

Abstract
We report the design and engineering of a robust, reagentless fluorescent glucose biosensor based on the periplasmic glucose-binding protein obtained from Thermotoga maritima (tmGBP). The gene for this protein was cloned from genomic DNA and overexpressed in Escherichia coli, the identity of its cognate sugar was confirmed, ligand binding was studied, and the structure of its glucose complex was solved to 1.7 Å resolution by X-ray crystallography. TmGBP is specific for glucose and exhibits high thermostability (midpoint of thermal denaturation is 119 ± 1°C and 144 ± 2°C in the absence and presence of 1 mM glucose, respectively). A series of fluorescent conjugates was constructed by coupling single, environmentally sensitive fluorophores to unique cysteines introduced by site-specific mutagenesis at positions predicted to be responsive to ligand-induced conformational changes based on the structure. These conjugates were screened to identify engineered tmGBPs that function as reagentless fluorescent glucose biosensors. The Y13C•Cy5 conjugate is bright, gives a large response to glucose over concentration ranges appropriate for in vivo monitoring of blood glucose levels (1–30 mM), and can be immobilized in an orientation-specific manner in microtiter plates to give a reversible response to glucose. The immobilized protein retains its response after long-term storage at room temperature.
Keywords: glucose-binding protein, structure, thermostable, fluorescent biosensor
Bacterial periplasmic binding proteins (PBPs) are members of a superfamily that mediate chemotaxis and uptake of sugars, amino acids, oligopeptides, and metals (Ames 1986). The binding pocket of each PBP is located between two domains, which are linked by a hinge region (Quiocho and Ledvina 1996). Upon ligand binding, PBPs undergo a conformational change mediated by a hinge-bending event to adopt a closed form. Mesophilic PBPs have been successfully engineered as reagentless fluorescent (Gilardi et al. 1994; Tolosa et al. 1999; Dattelbaum and Lakowicz 2001; Marvin and Hellinga 2001a) and electrochemical (Benson et al. 2001) biosensors. Their specificity has been manipulated extensively by rational design (Dwyer et al. 2003, 2004; Looger et al. 2003; Allert et al. 2004). Here, we have identified, characterized, and determined the crystal structure of a thermostable periplasmic glucose-binding protein (tmGBP) from the thermophile Thermotoga maritima (Huber 1986) and engineered it into a reagentless fluorescent glucose biosensor.
Close monitoring of blood glucose is essential to prevent chronic diabetic complications caused by hypo- and hyperglycemic conditions (Hoffman 2004; Xu et al. 2005). Development of glucose sensors to monitor blood glucose levels in a reliable, continuous, rapid, and automated fashion remains a major goal for the management of diabetes (Moschou et al. 2004). Current glucose monitoring technologies include fluorescent sensors (de Lorimier et al. 2002; Karnati et al. 2002; Ye and Schultz 2003), colorimetric sensors (Yamazaki et al. 2000), microdialysis probes (Keck et al. 1991), electroenzymatic sensors (Clark Jr. and Duggan 1982), and polymer sensors (Chen et al. 1997). The most successful commercial blood glucose sensors so far are colorimetric sensors based on the glucose oxidase (Manstein et al. 1986). However, these sensors require aggressive finger sticks, which limit their application (Ervin and Kiser 1999). The development of reliable in vivo glucose sensors therefore remains an important challenge.
Reagentless fluorescent biosensors may offer advantages over enzyme-based biosensors for glucose, because no component is depleted or produced during sensing and the sensor composition does not change (Hellinga and Marvin 1998). A variety of proteins have been engineered to develop reagentless fluorescent glucose sensors (Scognamiglio et al. 2004), including bacterial glucose oxidase (D'Auria et al. 1999), glucokinase (D'Auria et al. 2002), glucose dehydrogenase (D'Auria et al. 2000), and periplasmic glucose-binding protein (Salins et al. 2001; de Lorimier et al. 2002).
An optimal biosensor requires several features such as high specificity, response within the dynamic range of blood glucose fluctuation (1–20 mM), fast response, high reproducibility, and robustness under operating conditions. In vivo use imposes additional requirements such as biocompatibility, immunogenicity, sterility, and long-term stability in physiological environments (Moschou et al. 2004). Although previously developed glucose sensors fulfill many of these requirements, none are wholly satisfactory (Kerner 2001). Owing to their robust nature, proteins from thermophiles have long been widely applied in industry (Antranikian 2005). Recently, glucose dehydrogenase from thermophilic Thermoplasma acidophilum and glucokinase from thermophilic Bacillus stearothermophilus have been utilized to build thermostable fluorescent glucose sensors (D'Auria et al. 2000, 2002). The tmGBP-based glucose biosensor presented here provides suitable improvements (including thermal and long-term stability) over reagentless fluorescent sensor technologies based on Escherichia coli GBP (Salins et al. 2001; de Lorimier et al. 2002).
Results and Discussion
Cloning of glucose-binding protein from Thermotoga maritima
T. maritima is a gram-negative bacterium that grows optimally at 80°C and was originally isolated from hot marine mud at Vulcano, Italy (Huber 1986). The genomic sequence of T. maritima (Nelson et al. 1999) contains seven open reading frames (ORFs) annotated as putative periplasmic sugar-binding proteins, among which tm0114 encodes a protein showing the highest sequence homology with E. coli glucose/galactose-binding protein (ecGBP, 25% sequence identity and 42% sequence similarity for the 201 residues that can be aligned). The tm0114 ORF from the genomic DNA of T. maritima with its N-terminal signal peptide predicted (the first 31 residues) and removed was cloned into the pET21a (Novagen) expression vector, with a C-terminal in-frame hexahistidine oligopeptide tag for immobilized metal affinity chromatography (de Lorimier et al. 2006). The protein product of this ORF, tmGBP, was overexpressed in BL21-DE3-Rosetta cells (Novagen) at 2 mg/L and purified.
Identification of the cognate ligand
Ligand binding can be detected readily by measuring the effect of ligand on protein thermostability (Schellman 1975; Simpson et al. 1991). The thermostability of tmGBP in the absence of ligand and in the presence of different monosaccharides was measured to explore tmGBP binding specificity, using circular dichroism (CD) to follow the native state as a function of temperature (Schellman 1975). In the absence of chemical denaturant, tmGBP does not unfold below 100°C. However, thermal denaturation transitions can be observed in the presence of guanidine hydrogen chloride (GuHCl; 3–6 M, incubated for 48 h at room temperature prior to measuring thermal melts). The T m values exhibit a linear dependence on GuHCl concentration (Fig. 1B), permitting the T m value in the absence of GuHCl to be estimated by extrapolation. TmGBP stabilities were determined in the presence of several candidate monosaccharides (glucose, mannose, galactose, ribose, and allose). In the presence of 5 M GuHCl, 1 mM glucose, 10 mM mannose, and 50 mM galactose stabilize tmGBP by 19°C, 10°C, and 3°C, respectively (data not shown). Ribose and allose do not stabilize tmGBP. TmGBP is therefore characterized as a glucose-binding protein, since glucose is the most stabilizing ligand. The T m values of tmGBP in the presence of 1 mM glucose also exhibit linear dependence on GuHCl concentrations (Fig. 1B).
