

Abstract
CD8+ cytotoxic T lymphocytes (CTLs) play an important role in containment of virus replication in primary human immunodeficiency virus (HIV) infection. HIV's ability to mutate to escape from CTL pressure is increasingly recognized; but comprehensive studies of escape from the CD8 T cell response in primary HIV infection are currently lacking. Here, we have fully characterized the primary CTL response to autologous virus Env, Gag, and Tat proteins in three patients, and investigated the extent, kinetics, and mechanisms of viral escape from epitope-specific components of the response. In all three individuals, we observed variation beginning within weeks of infection at epitope-containing sites in the viral quasispecies, which conferred escape by mechanisms including altered peptide presentation/recognition and altered antigen processing. The number of epitope-containing regions exhibiting evidence of early CTL escape ranged from 1 out of 21 in a subject who controlled viral replication effectively to 5 out of 7 in a subject who did not. Evaluation of the extent and kinetics of HIV-1 escape from >40 different epitope-specific CD8 T cell responses enabled analysis of factors determining escape and suggested that escape is restricted by costs to intrinsic viral fitness and by broad, codominant distribution of CTL-mediated pressure on viral replication.
Keywords: cellular immunity, antigenic variation, HIV-1, viremia, biological models
Introduction
HIV-1–specific CD8+ CTL responses are induced in primary infection (1, 2) and multiple lines of evidence suggest that they make an important contribution to control of acute and early viral replication (for review see reference 3). Prophylactic and therapeutic vaccination strategies are being designed to target this arm of the immune response; however, there are several factors that may limit their success (4). Among these is viral sequence variation leading to immune escape (5, 6). A better understanding of the mechanisms by which HIV-1 evades control by the primary CTL response may thus help to facilitate rational vaccine design.
Both we and others have demonstrated selection for viral variants able to escape control by epitope-specific CTL responses during acute and early HIV-1 infection (7, 8); however, problems associated with studying T cell responses in natural acute HIV-1 infection, including limited availability of suitable samples and the sequence variability among infecting virus strains, have made it difficult to generalize these findings, leaving many important questions unanswered. CTL escape has been shown to be a hallmark of acute/early simian immunodeficiency virus infection (9). Is it also typical of acute HIV infection? To what extent and with what kinetics are epitope-specific responses evaded, and what are the factors that control this? And how do the primary CTL response and associated viral escape relate to the persisting viral load established?
To begin to address these questions, we analyzed the epitope specificity, breadth, and pattern of immunodominance within the primary CTL response to autologous virus Env, Gag, and Tat proteins in addition to the extent, kinetics, and pathways of virus escape during acute and early HIV-1 infection in three patients representative of groups establishing persisting viral loads in the highest and lowest ranges (10). In all three patients, we observed very rapid escape from components of the primary HIV-specific CD8 T cell response, demonstrating the potency of CTL pressure on viral replication. However, there were marked differences in the subsequent extent and kinetics of escape in these individuals. By analyzing features of epitope-specific CD8 T cell responses escaped to differing extents/with differing kinetics, we were able to gain insight into the interacting factors that determine CTL escape, providing evidence that escape is restricted by viral fitness constraints and is promoted when a response makes a particularly dominant contribution to control of viral replication (due to its magnitude and/or efficacy). These results have implications for HIV vaccine design, supporting the importance of restriction of viral escape from CD8 T cell control and suggesting means by which this may be achieved.
Materials and Methods
Patient Samples.
All patients presented to hospital emergency rooms affiliated with the University of Alabama at Birmingham and were studied, with written informed consent, according to protocols approved by the University's Institutional Review Board. The clinical profiles and viral loads of the three study subjects have been reported previously: WEAU and BORI are patients 1 and 3, respectively (11), and SUMA is patient 4 (12); additional viral load data has been described previously (13). Peripheral blood samples were collected in acid citrate dextrose. PBMCs were isolated and cryopreserved as described previously (1); plasma was also cryopreserved.
Recombinant Vaccinia Viruses (rVVs).
rVV vSC8, which expresses only Escherichia coli β-galactosidase (β-gal) was obtained from B. Moss (National Institutes of Health, Bethesda, MD). rVVs coexpressing β-gal and full-length gp160 or Gag proteins or sections thereof derived from the autologous HIV-1 in patient BORI at 9 days following onset of symptoms (DFOSx), and viruses expressing the Tat protein from patient SUMA's autologous virus at 8 or 69 DFOSx were produced by homologous recombination into the thymidine kinase gene of VV strain WR as described previously (7). Protein expression was confirmed by Western blotting.
Peptides.
Synthetic peptides were purchased as Pepsets produced in a peptide–amino acid format (Mimotopes) or were synthesized by the protein chemistry unit (Institute for Animal Health) using Fmoc or TBoc chemistry.
IFN-γ ELISPOT Assays.
IFN-γ ELISPOT assays were performed as described previously (14). In brief, MultiScreen plates (MAHA S45; Millipore) were coated overnight with anti–IFN-γ capture antibody 1-D1K (5 μg/ml; Mabtech), washed four times, and blocked. Patient PBMCs were added at 105–2 × 105 cells/well and incubated for 18 h with peptides (at 10−5 M) or 10 μg/ml phytohaemagglutinin (Sigma-Aldrich) as a positive control. Duplicate wells in which cells were incubated with medium only were included on each plate as a negative control. Plates were washed again, and IFN-γ spots were detected using 1 μg/ml biotin-conjugated anti–IFN-γ mAb clone 7-B6-1 (Mabtech), 1 μg/ml anti-biotin–ALP (Vector Laboratories), and a chromogenic alkaline phosphatase substrate (Bio-Rad Laboratories). Spots were enumerated using an AID automated image analysis system with AID ELISPOT version 2.5 software (Autoimmun Diagnostika GmbH). Results are expressed as mean (of duplicate or triplicate wells) spot-forming cells per 106 PBMCs. In epitope mapping experiments, responses were considered positive if they exceeded the background counts (spots formed after stimulation of PBMCs with medium alone) by >50 spot-forming cells/106 PBMCs. All positive responses were confirmed in a minimum of three independent experiments.
51Cr Release Assays.
51Cr release assays were performed as described previously (7). Target cells were autologous or allogeneic EBV-B-LCL, either infected ∼18 h before the assay with rVVs (at a multiplicity of infection of 10 PFU/cell) or left uninfected and pulsed with synthetic peptides during the assay. rVV infection of target cells was confirmed by fluorescein di-β-d-galactopyranoside (FDG; BDH Laboratory Supplies) staining for β-gal expression as described previously (15). Target cells were used at 1.5 × 104 cells/well. Effector cells were either polyclonal CTL, produced by culturing patient PBMCs for 10 d with 10 U/ml IL-2 (Glaxo SmithKline) and anti-CD3 antibody (produced from hybridoma OKT3) or short-term CTL lines generated by limiting dilution culture. These were used at effector:target ratios of at least 50:1 for polyclonal CTLs or 5:1 for lines. All variables were assayed in triplicate. Results are expressed as the percent specific 51Cr release, calculated (as mean test counts − mean spontaneous counts/mean maximum counts − mean spontaneous counts) × 100.
Viral Sequencing.
Gp160, Gag, and Tat genes were amplified by nested PCR from plasma HIV-1 RNA and sequenced. In brief, HIV-1 RNA was isolated from virions in plasma using a QIAmp Viral RNA Mini Kit (QIAGEN), and cDNA was prepared from replicate plasma virus RNA preparations (4,000–8,000 RNA molecules/reaction) using SuperScript II (Invitrogen). Replicate cDNA samples (1, 10, 100, or 1,000 molecules each) were subjected to nested PCR amplification as described previously (7), using primer sequences listed in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20040511/DC1). The PCR products were cloned into pCR-XL-TOPO (Invitrogen), and double-strand sequence analysis was performed using an ABI 373A Sequenator and the Taq Dye Primer Cycle Sequencing Kit (ABI). The sequences were analyzed using Sequencher (Gene Codes Corp) and Microgenie (Beckman Coulter) software packages.
