

Abstract
Krüppel-like factor 5 (KLF5) is a zinc finger-containing transcription factor that regulates proliferation of various cell types, including fibroblasts, smooth muscle cells, and intestinal epithelial cells. To identify proteins that interact with KLF5, we performed a yeast two-hybrid screen of a 17-day mouse embryo cDNA library with KLF5 as bait. The screen revealed 21 preys clustered in four groups as follows: proteins mediating gene expression, metabolism, trafficking, and signaling. Among them was protein inhibitor of activated STAT1 (PIAS1), a small ubiquitin-like modifier (SUMO) ligase that regulates transcription factors through SUMOylation or physical interaction. Association between PIAS1 and KLF5 was verified by co-immunoprecipitation. Structural determination showed that the acidic domain of PIAS1 bound to both the amino- and carboxyl-terminal regions of KLF5 and that this interaction was inhibited by the amino terminus of PIAS1. Indirect immunofluorescence demonstrated that PIAS1 and KLF5 co-localized to the nucleus. Furthermore, the PIAS1-KLF5 complex was co-localized with the TATA-binding protein and was enriched in RNA polymerase II foci. Transient transfection of COS-7 cells by PIAS1 and KLF5 significantly increased the steady-state protein levels of each other. Luciferase reporter and chromatin immunoprecipitation assays showed that PIAS1 significantly activated the promoters of KLF5 and PIAS1 and synergistically increased the transcriptional activity of KLF5 in activating the cyclin D1 and Cdc2 promoters. Importantly, PIAS1 increased the ability of KLF5 to enhance cell proliferation in transfected cells. These results indicate that PIAS1 is a functional partner of KLF5 and enhances the ability of KLF5 to promote proliferation.
Understanding the molecular basis of cellular proliferation and signaling is an important goal in studying various disease conditions. Regulation of gene transcription plays an essential role in these processes. A number of Krüppel-like transcription factors play important roles in regulating cell proliferation and differentiation or in the pathogenesis of many diseases. Among them is Krüppel-like factor (KLF)2 5, which belongs to the Sp/KLF family of transcription factors (1–4). KLF5 (also known as intestine-enriched Krüppel-like factor or IKLF) (5, 6) is an important regulator of cell proliferation (7). Expression of KLF5 is enriched in the proliferating crypt cells of the intestinal epithelium (5, 8). In nontransformed intestinal epithelial cells and fibroblasts, overexpression of KLF5 promotes proliferation (9–13). A mechanism by which KLF5 accomplishes its pro-proliferative effect is to transcriptionally activate several cell cycle-promoting genes, including cyclin D1, cyclin B1, and Cdc2 (9, 11, 12). As such, KLF5 has been shown to be an important mediator for the transforming effect of oncogenic H-Ras (11, 12). Upon overexpression, KLF5 itself leads to cellular transformation (13). These studies illustrate an important function of KLF5 in regulating cellular proliferation.
To further elucidate the biochemical mechanisms of action of KLF5 in regulating physiologically relevant processes such as growth and proliferation, we conducted a yeast two-hybrid screen to identify its interacting partners. Among the preys identified was protein inhibitor of activated STAT1 (PIAS1). The interaction between KLF5 and PIAS1 was further characterized. PIAS1 is a member of the PIAS family of proteins consisting of PIAS1, PIAS3, PIASx, and PIASy (14). Mammalian PIAS proteins were initially identified as negative regulators of STAT signaling (15, 16). PIAS proteins contain a RING domain and exhibit small ubiquitin-related modifier (SUMO) E3 ligase activity (17, 18). SUMO is a small protein with 32% identity to ubiquitin and is covalently attached to substrate proteins (19–24). SUMOylation typically occurs at the lysine residue of a consensus motif, ΨKXE (where Ψ is hydrophobic and X is any amino acid) (19–22, 24, 25). Although PIAS proteins SUMOylate transcription factors such as p53, SMAD4, and STAT1 (26–29), they can also regulate activities of their targets by protein-protein interaction without SUMOylation (14).
Recent studies indicate that PIAS1 directly binds to A/T-rich DNA segments through its SAP (SAF-A/B, Acinus, and PIAS) domain (30). PIAS1 also interacts with TATA-binding protein (TBP), a rate-limiting factor for initiation of transcription (31). TBP is a subunit of TFIID, which is the first protein to bind to DNA during the formation of the preinitiation complex of RNA polymerase II (pol II). TFIID binding initiates the recruitment of other factors required for pol II to begin transcription (32). pol II is mainly localized to nuclear speckles termed pol II foci (33, 34), which overlie three spatially connected nuclear compartments, perichromatin fibrils, interchromatin granule clusters (ICGCs), and Cajal bodies (35). Cajal bodies are initial assembly sites for pol II transcription machineries (35), which are delivered to ICGCs for storage and subsequently transported to active transcription sites located at the periphery of perichromatin fibrils, which represent nascent transcripts (35, 36). Rapid shuttle of proteins between these compartments constantly occurs (35).
