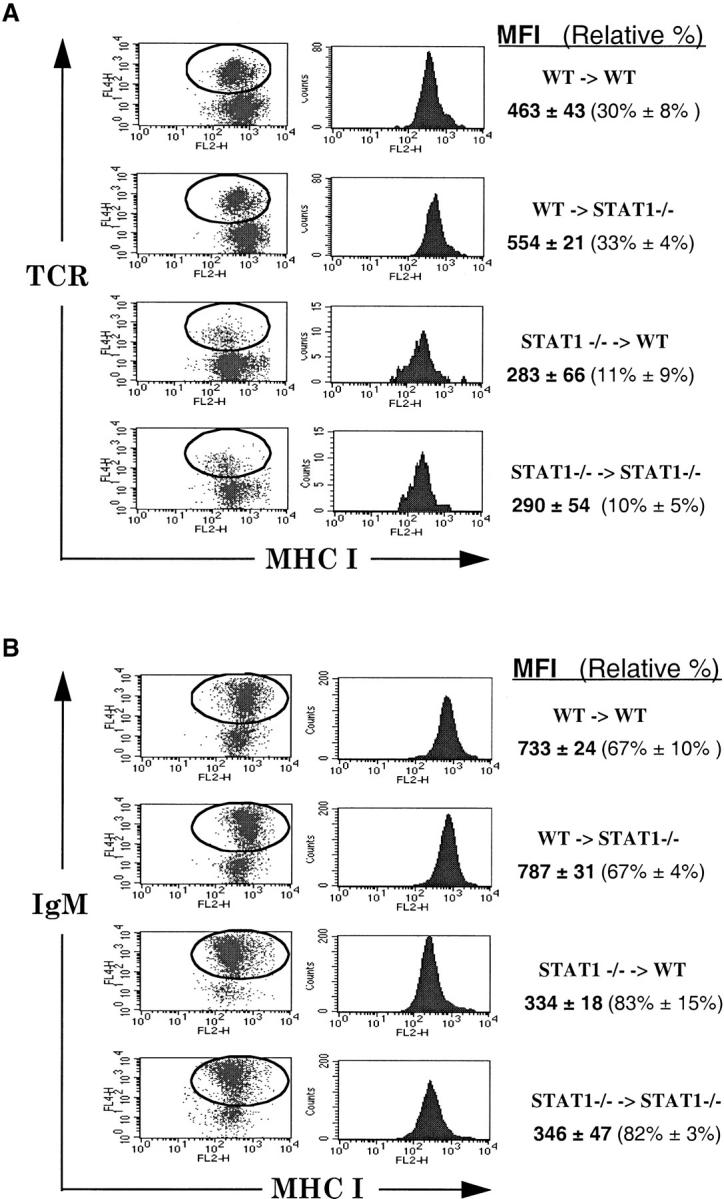
Expression of MHC class I is cell autonomous. Peripheral blood lymphocytes prepared after BMT were double stained with anti–H-2Kb–PE and anti–TCR-allophycocyanin (A) or anti–IgM-allophycocyanin (B), followed by FACS™ analysis gating on GFP-positive lymphocytes. Mean fluorescence intensity (MFI) is the average MHC class I intensity of GFP–TCR or GFP–IgM DP lymphocytes (circled regions) from three to four chimeric mice. The relative percentages of TCR+ or IgM+ cells among GFP+ lymphocytes are shown.
STAT1-dependent but IFN-independent Regulation of MHC Class I Expression in Mature T Lymphocytes.
The greater reduction of class I heavy and light chain mRNA in STAT1−/− cells relative to IFN receptor mutants (Fig. 2) suggested that STAT1 may be important even in the absence of IFN. To further examine the role of constitutive IFN signaling in MHC expression patterns on different subsets of lymphocytes, we compared MHC class I antigen levels on lymphocytes from mice devoid of IFN signaling by combined loss of receptors for both IFN-α and IFN-γ (Fig. 4). As noted above, higher levels of MHC class I expression were detected on lymphocytes from peripheral organs than on thymocytes. Different subsets of T lymphocytes also displayed different levels of class I. CD4+CD8+ DP thymocytes expressed very limited amounts of surface MHC class I compared with CD4−CD8− DN or CD4− CD8+ and CD4+CD8− SP thymocytes (Table ). In peripheral organs, the levels of MHC class I on both CD4 and CD8 T lymphocytes were further elevated compared with those of SP thymocytes.
Figure 4.

Reduced surface expression of MHC class I antigens in different subsets of STAT1−/− lymphocytes. Freshly isolated lymphocytes from thymi (A) or lymph nodes (B) of wild-type (dashed lines), STAT1−/− (bold lines), or (AR+GR)−/− (thin lines) mice were triple stained with CD4–TC, CD8–PE, and MHC class I–FITC, followed by FACS™ analysis. CD4−CD8− lymphocytes from lymph nodes were regarded as B cells.
Table 1.
Mean Fluorescence Intensity of MHC Class I in Different Subsets of Lymphocytes
| Thymocytes | Splenocytes | Lymph nodes | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CD4−8− | CD4−8+ | CD4+8+ | CD4+8− | B | CD8 | CD4 | B | CD8 | CD4 | |
| Wild type | 974 | 706 | 69 | 531 | 974 | 1,562 | 1,368 | 1,097 | 1,709 | 1,520 |
| STAT1−/− | 374 | 186 | 30 | 107 | 585 | 396 | 379 | 632 | 358 | 252 |
| IFNAR−/− | 689 | 614 | 52 | 463 | 663 | 1,273 | 926 | 703 | 1,230 | 904 |
| IFNGR−/− | 530 | 538 | 40 | 346 | 599 | 874 | 644 | 665 | 1,099 | 664 |
| (AR+GR)−/− | 464 | 489 | 43 | 321 | 597 | 793 | 577 | 586 | 883 | 537 |
In spite of the differential expression of MHC class I on different lymphocyte populations, when the levels of MHC class I were compared between STAT1−/− and wild-type animals, all subsets of lymphocytes, whether from thymus, spleen, or lymph nodes, showed reduced expression of MHC class I in the absence of STAT1 (Table and Fig. 4). Whereas DP and DN thymocytes and B lymphocytes from spleen and lymph nodes showed 40–50% reduction of MHC class I, SP thymocytes (CD4−CD8+ and CD4+CD8−) and peripheral T lymphocytes (both CD4 and CD8) showed a more pronounced reduction of MHC class I expression (only 16–26% of wild-type levels). MHC class I expression was also reduced on other cells types, including monocytes, macrophages, NK cells, and mouse embryonic fibroblasts (data not shown), suggesting that STAT1 is generally required for maintaining constitutive levels of MHC I expression.