Online Supplemental Material
Fig. S1 shows computer-generated models of index HIV-1 epitope peptides and variants thereof bound to their restricting HLA class I molecules. Table S1 depicts primers used for nested PCR amplification of HIV-1 genes from plasma viral RNA. Table S2 shows the number of CD8 T cells producing IFN-γ in response to stimulation with epitope peptides at time points in acute, subacute, and early infection in patients BORI, WEAU, and SUMA. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20040511/DC1.
Results
Characterization of the Primary CD8+ T Cell Response to Autologous Virus Env, Gag, and Tat Proteins.
Subjects WEAU, BORI, and SUMA each presented with acute symptomatic HIV-1 infection, with high titres of circulating virus and undetectable HIV-1 antibodies (11, 12). None of these patients received early antiretroviral therapy. After clinical resolution of primary HIV infection, WEAU and BORI established high persisting viral loads (90,109 HIV-1 RNA copies/ml at 212 d after onset of symptoms [DFOSx] and 186,305 HIV-1 RNA copies/ml at 218 DFOSx, respectively) and both experienced marked declines in peripheral blood CD4 cells within 2 yr of infection (to 30 and 393 cells/mm3, respectively). Conversely, SUMA established a very low persisting viral load (2,268 HIV-1 RNA copies/ml at 278 DFOSx) and maintained a high CD4 count (722 cells/mm3) throughout 7 yr of follow-up (13).
Initial analysis of the primary CTL response in these subjects indicated that responses to Env, Gag, and/or Tat were particularly immunodominant (reference 1 and unpublished data); thus, we focused our analysis on these proteins. We began by mapping the epitopes recognized by CD8+ T cells derived from each patient during the first 3 mo of infection. To avoid missing T cell responses due to sequence differences between the patients' autologous virus proteins and heterologous reagents, epitope mapping was performed using reagents corresponding to the earliest available sequence of each patient's autologous virus (15, 9, and 4 DFOSx in WEAU, BORI, and SUMA, respectively). In WEAU and SUMA, epitopes were located by screening overlapping synthetic peptides corresponding to their autologous viral gp160, Gag (p24/p17), and Tat sequences, whereas in BORI, epitope-containing regions in gp160 and Gag were initially located by testing the recognition of rVVs expressing sections of the autologous virus gp160 and Gag sequences followed by more precise epitope mapping within these regions and also in Tat using synthetic peptides.
The epitopes to which responses were identified in each patient, together with their HLA restriction where characterized, are shown in Table I. Responses to between 8 and 24 epitopes were identified in each patient, emphasizing the potential breadth of the virus-specific CD8+ T cell response in primary HIV-1 infection (16). The HIV-specific CD8 response in patient SUMA was directed against a minimum of 24 epitopes in gp160, Gag, and Tat alone, with limited analysis of the HLA restriction of these responses revealing usage of all four HLA-A and -B alleles, and at least one HLA-C allele. Notably, the epitope breadth of the CD8+ T cell response to gp160, Gag, and Tat in SUMA was almost twice that in WEAU, and more than twice that identified in BORI. Furthermore, individual epitope-specific responses in patient SUMA also used a broad array of T cell clones (unpublished data).
Table I.
Epitopes/Epitope-containing Regions Identified as Being Recognized by the CD8+ T Cell Response during Acute/Early HIV-1 Infection in Subjects BORI, WEAU, and SUMA
| Patient | Viral protein | Epitope/regiona | Designation | MHC restrictionb |
|---|---|---|---|---|
| BORI | gp160 | 207-S FEPIPIH Y-215 | gp160 SY9 | A*2902 |
| gp160 | 237-CK NVSTVQC-245 | gp160 CC9 | C*0802 | |
| gp160 | 589-ERYLKDQQL-597 | gp160 EL9 | B*1402 | |
| Gag | 119-AA D TGN SS Q-127 | Gag AQ9 | ND | |
| Gag | 124-N SS Q VSQNYP-133 | Gag NP10 | ND | |
| Gag | 295-DRFYKTLRAEQ-305 | Gag DQ11 | B*1402 | |
| Gag | 138–156 | Gag 138–156 | ND | |
| Tat | 24-NCYCKKCCY-32 | Tat NY9 | A*2902 | |
| WEAU | gp160 | 30-A E N LWVTVY-38 | gp160 AY9 | B*4403 |
| gp160 | 214-S FEPIPIH Y-222 | gp160 SY9 | A*2902 | |
| gp160 | 313-TL G P G R V L Y-321 | gp160 TY9 | ND | |
| gp160 | 348–366 | gp160 348–366 | ND | |
| gp160 | 828-EGTDR IV I EI-836 | gp160 EI9 | ND | |
| gp160 | 837-V QR TC RA I L-845 | gp160 VL9 | ND | |
| Gag | 71–90 | Gag 71–90 | ND | |
| Gag | 99-EV KDTKEAL-107 | Gag EVL9 | B*0801 | |
| Gag | 153-NAWVKIEEK-162 | Gag NK9 | ND | |
| Gag | 175-LSEGATPQDL-184 | Gag LL10 | B*4403 | |
| Gag | 231–270 | Gag 231–270 | ND | |
| Gag | 380–410 | Gag 380–410 | ND | |
| Tat | 24-NCYCKRCCF-32 | Tat NF9 | A*2902 | |
| Tat | 80–101 | Tat 80–101 | ND | |
| SUMA | gp160 | 29-EN L WVTVYY-37 | gp160 EY9 | ND |
| gp160 | 160–180 | gp160 160–180 | ND | |
| gp160 | 374-NCGGEFFYCN-383 | gp160 NN10 | ND | |
| gp160 | 380-FYCN T T QLF-389 | gp160 FF9 | A*2402 | |
| gp160 | 390–401 | gp160 390–401 | ND | |
| gp160 | 402–413 | gp160 402–413 | ND | |
| gp160 | 410-425 | gp160 410–425 | ND | |
| gp160 | 426-441 | gp160 426–441 | ND | |
| gp160 | 439-I RCSSNITGL-448 | gp160 IL10 | ND | |
| gp160 | 485-YKVV KI EPL-493 | gp160 YL9 | ND | |
| gp160 | 583-ERYLKDQQL-591 | gp160 EL9 | B*1402 | |
| Gag | 5-ASVLSG G EL-13 | Gag AL9 | ND | |
| Gag | 83-TLYCVHQ K I-91 | Gag TI9 | A*1103 | |
| Gag | 260-EIYKRWIIL-268 | Gag EIL9 | A*2402 | |
| Gag | 269-GLNKIVRMY-277 | Gag GY9 | B*1501 | |
| Gag | 295-DYVDRFYKT-303 | Gag DT9 | A*2402 | |
| Gag | 305-RAEQASQEV-313 | Gag RV9 | C*0802 | |
| Gag | 335-KALGPAATL-343 | Gag KL9 | ND | |
| Gag | 349-ACQGVGGPGHK-359 | Gag AK11 | A*1103 | |
| Gag | 410-440 | Gag 410–440 | ND | |
| Tat | 12-KHPGSQPK T A-21 | Tat KA10 | ND | |
| Tat | 32-FHCQ VCFMT K-41 | Tat FK10 | ND | |
| Tat | 36-VCFMT K GLGI-45 | Tat VI10 | B*1501 | |
| Tat | 39-MT K GLGISY-47 | Tat MY9 | B*1501 |
The NH2- and COOH-terminal amino acid numbers of each epitope/epitope-containing region within the subject's autologous virus Env, Gag, or Tat protein sequence are shown. Where the optimal epitope sequence was determined, the amino acid sequence of the epitope (as present in the subject's autologous virus at the earliest time point analyzed) is also given. The residues within each optimal epitope sequence are coded to indicate the extent of amino acid variability at that position amongst the HIV-1 clade B sequences listed in the HIV sequence database (reference 21). Residues shown in regular text were invariant amongst all sequences compared; at bold positions, there was very low variability, with ≥95% of sequences sharing a common amino acid; at italicized positions, there was more variability, with <95 but >79% of sequences sharing a common amino acid; and at underlined residues, there was greatest variability, with ≤79% of sequences sharing a common amino acid.