Remarkably, many of the TBP-interacting proteins isolated in yeast two-hybrid screens encoded either PIAS1 or PIAS3 (31). Both PIAS1 and TBP bind to the minor groove of the A/T-rich DNA (30, 37). Thus, PIAS1 may be important in regulating TBP. TBP is presumably localized to chromatin where transcription is initiated. However, it can also be localized to the storage/assembly sites for pol II transcription machineries such as Cajal bodies (38). Previously, the role of PIAS1 as a SUMO E3 ligase was extensively studied. However, how PIAS1 acts as a transcriptional co-regulator was less well established, in part because the effects of PIAS1 in regulating transcription are highly context- and target-dependent.
This study reports the identification of 21 preys that interact with KLF5 in a yeast two-hybrid screen. They include proteins involved in gene expression, metabolism, trafficking, and signaling. A specific interaction is demonstrated between KLF5 and PIAS1. A consequence of this interaction is enhanced steady-state levels of both proteins. This leads to a synergistic increase in the ability of KLF5 to transcriptionally activate a number of target genes that are important for cell cycle progression and to stimulate cell proliferation. Consistent with the up-regulation of KLF5 transcriptional activity by PIAS1, KLF5 and PIAS1 co-localize with TBP to pol II nuclear foci. These results indicate that PIAS1 is a positive regulator that enhances the transcriptional ability of KLF5 to promote proliferation.
Experimental Procedures
Plasmid Constructs
The bait plasmid, pGBKT7-KLF5, was constructed by fusing the coding region of mouse KLF5 (5) to the GAL4 DNA-binding domain present in the pGBKT7 plasmid (Clontech). Several constructs were generated that contained partial deletions in the KLF5 sequence using appropriate restriction endonucleases. The pGADT7-Rec-PIAS1 plasmid was recovered from the yeast two-hybrid screen as a prey that specifically interacted with KLF5. Several deletion constructs were generated from this plasmid to produce truncated PIAS1 sequences. The pGL3-mPIAS1 luciferase reporter plasmid was generated by cloning 2 kb of the mouse PIAS1 promoter from genomic DNA using PCR. To generate the KLF5 luciferase reporter plasmid, pGL3-mKLF5, a 1.6-kb mouse KLF5 promoter was amplified from a bacterial artificial chromosome clone, RPCI-23 302C3 (ResGen, Invitrogen), and inserted into pGL3-Basic (Promega). Expression constructs containing 3xFLAG-PIAS1, 3xFLAG-PIAS3, and the PDGF-A luciferase reporter plasmids were generous gifts of Drs. K. Morohashi and D. Kaetzel (39–41). PMT3-KLF5, pMT3-KLF5-HA, and the cyclin D1 and Cdc2 luciferase reporter plasmids were described previously (11, 12).
Yeast Two-hybrid Screening
To perform yeast two-hybrid screening, pGBKT7-KLF5 was used as the bait. This plasmid was transformed into the yeast strain AH109. The resulting strain was mated with the yeast strain Y187 pre-transformed with a MATCHMAKER 17-day mouse embryo cDNA library (Clontech) and selected on synthetic dropout (SD) plates lacking adenine, histidine, leucine, and tryptophan (SD/–ade/–his/–leu/–trp). Colonies obtained from the screen were confirmed by re-streaking on SD/–ade/–his/–leu/–trp containing X-gal. Plasmids from the positive clones were isolated, rescued in Escherichia coli, and sequenced to identify their cDNA inserts. The bait and prey were then co-transformed into AH109, and the specificity of the interactions was confirmed by growth on SD/–ade/–his/–leu/–trp plates. For deletion analysis, the various KLF5 bait and PIAS1 prey plasmids were co-transformed into AH109, and interactions were confirmed by growth on SD/–ade/–his/–leu/–trp in the absence or presence of X-gal.
Transfection, Co-immunoprecipitation, and Western Blotting
COS-7 cells were transfected using Lipofectamine 2000 (Invitrogen) with pMT3 vector alone, pMT3-KLF5-HA, 3xFLAG-PIAS1, or pMT3-KLF5-HA and 3xFLAG-PIAS1. Twenty four hours following transfection, the cells were lysed by boiling in SDS sample buffer (0.625 m Tris-HCl, pH 6.8, 2% SDS, 8.75% glycerol containing 20 mm N-ethylmaleimide (NEM), 5% of 2-mercaptoethanol, 0.5 mm phenylmethylsulfonyl fluoride (PMSF), and 5 μg/ml aprotinin) for 10 min. Then acid-washed glass beads (Sigma) were added, and the mixture was vigorously vortexed for 1 min to shear the DNA and reduce viscosity. The lysates were centrifuged at 10,000 × g for 1 min, and the supernatant was saved for further analysis.