Figure 1.
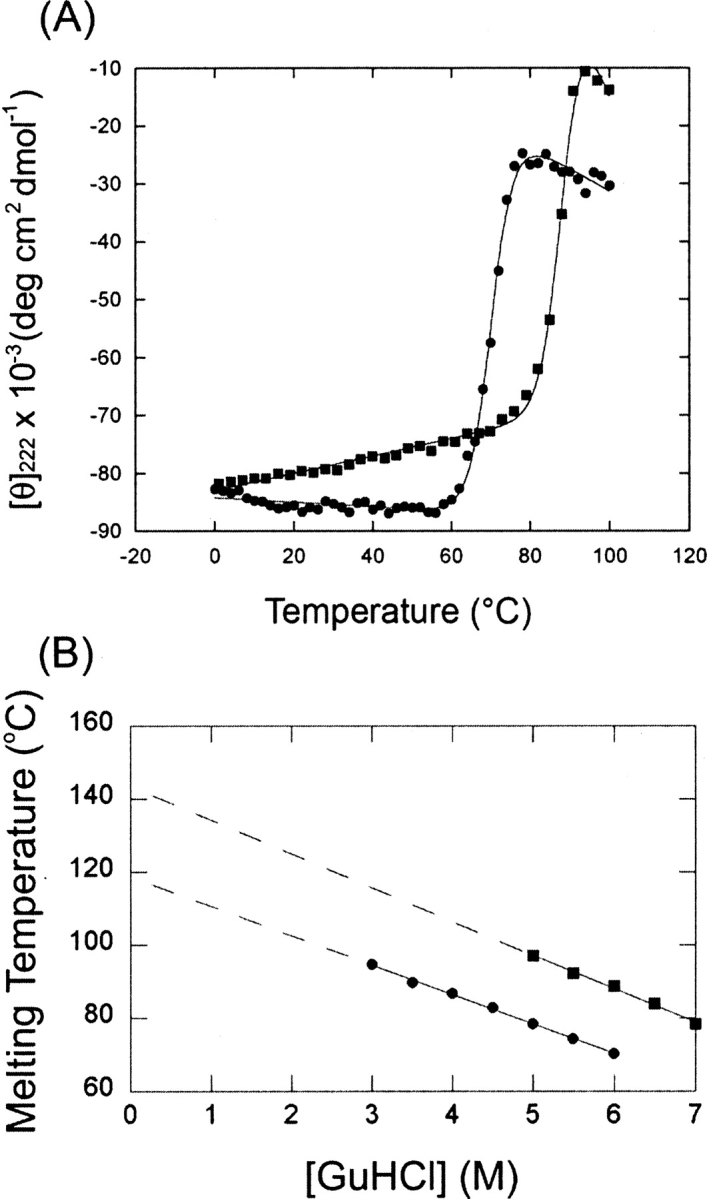
Thermostability of tmGBP. (A) Thermal denaturation of tmGBP in 6 M guanidine hydrogen chloride: (•) in the absence of glucose; (▪) in the presence of 1 mM glucose. Continuous lines are fit to a two-state transition model (Schellman 1987). (B) Thermostability in buffer was obtained by linear extrapolation of melting transition midpoint determined: (•) in the absence of glucose; (▪) in the presence of 1 mM glucose. Straight lines represent linear fits to the data.
Crystal structure of tmGBP
The X-ray crystal structure of tmGBP-glucose complex was determined to 1.7 Å resolution. The structure was solved by molecular replacement using the ligand-bound E. coli ribose-binding protein (ecRBP, pdb code 2DRI) as a model and refined to an Rfree = 23.8% and an Rwork = 19.7% with good stereochemistry (Table 1). The model contains two monomers, two glucose molecules, and 668 water molecules. Monomers A and B are intact (residues 1–305), with monomer A displaying additional electron density for the C-terminal histidine tag encoded by the expression vector. Monomer B exhibits higher average temperature factors and is not as well-ordered as monomer A, perhaps due to differences in crystal packing.
Table 1.
Data collection and refinement statistics of tmGBP

The overall structure of tmGBP is similar to that of other ligand-bound Class I periplasmic binding proteins (Fukami-Kobayashi et al. 1999), consisting of two domains, each comprised of a central parallel β-sheet surrounded by several α-helices, connected by a three-stranded hinge (Fig. 2A). The glucose-binding pocket is buried at the interface of the two domains. Based on the DALI server (Holm and Sander 1993), tmGBP exhibits the highest structural similarity to ecRBP with 1.5 Å RMSD for Cα atoms. Superposition of the tmGBP structure with the crystal structures of ecGBP (pdb code 2GBP) revealed 1.9 Å RMSD for Cα atoms. The tmGBP structure diverges from ecRBP and ecGBP mostly in variable surface loop regions and lacks the calcium-binding site present in ecGBP.
Figure 2.

Crystal structure of tmGBP. (A) The overall structure complexed with glucose (ball and stick) including sites of cysteine mutations for fluorophore attachment (magenta spheres). (B) Close-up view of the binding pocket: (green: glucose; dashed line: hydrogen bonds). The figure was generated using MOLSCRIPT (Kraulis 1991) and RASTER3D (Merritt and Murphy1994).
Unambiguous electron density in the ligand-binding pocket of the tmGBP structure reveals that the β-D-glucose molecule is bound in a single conformation in the chair configuration, as in other glucose-binding proteins. As in all sugar-binding PBPs, the glucose forms extensive van der Waals contacts with aromatic rings of Y13 and Y189 located at either side of the sugar ring. An extensive hydrogen-bond network between the sugar hydroxyl groups and binding-site residues forms an annulus of interactions approximately coincident with the sugar plane (Fig. 2B), as observed in other glucose-binding proteins (Vyas et al. 1994; Cuneo et al. 2006). W14 lies perpendicular to the glucose molecule and forms a hydrogen-bond interaction with the O4 hydroxyl of the sugar. The arrangement of aromatic residues in tmGBP around its ligand is similar to that observed in ecRBP (Bjorkman et al. 1994), E. coli allose-binding protein (Chaudhuri et al. 1999), and ecGBP (Vyas et al. 1994). Modeling of a galactose molecule in the tmGBP binding pocket suggests that having the O4 hydroxyl in an axial position results in a loss of hydrogen-bonding potential with the imino nitrogen of W14 and would interfere with optimal stacking interactions with the aromatic ring of Y189. Every hydroxyl group of the sugar except for the O6 moiety is involved in cooperative hydrogen-bond interactions while the ring oxygen is within hydrogen-bonding distance to the main-chain amide of A190. Binding-site residues K8, D88, N136, R140, and E163 serve as bidentate ligands to the bound glucose. There are no direct water-mediated contacts to the glucose molecule.