The HLA class I allele presenting each epitope is shown, where known. In some cases, the restriction of the epitope-specific response was determined experimentally, whereas in others the presenting HLA allele was deduced based on previous reports of the HLA restriction of the epitope concerned (reference 46).
Next, we compared the relative magnitude of the T cell response to the different epitopes each patient recognized during acute (HIV-1 ELISA and immunoblot negative), subacute (antibody indeterminant), and early (antibody positive) infection. This analysis (Fig. 1 and Table S2) revealed marked differences in the relative immunodominance of epitope-specific responses within the primary HIV-specific CD8+ T cell response in the three individuals. Most notably, in the two patients who subsequently established high persisting HIV-1 loads, the virus-specific CD8+ T cell response during acute HIV-1 infection was highly biased toward one (WEAU) or several (BORI) viral epitopes. Although the response in these patients evolved after control of the acute viral burst to become somewhat less heavily weighted toward the initially immunodominant epitopes, it still remained very unevenly biased: in WEAU, >90% of the T cell response analyzed was directed against just five viral epitopes in early infection, and there was a similar picture in BORI. In contrast, although there was also a hierarchy of epitope immunodominance in SUMA's primary HIV-specific CD8+ T cell response, with responses to two epitopes (gp160 EL9 and Tat MY9) being particularly prominent during acute infection, the CTL response rapidly became more evenly balanced, so that in early infection, only ∼20% of the response was directed toward these epitopes and ∼80% was spread over the remaining epitopes.
Figure 1.

Relative immunodominance and functional avidity of T cell responses to different viral epitopes during acute, subacute, and early HIV-1 infection in subjects BORI, WEAU, and SUMA. The T cell response to each of the gp160, Gag, and Tat epitopes recognized by subjects BORI, WEAU, and SUMA was measured at time points during acute HIV-1 infection (9–16 DFOSx and before detection of HIV-1 antibodies by ELISA or Western immunoblot), subacute infection (3–4 wk FOSx and before full immunoblot seroconversion), and early infection (between 5 and 10 wk FOSx) using IFN-γ ELISPOT assays. The relative immunodominance of the responses to different epitope peptides at each time point is shown, calculated by expressing the number of epitope–peptide-specific IFN-γ–producing cells as a percentage of the overall response detected to all the peptides tested at that time point. The magnitude of individual epitope-specific responses is shown in Table S2. The functional avidity of patient T cell responses to each of the epitopes they recognized was also compared by assessing the ability of polyclonal patient CTL or epitope-specific CTL lines to mediate lysis of autologous EBV-B-LCL target cells pulsed with different concentrations of the epitope peptide in 51Cr release assays. The values shown represent the peptide concentration required to sensitize target cells for half-maximal CTL lysis.
The avidity of the T cell response to certain of the epitopes recognized in each patient was also evaluated (Fig. 1). Analysis of the relationship between the avidity and immunodominance of T cell responses to different epitopes during acute and early infection revealed that the most immunodominant responses in all three patients were frequently of moderate-to-high avidity (≥10−7/8 M), although there was not a significant correlation between avidity and immunodominance.
Analysis of Selection for Sequence Changes within CTL Epitope-containing Regions.
Next, we investigated the extent, kinetics, and mechanisms of viral escape from each of the epitope-specific CD8+ T cell responses identified in the three study subjects. Direct population sequencing of gag, env, and tat genes from uncultured plasma virus RNA (cDNA) from serial time points was performed, as described previously (7, 17). The same genes were molecularly cloned from plasma viral RNA (cDNA) and individual clones were sequenced. Both approaches were used to identify changes in the plasma virus quasispecies of the three subjects over the course of 556–772 d of follow-up. Particular attention was given to sequences encompassing the epitopes shown to be recognized by each patient's CTL response. In all, 298 complete genes and 1,044 subgenomic fragments, together totalling 840,000 nucleotides, were analyzed (GenBank/EMBL/DDBJ accession nos. AY281863-AY282367, AY223719-AY223754, AY223761-AY223790, and U21135). Changes that emerged within stretches of sequence containing one or more CTL epitopes (epitope-containing regions) are shown in Fig. 2. Complete replacement of the initial (highly homogeneous) virus population with viruses bearing nonsynonymous sequence changes within one or more epitope-containing regions was found to occur with very rapid kinetics in all three patients (Fig. 2). In BORI and WEAU, there was rapid selection for amino acid changes in multiple epitope-containing regions (6 out of 7 in BORI and 4 out of 14 in WEAU) within the first year of infection. Conversely, of the 24 epitopes recognized by SUMA, there were changes during the same time frame in only a single region of Tat that was targeted by CTL responses against three overlapping CTL epitopes. Only much later, at 736 DFOSx, were changes detected in a fourth epitope in the env gene.
Figure 2.

Summary of sequence changes detected over time in each subject's plasma virus quasispecies within regions recognized by the CD8+ T cell response. Sequence changes occurring within epitope-containing regions in the in vivo viral quasispecies over time in (a) BORI, (b) WEAU, and (c) SUMA are shown. A panel of molecular clones spanning each epitope-containing region was derived from plasma viral RNA (cDNA) at serial time points in infection (DFOSx), and the sequence of independent clones was determined. Each panel shows the deduced amino acid sequence of clones from a single epitope/epitope-containing region. Where overlapping CD8 T cell epitopes were identified within one region, their relative location is indicated. For each epitope/epitope-containing region, the deduced amino acid sequence of the predominant viral species present at the earliest time point tested (the index sequence) is shown in full. Any variant sequences present at this time point, plus all sequences determined at subsequent time points were compared with the index sequence; variant amino acids are indicated, with a dash (-) denoting amino acid identity. The slash (/) denotes an amino acid deletion, and insertions are indicated by vertical pipes. At each time point, the proportion of clones analyzed that had a given sequence is shown, and the total percentage of clones analyzed with variant sequences is also indicated. Sequence analysis of the WEAU gp160 AY9 epitope at time points from D16 to D136 was reported previously (reference 7).
Effects of Amino Acid Changes Selected In Vivo on Epitope Recognition or Processing.
Next, we examined the phenotypic effects of these changes on virus susceptibility to CTL recognition. First, the ability of synthetic peptides corresponding to the index and variant sequences of each variable epitope to sensitize autologous target cells for lysis by polyclonal patient CTLs was evaluated. In the vast majority of instances where the frequency of epitope-specific CTLs was high enough to permit this analysis, the amino acid changes that were selected for within epitope sequences were found to reduce the ability of the epitope peptide to sensitize target cells for CTL lysis by at least 10-fold, and in some cases to undetectable levels (Fig. 3). We did not investigate directly whether this was due to impairment of peptide binding to MHC and/or to alteration of recognition of the peptide–MHC complex by T cells after binding to MHC. However, computer-generated peptide–MHC models of the index and predominant mutant forms of epitope peptides whose HLA restriction was known (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20040511/DC1) suggested that in the majority of cases (7/8 examples analyzed), the amino acids that were altered did not form part of the TCR contact surface. Conversely, alterations in MHC anchor residues (e.g., positions 2, 3, and/or 9 of the HLA-A29–restricted epitope SFEPIPIHY; position 5 of the HLA-B14–restricted epitope ERYLKDQQL; and position 2 of the HLA-B44–restricted epitope AENLWVTVY) were relatively common.