RNA expression studies (Fig. 3) suggested that class I levels regulated by STAT1 were at least partially independent of the presence of either IFN receptor. This observation left open the possibility that the more severe defect observed in the absence of STAT1 could be due to the combined loss of IFN-α and IFN-γ signaling. We therefore tested class I protein levels in the absence of both IFN-α and IFN-γ receptors using cells from (AR+GR)−/− mice. To exclude the possibility that any variation in MHC class I expression was due to differences in genetic background, wild-type, STAT1−/−, IFNAR−/−, and (AR+GR)−/− mice were maintained on the 129 (H-2b) strain background.
A similar reduced level of MHC class I was observed on B cells of both spleens and lymph nodes from (AR+GR)−/− and STAT1−/− mice (Table and Fig. 4 B). Therefore, the STAT1 requirement in B cells is likely to be entirely downstream of IFN signaling. Surprisingly, however, SP thymocytes and CD4 and CD8 T lymphocytes from STAT1−/− mice displayed a more pronounced reduction of MHC class I (∼50%) than those from (AR+GR)−/− mice. Therefore, STAT1 was required for the expression of MHC class I antigens on maturing and peripheral T lymphocytes even in the absence of any IFN signaling.
IFN-independent Phosphorylation and Nuclear Translocation of STAT1 in Resting T but not B Lymphocytes.
The preceding results showed that endogenous IFNs were present and functional in normal animals but also indicated that STAT1-dependent gene expression could be independent of IFN signaling. These results prompted us to test the tyrosine phosphorylation and nuclear accumulation of STAT1, an indication of its activation by cytokines. Nuclear extracts prepared from freshly isolated splenocytes of wild-type, STAT1−/−, and (AR+GR)−/− mice were subjected to immunoblot with antibody specific for tyrosine-phosphorylated STAT1. As shown in Fig. 5 A, top panel, phospho-STAT1α and phospho-STAT1β were indeed detected in nuclear extracts of wild-type resting splenocytes, presumably indicative of the action of IFN. Strikingly, a lower but still significant level of phospho-STAT1 was also detected in the (AR+GR)−/− splenocytes. Whereas the decreased activation of STAT1 in (AR+GR)−/− lymphocytes relative to wild-type cells was presumably due to the loss of IFN responsiveness, the residual phospho-STAT1 in (AR+GR)−/− lymphocytes must result from an IFN-independent mechanism. These results demonstrate that both IFN-dependent and -independent pathways contributed to the basal activation of STAT1 in lymphocytes.
Figure 5.

(A) Basal phosphorylation of STAT1 in splenocytes of wild-type and (AR+GR)−/− mice. Nuclear extracts prepared from 1.5 × 107 freshly isolated splenocytes of wild-type, STAT1−/−, and (AR+GR)−/− mice were resolved in 7% SDS-PAGE and transferred to nitrocellulose membrane, followed by immunoblot with anti–phosphotyrosyl-STAT1 (STAT1-PTyr) antibody (top panel). The blot was then stripped and reblotted with anti-STAT1C antibody that only recognizes full length STAT1α (bottom panel). (B) Basal phosphorylation of STAT1 in B but not T cells completely depends on IFN signaling. Western blot as described for A was performed using nuclear extracts prepared from 107 splenic T or B cells or thymocytes of wild-type and (AR+GR)−/− mice.
MHC class I expression was differentially regulated in T and B cells; specifically, STAT1-mediated expression in B cells was entirely IFN dependent, yet both IFN-dependent and -independent pathways were involved in STAT1-mediated expression in T cells (Fig. 4). Therefore, we tested whether the regulation of basal activation of STAT1 also varied in these two subsets of lymphocytes. Nuclear extracts from isolated splenic T and B lymphocytes of wild-type and (AR+GR)−/− mice were analyzed for phospho-STAT1. As shown in Fig. 5 B, activated STAT1α and STAT1β were detected in both T and B lymphocytes of wild-type mice. In contrast to wild-type lymphocytes, phospho-STAT1 levels were undetectable in B cells from (AR+GR)−/− mice but could still be detected in T cells. Therefore, STAT1 activity in B cells was entirely dependent on IFN signaling, whereas both IFN-dependent and -independent pathways mediated STAT1 activation selectively in T cells. This dual mode of STAT1 activation was not limited to splenic T lymphocytes; basal activation of STAT1 was also present in (AR+GR)−/− thymocytes (Fig. 5 B). This pattern of STAT1 activation mirrors the regulation we found for MHC class I gene expression.
Selective Activation of STAT1 by IL-7 in Mature T but not B Lymphocytes.
The previous results defined an IFN-independent activation of STAT1 in T but not B cells. We reasoned that factors involved in selective activation of STAT1 might be cytokines preferentially acting on thymic and splenic T cells but not on mature B cells. One such candidate is IL-7, whose receptor is expressed on both T and B lineage progenitors 52 53 54 but not on DP thymocytes or mature peripheral B lymphocytes 55. STAT1 phosphorylation in freshly isolated splenocytes was blocked by the kinase inhibitor staurosporine (data not shown), suggestive of the action of a cytokine signaling pathway. Therefore, we first examined whether IL-7 could activate STAT1 in lymphocytes. STAT1 activation was significantly enhanced after either IL-7 or IFN-γ treatment of splenic T cells from wild-type mice (Fig. 6 A, top panel). A comparable level of activated STAT1 was present in T cells of (AR+GR)−/− mice after IL-7 treatment but not after IFN-γ treatment, due to the absence of IFN-γ receptors. STAT1 was also activated in thymocytes of wild-type and (AR+GR)−/− mice in response to IL-7 (data not shown). In marked contrast to T cells, no activation of STAT1 was observed in IL-7–treated B lymphocytes, although IFN-γ was still capable of activating STAT1 in B cells of wild-type though not (AR+GR)−/− mice (Fig. 6 A, bottom panel).