The X-ray crystal structures of three periplasmic glucose-binding proteins have been solved: mesophilic ecGBP (Vyas et al. 1994), thermophilic tmGBP, and Thermus thermophilus GBP (ttGBP) (Cuneo et al. 2006). A number of commonalities and differences in sugar recognition and modes of structural adaptation to accommodate different sugars emerge. First, three water-mediated hydrogen bonds in ecGBP are not observed in the thermophilic GBPs. Second, all of the hydrogen-bonding potential of the sugar hydroxyls is fully satisfied in both ecGBP and ttGBP, while the O6 hydroxyl of tmGBP is unsatisfied, consistent with the observation that tmGBP binds xylose (Nanavati et al. 2006). Third, tmGBP is most closely related in sequence and structure to ecRBP, a class I PBP, suggesting that ribose- and glucose-binding sites in this subclass are readily interconverted. TtGBP belongs to the class II PBP family and is closely related to ecMBP, a disaccharide-binding protein, and reveals adaptations that interconvert to mono- and disaccharide-binding proteins. Fourth, ecGBP has a calcium-binding site, which is absent in both tmGBP and ttGBP. These results demonstrate that there are multiple pathways for evolving glucose-binding sites within the PBP superfamily.
Structure-based design of fluorescent glucose biosensors
Structure-based protein design has been used to introduce covalently coupled fluorescent reporter groups into binding proteins (Dwyer and Hellinga 2004; Medintz and Deschamps 2006) and manipulate the dynamic range of the ligand-dependent response of the resulting sensors (Marvin et al. 1997; de Lorimier et al. 2002). PBPs can be converted into reagentless fluorescent biosensors by covalently coupling single, environmentally sensitive, thiol-reactive fluorophores to cysteines introduced at critical positions that couple ligand-mediated conformational transitions to changes in fluorescence emission intensity by a variety of mechanisms (Marvin et al. 1997; Dattelbaum and Lakowicz 2001; de Lorimier et al. 2002). Three types of fluorophore attachment positions have been identified (de Lorimier et al. 2002): endosteric (ligand and fluorophore are in direct contact), peristeric (fluorophore is located at the periphery of the binding site), and allosteric (fluorophore is positioned some distance away from the binding site and responds to long-range conformational changes).
Endosteric (Y13C, W14C, and Y189C), peristeric (S131C), and allosteric (M239C) sites were identified by inspection of the tmGBP structure (Fig. 2A). Cysteine mutations were introduced at these positions by the PCR overlap method (Ho et al. 1989). The glucose-induced change of fluorescence emission intensity of environmentally sensitive thiol-reactive fluorophores (N,N′-dimethyl-N-[iodoacetyl]-N′-[7-nitrobenz-2-oxa-1,3-diazol-4-yl] ethylenediamine [IANBD], acrylodan, Cy5, and Cy3) conjugated at each cysteine mutant was determined (Table 2). Changes in the emission intensity were characterized by three parameters (de Lorimier et al. 2002): the wavelength shift of the emission maximum, the direction of intensity change (increase or decrease), and the magnitude of the change (ΔF max = | [F sat − F apo] / F apo | × 100%, where F sat and F apo are fluorescence intensity of saturated and apo form at the wavelength with maximal intensity change, respectively). All cysteine mutants except M239C exhibited significant glucose-induced signal changes (ΔF max > 50%) with at least one fluorophore. The glucose affinity for each conjugate that exhibits a measurable change (ΔF max > 10%) was determined by titration (Table 2).
Table 2.
Spectral and binding properties of tmGBP conjugates at 25°C, with 150 mM NaCl, 20 mM MOPS, and pH 7.0

The glucose-binding affinity of wild-type tmGBP could not be determined since no change in tryptophan fluorescence was observed upon glucose addition. The affinities of the conjugates follow the trends expected for the attachment points: allosteric > peristeric > endosteric. The allosteric M239C•acrylodan conjugate showed the highest affinity to glucose (K d = 12.7 ± 0.9 nM). M239 is located under the C-terminal “flap” (Fig. 2A) of tmGBP. The flap region may undergo rearrangements upon glucose binding that induce the environmental change around M239C. The binding affinity of wild-type PBPs is generally determined as 0.1–1 μM (de Lorimier et al. 2002); so most likely M239C•acrylodan leads to a tighter glucose binding than wild-type tmGBP. Allosteric fluorescent conjugates positioned at or close to the hinge region of PBPs often exhibit higher binding affinity than wild-type proteins, because bulky fluorophores at these positions perturb the ligand-free open form of PBPs and hence stabilize the ligand-bound closed form (Marvin et al. 1997; Marvin and Hellinga 2001b).
Located in the “jaw” of the binding pocket (Fig. 2A), the K d value (18.1 ± 1.4 μM) of the S131C•IANBD peristeric conjugate is most likely larger than that of wild-type tmGBP (estimated to be 0.1–1 μM), probably because the bulky conjugated fluorophore destabilizes the ligand-bound closed form of tmGBP. S131C•IANBD exhibits the largest signal change (▵F max = 790%) among all conjugates tested. A Stern-Volmer (SV) iodine quenching experiment was performed to further probe the origin of the change in emission intensity of this conjugate. The quenching constant, K SV, was determined as 2.4 M−1 and 1.6 M−1 in the apo- and ligand-saturated forms, respectively (data not shown). The observations suggest that IANBD is less accessible to solvent in the glucose-binding closed form than in the glucose-free open form. However, the 7.9-fold increase of fluorescence intensity is probably not due solely to a change in solvent accessibility. The D95C•NBD conjugate of E. coli maltose-binding protein (ecMBP) also shows a large change upon ligand binding. Computational modeling in combination with mutagenesis experiments suggests that a ligand-induced change in hydrogen-bonding interaction between NBD and ecMBP is the dominant factor accounting for the magnitude of the fluorescence intensity change (Dattelbaum et al. 2005). The mechanism of fluorescence change of tmGBP S131C•IANBD also may involve changes in hydrogen-bonding interactions.
The endosteric mutations Y13C, W14C, and Y189C are located within the binding pocket of tmGBP. Y13 and Y189 sandwich glucose; W14 is involved in a hydrogen-bonding interaction with glucose. All endosteric conjugates have K d values in the millimolar range, indicating that glucose binding is strongly perturbed by the introduction of the fluorophore into the binding pocket, as expected (Marvin et al. 1997; de Lorimier et al. 2002).