Figure 3.
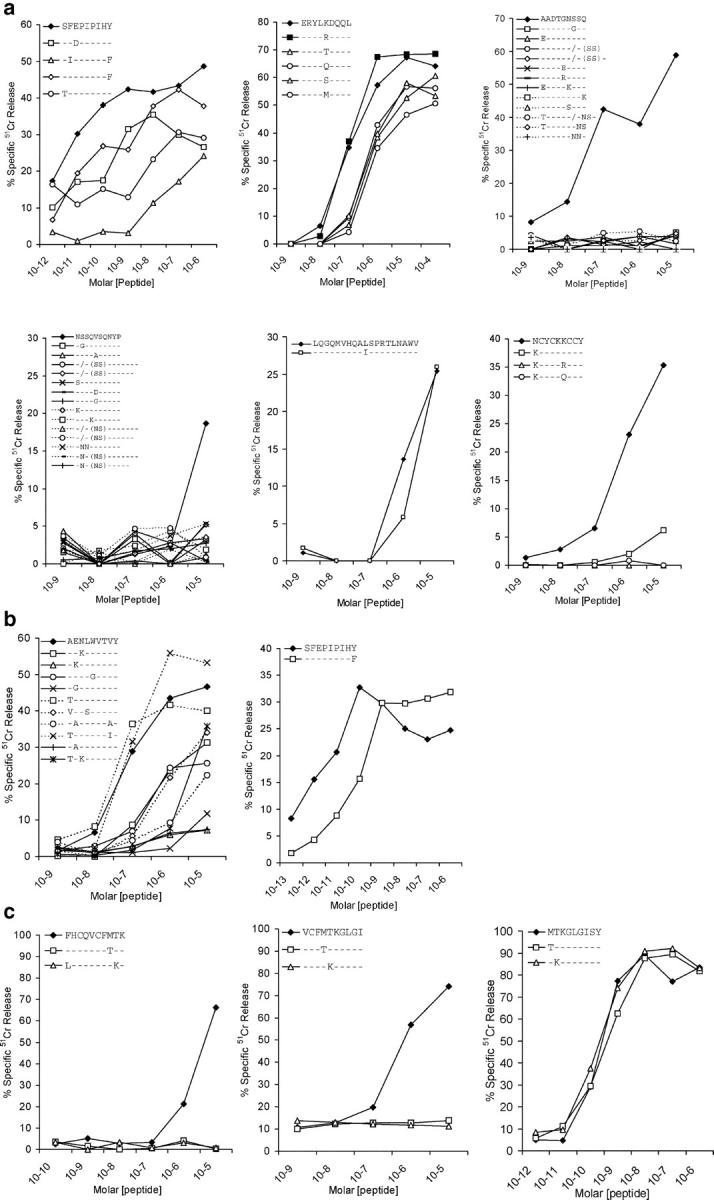
Effect of the amino acid changes selected for in each subject's in vivo viral quasispecies on epitope-specific CD8+ CTL responses. The relative ability of synthetic peptides corresponding to the indicated index and variant epitope sequences to sensitize autologous target cell lines for lysis by polyclonal CTLs derived from subjects BORI (a), WEAU (b), and SUMA (c) early after infection was compared using in vitro cytotoxicity assays. The results shown are the specific lysis (% specific 51Cr release) of target cells incubated with the indicated concentrations of each peptide.
Although in the majority of cases tested, the amino acid changes selected for within epitope sequences reduced the ability of the epitope peptide to sensitize target cells for lysis in vitro, one of the exceptions to this was the position 2 threonine (T) to lysine (K) change in the Tat MY9 epitope in patient SUMA (Fig. 3 c). The region of Tat where this epitope was located also contained two other overlapping CTL epitopes (Fig. 2 c), whose recognition was ablated by the amino acid changes selected for in this region. Although the T cell response to the latter epitopes may have driven the emergence of these amino acid changes, this seemed unlikely given that these responses were not at all dominant in the patient's primary CD8+ T cell response and were of very low avidity (Fig. 1). This suggested that the amino acid changes that emerged in this region of Tat may have been selected for because they conferred escape from the Tat MY9 response by affecting the processing of this epitope. To investigate this possibility, we generated two rVVs encoding the index (D8) and variant (D69) patient virus Tat sequences (which differed only in the F to L and T to K substitutions at positions 32 and 40), and compared their ability to sensitize autologous target cells for lysis by a CTL line specific for the Tat MY9 epitope. Both viruses expressed similar levels of Tat (Fig. 4 a) and infected target cells with high efficiency (Fig. 4 b); but only the virus expressing the index Tat sequence sensitized target cells for lysis by Tat MY9-specific CTLs (Fig. 4 c). Together with the observation that the mutant Tat MY9 epitope peptide was able to sensitize target cells for CTL lysis just as efficiently as the index peptide (Fig. 3 c), this result suggested that the amino acid changes at Tat residues 32 and/or 40 in the D69 Tat sequence prevented processing of the Tat MY9 epitope for presentation with HLA-B15. Other instances of HIV-1 escape from epitope-specific CD8 responses via acquisition of mutations affecting antigen processing have been reported recently (18).
Figure 4.

Effect of amino acid changes selected for in SUMA's Tat sequence on processing of the Tat MY9 epitope. (a) Confirmation of Tat expression in cells infected with rVVs encoding Tat proteins cloned from the plasma viral population present in SUMA 8 (vSUMAtat-D8) or 69 (vSUMAtat-D69) DFOSx. Western blotting was performed on lysates from cells infected 48 h previously with vSC8 (a control rVV expressing β-gal only), vSUMAtat-D8, or vSUMAtat-D69, using the HIV-1 Tat-specific monoclonal antibody EVA3021. The positions of molecular weight markers (daltons) are shown, and the Tat band is indicated by an arrow. (b) Confirmation of efficient infection of target cells by vSC8, vSUMAtat-D8, and vSUMAtat-D69. EBV-B-LCL target cells were infected at an multiplicity of infection of 10 with the indicated rVVs (all of which expressed the marker protein β-gal), and the proportion of infected cells was assessed 18 h later by staining with FDG, a fluorescent substrate for β-gal. Unshaded histograms represent FDG staining of uninfected cells, and shaded histograms represent staining of infected cells. (c) Analysis of the ability of a Tat MY9-specific CTL line derived from subject SUMA early after infection to mediate lysis (% specific 51Cr release) of autologous (auto) and allogeneic (allo) EBV-B-LCL target cells infected with vSC8, vSUMAtat-D8, or vSUMAtat-D69, or pulsed with the MY9 epitope peptide at 10−5 M.
A notable example of selection for amino acid change in an epitope-containing region to which a subdominant CTL response was mounted was at gp160 348–366 in WEAU. Sequence analysis of this region (Fig. 2) suggested selection pressure beginning as early as day 44, with fixation of a lysine (K) to glutamic acid (E) substitution by day 136. CTL reactivity to this region was too low to enable the effects of this mutation on the CTL response to be investigated, but in other studies involving the same subject, we found that this K to E substitution contributed to virus escape from autologous neutralizing antibodies (NAbs; reference 19).