Figure 6.

Differential activation of STAT1 by IL-7 in T and B cells. (A) Nuclear extracts prepared from selected T (top panel) or B (bottom panel) cells were left untreated or treated with IL-7 or IFN-γ for 30 min, followed by electrophoresis and immunoblot with anti–phosphotyrosyl-STAT1 (STAT1-PTyr) as described in the Fig. 5 legend. Basal phosphorylation of STAT1 was readily detected upon longer exposure (not shown). (B) EMSA was performed with nuclear extracts prepared as described for A in the presence or absence of antibody to STAT1C.
To further confirm the selective response to IL-7 in T versus B cells, electrophoretic mobility gel shift (EMSA) was performed using nuclear extracts from purified lymphocyte populations. Whereas IL-7 treatment induced the formation of two complexes in T cells from wild-type mice, IFN-γ only activated a complex with mobility corresponding to the lower band of the IL-7–activated complexes (Fig. 6 B). Antibodies to STAT1 (Fig. 6 B, fourth lane from left) and STAT5 (not shown) identified these complexes as STAT1 (Fig. 6 B, bottom panel) and STAT5 (Fig. 6 B, top panel). However, whereas a STAT1 complex was activated by IFN-γ in B cells from wild-type mice, no complexes were activated in response to IL-7 in these cells (Fig. 6 B, bottom panel). IL-2 and IL-4, other cytokines capable of activating STAT5, failed to activate STAT1 (data not shown). Therefore, IL-7 selectively activated STAT1 in T but not B cells and functioned independently from IFN, consistent with the pattern previously observed for MHC class I expression.
IL-7–dependent Induction of MHC Class I Expression on T Cells.
As STAT1 is essential for MHC class I expression, the differential activation of STAT1 in T and B cells by IL-7 prompted us to examine IL-7–dependent modulation of class I expression. Splenocytes and thymocytes of wild-type, (AR+GR)−/−, and STAT1−/− mice were treated with IL-7 or IFN-γ for 48 h, and induction of class I protein expression was followed by FACS™ analysis (Table ). Whereas IFN-γ induced MHC class I expression on both T and B cells of wild-type mice, IL-7 selectively induced class I expression only on T cells (Table , top). This IL-7–dependent induction of MHC class I was not secondary to IFN signaling, as a comparable induction of MHC class I expression occurred on T cells from (AR+GR)−/− mice. IL-7 also induced class I on DN and SP thymocytes from wild-type mice but not on DP thymocytes that lack IL-7Rα (Table , bottom). In contrast, neither IL-7 nor IFN-γ caused significant class I induction on cells deficient in STAT1, similar to the lack of effect of IL-7 on receptor-negative B cells. Analogous results were obtained with IL-7R–positive pre-B cell lines derived from bone marrow; MHC class I was induced in response to IL-7 on wild-type but not STAT1−/− cells (data not shown). Taken together, these results demonstrate that STAT1 is capable of mediating MHC class I induction in response to IL-7, and we conclude that STAT1 and IL-7 contribute to the basal level of MHC class I on mature T lymphocytes.
Table 2.
Mean Channel Shift of MHC Class I Expression
| Treatment | Splenocytes: | Wild type | (AR+GR)−/− | STAT1−/− | |||||
|---|---|---|---|---|---|---|---|---|---|
| T | B | T | B | T | B | ||||
| IFN-γ | 76 | 379 | 0 | 0 | 6 | 0 | |||
| IL-7 | 103 | 27 | 101 | 12 | 18 | 7 | |||
| Treatment | Thymocytes: | Wild type | STAT1−/− | ||||||
| CD4+8− | CD4−8+ | CD4−8− | CD4+8+ | CD4+8− | CD4−8+ | CD4−8− | CD4+8+ | ||
| IFN-γ | 97 | 52 | 69 | 60 | 1 | 0 | 1 | 1 | |
| IL-7 | 60 | 50 | 30 | 1 | 9 | 7 | 8 | 2 | |
Splenocytes and thymocytes were treated with IFN-γ (100 U/ml) or IL-7 (10 ng/ml) for 48 h.
Discussion
We have demonstrated a role for constitutive IFN signaling in the absence of inflammation for homeostatic gene regulation in normal lymphocytes. This genetic and biochemical analysis has also revealed underlying differences in the regulation of MHC class I expression in T and B cells. Moreover, these data uncovered a novel IFN-independent but IL-7–dependent STAT1 activity that operates selectively in T lymphocytes. A model for these different modes of regulation is illustrated in Fig. 7. In both T and B cells, constitutive IFN signaling activates STAT1 to maintain MHC class I gene expression either directly in combination with STAT2 and IRF-9 or possibly indirectly through its downstream target, IRF-1. In addition, both cell types have IFN-independent mechanisms for regulation of class I levels. In T cells, an IL-7–dependent but IFN-independent mechanism activates STAT1 phosphorylation, and this STAT1-dependent signaling is necessary for full class I expression. In B cells, the IFN-independent mechanism is also STAT1 independent, possibly acting through the B cell–specific protein CIITA that has been shown to modulate MHC class I gene expression 12 13.
Figure 7.

Differential regulation of constitutive MHC class I expression in T and B cells. Two pathways contribute to STAT1 activation that are essential for basal MHC class I expression in T cells. The first pathway is IFN dependent, whereas the second does not require IFN receptors and may depend on a cytokine, such as IL-7, to maintain MHC class I expression. In B cells, STAT1 activation is entirely dependent on IFN, whereas additional B cell–specific transcription factors, such as CIITA, contribute to the maintenance of MHC class I expression in a STAT1-independent manner.