The effect of temperature on glucose-binding affinity was studied for selected conjugates (Fig. 3). Remarkably, Y13C•IANBD exhibits little change in affinity from 25°C to 65°C. Y13C•Cy5 and Y189C•acrylodan both show decreases in K d values with an increase of temperature. W14C•IANBD and S131C•IANBD both have a binding optimum at 45°C. The temperature-dependent changes of the apparent K d of these conjugates are impossible to predict a priori or to interpret by simple physicochemical models because many factors are involved, including the effects on ligand binding and solvation, intrinsic fluorescence of the fluorophore, and intrinsic equilibrium between the open and closed states. The differences observed for the same fluorophore in different conjugates strongly suggests that among these mechanisms, the interaction between fluorophore and protein plays the dominant role in the temperature-dependent effects.
Figure 3.

Temperature dependence of glucose affinity for selected fluorescent conjugates (20 mM MOPS and 150 mM NaCl, pH 7.0). (•) Y13C•Cy5, (▪) Y13C•IANBD, (♦) W14C•IANBD, (▴) S131C•IANBD, (▾) Y189C•Acrylodan. Data points are connected by lines.
Hydrogen-bonding interactions between the sugar and protein
The interactions between glucose and tmGBP were studied with the peristeric S131C•IANBD conjugate because it has reasonable binding affinity and a large ligand-induced fluorescent signal increase. Hydrogen-bonding contributions were probed using epimers that have a ring hydroxyl in the opposite configuration of glucose (mannose, allose, and galactose), and deoxy sugars (2-deoxyglucose, 3-deoxyglucose, and 6-deoxyglucose). The five-carbon sugar xylose was used to probe the contributions of the C6 carbon and hydroxyl group. In addition to the effect on affinity, different sugars also exhibited differences in the measured magnitudes of fluorescence intensity change (Table 3).
Table 3.
Binding affinity and fluorescence change of S131C•IANBD responding to different sugars (25°C, 150 mM NaCl, 20 mM MOPS, and pH 7.0)

The deoxysugar 2-deoxyglucose exhibits a 100-fold decrease in binding affinity, consistent with a loss of hydrogen-bonding interactions between the glucose O2 hydroxyl group and D88, R140, and N235. The 2-OH epimer mannose binds tmGBP with similar affinity as 2-deoxyglucose, suggesting that all the hydrogen-binding interactions at the O2 hydroxyl are also missing. Similarly, 3-deoxyglucose and the 3-OH epimer allose both exhibit a >2000-fold decrease in affinity, consistent with the observed hydrogen bonds between K8, D88, N136, and the 3-hydroxyl. The 4-OH epimer galactose exhibits a 100-fold decrease in affinity consistent with a loss of a hydrogen bond to W14, whereas ecGBP and ttGBP both bind glucose and galactose. The loss of O6 in 6-deoxyglucose results in a tighter binding, presumably because the glucose O6 does not interact with neighboring residues, and 6-deoxyglucose is less bulky than glucose. The removal of both the C6 carbon and O6 hydroxyl in xylose results in a twofold decrease in binding affinity, indicating that tmGBP binds xylose tightly, consistent with previous observations (Nanavati et al. 2006).
A glucose biosensor based on the Y13C•Cy5 conjugate
Y13C•Cy5 exhibits a 50% decrease in emission intensity and a 7-nm wavelength shift in response to glucose. This conjugate is a promising candidate for developing a deployable glucose sensor: at 37°C, Y13C•Cy5 binds glucose with a K d of 5.6 ± 0.7 mM (Fig. 4), permitting determination in the 1–30 mM range within 5% accuracy (Marvin et al. 1997), which matches the dynamic range of physiological blood glucose concentration fluctuation (euglycemia, 4–6 mM [Nasraway Jr. 2006]; hypoglycemia, <3 mM [Warren and Frier 2005]; hyperglycemia, >7 mM [Picardi and Pozzilli 2003]); the fluorescent Cy5 probe has near-infrared emission and excitation wavelengths, which fall within the transparency window where autofluorescence from blood and tissue do not interfere with its signal (Pickup et al. 2005); its emission wavelength shift enables ratiometric measurement (app K d = 2.7 ± 0.4 mM, determined with the ratio of emissions at 666 nm over emissions at 700 nm), which permits self-calibration (Lakowicz 1999); and it is very bright (extinction coefficient = 25,000 M−1 cm−1). One limitation of Y13C•Cy5 is the temperature dependence of glucose affinity (data were fit as lnK d = −16.82 + 5888.5 × (1/T), so δK d/δT = −0.54 mM K−1 at 37°C), which requires temperature monitoring to ensure accuracy.
Figure 4.

Fluorescent response of the Y13C•Cy5 conjugate upon glucose titration (20 mM MOPS and 150 mM NaCl, pH 7.0, 37°C). (Inset) Emission spectra of this conjugate in the absence (solid line) and presence (dashed line) of 140 mM glucose. (Bottom) Fit to a hyperbolic binding isotherm.
Immobilization of Y13C•Cy5
Protein immobilization is critical for biosensor device construction (Cao 2005). The effects of semi-specific and orientation-specific immobilization on the sensing properties of the Y13C•Cy5 conjugate were investigated. For semi-specific immobilization, the Y13C•Cy5 conjugate was reacted with an NHS ester derivative of biotin that couples to the primary amine of exposed lysine. Mass spectrometry showed that on average each protein was modified by two biotins. Conjugation of biotin had no effect on K d and ΔF max in solution (data not shown). The biotinylated protein was then immobilized on 96-well streptavidin-coated microtiter plates. The immobilized protein exhibited an 18% maximal decrease in fluorescence intensity upon addition of glucose compared with the 47% maximal decrease observed in solution. Glucose titrations revealed that the immobilized protein has K d values of 2.7 ± 0.3 mM at 25°C and 0.9 ± 0.2 mM at 37°C (data not shown), the same trend of the temperature dependence as observed in solution (K d = 15.3 ± 1.8 mM at 25°C, and K d = 5.6 ± 0.7 mM at 37°C). Taken together, these observations are consistent with a model in which immobilization slightly affects the intrinsic equilibrium between the open and closed states of binding proteins, favoring the closed state and thus increasing the affinity for glucose and decreasing the fluorescence response.