Altogether, six out of eight epitopes in BORI had changes consistent with CTL escape, of which changes in five (gp160 SY9, gp160 EL9, Gag AQ9, Gag NP10, and Tat NY9) were confirmed to affect CTL recognition (Fig. 3 a), with a sixth not tested. In WEAU, changes in 4 out of 14 epitopes suggested possible CTL escape with 2 (gp160 AY9 and gp160 SY9) confirmed as such (Fig. 3 b). A third (Tat NF9) likely also represented escape by analogy with escape at the same Tat epitope in BORI (Fig. 3 a). In SUMA, changes in 4 out of 24 epitopes, 3 of which were overlapping, all suggested escape. Three were confirmed as such (Figs. 3 c and 4), and the fourth (gp160 EL9) was suggested by analogy with escape at the same gp160 epitope in BORI.
Features of Epitope-specific Responses Associated with Selection for Escape Mutations.
In total, 14 out of the 46 CTL responses studied here were associated with evolution of viral sequence changes suggesting possible CTL escape. Escape from responses directed against certain epitopes may be restricted by the costs of mutation to viral fitness (see next paragraph). Nonetheless, comparison of features of responses that were differentially escaped suggested that both quantitative and qualitative factors play a role in determining the pressure exerted by individual epitope-specific CTL responses on in vivo viral replication. The responses that were of highest magnitude during acute infection in each patient were among those that were escaped; but there was also evidence of rapid escape from relatively subdominant responses. Although many immunodominant responses were of high functional avidity (Fig. 1), there was not strong evidence to support functional avidity being an important independent determinant of CTL escape. However, the protein specificity of responses did seem to play a role, as 5/7 (71%) responses to epitopes in Tat, a protein expressed early in the viral lifecycle, were escaped, whereas only 10/39 (26%) responses directed against gp160 and Gag were escaped in a similar time frame, a significantly lower proportion (P = 0.026 as determined using Fisher's exact test). A recent in vitro paper lends further support to specificity being more important than avidity in determining the antiviral efficacy of CTL (20).
Constraints on Viral Sequence Variability Restrict CTL Escape.
To investigate the impact of sequence constraints imposed by the need to maintain viral replicative functions, we compared the extent and kinetics of escape from CTL responses directed against epitopes located in conserved and more variable viral sequences. In Table I, the epitope sequences are coded to reflect the degree of conservation of each amino acid among clade B HIV-1 sequences (21). Some of the most highly conserved epitope sequences were those in Gag p24, including the HLA-B14–restricted Gag DQ11 epitope recognized by patient BORI, where constraints on immune escape due to the essential role of invariant amino acids have been reported previously (22). No escape from responses to these highly conserved Gag p24 epitopes was observed. Certain gp160 epitopes were also relatively conserved, such as the HLA-A29–restricted gp160 SY9 epitope recognized by BORI and WEAU and the B14-restricted gp160 EL9 epitope recognized by BORI and SUMA. There was slow selection for amino acid changes that conferred some resistance to recognition by the T cell response directed against these epitopes; but these changes did not ablate epitope recognition altogether (Fig. 3), likely reflecting constraints on the positions within the epitope at which even conservative amino acid changes could be tolerated (e.g., possibly just at position 5 in the gp160 EL9 epitope). In contrast, other epitopes, such as the overlapping Gag p17 epitopes (Gag AQ9 and Gag NP10) recognized by patient BORI, were located in more variable sequences where rapid acquisition of amino acid changes that conferred a high degree of CTL escape could likely be accommodated more readily.
Comparison of the Overall Extent and Kinetics of Escape from the Primary HIV-specific CTL Response in Patients BORI, WEAU, and SUMA.
The overall extent and kinetics of virus escape from epitope-specific CTL responses in each subject are illustrated in Fig. 5. In every case, there was evidence for complete or near-complete fixation of escape mutants in at least one CTL epitope within 65–72 DFOSx. However, in BORI and WEAU, there was evidence of sequential fixation of multiple escape mutations within the first ∼1 yr of infection (Fig. 5, a and b). Conversely, there was much more limited CTL escape in SUMA during the same time frame (Fig. 5 c). To confirm the validity of the latter observation (and exclude the possibility that there was extensive escape from responses directed against epitopes in other viral proteins during early infection in patient SUMA), we have recently extended similar analysis to SUMA's primary CD8 T cell response against the entire autologous virus proteome. This revealed that there was no selection for escape-conferring amino acid changes within or around any other epitopes in the virus throughout the first year of infection (unpublished data).
Figure 5.

Extent and kinetics of escape-conferring mutation in epitope-containing regions in Env, Gag, and Tat in subjects BORI, WEAU, and SUMA. Panels a and e; b; and c illustrate (for subjects BORI, WEAU, and SUMA, respectively) the extent and kinetics of accumulation of mutations (with the exception of those shown not to confer CTL escape in Fig. 3) within epitope-containing regions in gp160, Gag, and Tat (% mutant) at time points analyzed during the first 400 DFOSx. In BORI, the five mutating regions shown constituted five out of seven epitope-containing regions identified in this patient; in WEAU, the 4 mutating epitope-containing regions constituted 4 of 14 regions to which responses were identified, and in SUMA the single mutating epitope-containing region (which contained the Tat FK10/VI10/MY9 epitopes) constituted 1 of 21 regions to which responses were identified. Panels d and f show the relative immunodominance (calculated as described in the legend to Fig. 1) of the response to the gp160 EL9 epitope in subjects BORI and SUMA respectively. Panels e and g illustrate the kinetics of accumulation of escape-conferring mutations (% mutant) in this epitope within the viral quasispecies in subjects BORI and SUMA, respectively.
The limited early viral mutational escape in patient SUMA ensued in the context of a CD8 T cell response that was broad and relatively evenly directed. In contrast, in patients BORI and WEAU, the primary CTL response was more heavily biased against a limited number of viral epitopes. This suggests the importance of broad, even distribution of CD8 T cell pressure in restricting viral escape, a hypothesis further supported by observation that the response to an epitope (gp160 EL9) that was recognized by both BORI and SUMA was escaped more rapidly in the former patient, where it formed part of a relatively unbalanced CTL response, than in the latter patient where it formed part of a broader, more evenly directed response (Fig. 5, d–g).
Discussion
Understanding the dynamic virus–immune system interactions that take place during acute HIV-1 infection and how they may impact on the persisting viral load is important but technically challenging. In this paper, we characterized the primary CD8+ T cell response to autologous virus Env, Gag, and Tat proteins (which together compromise ∼45% of the viral proteome) and escape from this response in three HIV-1–infected individuals. We observed rapid escape from components of the primary HIV-specific CTL response in all three subjects, but found a marked difference in the subsequent extent and kinetics of escape in these individuals. By analyzing quantitative and qualitative features of CD8 T cell responses directed against >40 different epitopes and their escape from CTL control, we were able to gain insight into the interacting factors that determine CTL escape. Our results suggest that costs to intrinsic viral fitness and broad, codominant distribution of CTL-mediated pressure on viral replication restrict escape and imply that this may in turn influence the extent to which virus containment is achieved.
In all three of our study subjects, there was complete or near-complete replacement of the replicating viral quasispecies by mutants able to escape from components of the primary CTL response within 65–72 DFOSx, demonstrating the controlling pressure exerted by CD8 T cells on HIV replication during primary infection. In contrast, autologous HIV-1 NAbs were not detectable in the same subjects until 72 DFOSx or later, and NAb escape mutants were generally not found until later still (19). These and other results (1–3, 7, 23) emphasize the difference in kinetics of the CD8 T cell and NAb responses in primary HIV-1 infection, and clarify that the virus-specific CD8 T cell response plays the dominant role in containing virus replication in the earliest stages of natural infection.