For co-immunoprecipitation, COS-7 cells were transfected with the indicated plasmids. Vector alone was also transfected whenever necessary to compensate for the total amount of plasmids transfected. One or 2 days after transfection, the cells were lysed in 20 mm Tris-HCl, pH 7.4, 1% Nonidet P-40, 135 mm NaCl, and 20 mm NEM containing a protease inhibitor mixture (Roche Applied Science). The lysate was incubated with a polyclonal anti-HA or monoclonal anti-FLAG antibody (Sigma) at 4 °C for 1 h. Pre-washed EZview Red protein G affinity gel (Sigma) was then added, and the mixture was rocked for an additional hour. The immune complexes were washed four times, and the precipitated proteins were detected by Western blotting with a monoclonal anti-HA (Covance), anti-FLAG (Sigma), or rabbit polyclonal anti-KLF5 antibody (raised against residues 95–111 of mouse KLF5). For immunoprecipitation of endogenous KLF5 and PIAS1, nuclear extracts from COS-7 cells were prepared, and immunoprecipitation was performed with a nuclear extract/co-immunoprecipitation kit (Active Motif). The extract was divided equally in half. Half of the extract was immunoprecipitated with a mouse monoclonal anti-PIAS1 antibody (Invitrogen), and the other half was immunoprecipitated with a mouse anti-HA antibody (Covance) as the control. This was followed by incubation with protein A-agarose beads (Upstate) in the presence of 5 mm NEM according to the manufacturer's instructions. The precipitated proteins were detected by Western blotting with the rabbit anti-KLF5 and mouse anti-PIAS1.
Immunofluorescence
COS-7 cells were transfected with vector alone, 3xFLAG-PIAS1, pMT3-KLF5-HA, or 3xFLAG-PIAS1 and pMT3-KLF5-HA. Twenty four hours following transfection, the cells were fixed with methanol and stained with mouse anti-FLAG and rabbit anti-HA antibodies (Sigma) followed by fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse and rhodamine red-X (RRX)-conjugated goat anti-rabbit secondary antibodies (Jackson ImmunoResearch). The cells were then stained with Hoechst 33258 dye to visualize the nuclei. For bromodeoxyuridine (BrdUrd) incorporation, the transfected cells were pulse-labeled with 100 μm BrdUrd for 30 min. After fixation in methanol, the cells were treated with 2 n HCl at room temperature for 30 min to denature the DNA, neutralized with 0.1 m sodium borate, pH 8.5, washed with phosphate-buffered saline, and blocked with 2% bovine serum albumin in phosphate-buffered saline for 1 h. The cells were stained sequentially with the following combination of antibodies: mouse anti-BrdUrd followed by a Cy5-conjugated goat anti-mouse secondary antibody (Jackson ImmunoResearch), rabbit anti-HA (Sigma) followed by the RRX-conjugated goat anti-rabbit secondary antibody, and FITC-conjugated chicken anti-FLAG (Immunology Consultants Laboratory). The cells were then stained with Hoechst dye to reveal the nuclei. Images were captured with an Axioskop 2 plus microscope (Zeiss). To demonstrate co-localization of KLF5 and PIAS1 with TBP, the HA-KLF5 and/or FLAG-PIAS1-transfected cells were stained with mouse anti-FLAG, chicken anti-HA (Chemicon), and rabbit anti-TBP (Santa Cruz Biotechnology), followed by FITC-conjugated donkey anti-mouse, Rhodamine Red-X (RRX)-conjugated donkey anti-chicken, and aminomethylcoumarin-conjugated donkey anti-rabbit secondary antibodies (Jackson ImmunoResearch). As a control, the vector alone-transfected cells were stained with the same primary and secondary antibodies except anti-TBP. To demonstrate co-localization of KLF5 and PIAS1 with pol II, the HA-KLF5- and/or FLAG-PIAS1-transfected cells were stained with mouse anti-FLAG, chicken anti-HA (Chemicon), and a rabbit antibody raised against the large subunit of RNA polymerase II (Santa Cruz Biotechnology), followed by FITC-conjugated donkey anti-mouse, Rhodamine Red-X (RRX)-conjugated donkey anti-chicken, and Cy5-conjugated donkey anti-rabbit secondary antibodies (Jackson ImmunoResearch). The vector alone-transfected cells were stained with the same primary and secondary antibodies except anti-pol II. The cells were also stained with Hoechst dye to reveal the nuclei.
Luciferase Assays
COS-7 cells were transfected with equal amounts of vector alone, pMT3-KLF5, 3xFLAG-PIAS1, or pMT3-KLF5 and 3xFLAG-PIAS1, together with the KLF5, PIAS1, PDGF-A, cyclin D1, or Cdc2-luciferase reporter. A Renilla luciferase control vector was co-transfected to normalize the transfection efficiency. Cell lysis and reporter assay were performed 2 days after the transfection with dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions.