The role of IL-7 has been mainly characterized during lymphoid development, where it is required to prevent apoptosis in lymphoid precursors 34 and to serve as a cofactor for V(D)J recombination in both T and B lymphocytes 53 54. These actions of IL-7 depend on JAK1, 56 JAK3 57, and STAT5, though the requirement for STAT1 signaling is less clear 58 59. Here we provide evidence that IL-7 is also important in mature T cells for maintenance of constitutive expression of MHC class I through activated STAT1. However, the role of STAT1 activation in the upregulation of MHC class I in response to IL-7 is likely indirect, possibly through IRF-1, a downstream target of STAT1. STAT1 can be activated in response to cytokines and growth factors other than IFN, at least in cell culture, but initial evidence from in vivo studies largely failed to support these alternative activators, suggesting instead that STAT1 was dedicated to IFN signaling 21 36 60. Recent evidence has suggested that fibroblast growth factor also functions through STAT1 in developing chondrocytes, at least in organ culture 28. IL-7–mediated STAT1 activation demonstrates a novel role for STAT1 outside the scope of IFN.
For B cells, no phospho-STAT1 accumulated in the absence of IFN signaling, and MHC class I levels were equivalent between STAT1−/− and (AR+GR)−/− mice. A STAT1-independent mechanism was operative for maintaining basal expression (Fig. 7), possibly through B cell–specific proteins such as CIITA, first characterized as regulators of MHC class II 12 13. Indeed, overexpression of CIITA in human G3A cells, which are defective in CIITA, enhances the expression of MHC class I. It is likely that the action of CIITA compensates for the loss of IFN and STAT1 signaling in B cells derived from (AR+GR)−/− and STAT1−/− mice, allowing them to retain modest expression of MHC class I. The unchanged patterns of surface MHC class II (Fig. 1 B) and equal levels of CIITA mRNA in all mouse strains tested (data not shown) suggest that CIITA-dependent pathways are intact in the absence of STAT1, although their responsiveness to IFN is lost. Interestingly, expression of MHC class I on peripheral macrophages from STAT1−/− and (AR+GR)−/− mice were similar (data not shown). Macrophages also express CIITA and display high constitutive levels of MHC class II, consistent with a class I phenotype similar to that of B cells. CIITA is unable to perform this basal function in most tissues, as its expression outside the antigen presentation system is dependent on IFN induction. The important role of B lymphocytes and macrophages as APCs may explain why they have evolved independent mechanisms to maintain MHC expression.
We have demonstrated that basal activation of STAT1 in resting lymphocytes relies on the action of at least two different cytokines, namely IFN and probably IL-7. Although both IFN and IL-7 are involved in activating STAT1 in T lymphocytes, this action is entirely IFN dependent in B lymphocytes. Similarly, STAT1 basal phosphorylation is maintained by both IFN and IL-7 in SP and DN thymocytes but not in DP thymocytes that lack IL-7Rα and display very low levels of MHC class I. One of the functions of the constitutively activated STAT1 is to maintain MHC class I expression, although it is also likely that other targets exist as well. Interestingly, it has recently been shown that constitutively phosphorylated STAT1 accumulates in lung epithelium of asthmatic patients in the absence of IFN 61, suggesting that the tight regulation of STAT1 phosphorylation is important for normal physiology. Although it has been known that IFN is responsible for increased class I expression during immune responses, we provide evidence for the continuous presence of IFNs in vivo in the absence of overt infection in the maintenance of basal levels of expression. The presence of endogenous IFNs in vivo has been suggested from low levels of IFN mRNA detected in various human organs 62, but the presence or significance of IFN protein in the absence of an inflammatory response has been less clear. Here we provide genetic and biochemical evidence for IFNs exerting a constitutive biological response. The source of basal IFN has not been determined, and we cannot exclude a role for subclinical infection or possibly normal intestinal flora. However, we suggest that basal IFN production, whether spontaneously produced or induced by common environmental stimuli, contributes to normal immune homeostasis. Consistent with this notion, gene-targeted loss of the inhibitor SOCS1 produced severe pathology 63 that appeared to be entirely due to unopposed IFN signaling 64a, again suggesting the presence of biologically active IFN in the absence of overt infection. Likewise, the transcriptional repressor IRF-2 appears to be essential to inhibit basal IFN responses, and ablation of IRF-2 leads to IFN-dependent pathology 64. These findings suggest a novel function for IFN as a homeostatic cytokine in addition to its role as an inflammatory mediator. Constitutive IFN signaling is required for beneficial responses, such as maintenance of MHC class I expression and perhaps priming a state of readiness for combating viral infection 65, whereas the less benign effects required only during inflammation are held in check by negative regulators, such as IRF-2 and SOCS1. It seems likely that other cytokines would have similar dual roles in both homeostatic and stress situations.
The mechanisms of developmental control of MHC class I expression are still largely uncharacterized. We show that expression of MHC class I is downregulated during the transition from DN to DP thymocytes, followed by reexpression on SP thymocytes. It was further enhanced when SP thymocytes matured to peripheral CD4 and CD8 T lymphocytes. Relative changes of MHC class I levels from DN to DP and from DP to SP thymocytes or SP thymocytes to peripheral T lymphocytes were comparable in wild-type and IFN receptor and STAT1 mutant mice (Table ), suggesting that neither IFN nor STAT1 is responsible for this developmental control. Interestingly, PML expression, a possible regulator of MHC class I downstream of STAT1 51, was also higher in spleen than thymus; therefore, it may be one of the factors underlying developmental control.