From the kinetics of replacement of wild-type virus in the plasma by escape mutants, we could calculate the selective advantage that escape from epitope-specific responses conferred on the virus, and hence gain a minimum estimate of the impact of the CTL response on viral replication. Based on frequency estimates of single point mutations of 10−2–10−4 (24) (compatible with the reported HIV mutation rate of 3.4 × 10−5 mutations per base pair per single cycle of replication; reference 25), a virus generation time of ∼2 d (26), and our observation that the earliest escape-conferring mutations reached fixation in the plasma viral population within 60–80 d, we calculated (24) the relative fitness advantage of mutant viruses over wild-type virus (in the face of CTL pressure) to be 1.12–1.36. Thus, individual epitope-specific CTL responses reduced viral replication by at least 10–30%; i.e., if a cell infected with wild-type virus has the capacity to produce 100 infectious virions in its lifetime (burst size), a CTL response to only a single epitope would reduce the burst size to between 70 and 90 infectious virions. We have shown previously how modest reductions in the lifespan of productively infected cells could elicit such an effect on virus burst size, and by inference, on plasma virus load (27, 28). Notably, the CTL responses that drove complete selection for escape-conferring mutations most rapidly were directed against epitopes in Tat (the Tat NY9 response in BORI and the Tat MY9 response in SUMA, which were escaped to completion within 65/69 DFOSx, respectively). There is also other evidence for responses to Tat (and Nef and Rev) being particularly effective at controlling in vivo virus replication (9, 29, 30). These proteins are all expressed early in the viral life cycle, allowing rapid epitope presentation on infected cells and providing a longer “window of opportunity” for CTL action, which in vitro studies have shown can considerably enhance the efficiency of CTL control of viral replication (20, 31).
Although components of the primary HIV-specific CTL response were initially escaped with similar kinetics in all three subjects, there was a marked difference in the subsequent extent and kinetics of CTL escape in these individuals. In both BORI and WEAU, there was selection for mutations in multiple (four to six) epitope-containing regions within 218 DFOSx, whereas in SUMA there was acute phase escape in just a single epitope-containing region in the entire virus, and no further CTL escape until much later into infection (Fig. 5). In all three subjects, CTL escape occurred as primary virus replication was being contained. Given that the escape-conferring mutations must have conferred a fitness advantage on the virus to have been selected for in the in vivo viral quasispecies, escape variant selection must, by definition, have affected the efficiency of control of virus replication. It is notable that SUMA, whose virus underwent very limited escape from the primary CD8 T cell response, established a low set-point persisting viral load, whereas the other two patients, in whom there was much more extensive escape during the first 6 mo of infection, established high persisting viral loads. These observations are consistent with the hypothesis that the extent of acute-phase viral escape from the CD8 T cell response may be one of the factors that determines the persisting viral load established in early HIV-1 infection. In turn, the level of viral replication may impact on the kinetics of escape viral variant selection. However, the multiple observations (32) of rapid viral evolution to acquire resistance to antiretroviral agents in patients where the level of ongoing viral replication had been reduced to levels much lower than that in patient SUMA would argue against the viral load being the key factor limiting escape in this patient.
The differing levels of escape in these three patients occurred in the context of CTL responses that differed in both epitope breadth and in the relative immunodominance of responses to individual epitopes, with CTL pressure in patient SUMA being more evenly spread across a greater number of epitopes during early infection than in patients WEAU and BORI (Fig. 1). We also observed that in all three patients, every epitope-specific CTL response that had a particularly high relative immunodominance in acute infection (comprising >25% of the overall T cell response to the Env, Gag, and Tat epitopes identified in the patient concerned) was escaped within ∼6-7 mo FOSx. Subdominant responses directed against epitopes in Tat, which likely also played a prominent role in in vivo control of viral replication, were also rapidly escaped. Based on these observations, we hypothesized that escape may be promoted by biasing of CTL pressure toward particular epitopes, and restricted by broad, codominant distribution of CTL pressure; and correspondingly that qualitative features of the HIV-specific CTL response may be important determinants of the efficiency of control of viral replication (perhaps more so than the magnitude of the response).
Consistent with this hypothesis, several studies have found no relationship between the total magnitude of the HIV-specific CD8+ T cell response and the viral load at different stages of infection (16, 33, 34). However, analyses of substantial distortion of the T cell repertoire by mono/oligoclonal T cell expansions during primary HIV infection in patients who subsequently established high but not low persisting viral loads (35, 36) support there being an association between qualitative aspects of the primary HIV-specific T cell response and the efficiency of control of viral replication, and further suggest that biasing of the response toward immunodominant epitopes recognized by a limited number of T cell clones may be detrimental. Interestingly, two recent papers found no relationship between the number of epitopes recognized by the HIV-specific CD8+ T cell response and the viral load (16, 34). Neither paper compared the breadth of the primary CD8 response with the persisting viral load established in early infection; however, this observation could indicate that the relative immunodominance of epitope-specific responses (not reported in these papers) is more critical in dictating the efficiency of control of viral replication than the absolute number of epitopes recognized, or that other factors have a dominant influence on the efficiency of viral containment.
Another factor that likely has an important impact on the extent and kinetics of viral escape from epitope-specific CTL responses is the cost of escape to intrinsic viral fitness. Although we did not perform any direct comparison of the intrinsic fitness of viral isolates bearing index and mutant epitope sequences, the impact of viral fitness costs on escape was strongly suggested by comparison of the extent and kinetics of escape from CTL responses directed against epitopes in conserved versus more variable viral sequences. Several other papers (22, 37) also provide examples of high costs to intrinsic viral fitness restricting escape from CTL responses to particular epitopes. Although our results suggest that escape is less likely to occur if CTL pressure is codominantly directed against multiple viral epitopes (i.e., if many epitopes are recognized, and both the relative immunodominance and the in vivo efficacy of all responses are similar), selection for escape will also depend on the interplay of these variables with costs of escape to intrinsic viral fitness. Thus, for example, a response biased against a limited number of epitopes with very high escape-related intrinsic fitness costs may be escaped to a lesser extent than a broader response directed against epitopes where amino acid change is readily accommodated. The complexity of factors that interact to determine the extent and kinetics of acute-phase CTL escape may mean that the persisting viral load is not adequately reflected by consideration of any single parameter in isolation.
If escape from an epitope-specific CTL response is promoted as its contribution to overall control of viral replication increases, escape will be reduced not only as the breadth and codominance of epitope-specific components of the CTL response increases, but also as the contribution made by other arms of the immune response (e.g., antibody and CD4+ T cell responses) to control of viral replication increases. In support of this, studies in murine virus infection models have illustrated not only that CTL escape is enhanced by biasing of T cell pressure toward a limited number of viral epitopes (for review see reference 38), but also that immune escape is promoted in situations where one arm of the immune response makes a particularly overriding contribution to overall control of viral replication (39). Interestingly, we were able to detect a strong HIV-specific CD4+ T cell response during primary infection in patient SUMA but not in patients WEAU or BORI (40), which may have helped to increase the breadth of the effector T cell response.
Our observation of CTL escape in patients who were representative of individuals who naturally established persisting viral loads in the uppermost and lowest ranges (10) adds to a growing body of evidence suggesting that CTL escape is prevalent in HIV-1 infection (9, 41, 42). If CTL escape constitutes a common and significant means of immune evasion in early HIV-1 infection, vaccination strategies should be designed to elicit a response that will have the minimal chance of being escaped after infection. Our findings indicate that one way in which escape can be reduced is by induction of a T cell response that exerts even pressure against multiple viral epitopes (e.g., composed of multiple epitope-specific responses of similar magnitude and efficacy). Experience has already shown that efficacious vaccine-induced CD8+ T cell responses can be rapidly evaded if they are too narrowly directed (43–45). However, our results further indicate that to reduce escape it will also be important to induce CD8+ T cell responses to invariant sites in the virus, where escape is likely to be restricted by costs to intrinsic viral fitness. Thus, we predict that a CD8 T cell response-inducing vaccine is likely to achieve optimal control of virus replication by eliciting CTL responses of evenly high magnitude and efficacy to a series of viral epitopes that exhibit high escape-related costs to intrinsic viral fitness.