Chromatin Immunoprecipitation (ChIP) Assays
To examine the binding of KLF5 and PIAS1 to target promoters, COS-7 cells were transfected as described in the luciferase assays, except that pMT3-KLF5 was replaced with pMT3-KLF5-HA whenever necessary to efficiently pull down KLF5. The HA tag does not affect KLF5 activity. ChIP assays were performed with DNA purified from the corresponding cells, using chromatin immunoprecipitation assay kit (Upstate) according to the manufacturer's instructions. Immunoprecipitation was conducted using mouse anti-FLAG or rabbit anti-HA, KLF5, or FLAG antibody (Sigma), whereas mouse anti-His (Santa Cruz Biotechnology) or normal rabbit serum (IgG) was used as the negative control. Nested PCR was performed after each immunoprecipitation. For the KLF5 promoter, the first-round PCR primers were GGAGAGATCTCCCTCCGCGTC and AGCGATCGCGATCCCACCTCGC. The second-round (nested) primers were GGAGAGATCTCCCTCCGCGTC and CGTCACCGCCGAGGTCCTC. For the PIAS1 promoter, the first-round primers were CGAGGGGGTAGTAACTGTCAAAC and TTGCGTCTGTCAGCGCAAGCG. The nested primers were CCGCTTGGCGCCATTAT and GTCCGCAAGCTTGCGTCTGTCAGCGCAAGCGC. For the PDGF-A promoter, the first-round primers were GGGGCTTTGATGGATTTAGCTGC and AGAGGGTTATAGCGCCGCC. The nested primers were ATTTAGCTGCTTGCGCG and AGAGGGTTATAGCGCCGCC. For the cyclin D1 promoter, the first-round primers were AGGAAATGCTGGCCACCATCTTG and GGTTTTCATAGAAATGCAAATCGC. The nested primers were CTGCTGGAATTTTCGGG and CGGCAGGCCACACGC. For the Cdc2 promoter, the first-round primers were TGAGGTAGAAACAAAGCACAGCG and AGAGCCAGCTTTGAAGCCAAGTG. The nested primers were ACATTTTTGAGGCGGTC and AGCCAAGTGCGAGCAG. A small fraction of each lysate prior to the immunoprecipitation was also amplified with nested PCR or with a GAPDH PCR mixture kit (Active Motif) as the positive and negative controls for input.
Results
Identification of Proteins That Interact with KLF5
To identify proteins interacting with KLF5, we screened a mouse 17-day embryonic cDNA library under high stringency with a KLF5-GAL4 DNA-binding domain fusion construct as the bait. We chose the embryonic library because KLF5 is highly expressed during this stage of development (8). After screening 106 independent clones with the bait, 30 positive clones that represented 21 genes were identified (Table 1). These preys can be divided into four groups. The largest group of the preys is involved or is predicted to be involved in the regulation of gene expression and contains ANKRD17, DnaJ homolog subfamily C member 4, HNRPF, PIAS1, PRIM2, RPS2, REC14, and SFRS15. The second group contains three known and one predicted metabolic enzymes, DCK, FXNa, PCCA, and PMM2, which catalyze nucleic acid, amino acid, lipid, and carbohydrate metabolism. The third group contains PI3KR1/p85α, the major regulatory subunit of phosphatidylinositol 3-kinase (PI 3-kinase). Strikingly, four proteins directly involved in intracellular trafficking, ARFGAP3, α-taxilin, SNAP23, and SNX3, were also isolated from the screen, and they make up the fourth group. Many of the preys are known nuclear proteins (Table 1).
TABLE 1. Proteins identified in the yeast two-hybrid screen that interacted with KLF5.
The symbols used are as follows: ++, the prey is primarily localized to the nucleus; +, the prey has been reported to be localized to the nucleus although the nucleus is not the primary location; (−), the prey is localized in the cytoplasm but in a perinuclear location.
| Gene Name | Gene ontology | Nuclear |
|---|---|---|
| PIAS1 | Transcriptional co-regulator and SUMO E3 ligase | ++ |
| HNRPF | Pre-mRNA splicing factor that interacts with TBP and RNA polymerase II | ++ |
| PRIM2 (DNA primase 2) | Primase essential to DNA replication | ++ |
| REC14 (recombination protein REC14)a | Transcriptional regulation | ++ |
| SFRS15a | Pre-mRNA splicing factor that interacts with RNA polymerase II carboxyl-terminal domain | ++ |
| RPS2 (ribosomal protein S2) | Ribosomal subunit | + |
| DNAJC4 (DnaJ homolog subfamily C member 4) | Putative chaperone activity | NDb |
| ANKRD17 | Marker for liver development | ND |
| DCK | Nucleic acid metabolism | + |
| FXNaa | Putative amino peptidase | ND |
| PCCA | Amino acid and fatty acid metabolism | + |
| PMM2 | GDP-mannose and dolichol-phosphate-mannose synthesis | (−) |
| PI3KR1/p85α (phosphatidylinositol 3-kinase, regulatory subunit, polypeptide 1) | PI 3-kinase signal pathways | + |
| α-Taxilin | Intracellular vesicle transport | + |
| ARFGAP3 | Vesicle budding | (−) |
| SNAP23a | Membrane fusion; vesicle targeting | + |
| SNX3 | Intracellular trafficking | (−) |
| KIAA1033 | Unknown | ND |
| LOC 380885 | Unknown | ND |
| RIKEN cDNA F830013N24 | Unknown | ND |
| RP23-418O21a | Unknown | ND |
Preys that were represented by more than one clones.