IRF-1 has also been shown to affect MHC class I expression, inducing mRNA levels for heavy chain, LMP2, and TAP1 66 67. IRF-1 is a STAT1 target and was substantially reduced in STAT1−/− mice and, therefore, is likely one of the downstream mediators of STAT1 in this process. However, IRF-1 is not essential for regulation of MHC class I expression, at least in response to IFN-α/β, as MHC class I expression was still induced in IRF-1−/− cells, presumably through the action of the ISGF3 complex 24. Nonetheless, STAT1−/− and IRF-1−/− mice do not display identical phenotypes. Despite the decreased expression of MHC class I on STAT1−/− cells, we have not observed any abnormalities in CD8 development (data not shown), suggesting that the levels of MHC class I on thymic epithelium were still sufficient to rescue CD8+ SP cells during positive selection. In contrast, IRF-1−/− mice show an increased ratio of CD4/CD8 cells due to an intrinsic requirement of IRF-1 for CD8 cell development 66.
One of the main functions of MHC class I is to facilitate CD8 T lymphocyte–mediated cell cytotoxicity during intracellular infection by viruses or bacteria. However, these pathogens have evolved many strategies to escape from immune responses by targeting the class I complex 68. For example, cytomegalovirus and adenovirus are able to retain MHC class I molecules in the endoplasmic reticulum or Golgi complex through a retrieval signal in the cytoplasmic tails of their viral proteins 69 70 71. HIV Nef protein modifies the endocytic machinery to downmodulate surface expression of MHC class I 72. Recently, JAK–STAT signaling was also shown to be disrupted during viral infection, resulting in suppression of MHC induction by IFN-γ 73 74. The multiple modes of regulation of class I genes in B cells and other APCs may provide a backup system for the host during viral infection. When IFN signaling is targeted by viruses, the professional APCs would be less sensitive to the loss of IFN responsiveness in MHC class I regulation by relying on STAT1-independent pathways. Indeed, STAT1−/− mice displayed comparable virus clearance compared with wild-type mice after influenza virus infection 75. In vitro CTL activity against autologous targets after infection with influenza virus was also indistinguishable in STAT1−/− and wild-type mice (data not shown).
Acknowledgments
We thank John Hirst for FACS® analysis, Michel Aguet, Robert Schreiber, and Joan Durbin for IFN receptor mutant mice, Yang Liu for the PML cDNA clone, Rachel Gertner for expert technical assistance, Juerg Schwaller for advice on retroviral infections, Drs. Nolan, Pear, Sun, Littman, and Schindler for gifts of reagents, and Stan Vukmanovic and Alan Frey for helpful discussions and comments on the manuscript.
R. Gimeno was supported by a postdoctoral fellowship from the Ministerio de Educacion y Ciencia (Spain). This work was supported by National Institutes of Health (NIH) grant R01AI28900. FACS® analysis was supported in part by NIH grant P30CA16087 to the Kaplan Cancer Center.
Footnotes
1used in this paper: BMT, bone marrow transplantation; DN, double-negative; DP, double-positive; EMSA, electrophoretic mobility gel shift; GFP, green fluorescent protein; ICS, IFN consensus sequence; IRF, IFN regulatory factor; NF, nuclear factor; PML, promyelocytic leukemia; SP, single-positive; STAT, signal transducer and activator of transcription; TAP, transporter associated with antigen processing
References
- David-Watine B., Israël A., Kourilsky P. The regulation and expression of MHC class I genes. Immunol. Today. 1990;11:286–292. doi: 10.1016/0167-5699(90)90114-o. [DOI] [PubMed] [Google Scholar]
- Ozato K., Wan Y.-J., Orrison B. Mouse major histocompatibility class I gene expression begins at midsomite stage and is inducible in earlier-stage embryos by interferon. Proc. Natl. Acad. Sci. USA. 1985;82:2427–2431. doi: 10.1073/pnas.82.8.2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe L., Jeannotte J., Bikoff E.K., Robertson E.J. Analysis of β2-microglobulin gene expression in the developing mouse embryo and placenta. J. Immunol. 1990;145:3474–3482. [PubMed] [Google Scholar]
- Drezen J.M., Nouvel P., Babinet C., Morello D. Different regulation of class I gene expression in the adult mouse and during development. J. Immunol. 1992;149:429–437. [PubMed] [Google Scholar]
- Segars J.H., Nagata T., Bours V., Medin J.A., Franzoso G., Blanco J.C., Drew P.D., Becker K.G., An J., Tang T. Retinoic acid induction of major histocompatibility complex class I genes in NTera-2 embryonal carcinoma cells involves induction of NF-kappa B (p50-p65) and retinoic acid receptor beta-retinoid X receptor beta heterodimers. Mol. Cell. Biol. 1993;13:6157–6169. doi: 10.1128/mcb.13.10.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney J.R., Sykulev Y., Eisen H.N., Tonegawa S. Differences in the level of expression of class I major histocompatibility complex proteins on thymic epithelial and dendritic cells influence the decision of immature thymocytes between positive and negative selection. Proc. Natl. Acad. Sci. USA. 1998;95:5235–5240. doi: 10.1073/pnas.95.9.5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting J.P., Baldwin A.S. Regulation of MHC gene expression. Curr. Opin. Immunol. 1993;5:8–16. doi: 10.1016/0952-7915(93)90074-3. [DOI] [PubMed] [Google Scholar]