Acknowledgments
We are grateful to the patients who gave samples for this work, B. Moss for providing rVV vSC8, the National Institute for Biological Standards and Control Centralized Facility for AIDS reagents and the EU Program EVA for antibodies ARP-315, 323, 360, 3038, 3078, 3025 and EVA-327, 328, 334, 332, 3021; and J. Tite for recombinant IL-2. We thank M. Oldstone, H. Lewicki, and J.-E. Gairin for their input during the initial stages of this work; J. Nelson and N. Peffer for assistance with the production of rVVs encoding sections of the gp160 and Gag proteins from patient BORI's autologous virus; L. Hunt for peptide synthesis; B. Korber, M. Maini, P. Sharp, and D. Tough for helpful discussions and critical appraisal of the paper; and W. Abbott for help with preparation of figures.
This work was supported by National Institutes of Health grant nos. AI 37430 and AI 41530 and by core funding from The Edward Jenner Institute. This is manuscript no. 58 from the Edward Jenner Institute.
The authors have no conflicting financial interests.
Abbreviations used in this paper: β-gal, β-galactosidase; DFOSx, days following onset of symptoms; NAb, neutralizing antibody; rVV, recombinant vaccinia virus.
References
- 1.Borrow, P., H. Lewicki, B.H. Hahn, G.M. Shaw, and M.B. Oldstone. 1994. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J. Virol. 68:6103–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koup, R.A., J.T. Safrit, Y. Cao, C.A. Andrews, G. McLeod, W. Borkowsky, C. Farthing, and D.D. Ho. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McMichael, A.J., and S.L. Rowland-Jones. 2001. Cellular immune responses to HIV. Nature. 410:980–987. [DOI] [PubMed] [Google Scholar]
- 4.McMichael, A., and T. Hanke. 2002. The quest for an AIDS vaccine: is the CD8+ T-cell approach feasible? Nat. Rev. Immunol. 2:283–291. [DOI] [PubMed] [Google Scholar]
- 5.Barouch, D.H., and N.L. Letvin. 2002. Viral evolution and challenges in the development of HIV vaccines. Vaccine. 20:A66–A68. [DOI] [PubMed] [Google Scholar]
- 6.O'Connor, D.H., T.M. Allen, and D.I. Watkins. 2002. Cytotoxic T-lymphocyte escape monitoring in simian immunodeficiency virus vaccine challenge studies. DNA Cell Biol. 21:659–664. [DOI] [PubMed] [Google Scholar]
- 7.Borrow, P., H. Lewicki, X. Wei, M.S. Horwitz, N. Peffer, H. Meyers, J.A. Nelson, J.E. Gairin, B.H. Hahn, M.B.A. Oldstone, and G.M. Shaw. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat. Med. 3:205–211. [DOI] [PubMed] [Google Scholar]
- 8.Geels, M.J., M. Cornelissen, H. Schuitemaker, K. Anderson, D. Kwa, J. Maas, J.T. Dekker, E. Baan, F. Zorgdrager, R. van den Burg, et al. 2003. Identification of sequential viral escape mutants associated with altered T cell responses in a human immunodeficiency virus type 1-infected individual. J. Virol. 77:12430–12440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O'Connor, D.H., T.M. Allen, T.U. Vogel, P. Jing, I.P. DeSouza, E.L. Dodds, E.J. Dunphy, C. Melsaether, B. Mothe, H. Yamamoto, et al. 2002. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat. Med. 8:493–499. [DOI] [PubMed] [Google Scholar]
- 10.Mellors, J.W., C.R. Rinaldo Jr., P. Gupta, R.M. White, J.A. Todd, and L.A. Kingsley. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 272:1167–1170. [DOI] [PubMed] [Google Scholar]
- 11.Clark, S.J., M.S. Saag, W.D. Decker, S. Campbell-Hill, J.L. Roberson, P.J. Veldkamp, J.C. Kappes, B.H. Hahn, and G.M. Shaw. 1991. High titers of cytopathic virus in plasma of patients with symptomatic primary HIV-1 infection. N. Engl. J. Med. 324:954–960. [DOI] [PubMed] [Google Scholar]
- 12.Graziosi, C., G. Pantaleo, L. Butini, J.F. Demarest, M.S. Saag, G.M. Shaw, and A.S. Fauci. 1993. Kinetics of human immunodeficiency virus type 1 (HIV-1) DNA and RNA synthesis during primary HIV-1 infection. Proc. Natl. Acad. Sci. USA. 90:6405–6409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piatak, M.J., M.S. Saag, L.C. Yang, S.J. Clark, J.C. Kappes, K.-C. Luk, B.H. Hahn, G.M. Shaw, and J.D. Lifson. 1993. High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science. 259:1749–1754. [DOI] [PubMed] [Google Scholar]
- 14.Lalvani, A., R. Brookes, S. Hambleton, W.J. Britton, A.V. Hill, and A.J. McMichael. 1997. Rapid effector function in CD8+ memory T cells. J. Exp. Med. 186:859–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nolan, G.P., S. Fiering, J.F. Nicolas, and L.A. Herzenberg. 1988. Fluorescence-activated cell analysis and sorting of viable mammalian cells based on beta-D-galactosidase activity after transduction of Escherichia coli lacZ. Proc. Natl. Acad. Sci. USA. 85:2603–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Addo, M.M., X.G. Yu, A. Rathod, D.I. Cohen, R.L. Eldridge, D. Strick, M.N. Johnstone, C. Corcoran, A.G. Wurcel, C.A. Fitzpatrick, et al. 2003. Comprehensive epitope analysis of human immunodeficiency virus type 1 (HIV-1)-specific T-cell responses directed against the entire expressed HIV-1 genome demonstrate broadly directed responses, but no correlation to viral load. J. Virol. 77:2081–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei, X., S.K. Ghosh, M.E. Taylor, V.A. Johnson, E.A. Emini, P. Deutsch, J.D. Lifson, S. Bonhoeffer, M.A. Nowak, B.H. Hahn, et al. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 373:117–122. [DOI] [PubMed] [Google Scholar]
- 18.Draenert, R., S. Le Gall, K.J. Pfafferott, A.J. Leslie, P. Chetty, C. Brander, E.C. Holmes, S.C. Chang, M.E. Feeney, M.M. Addo, et al. 2004. Immune selection for altered antigen processing leads to cytotoxic T lymphocyte escape in chronic HIV-1 infection. J. Exp. Med. 199:905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei, X., J.M. Decker, S. Wang, H. Hui, J.C. Kappes, X. Wu, J.F. Salazar-Gonzalez, M.G. Salazar, J.M. Kilby, M.S. Saag, et al. 2003. Antibody neutralization and escape by HIV-1. Nature. 422:307–312. [DOI] [PubMed] [Google Scholar]
- 20.Yang, O.O., P.T.N. Sarkis, A.K. Trocha, S.A. Kalams, R.P. Johnson, and B.D. Walker. 2003. Impacts of avidity and specificity on the antiviral efficacy of HIV-1-specific CTL. J. Immunol. 171:3718–3724. [DOI] [PubMed] [Google Scholar]
- 21.HIV Sequence Compendium. 2003. C.L. Kuika, B. Foley, E. Freed, B. Hahn, B. Korber, P.A. Marx, F. McCutcher, J.W. Mellors, and S. Wolinsky, eds. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM.