ND indicates not determined.
KLF5 and PIAS1 Interact with Each Other
We further investigated the role of PIAS1 in regulating KLF5 because PIAS1 is a nuclear protein known to play an important role in regulating transcription factors. To confirm their interaction, the KLF5 bait and PIAS1 prey were co-transformed into yeast, and specific interaction was demonstrated by the ability of the yeast colonies to grow in the absence of selective nutrients (Fig. 1A). This interaction was independently verified by co-immunoprecipitation in COS-7 cells transfected with mammalian expression constructs containing HA-tagged KLF5 and FLAG-tagged PIAS1 (Fig. 1B). FLAG-PIAS1 also co-immunoprecipitated endogenous KLF5 (Fig. 1C). Finally, endogenous PIAS1 and KLF5 in COS-7 nuclear extract co-immunoprecipitated (Fig. 1D). Therefore, KLF5 and PIAS1 physically interact with each other in both yeast and mammalian cells.
FIGURE 1. KLF5 and PIAS1 interact with each other.

A, yeast two-hybrid assay shows that KLF5 interacts with PIAS1 but not UKP68, an RNA-binding protein used as a negative control. The bait and prey plasmids were co-transformed into host strain AH109, and the specificity of their interactions was demonstrated by growth on SD/–ade/–his/–leu/–trp. B, COS-7 cells were transfected with vector alone (lane 1), expression construct containing HA-tagged KLF5 (lane 2), or expression constructs containing HA-KLF5 and FLAG-tagged PIAS1 (lane 3). Immunoprecipitation was conducted with a rabbit HA antibody, and the precipitated complexes were analyzed by Western blotting using mouse HA and FLAG antibodies, respectively. Note that cells transfected with HA-KLF5 alone contained three times as much input DNA as those transfected with both plasmids. C, COS-7 cells were transfected with FLAG-tagged PIAS1 (lane 1) or vector alone (lane 2). Immunoprecipitation (IP) was conducted with a mouse FLAG antibody, and the precipitated complexes were analyzed by Western blotting using a rabbit KLF5 antibody. D, untransfected COS-7 cells were lysed, and the nuclear extract was prepared. The extract was immunoprecipitated with a mouse PIAS1 antibody (lane 1) or mouse HA antibody (lane 2) as the control, and the immunoprecipitates were analyzed by Western blotting with a rabbit KLF5 antibody.
KLF5 and PIAS1 Co-localize with TBP to pol II Nuclear Foci
Consistent with the physical interaction between KLF5 and PIAS1, indirect immunofluorescence microscopy showed that a significant fraction of KLF5 co-localized with PIAS1 within the nucleus (Fig. 2). Although both proteins were dispersed throughout the nucleus, the staining was more concentrated in subnuclear speckles in co-transfected cells (Fig. 2, bottom row). Because KLF5 is a transcription factor and the pattern of distribution of these speckles resembles those of factors involved in coupled transcription and pre-mRNA splicing, we sought to determine whether additional factors might be included in the speckles. We chose to examine TBP and pol II, the latter serving as a marker for pol II nuclear foci (33, 34). Results of the study demonstrated that KLF5 and PIAS1 co-localized with TBP and pol II in co-transfected cells (Figs. 3 and 4, respectively). In contrast, in singly transfected cells, PIAS1 co-localized with TBP but not pol II, and KLF5 only partially co-localized with TBP and pol II (Figs. 3 and 4). These results suggest that the complex formed between KLF5 and PIAS1 facilitated each other's co-localization to TBP-enriched pol II nuclear foci.
FIGURE 2. KLF5 and PIAS1 co-localize to the nucleus.

COS-7 cells were transfected with vector alone, FLAG-PIAS1, HA-KLF5, or both. Cells were stained with mouse FLAG antibody and rabbit HA antibody followed by FITC-conjugated anti-mouse (green) and RRX-conjugated anti-rabbit (red) secondary antibodies. Cells were counter-stained with Hoechst 33258 (blue) to visualize the nuclei. Images on the bottom row are enlarged from the co-transfected cells. The boxes show co-localization of the two proteins to nuclear speckles.
FIGURE 3. KLF5 and PIAS1 co-localize with TBP.