As reported previously, N- or C-terminal fusions of Y13C•Cy5 with a His2Cys2 zinc finger domain (Zif) were used for orientation-specific immobilization (de Lorimier et al. 2006). In solution, tmGBP-Y13C-NZif exhibits almost the same K d and ΔF max values as tmGBP without a zinc finger fusion, whereas tmGBP-Y13C-CZif shows a 1.5-fold decrease in glucose affinity. Upon immobilization, both tmGBP-Y13C-NZif and tmGBP-Y13C-CZif exhibit a twofold decrease in ΔF max and two- and fourfold increases in affinity compared with fusion proteins in solution, respectively (de Lorimier et al. 2006).
Robustness and reversibility of the Y13C•Cy5 glucose sensor
The thermostability of Y13C•Cy5 and long-term stability in solution or on surfaces were determined to study the robustness of this sensor. Thermal melts of Y13C•Cy5 determined in the presence of 3–5 M GuHCl by CD revealed that the T m value in buffer is 114(±2)°C (data not shown), only 5°C less stable than the wild-type tmGBP. To test the long-term stability in solution, Y13C•Cy5 was stored for 2 mo at 4°C, followed by fluorescence titration at 37°C. This stored protein exhibited a K d = 6.0 ± 0.6 mM, ΔF max = −43%, compared with K d = 5.6 ± 0.6 mM, ΔF max = −51% for freshly prepared sample. Long-term stability on surfaces was tested with immobilized Y13C•Cy5 that had been stored at room temperature for 1 mo. Stored Y13C•Cy5 behaved nearly identically to freshly immobilized protein (Fig. 5: K d = 2.7 ± 0.3 mM, ΔF max = −18% for fresh plate; K d = 3.3 ± 0.7 mM, ΔF max = −16% for 1-mo-old plate).
Figure 5.

Fluorescence titration of semi-specifically immobilized Y13C•Cy5 (20 mM MOPS and 150 mM NaCl, pH 7.0, 25°C). (▪) Freshly immobilized protein, (•) immobilized proteins with 1 mo of storage at room temperature. Continuous lines are fit to a hyperbolic binding isotherm on a semi-log plot.
The reversibility of glucose binding was tested for orientation-specifically immobilized Y13C•Cy5 (tmGBP-Y13C-CZif) on maleimide-derivatized plates (de Lorimier et al. 2006). Immobilized tmGBP-Y13C-CZif was titrated with glucose, repeatedly rinsed with buffer to remove bound glucose, and followed by another glucose titration. Five such cycles showed that glucose binding of immobilized Y13C•Cy5 is completely reversible (Fig. 6).
Figure 6.

Successive cycles of glucose titration and buffer washes of tmGBP-Y13C-Czif immobilized on microtiter plates (20 mM MOPS and 150 mM NaCl, pH 7.0, 25°C). Data of apo and saturated ligand binding states for five cycles are connected by straight lines.
Conclusions
Here, we report the cloning, expression, structural study, and biochemical characterization of tmGBP. The thermostable tmGBP protein was successfully converted into a series of fluorescent glucose biosensors with different fluorescent responses and dynamic ranges. These tmGBP-based fluorescent biosensors are sensitive, rapid, continuous, reproducible, and reversible. In contrast to enzymatic glucose sensors constructed from glucose oxidase (Abel and von Woedtke 2002), these tmGBP-based fluorescent sensors are reagentless. The Y13C•Cy5 conjugate exhibits a high specificity for glucose, ratiometric fluorescent change in the near-infrared excitation and emission wavelength range, and responds over a glucose concentration range (1–30 mM) that covers the clinically relevant blood glucose levels; hence, it is the best candidate as a glucose biosensor.
Materials and Methods
Molecular cloning and protein purification
The tm0114 gene of Thermotoga maritima was cloned from genomic DNA. The N-terminal periplasmic signal peptide sequence of tm0114 was predicted using the software SignalP (Bendtsen et al. 2004). The tm0114 gene without its signal peptide and an additional C-terminal Gly-Ser-(His)6 tag was amplified from T. maritima genomic DNA (American Type Culture Collection [ATCC]) by PCR. The whole gene was designated wild-type tmGBP. Amplified product was cloned into a pET21a vector (Novagen) using 5′ NdeI and 3′ EcoRI restriction sites and verified by DNA sequencing. Cysteine mutations were generated by overlapping PCR mutagenesis (Ho et al. 1989). Proteins were produced by overexpression in the Rosetta-gami(DE3)pLysS strain (Novagen) that contains rare tRNA genes. Transformed cells were fermented in Terrific Broth medium (Research Products International Corp.) at 37°C and induced with 1 mM IPTG at OD600 = 0.6, and grown for 3 h at 44°C prior to harvesting by centrifugation. Proteins were purified from a cleared lysate by immobilized metal affinity chromatography (IMAC) as described (de Lorimier et al. 2002). Following affinity purification, the wild-type tmGBP protein was further purified on a HiLoad 26/60 Superdex 75 gel filtration column (Amersham Biosciences) equilibrated with 0.3 M NaCl, 10 mM Tris, and 5 mM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP; pH 7.0) and then concentrated to 10 mg/mL for crystallization. Concentrations of proteins were determined spectrophotometrically by the von Hippel method (Gill and von Hippel 1989), using calibrated ɛ280 = 26,000 M−1 cm−1.
Determination of thermostability
Thermostabilities were determined on an Aviv model 202 Circular Dichroism Spectrometer (5 μM protein samples, buffered with 50 mM sodium phosphate and 100 mM NaCl, pH 7.0). GuHCl was added to samples to denature the protein. The concentration of GuHCl stock solutions was determined by refractive index measurements (Pace 1986). For each thermal melt curve, measurements were made at 222 nm with three-time averages from 2°C to 98°C. The CD signal was converted to the mean residue ellipticity ([θ]222) which was fit as a two-state transition to determine the melting temperature, T m, as described (Schellman 1987).
Crystallization, data collection, structure determination, and refinement
Single crystals (0.1 mm × 0.03 mm × 0.4 mm) of tmGBP in complex with glucose were obtained in 1 mo at 18°C by the vapor diffusion hanging-drop method in a solution containing 0.2 M sodium formate and 17% PEG 3350. For cryo-protection, the crystals were dehydrated overnight by equilibration against 0.2 M sodium formate and 25% PEG 3350, and then were dragged quickly through the same solution supplemented with 10% ethylene glycol, followed by flash-freezing in liquid nitrogen.