- 22.Wagner, R., B. Leschonsky, E. Harrer, C. Paulus, C. Weber, B.D. Walker, S. Buchbinder, H. Wolf, J.R. Kalden, and T. Harrer. 1999. Molecular and functional analysis of a conserved CTL epitope in HIV-1 p24 recognized from a long-term nonprogressor: constraints on immune escape associated with targeting a sequence essential for viral replication. J. Immunol. 162:3727–3734. [PubMed] [Google Scholar]
- 23.Richman, D.D., T. Wrin, S.J. Little, and C.J. Petropoulos. 2003. Rapid evolution of the neutralizing antibody response in HIV type 1 infection. Proc. Natl. Acad. Sci. USA. 100:4144–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak, M.A., and R.M. May. 2000. Virus dynamics. Oxford University Press, Oxford, UK. 237 pp.
- 25.Mansky, L.M., and H.M. Temin. 1995. Lower in vivo mutation rate of human immunodeficiency virus-type 1 than that predicted from the fidelity of reverse transcriptase. J. Virol. 69:5087–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Markowitz, M., M. Louie, A. Hurley, E. Sun, M. DiMascio, A.S. Perelson, and D.D. Ho. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 77:5037–5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klenerman, P., R.E. Phillips, C.R. Rinaldo, L.M. Wahl, G. Ogg, R.M. May, A.J. McMichael, and M.A. Nowak. 1996. Cytotoxic T lymphocytes and viral turnover in HIV type 1 infection. Proc. Natl. Acad. Sci. USA. 93:15323–15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goh, W.C., M.E. Rogel, C.M. Kinsey, S.F. Michael, P.N. Fultz, M.A. Nowak, B.H. Hahn, and M. Emerman. 1998. HIV-1 Vpr increases viral expression by manipulation of the cell cycle: a mechanism for selection of Vpr in vivo. Nat. Med. 4:65–71. [DOI] [PubMed] [Google Scholar]
- 29.Allen, T.M., D.H. O'Connor, P. Jing, J.L. Dzuris, B.R. Mothe, T.U. Vogel, E. Dunphy, M.E. Liebl, C. Emerson, N. Wilson, et al. 2000. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 407:386–390. [DOI] [PubMed] [Google Scholar]
- 30.Stittelaar, K.J., R.A. Gruters, M. Schutten, C.A. van Baalen, G. van Amerongen, M. Cranage, P. Liljestrom, G. Sutter, and A.D. Osterhaus. 2002. Comparison of the efficacy of early versus late viral proteins in vaccination against SIV. Vaccine. 20:2921–2927. [DOI] [PubMed] [Google Scholar]
- 31.Baalen, C.A., C. Guillon, M.M. Baalen, E.J. Verschuren, P.H. Boers, A.D. Osterhaus, and R.A. Gruters. 2002. Impact of antigen expression kinetics on the effectiveness of HIV-specific cytotoxic T lymphocytes. Eur. J. Immunol. 32:2644–2652. [DOI] [PubMed] [Google Scholar]
- 32.Richman, D.D. 2001. HIV chemotherapy. Nature. 410:995–1001. [DOI] [PubMed] [Google Scholar]
- 33.Betts, M.R., D.R. Ambrozak, D.C. Douek, S. Bonhoeffer, J.M. Brenchley, J.P. Casazza, R.A. Koup, and L.J. Picker. 2001. Analysis of total human immunodeficiency virus (HIV)-specific CD4+ and CD8+ T-cell responses: relationship to viral load in untreated HIV infection. J. Virol. 75:11983–11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao, J., J. McNevin, S. Holte, L. Fink, L. Corey, and M.J. McElrath. 2003. Comprehensive analysis of human immunodeficiency virus type 1 (HIV-1)-specific gamma interferon-secreting CD8+ T cells in primary HIV-1 infection. J. Virol. 77:6867–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pantaleo, G., J.F. Demarest, H. Soudeyns, C. Graziosi, F. Denis, J.W. Adelsberger, P. Borrow, M.S. Saag, G.M. Shaw, R.P. Sekaly, and A.S. Fauci. 1994. Major expansion of CD8+ T cells with a predominant Vβ usage during the primary immune response to HIV. Nature. 370:463–467. [DOI] [PubMed] [Google Scholar]
- 36.Pantaleo, G., J.F. Demarest, T. Schacker, M. Vaccarezza, O.J. Cohen, M. Daucher, C. Graziosi, S.S. Schnittman, T.C. Quinn, G.M. Shaw, et al. 1997. The qualitative nature of the primary immune response to HIV infection is a prognosticator of disease progression independent of the initial level of plasma viremia. Proc. Natl. Acad. Sci. USA. 94:254–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kelleher, A.D., C. Long, E.C. Holmes, R.L. Allen, J. Wilson, C. Conlon, C. Workman, S. Shaunak, K. Olson, P. Goulder, et al. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borrow, P., and G.M. Shaw. 1998. Cytotoxic T-lymphocyte escape viral variants: how important are they in viral evasion of immune clearance in vivo? Immunol. Rev. 164:37–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Selier, P., B.M. Senn, P. Klenerman, U. Kalinke, H. Hengartner, and R.M. Zinkernagel. 2000. Additive effect of neutralizing antibody and antiviral drug treatment in preventing virus escape and persistence. J. Virol. 74:5896–5901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gloster, S.E., P. Newton, D. Cornforth, J.D. Lifson, I. Williams, G.M. Shaw, and P. Borrow. 2004. Association of strong virus-specific CD4+ T cell responses with efficient natural control of primary HIV-1 infection. AIDS. 18:749–755. [DOI] [PubMed] [Google Scholar]
- 41.Moore, C.B., M. John, I.R. James, F.T. Christiansen, C.S. Witt, and S.A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science. 296:1439–1443. [DOI] [PubMed] [Google Scholar]
- 42.Yusim, K., C. Kesmir, B. Gaschen, M.M. Addo, M. Altfeld, S. Brunak, A. Chigaev, V. Detours, and B.T. Korber. 2002. Clustering patterns of cytotoxic T-lymphocyte epitopes in human immunodeficiency virus type 1 (HIV-1) proteins reveal imprints of immune evasion of HIV-1 global variation. J. Virol. 76:8757–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mortara, L., F. Letourneur, H. Gras-Masse, A. Venet, J.-G. Guillet, and I. Bourgault-Villada. 1998. Selection of virus variants and emergence of virus escape muatants after immunization with an epitope vaccine. J. Virol. 72:1403–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barouch, D.H., J. Kunstman, M.J. Kuroda, J.E. Schmitz, S. Santra, F.W. Peyerl, G.R. Krivulka, K. Beaudry, M.A. Lifton, D.A. Gorgone, et al. 2002. Eventual AIDS vaccine failure in a rhesus monkey by viral escape from cytotoxic T lymphocytes. Nature. 415:335–339. [DOI] [PubMed] [Google Scholar]
- 45.Allen, T.M., L. Mortara, B.R. Mothe, M. Liebl, P. Jing, B. Calore, M. Piekarczyk, R. Ruddersdorf, D.H. O'Connor, X. Wang, et al. 2002. Tat-vaccinated macaques do not control simian immunodeficiency virus SIVmac239 replication. J. Virol. 76:4108–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.HIV Molecular Immunology. 2003. B.T.M. Korber, C. Brander, B.F. Haynes, R. Koup, C. Kuika, J.P. Moore, B.D. Walker, and D.I. Watkins, eds. Theoretical Biology and Biophysics Group, Los Alamos National Laboratory, Los Alamos, NM.