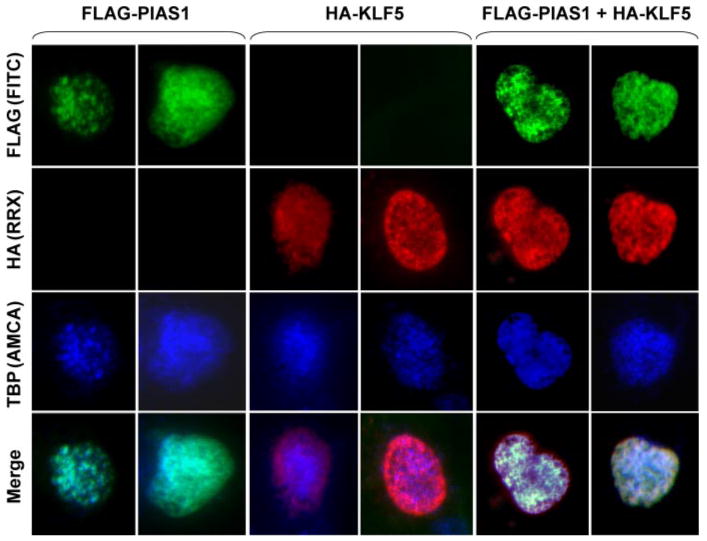
COS-7 cells were transfected with FLAG-PIAS1, HA-KLF5, or both. Cells were stained with mouse FLAG antibody, chicken HA antibody, and rabbit TBP antibody followed by FITC-conjugated anti-mouse (green), RRX-conjugated anti-chicken (red), and aminomethylcoumarin (AMCA)-conjugated anti-rabbit (blue) secondary antibodies. Two transfected cells each are shown.
FIGURE 4. KLF5 and PIAS1 co-localize with RNA polymerase II.

COS-7 cells were transfected with FLAG-PIAS1, HA-KLF5, or both. Cells were stained with mouse FLAG antibody, chicken HA antibody, and rabbit pol II antibody followed by FITC-conjugated anti-mouse (green), RRX-conjugated anti-chicken (red), and Cy5-conjugated anti-rabbit (yellow) secondary antibodies. Cells were counter-stained with Hoechst 33258 (blue) to visualize the nuclei. Two transfected cells each are shown.
Structural Determination of the Interaction between KLF5 and PIAS1
We next determined the regions of KLF5 and PIAS1 that are required for their interaction by performing a deletion analysis of the two proteins using yeast two-hybrid. The various deletion constructs studied are summarized in Fig. 6A. Results shown in Fig. 5A indicate that the carboxyl-terminal region of PIAS1 (316–651) is sufficient for the interaction with KLF5. In contrast, the amino-terminal region of PIAS1 (1–309 and 1–438) does not interact with either full-length or truncated KLF5. These results also suggest that the MIZ-ZF motif of PIAS1 (Fig. 6A), which contains a RING domain necessary for SUMO ligase activity (14), is not required for the interaction with KLF5.
FIGURE 6. Summary of the interaction between KLF5 and PIAS1.

A, diagram of the deletion constructs of PIAS1 and KLF5 used in this study. Numbers refer to amino acid residues. The potential motifs and their locations are illustrated as well. Upper panel, in PIAS1, SAP represents SAF-A/B, acinus and PIAS, a DNA-binding domain (14); MIZ-ZF represents Msx-interacting zinc finger, a RING domain that is required for SUMO ligase activity (14); AD represents acidic domain (14); and S/T stands for serine/threonine-rich domain (14). Lower panel, in KLF5, the amino acid sequence VPII represents a potential SUMO-binding motif (85); amino acid sequences IKTE and IKQE represent potential SUMOylation motifs, ΨKXE (19–22, 24, 25); and ZF is the C2H2 zinc finger DNA-binding domain (5). B, summary of regions of KLF5 and PIAS1 involved in their interaction. The dark gray bars indicate regions of interaction. Double arrows indicate positive interactions. The blocked line indicates inhibition of the interaction.
FIGURE 5. The acidic domain of PIAS1 interacts with amino- and carboxyl-terminal regions of KLF5.

A, carboxyl-terminal but not amino-terminal region of PIAS1 interacts with KLF5. Yeasts transformed with the indicated KLF5 baits and PIAS1 preys were streaked onto plates containing selective media and maintained at 30 °C for 3 days. B and C, the amino terminus of PIAS1 inhibits KLF5-PIAS1 interaction. Yeasts transformed with the indicated baits and preys were streaked onto plates with selective media at 30 °C for 3 days (B). Growth of colonies on selective media containing X-gal was observed up to 3 days after plating. The percentages of colonies that grew and turned blue are noted. N = total number of colonies counted (C). D, zinc finger DNA-binding domain of KLF5 is involved in interaction with PIAS1. Yeasts transformed with the indicated baits and preys were streaked onto plates with selective media and grown at 30 °C for 3 days. E, the acidic domain of PIAS1 interacts with KLF5. Yeasts transformed with the indicated baits and preys were streaked onto plates with selective media and grown at 30 °C for 3 days.
Additional deletion studies suggest that the amino-terminal region of PIAS1 (1–316) not only is dispensable for the interaction between PIAS1 and KLF5 but appears to inhibit their interaction. This is based on results in Fig. 5B showing that although the carboxyl terminus of PIAS1 (316–651) interacted with all of the four KLF5 deletion constructs (1–161, 1–165, 1–251, and 1–309), full-length PIAS1 interacted poorly, if at all, with the two most amino-terminal KLF5 fragments (1–161 and 1–165). Fig. 5C shows that among 23 PIAS1/KLF5 (1–161) colonies and 22 PIAS1/KLF5 (1–165) colonies derived from the two-hybrid assays, few grew on selective media and turned blue. These results suggest that the amino terminus of PIAS1 may interfere with the ability of its carboxyl terminus to interact with the amino-terminal 165 amino acids of KLF5.