- Girdlestone J. Transcriptional regulation of MHC class I genes. Eur. J. Immunogenet. 1996;23:395–413. doi: 10.1111/j.1744-313x.1996.tb00015.x. [DOI] [PubMed] [Google Scholar]
- Israel A., Kimura A., Kieran M., Yano O., Kanellopoulus J., Le B.O., Kourilsky P. A common positive trans acting factor binds to enhancer sequences in the promoters of mouse H-2 and β2-microglobulin genes. Proc. Natl. Acad. Sci. USA. 1987;84:2653–2657. doi: 10.1073/pnas.84.9.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayoshi Y., Miyazaki J.I., Burke P.A., Hamada K., Appella E., Ozato K. Binding of multiple nuclear factors to the 5′ upstream regulatory element of the murine major histocompatibility class I gene. Mol. Cell. Biol. 1987;7:4542–4548. doi: 10.1128/mcb.7.12.4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D.R., Pober J.S. HLA class I heavy-chain gene promoter elements mediating synergy between tumor necrosis factor and interferons. Mol. Cell. Biol. 1994;14:1322–1332. doi: 10.1128/mcb.14.2.1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin B.K., Chin K.C., Olsen J.C., Skinner C.A., Dey A., Ozato K., Ting J.P. Induction of MHC class I expression by the MHC class II transactivator CIITA. Immunity. 1997;6:591–600. doi: 10.1016/s1074-7613(00)80347-7. [DOI] [PubMed] [Google Scholar]
- Gobin S.J., Peijnenburg A., Keijsers V., van den Elsen P.J. Site alpha is crucial for two routes of IFN gamma-induced MHC class I transactivationthe ISRE-mediated route and a novel pathway involving CIITA. Immunity. 1997;6:601–611. doi: 10.1016/s1074-7613(00)80348-9. [DOI] [PubMed] [Google Scholar]
- York I.A., Rock K.L. Antigen processing and presentation by the class I major histocompatibility complex. Annu. Rev. Immunol. 1996;14:369–396. doi: 10.1146/annurev.immunol.14.1.369. [DOI] [PubMed] [Google Scholar]
- Lehner P.J., Cresswel P. Processing and delivery of peptides presented by MHC class I molecules. Curr. Opin. Immunol. 1996;8:59–67. doi: 10.1016/s0952-7915(96)80106-3. [DOI] [PubMed] [Google Scholar]
- Gaczynska M., Rock K.L., Goldberg A.L. Gamma-interferon and expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature. 1993;365:264–267. doi: 10.1038/365264a0. [DOI] [PubMed] [Google Scholar]
- Van Kaer L., Ashton-Rickardt P.G., Eichelberger M., Gaczynska M., Nagashima K., Rock K.L., Goldberg A.L., Doherty P.C., Tonegawa S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity. 1994;1:533–541. doi: 10.1016/1074-7613(94)90043-4. [DOI] [PubMed] [Google Scholar]
- Fehling H.J., Swat W., Laplace C., Kuhn R., Rajewsky K., Muller U., von Boehmer H. MHC class I expression in mice lacking the proteasome subunit LMP-7. Science. 1994;265:1234–1237. doi: 10.1126/science.8066463. [DOI] [PubMed] [Google Scholar]
- Neefjes J.J., Momburg F., Hammerling G.J. Selective and ATP-dependent translocation of peptides by the MHC-encoded transporter. Science. 1993;261:769–771. doi: 10.1126/science.8342042. [DOI] [PubMed] [Google Scholar]
- Attaya M., Jameson S., Martinez C.K., Hermel E., Aldrich C., Forman J., Lindahl K.F., Bevan M.J., Monaco J.J. Ham-2 corrects the class I antigen-processing defect in RMA-S cells. Nature. 1992;355:647–649. doi: 10.1038/355647a0. [DOI] [PubMed] [Google Scholar]
- Meraz M.A., White M.J., Sheehan K.C.F., Bach E.A., Rodig S.J., Dighe A.S., Kaplan D.H., Riely J.K., Greenlund A.C., Campbell D. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Müller M., Laxton C., Briscoe J., Schindler C., Improta T., Darnell J.E., Stark G.R., Kerr I.M. Complementation of a mutant cell linecentral role of the 91 kDa polypeptide of ISGF3 in the interferon-α and -γ signal transduction pathways. EMBO (Eur. Mol. Biol. Organ.) J. 1993;12:4221–4228. doi: 10.1002/j.1460-2075.1993.tb06106.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D.E., Kessler D.S., Pine R., Reich N., Darnell J.E. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative control. Genes Dev. 1988;2:383–393. doi: 10.1101/gad.2.4.383. [DOI] [PubMed] [Google Scholar]
- Matsuyama T., Kimura T., Kitagawa M., Pfeffer K., Kawakami T., Watanabe N., Kündig T.M., Amakawa R., Kishihara K., Wakeham A. Targeted disruption of IRF-1 and IRF-2 results in abnormal type I IFN gene induction and aberrant lymphocyte development. Cell. 1993;75:83–97. [PubMed] [Google Scholar]
- Stark G.R., Kerr I.M., Williams B.R., Silverman R.H., Schreiber R.D. How cells respond to interferons. Annu. Rev. Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Darnell J.E. STATs and gene regulation. Science. 1997;277:1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Ihle J.N. STATssignal transducers and activators of transcription. Cell. 1996;84:331–334. doi: 10.1016/s0092-8674(00)81277-5. [DOI] [PubMed] [Google Scholar]
- Sahni M., Ambrosetti D.C., Mansukhani A., Gertner R., Levy D.E., Basilico C. FGF signaling inhibits chondrocyte proliferation and regulates bone development through the Stat1 pathway. Genes Dev. 1999;13:1361–1366. doi: 10.1101/gad.13.11.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K., Kondo M., Weissman I.L. Role of interleukin-7 in T-cell development from hematopoietic stem cells. Immunol. Rev. 1998;165:13–28. doi: 10.1111/j.1600-065x.1998.tb01226.x. [DOI] [PubMed] [Google Scholar]