The initial 2.6 Å resolution diffraction data set was collected on a Rigaku RaxisIV image plate detector and rotating anode system; the 1.7 Å resolution data were collected from the same crystal at NSLS beamline X25 on an ADSC Q315 CCD detector (both at 100 K). Data were integrated with XDS (Kabsch 1993) and scaled with SCALA (Collaborative Computational Project Number 4 1994). TmGBP crystals belong to space group C2 (a = 148 Å, b = 46 Å, c = 118 Å, β = 108°, two molecules in the asymmetric unit). The crystal structure was determined by molecular replacement with the program AMORE (Navaza 2001), using the ligand-bound form of E. coli ribose-binding protein (PDB code 2DRI); (Mowbray and Cole 1992) as a search model. A clear solution for both molecules was found yielding an R-factor of 52% and a correlation coefficient of 25.9. Manual model building was carried out in O (Jones et al. 1991), and all refinement was performed in REFMAC5 (Murshudov et al. 1997). Electron density maps showed unambiguous density for the bound glucose in each monomer, and the ligand was built and refined at full occupancy. The stereochemistry of the final model was determined by PROCHECK and MOLPROBITY (Laskowski et al.1993; Davis et al. 2004). Data collection and refinement statistics are summarized in Table 1. PDB coordinates and structure factors have been deposited in the RCSB Protein Data Bank (accession code 2H3H).
Fluorophore labeling and ligand-binding measurements
Purified cysteine mutants of tmGBP were dialyzed (150 mM NaCl, 20 mM MOPS, and 2 mM TCEP, pH 6.0) to deplete bound ligand. TCEP was removed by gel filtration (BioRad 10DG column) after dialysis. Each dialyzed protein was labeled (2–4 h, room temperature) with thiol-reactive acrylodan, IANBD (Invitrogen), Cy5, and Cy3 (Amersham Biosciences). The conjugation step was performed for 2–4 h at room temperature. Unreacted fluorophore was removed by gel filtration (BioRad 10DG columns). Fluorescence labeling efficiency was determined as >80% for all conjugates, as previously described (Smith et al. 2005).
Ligand-induced changes of fluorescence intensities were measured on a SLM Aminco-Bowman series 2 fluorimeter (acrylodan: λ ex = 390 nm, λ em = 500 nm; IANBD: λ ex = 480 nm, λ em = 550 nm; Cy5: λ ex = 625 nm, λ em = 670 nm; Cy3: λ ex = 530 nm, λ em = 570 nm; slit widths: excitation, 4 nm; emission, 8 nm) at different temperatures (controlled by a water bath). Data were fit to a single-site binding isotherm (de Lorimier et al. 2002). Stern-Volmer (SV) iodine quenching experiments were carried out with 0–300 mM NaI, 200–500 mM NaCl to maintain a constant 500 mM ionic strength and 1 mM Na2S2O3 to prevent iodine formation (Dattelbaum et al. 2005).
Immobilization of tmGBP and plate-based fluorescence titration
The surface lysine residues of Y13C•Cy5 were nonspecifically modified (pH 7.5, 30 min, 25°C) by No-Weigh NHS-PEO4-Biotin (Pierce) using a 0.11:1.0 reagent:protein reaction stoichiometry (weight:weight). Unreacted NHS-PEO4-Biotin was removed by gel filtration (BioRad 10DG columns). On average, each Y13C•Cy5 conjugate was modified with two biotin molecules according to MALDI-TOF mass spectrometry. One hundred microliters of 4 μM biotinylated Y13C•Cy5 were loaded into SigmaScreen streptavidin-coated 96-well microtiter plates (Sigma-Aldrich), incubated for 2–4 h in the dark at room temperature, followed by rinsing with buffer to remove free protein. Fluorescence in each well was measured in the absence or in the presence of 1 nM–1 M glucose on a Spectra Max Gemini XS plate reader (Molecular Devices; 25°C or 37°C, excitation: 640 nm, emission: 680 nm, cutoff: 665 nm). The fluorescence of each observation was averaged from three readings, to get F apo and F glucose, respectively. Each condition was repeated in three wells, and the relative change in signal (F glucose / F apo) for each glucose concentration was averaged from these three wells. The averaged relative change of signal was fit into a single-site binding isotherm.
Acknowledgments
This work was funded by the NIH Director's Pioneer Award (5 DP1 OD000122-02) and a grant from the U.S. Department of Homeland Security (W81XWH-05-C-0161). Research was carried out in part at beamline X25 of the National Synchrotron Light Source, Brookhaven National Laboratory, which is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886.
Footnotes
Reprint requests to: Homme W. Hellinga, Duke University, Box 3711, Durham, NC 27710, USA; e-mail: hwh@biochem.duke.edu; fax: (919) 684-8885.
Abbreviations: PBP, periplasmic binding protein; tmGBP, Thermotoga maritima glucose-binding protein; ecRBP, Escherichia coli ribose-binding protein; ecGBP, E. coli glucose-binding protein; ttGBP, Thermus thermophilus glucose-binding protein; ecMBP, E. coli maltose-binding protein; ORF, open reading frame; CD, circular dichroism; GuHCl, guanidine hydrogen chloride; IANBD, N,N′-dimethyl-N-(iodoacetyl)-N′-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) ethylenediamine; Zif, zinc finger domain; TCEP, Tris(2-carboxyethyl)phosphine hydrochloride; IMAC, immobilized metal affinity chromatography; SV, Stern-Volmer.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.072969407.