Finally, results in Fig. 5, D and E, indicate that the glutamate/aspartate-rich acidic domain of PIAS1 interacted with the amino- and carboxyl-terminal zinc finger DNA-binding domain of KLF5. Fig. 5D shows that the carboxyl-terminal zinc finger DNA-binding domain of KLF5 (361–446) also interacted with PIAS1, as this KLF5 construct interacted with both the full-length and the carboxyl terminus of PIAS1. In contrast, the regions amino-terminal to the zinc finger motif of KLF5 (252–309) and (308–360) did not bind. Fig. 5E demonstrates that both the amino terminus of KLF5 (1–251) and the zinc finger DNA-binding domain of KLF5 (361–446) interacted with a region of PIAS1 (439–491) that contains its acidic domain. In contrast, the regions more carboxyl-terminal to the acidic domain of PIAS1 (492–568 and 569–651) did not bind. Fig. 6B summarizes the various regions in PIAS1 and KLF5 that are involved in their interaction.
Co-expression of KLF5 and PIAS1 Mutually Increases Steady-state Protein Levels of Each Other
Results of the co-immunoprecipitation experiments in Fig. 1B show that an equal amount of HA-KLF5 was precipitated from cells co-transfected with HA-KLF5 and FLAG-PIAS1 (lane 3) as compared with those transfected with HA-KLF5 alone (lane 2), even though the HA-KLF5 input in the latter was three times that of the former. This suggests that the presence of PIAS1 may increase the steady-state level of KLF5 in transfected cells. To verify this effect, we performed Western blot analysis of lysates from cells transfected with vector alone, HA-KLF5, FLAG-PIAS1, and both. As shown in Fig. 7A, the steady-state level of HA-KLF5 was significantly increased in PIAS1-co-transfected cells as compared with those transfected with HA-KLF5 alone (compare lane 4 with lane 2). Similarly, the level of PIAS1 was increased in co-transfected cells when compared with PIAS1 alone-transfected cells (Fig. 7A, compare lane 4 with lane 3). The ability of PIAS1 to increase KLF5 abundance is not limited to PIAS1, as PIAS3 also significantly increased the level of KLF5 in co-transfected cells (Fig. 7B).
FIGURE 7. KLF5 and PIAS mutually increase each other's abundance in co-transfected cells.

A, COS-7 cells were transiently transfected with vector alone, HA-KLF5, FLAG-PIAS1, or both. Twenty four hours following transfection, lysates were prepared for Western blot analyses with antibodies against HA, FLAG, or actin. B, COS-7 cells were transiently transfected with the indicated plasmids. Western blot analyses were performed 24 h later with the antibodies indicated.
Activation of the KLF5 and PIAS1 Promoters by PIAS1
PIAS1 not only increased the KLF5 protein abundance in the transfected cells but also stimulated activity of the KLF5 promoter. As shown by luciferase reporter assays, KLF5 inhibited the activity of its own promoter (Fig. 8A). In contrast, PIAS1 bound to and significantly activated the KLF5 promoter (Fig. 8, B and A, respectively). In addition, PIAS1 but not KLF5 activated the PIAS1 promoter (Fig. 8C). PIAS1 also bound to its own promoter as shown by the ChIP assay in Fig. 8D (lane 3). These results indicate that PIAS1 up-regulates KLF5 and PIAS1 expression at the promoter level.
FIGURE 8. Regulation of the KLF5 and PIAS1 promoters by PIAS1.

A, COS-7 cells were transfected with an empty vector (control), an expression vector containing HA-KLF5, or FLAG-PIAS1 and KLF5 promoter-luciferase reporter. Two days following transfection, dual luciferase assay was performed, and the relative luciferase unit (RLU) was determined after standardizing first with the activity of the internal control, Renilla luciferase. n = 7; **, p < 0.01; ***, p < 0.001 by two-tailed t test. B, in parallel, ChIP assays were performed with a mouse FLAG antibody (α FLAG) or a mouse nonspecific antibody (IgG) as control. The promoter and GAPDH in the input were also amplified as the controls for loading. C, COS-7 cells were transfected with an empty vector, an expression vector containing KLF5, or FLAG-PIAS1 and PIAS1 promoter-luciferase reporter. Two days following transfection, dual luciferase assay was performed, and the relative luciferase unit (60) was determined. n = 8; ***, p < 0.001 by two-tailed t test. D, in parallel, ChIP assays were performed with a mouse FLAG antibody (αFLAG) or a mouse nonspecific antibody (IgG) as control. Promoter input and GAPDH were also amplified. C = control; K = KLF5; P = PIAS1.