- von Freeden-Jeffry U., Vieira P., Lucian L.A., McNeil T., Burdach S.E., Murray R. Lymphopenia in interleukin (IL)-7 gene–deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 1995;181:1519–1526. doi: 10.1084/jem.181.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschon J.J., Morrissey P.J., Grabstein K.H., Ramsdell F.J., Maraskovshy E., Gliniak B.C., Park L.S., Ziegler S.F., Williams D.E., Ware C.B. Early lymphocyte expansion is severely impaired in interleukin 7 receptor–deficient mice. J. Exp. Med. 1994;180:1955–1960. doi: 10.1084/jem.180.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J.-X., Migone T.-S., Tsang M., Friedmann M., Weatherbee J.A., Zhou L., Yamauchi A., Bloom E.T., Mietz J., John S. The role of shared receptor motifs and common stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331–339. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- van der Plas D.C., Smiers F., Pouwels K., Hoefsloot L.H., Löwenberg B., Touw I.P. Interleukin-7 signaling in human B cell precursor acute lymphoblastic leukemia cells and murine BAF3 cells involves activation of STAT1 and STAT5 mediated via the interleukin-7 receptor α chain. Leukemia. 1996;10:1317–1325. [PubMed] [Google Scholar]
- Maraskovshy E., Teepe M., Morrissey P.J., Braddy S., Miller R.E., Lynch D.H., Peschon J.J. Impaired survival and proliferation in IL-7 receptor-deficient peripheral T cells. J. Immunol. 1996;157:5315–5323. [PubMed] [Google Scholar]
- Costello R.T., Mallet F., Chambost H., Sainty D., Gastaut J., Olive D. Differential modulation of immune recognition molecules by interleukin-7 in human acute leukaemias. Eur. Cytokine Netw. 1999;10:87–95. [PubMed] [Google Scholar]
- Durbin J.E., Hackenmiller R., Simon M.C., Levy D.E. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Müller U., Steinhoff U., Reis L.F.L., Hemmi S., Pavlovic J., Zinkernagel R.M., Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- Huang S., Hendriks W., Althage A., Hemmi S., Bluethmann H., Kamijo R., Vilcek J., Zinkernagel R.M., Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- Riley J.K., Takeda K., Akira S., Schreiber R.D. Interleukin-10 receptor signaling through the JAK-STAT pathway. Requirement for two distinct receptor-derived signals for anti-inflammatory action. J. Biol. Chem. 1999;274:16513–16521. doi: 10.1074/jbc.274.23.16513. [DOI] [PubMed] [Google Scholar]
- Maniatis T., Fritsh E., Sambrook J. Molecular Cloning. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- Pear W., Nolan G., Scott M., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller J., Frantsve J., Aster J., Williams I.R., Tomasson M.H., Ross T.S., Peeters P., van Rompaey L., van Etten R.A., Ilaria R., Jr. Transformation of hematopoietic cell lines to growth-factor independence and induction of a fatal myelo- and lymphoproliferative disease in mice by retrovirally transduced TEL/JAK2 fusion genes. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:5321–5333. doi: 10.1093/emboj/17.18.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-K., Bluyssen H.A.R., Levy D.E. Regulation of interferon-α responsiveness by the duration of Janus kinase activity. J. Biol. Chem. 1997;272:21872–21877. doi: 10.1074/jbc.272.35.21872. [DOI] [PubMed] [Google Scholar]
- Silvennoinen O., Schindler C., Schlessinger J., Levy D.E. Ras-independent signal transduction in response to growth factors and cytokines by tyrosine phosphorylation of a common transcription factor. Science. 1993;261:1736–1739. doi: 10.1126/science.8378775. [DOI] [PubMed] [Google Scholar]
- Revel M., Chebath J. Interferon-activated genes. Trends Biochem. Sci. 1986;11:166–170. [Google Scholar]
- Baeuerle P.A., Lenardo M.J., Pierce J.W., Baltimore D. Phorbol-ester-induced activation of the NF-κB transcription factor involves dissociation of an apparently cytoplasmic NF-κB/inhibitor complex. Cold Spring Harb. Symp. Quant. Biol. 1988;53:789–798. doi: 10.1101/sqb.1988.053.01.089. [DOI] [PubMed] [Google Scholar]
- Baldwin A.S.J. The NF kappa B and I kappa B proteinsnew discoveries and insights. Annu. Rev. Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Baeuerle P.A., Baltimore D. NF-kappa Bten years after. Cell. 1996;87:13–20. doi: 10.1016/s0092-8674(00)81318-5. [DOI] [PubMed] [Google Scholar]
- Hobart M., Ramassar V., Goes N., Urmson J., Halloran P.F. IFN regulatory factor-1 plays a central role in the regulation of the expression of class I and II MHC genes in vivo. J. Immunol. 1997;158:4260–4269. [PubMed] [Google Scholar]
- Drezen J.M., Babinet C., Morelo D. Transcriptional control of MHC class I and β2-microglobulin genes in vivo. J. Immunol. 1993;150:2805–2813. [PubMed] [Google Scholar]
- Zheng P., Guo Y., Niu Q., Levy D.E., Dyck J.A., Lu S., Sheiman L.A., Liu Y. Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature. 1998;396:373–376. doi: 10.1038/24628. [DOI] [PubMed] [Google Scholar]
- Hofmeister R., Khaled A.R., Benbernou N., Rajnavolgyi E., Muegge K., Durum S.K. Interleukin-7physiological roles and mechanisms of action. Cytokine Growth Factor Rev. 1999;10:41–60. doi: 10.1016/s1359-6101(98)00025-2. [DOI] [PubMed] [Google Scholar]
- Muegge K., Vila M.P., Durum S.K. Interleukin-7a cofactor for V(D)J rearrangement of the T cell receptor β gene. Science. 1993;261:93–95. doi: 10.1126/science.7686307. [DOI] [PubMed] [Google Scholar]
- Corcoran A.E., Riddell A., Krooshoop D., Venkitaraman A.R. Impaired immunoglobulin gene rearrangement in mice lacking the IL-7 receptor. Nature. 1998;391:904–907. doi: 10.1038/36122. [DOI] [PubMed] [Google Scholar]