References
- Abel P.U. and von Woedtke, T. 2002. Biosensors for in vivo glucose measurement: Can we cross the experimental stage? Biosens. Bioelectron. 17: 1059–1070. [DOI] [PubMed] [Google Scholar]
- Allert M., Rizk, S.S., Looger, L.L., and Hellinga, H.W. 2004. Computational design of receptors for an organophosphate surrogate of the nerve agent soman. Proc. Natl. Acad. Sci. 101: 7907–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ames G.F. 1986. Bacterial periplasmic transport systems: Structure, mechanism, and evolution. Annu. Rev. Biochem. 55: 397–425. [DOI] [PubMed] [Google Scholar]
- Antranikian K.E.a.G. 2005. Industrial relevance of thermophilic Archaea. Curr. Opin. Microbiol. 8: 649–655. [DOI] [PubMed] [Google Scholar]
- Bendtsen J.D., Nielsen, H., von Heijne, G., and Brunak, S. 2004. Improved prediction of signal peptides: SignalP 3.0. J. Mol. Biol. 340: 783–795. [DOI] [PubMed] [Google Scholar]
- Benson D.E., Conrad, D.W., de Lorimier, R.M., Trammell, S.A., and Hellinga, H.W. 2001. Design of bioelectronic interfaces by exploiting hinge-bending motions in proteins. Science 293: 1641–1644. [DOI] [PubMed] [Google Scholar]
- Bjorkman A.J., Binnie, R.A., Zhang, H., Cole, L.B., Hermodson, M.A., and Mowbray, S.L. 1994. Probing protein–protein interactions. The ribose-binding protein in bacterial transport and chemotaxis. J. Biol. Chem. 269: 30206–30211. [PubMed] [Google Scholar]
- Cao L. 2005. Immobilised enzymes: Science or art? Curr. Opin. Chem. Biol. 9: 217–226. [DOI] [PubMed] [Google Scholar]
- Chaudhuri B.N., Ko, J., Park, C., Jones, T.A., and Mowbray, S.L. 1999. Structure of D-allose binding protein from Escherichia coli bound to D-allose at 1.8 Å resolution. J. Mol. Biol. 286: 1519–1531. [DOI] [PubMed] [Google Scholar]
- Chen G., Guan, Z., Chen, C.T., Fu, L., Sundaresan, V., and Arnold, F.H. 1997. A glucose-sensing polymer. Nat. Biotechnol. 15: 354–357. [DOI] [PubMed] [Google Scholar]
- Clark L.C. and Duggan, C.A. 1982. Implanted electroenzymatic glucose sensors. Diabetes Care 5: 174–180. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4 1994. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50: 760–763. [DOI] [PubMed] [Google Scholar]
- Cuneo M.J., Changela, A., Warren, J.J., Beese, L.S., and Hellinga, H.W. 2006. The crystal structure of a thermophilic glucose binding protein reveals adaptations that interconvert mono and di-saccharide binding sites. J. Mol. Biol. 362: 259–270. [DOI] [PubMed] [Google Scholar]
- Dattelbaum J.D. and Lakowicz, J.R. 2001. Optical determination of glutamine using a genetically engineered protein. Anal. Biochem. 291: 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dattelbaum J.D., Looger, L.L., Benson, D.E., Sali, K.M., Thompson, R.B., and Hellinga, H.W. 2005. Analysis of allosteric signal transduction mechanisms in an engineered fluorescent maltose biosensor. Protein Sci. 14: 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Auria S., Herman, P., Rossi, M., and Lakowicz, J.R. 1999. The fluorescence emission of the apo-glucose oxidase from Aspergillus niger as probe to estimate glucose concentrations. Biochem. Biophys. Res. Commun. 263: 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Auria S., Di Cesare, N., Gryczynski, Z., Gryczynski, I., Rossi, M., and Lakowicz, J.R. 2000. A thermophilic apoglucose dehydrogenase as nonconsuming glucose sensor. Biochem. Biophys. Res. Commun. 274: 727–731. [DOI] [PubMed] [Google Scholar]
- D'Auria S., DiCesare, N., Staiano, M., Gryczynski, Z., Rossi, M., and Lakowicz, J.R. 2002. A novel fluorescence competitive assay for glucose determinations by using a thermostable glucokinase from the thermophilic microorganism Bacillus stearothermophilus . Anal. Biochem. 303: 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I.W., Murray, L.W., Richardson, J.S., and Richardson, D.C. 2004. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32: W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorimier R.M., Smith, J.J., Dwyer, M.A., Looger, L.L., Sali, K.M., Paavola, C.D., Rizk, S.S., Sadigov, S., Conrad, D.W., Loew, L., et al. 2002. Construction of a fluorescent biosensor family. Protein Sci. 11: 2655–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lorimier R.M., Tian, Y., and Hellinga, H.W. 2006. Binding and signaling of surface-immobilized reagentless fluorescent biosensors derived from periplasmic binding proteins. Protein Sci. 15: 1936–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diederichs K. and Karplus, P.A. 1997. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat. Struct. Biol. 4: 269–275. [DOI] [PubMed] [Google Scholar]
- Dwyer M.A. and Hellinga, H.W. 2004. Periplasmic binding proteins: A versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 14: 495–504. [DOI] [PubMed] [Google Scholar]
- Dwyer M.A., Looger, L.L., and Hellinga, H.W. 2003. Computational design of a Zn2+ receptor that controls bacterial gene expression. Proc. Natl. Acad. Sci. 100: 11255–11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer M.A., Looger, L.L., and Hellinga, H.W. 2004. Computational design of a biologically active enzyme. Science 304: 1967–1971. [DOI] [PubMed] [Google Scholar]
- Ervin K.R. and Kiser, E.J. 1999. Issues and implications in the selection of blood glucose monitoring technologies. Diabetes Technol. Ther. 1: 3–11. [DOI] [PubMed] [Google Scholar]
- Fukami-Kobayashi K., Tateno, Y., and Nishikawa, K. 1999. Domain dislocation: A change of core structure in periplasmic binding proteins in their evolutionary history. J. Mol. Biol. 286: 279–290. [DOI] [PubMed] [Google Scholar]
- Gilardi G., Zhou, L.Q., Hibbert, L., and Cass, A.E. 1994. Engineering the maltose binding protein for reagentless fluorescence sensing. Anal. Chem. 66: 3840–3847. [DOI] [PubMed] [Google Scholar]
- Gill S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182: 319–326. [DOI] [PubMed] [Google Scholar]
- Hellinga H.W. and Marvin, J.S. 1998. Protein engineering and the development of generic biosensors. Trends Biotechnol. 16: 183–189. [DOI] [PubMed] [Google Scholar]
- Ho S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77: 51–59. [DOI] [PubMed] [Google Scholar]
- Hoffman R.P. 2004. Practical management of type 1 diabetes mellitus in adolescent patients: Challenges and goals. Treat. Endocrinol. 3: 27–39. [DOI] [PubMed] [Google Scholar]
- Holm L. and Sander, C. 1993. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 233: 123–138. [DOI] [PubMed] [Google Scholar]
- Huber R.E.A. 1986. Thermotoga maritima sp. nov. represents a new genus of unique extremely thermophilic eubacteria growing up to 90°C. Arch. Microbiol. 144: 324–333. [Google Scholar]
- Jones T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A47: 110–119. [DOI] [PubMed] [Google Scholar]
- Kabsch W. 1993. Automatic processing of rotation diffraction data from crystals of iniitially unknown symmetry and cell constants. J. Appl. Crystallogr. 26: 795–800. [Google Scholar]
- Karnati V.V., Gao, X., Gao, S., Yang, W., Ni, W., Sankar, S., and Wang, B. 2002. A glucose-selective fluorescence sensor based on boronic acid–diol recognition. Bioorg. Med. Chem. Lett. 12: 3373–3377. [DOI] [PubMed] [Google Scholar]
- Keck F.S., Kerner, W., Meyerhoff, C., Zier, H., and Pfeiffer, E.F. 1991. Combination of microdialysis and Glucosensor permits continuous (on line) s.c. glucose monitoring in a patient operated device: I. In vitro evaluation. Horm. Metab. Res. 23: 617–618. [DOI] [PubMed] [Google Scholar]
- Kerner W. 2001. Implantable glucose sensors: Present status and future developments. Exp. Clin. Endocrinol. Diabetes Suppl. 2 109: S341–S346. doi: 0.1055/s-2001-18593. [DOI] [PubMed] [Google Scholar]
- Kraulis P.J. 1991. MOLSCRIPT: A program to produce both detailed and schematic plots of protein structures. J. Appl. Crystallogr. 24: 946–950. [Google Scholar]
- Lakowicz J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed, p. 698. Kluwer Academic Press, New York.