- Sudo T., Nishikawa S., Ohno N., Akiyama N., Tamakoshi M., Yoshida H., Nishikawa S. Expression and function of the interleukin 7 receptor in murine lymphocytes. Proc. Natl. Acad. Sci. USA. 1993;90:9125–9129. doi: 10.1073/pnas.90.19.9125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodig S.J., Meraz M.A., White J.M., Lampe P.A., Riley J.K., Arthur C.D., King K.L., Sheehan C.F., Yin L., Pennica D. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell. 1998;93:373–383. doi: 10.1016/s0092-8674(00)81166-6. [DOI] [PubMed] [Google Scholar]
- Nosaka T., van Deursen J., Tripp R.A., Thierfelder W.E., Witthuhn B.A., McMickle A.P., Doherty P.C., Grosveld G.C., Ihle J.N. Defective lymphoid development in mice lacking Jak3. Science. 1995;270:800–802. doi: 10.1126/science.270.5237.800. [DOI] [PubMed] [Google Scholar]
- Moriggl R., Topham D.J., Teglund S., Sexl V., McKay C., Wang D., Hoffmeyer A., van Deursen J., Sangster M.Y., Bunting K.D. Stat5 is required for IL-2-induced cell cycle progression of peripheral T cells. Immunity. 1999;10:249–259. doi: 10.1016/s1074-7613(00)80025-4. [DOI] [PubMed] [Google Scholar]
- Pallard C., Stegmann A.P., van Kleffens T., Smart F., Venkitaraman A., Spits H. Distinct roles of the phosphatidylinositol 3-kinase and STAT5 pathways in IL-7-mediated development of human thymocyte precursors. Immunity. 1999;10:525–535. doi: 10.1016/s1074-7613(00)80052-7. [DOI] [PubMed] [Google Scholar]
- O'Shea J.J. Jaks, STATs, cytokine signal transduction, and immunoregulationare we there yet? Immunity. 1997;7:1–11. doi: 10.1016/s1074-7613(00)80505-1. [DOI] [PubMed] [Google Scholar]
- Sampath D., Castro M., Look D.C., Holtzman M.J. Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma. J. Clin. Invest. 1999;103:1353–1361. doi: 10.1172/JCI6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovey M.G., Streuli M., Gresser I., Gugenheim J., Blanchard B., Guymarho J., Vignaux F., Gigou M. Interferon mRNA is produced constitutively in the organs of normal individuals. Proc. Natl. Acad. Sci. USA. 1987;84:5038–5042. doi: 10.1073/pnas.84.14.5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr R., Metcalf D., Elefanty A.G., Brysha M., Willson T.A., Nicola N.A., Hilton D.J., Alexander W.S. Liver degeneration and lymphoid deficiencies in mice lacking suppressor of cytokine signaling-1. Proc. Natl. Acad. Sci. USA. 1998;95:14395–14399. doi: 10.1073/pnas.95.24.14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander W.S., Starr R., Fenner J.E., Scott C.L., Handman E., Spriggs N.S., Corbin J.E., Cornish A.L., Darwiche R., Owczarek C.M. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- Harada H., Taniguchi T., Tanaka N. The role of interferon regulatory factors in the interferon system and cell growth control. Biochimie. 1998;80:641–650. doi: 10.1016/s0300-9084(99)80017-0. [DOI] [PubMed] [Google Scholar]
- Marié I., Durbin J.E., Levy D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO (Eur. Mol. Biol. Organ.) J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penniger J.M., Sirard C., Mittrucker H., Chidgey A., Kozieradzki I., Nghiem M., Boyd R., Taniguchi T., Matsuyama T., Mak T.W. The interferon regulatory transcription factor IRF-1 controls positive and negative selection of CD8+ thymocytes. Immunity. 1997;7:243–254. doi: 10.1016/s1074-7613(00)80527-0. [DOI] [PubMed] [Google Scholar]
- White L.C., Wright K.L., Felix N.J., Ruffner H., Reis L.F., Pine R., Ting J.P. Regulation of LMP2 and TAP1 genes by IRF-1 explains the paucity of CD8+ T cells in IRF-1−/− mice. Immunity. 1996;5:365–376. doi: 10.1016/s1074-7613(00)80262-9. [DOI] [PubMed] [Google Scholar]
- Ploegh H.L. Viral strategies of immune evasion. Science. 1998;280:248–253. doi: 10.1126/science.280.5361.248. [DOI] [PubMed] [Google Scholar]
- Ahn K., Angulo A., Ghazal P., Peterson P.A., Yang Y., Fruh K. Human cytomegalovirus inhibits antigen presentation by a sequential multistep process. Proc. Natl. Acad. Sci. USA. 1996;93:10990–10995. doi: 10.1073/pnas.93.20.10990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler H., Thale R., Lucin P., Muranyi W., Flohr T., Hengel H., Farrell H., Rawlinson W., Koszinowski U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity. 1997;6:57–66. doi: 10.1016/s1074-7613(00)80242-3. [DOI] [PubMed] [Google Scholar]
- Paabo S., Severinsson L., Andersson M., Martens I., Nilsson T., Peterson P.A. Adenovirus proteins and MHC expression. Adv. Cancer Res. 1989;52:151–163. doi: 10.1016/s0065-230x(08)60212-2. [DOI] [PubMed] [Google Scholar]
- Schwartz O., Marechal V., Le Gall S., Lemonnier F., Heard J.M. Endocytosis of major histocompatibility complex class I molecules is induced by the HIV-1 Nef protein. Nat. Med. 1996;2:338–342. doi: 10.1038/nm0396-338. [DOI] [PubMed] [Google Scholar]
- Miller D.M., Rahill B.M., Boss J.M., Lairmore M.D., Durbin J.E., Waldman W.J., Sedmak D.D. Human cytomegalovirus inhibits major histocompatibility complex class II expression by disruption of the Jak/Stat pathway. J. Exp. Med. 1998;187:675–683. doi: 10.1084/jem.187.5.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Look D.C., Roswit W.T., Frick A.G., Gris-Alevy Y., Dickhaus D.M., Walter M.J., Holtzman M.J. Direct suppression of Stat1 function during adenovirus infection. Immunity. 1998;9:871–880. doi: 10.1016/s1074-7613(00)80652-4. [DOI] [PubMed] [Google Scholar]
- Garcia-Sastre A., Durbin R.K., Zheng H., Palese P., Gertner R., Levy D., Durbin J.E. The role of interferon in influenza virus tissue tropism. J. Virol. 1998;72:8550–8558. doi: 10.1128/jvi.72.11.8550-8558.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]