- Laskowski R.A., MacArthur, M.W., Moss, D.S., and Thornton, J.M. 1993. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26: 283–291. [Google Scholar]
- Looger L.L., Dwyer, M.A., Smith, J.J., and Hellinga, H.W. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423: 185–190. [DOI] [PubMed] [Google Scholar]
- Manstein D.J., Pai, E.F., Schopfer, L.M., and Massey, V. 1986. Absolute stereochemistry of flavins in enzyme-catalyzed reactions. Biochemistry 25: 6807–6816. [DOI] [PubMed] [Google Scholar]
- Marvin J.S. and Hellinga, H.W. 2001a. Conversion of a maltose receptor into a zinc biosensor by computational design. Proc. Natl. Acad. Sci. 98: 4955–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin J.S. and Hellinga, H.W. 2001b. Manipulation of ligand binding affinity by exploitation of conformational coupling. Nat. Struct. Biol. 8: 795–798. [DOI] [PubMed] [Google Scholar]
- Marvin J.S., Corcoran, E.E., Hattangadi, N.A., Zhang, J.V., Gere, S.A., and Hellinga, H.W. 1997. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc. Natl. Acad. Sci. 94: 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medintz I.L. and Deschamps, J.R. 2006. Maltose-binding protein: A versatile platform for prototyping biosensing. Curr. Opin. Biotechnol. 17: 17–27. [DOI] [PubMed] [Google Scholar]
- Merritt E.A. and Murphy, M.E.P. 1994. Raster3D version 2.0. A program for photorealistic molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 50: 869–873. [DOI] [PubMed] [Google Scholar]
- Moschou E.A., Sharma, B.V., Deo, S.K., and Daunert, S. 2004. Fluorescence glucose detection: Advances toward the ideal in vivo biosensor. J. Fluoresc. 14: 535–547. [DOI] [PubMed] [Google Scholar]
- Mowbray S.L. and Cole, L.B. 1992. 1.7 Å X-ray structure of the periplasmic ribose receptor from Escherichia coli . J. Mol. Biol. 225: 155–175. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin, A.A., and Dodson, E.J. 1997. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. D53: 240–255. [DOI] [PubMed] [Google Scholar]
- Nanavati D.M., Thirangoon, K., and Noll, K.M. 2006. Several archaeal homologs of putative oligopeptide-binding proteins encoded by Thermotoga maritima bind sugars. Appl. Environ. Microbiol. 72: 1336–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasraway S.A. 2006. Hyperglycemia during critical illness. JPEN J. Parenter. Enteral Nutr. 30: 254–258. [DOI] [PubMed] [Google Scholar]
- Navaza J. 2001. Implementation of molecular replacement in AMoRe. Acta Crystallogr. D Biol. Crystallogr. 57: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Nelson K.E., Clayton, R.A., Gill, S.R., Gwinn, M.L., Dodson, R.J., Haft, D.H., Hickey, E.K., Peterson, J.D., Nelson, W.C., Ketchum, K.A., et al. 1999. Evidence for lateral gene transfer between Archaea and bacteria from genome sequence of Thermotoga maritima . Nature 399: 323–329. [DOI] [PubMed] [Google Scholar]
- Pace C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131: 266–280. [DOI] [PubMed] [Google Scholar]
- Picardi A. and Pozzilli, P. 2003. Dynamic tests in the clinical management of diabetes. J. Endocrinol. Invest. 26: 99–106. [PubMed] [Google Scholar]
- Pickup J.C., Hussain, F., Evans, N.D., and Sachedina, N. 2005. In vivo glucose monitoring: The clinical reality and the promise. Biosens. Bioelectron. 20: 1897–1902. [DOI] [PubMed] [Google Scholar]
- Quiocho F.A. and Ledvina, P.S. 1996. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: Variation of common themes. Mol. Microbiol. 20: 17–25. [DOI] [PubMed] [Google Scholar]
- Salins L.L., Ware, R.A., Ensor, C.M., and Daunert, S. 2001. A novel reagentless sensing system for measuring glucose based on the galactose/glucose-binding protein. Anal. Biochem. 294: 19–26. [DOI] [PubMed] [Google Scholar]
- Schellman J. 1975. Macromolecular binding. Biopolymers 14: 999–1018. [Google Scholar]
- Schellman J.A. 1987. The thermodynamic stability of proteins. Annu. Rev. Biophys. Biophys. Chem. 16: 115–137. [DOI] [PubMed] [Google Scholar]
- Scognamiglio V., Staiano, M., Rossi, M., and D'Auria, S. 2004. Protein-based biosensors for diabetic patients. J. Fluoresc. 14: 491–498. [DOI] [PubMed] [Google Scholar]
- Simpson H.D., Haufler, U.R., and Daniel, R.M. 1991. An extremely thermostable xylanase from the thermophilic eubacterium Thermotoga . Biochem. J. 277: 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.J., Conrad, D.W., Cuneo, M.J., and Hellinga, H.W. 2005. Orthogonal site-specific protein modification by engineering reversible thiol protection mechanisms. Protein Sci. 14: 64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa L., Gryczynski, I., Eichhorn, L.R., Dattelbaum, J.D., Castellano, F.N., Rao, G., and Lakowicz, J.R. 1999. Glucose sensor for low-cost lifetime-based sensing using a genetically engineered protein. Anal. Biochem. 267: 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas M.N., Vyas, N.K., and Quiocho, F.A. 1994. Crystallographic analysis of the epimeric and anomeric specificity of the periplasmic transport/chemosensory protein receptor for D-glucose and D-galactose. Biochemistry 33: 4762–4768. [DOI] [PubMed] [Google Scholar]
- Warren R.E. and Frier, B.M. 2005. Hypoglycaemia and cognitive function. Diabetes Obes. Metab. 7: 493–503. [DOI] [PubMed] [Google Scholar]
- Xu Y., He, Z., and King, G.L. 2005. Introduction of hyperglycemia and dyslipidemia in the pathogenesis of diabetic vascular complications. Curr. Diab. Rep. 5: 91–97. [DOI] [PubMed] [Google Scholar]
- Yamazaki T., Kojima, K., and Sode, K. 2000. Extended-range glucose sensor employing engineered glucose dehydrogenases. Anal. Chem. 72: 4689–4693. [DOI] [PubMed] [Google Scholar]
- Ye K. and Schultz, J.S. 2003. Genetic engineering of an allosterically based glucose indicator protein for continuous glucose monitoring by fluorescence resonance energy transfer. Anal. Chem. 75: 3451–3459. [DOI] [PubMed] [Google Scholar]