Stimulation of the Promoters of Growth-related Genes by KLF5 and PIAS1
KLF5 has been shown previously to stimulate expression of several genes involved in growth regulation, including platelet-derived growth factor A-chain (PDGF-A) (42), cyclin D1 (12), and Cdc2 (11). To determine whether PIAS1 is also involved in the regulation of expression of these genes, we performed co-transfection experiments using reporter constructs containing the promoters of the three respective genes and expression constructs containing KLF5 and PIAS1. As shown in Fig. 9A, both KLF5 and PIAS1 stimulated the PDGF-A promoter. In contrast, KLF5 but not PIAS1 transactivated the cyclin D1 and Cdc2 promoters (Fig. 9, C and E, respectively). However, when both KLF5 and PIAS1 were expressed, PIAS1 synergistically increased the promoter activities of cyclin D1 and Cdc2 (Fig. 9, C and E, respectively). ChIP assays showed that KLF5 bound to all three promoters (Fig. 9, B, D, and F, lane 2). When an antibody directed against KLF5 was used, the PDGF-A promoter also precipitated with the endogenous KLF5 (Fig. 9B, lane 3). In addition, PIAS1 bound to the cyclin D1 and Cdc2 promoters (Fig. 9, D and F, lane 3). In contrast, PIAS1 did not bind to the PDGF-A promoter and only precipitated with this promoter when KLF5 was co-expressed (Fig. 9B, lane 3 and 4, respectively). This is because the PDGF-A promoter used in this study contains only nucleotides −261 to +8 and is highly G/C-rich (82%). This region of the PDGF-A promoter contains a known KLF5-binding site but lacks an A/T-rich sequence to which PIAS1 binds (30, 41, 42). In cells co-transfected with KLF5 and PIAS1, KLF5 immunoprecipitated with the cyclin D1 but not the Cdc2 promoter (Fig. 9, D and F, lane 4, respectively). This was the case with either anti-HA (Fig. 9F) or anti-KLF5 (results not shown). A possible explanation for this is masking of the antigenic sites by the associated PIAS1, because PIAS1 binds to both the amino terminus and carboxyl-terminal DNA-binding domain of KLF5, and the closely packed transcriptional machinery may induce conformational change or interfere with binding to the antibodies used in the ChIP assays. Nevertheless, KLF5 enhanced PIAS1 binding to the Cdc2 promoter (Fig. 9F, compare lanes 4 and 3), and PIAS1 stimulated the activity of KLF5 toward the same promoter (Fig. 9E), supporting the notion that both KLF5 and PIAS1 interact with the Cdc2 promoter. Together, the results of luciferase reporter and ChIP assays indicate that PIAS1 enhances the ability of KLF5 to activate the promoters of the growth-related genes.
FIGURE 9. Induction of promoters of growth-related genes by KLF5 and PIAS1.
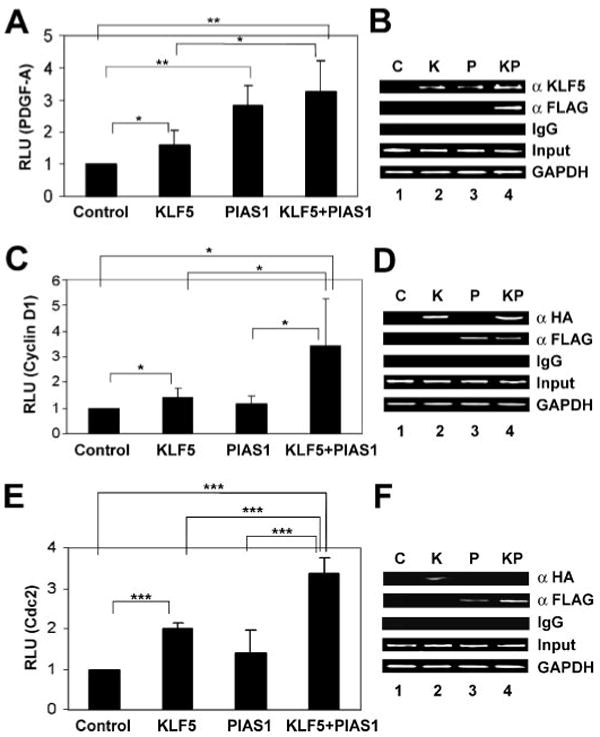
Co-transfection experiments were conducted in COS-7 cells with a luciferase reporter containing the PDGF-A (A), cyclin D1 (C), or Cdc2 (E) promoter, and an expression construct(s) containing KLF5, FLAG-PIAS1, or both. An empty vector was used as the control. Two days following transfection, dual luciferase assay was performed, and the relative luciferase unit (RLU) was determined after standardizing first with the activity of Renilla luciferase. n = 5 for A and n = 7 for C and E; *, p < 0.05; **, p < 0.01; ***, p < 0.001 by two-tailed t test. Similar co-transfections were conducted with a luciferase reporter containing the PDGF-A (B), cyclin D1 (D), or Cdc2 (F) promoter, and expression construct(s) containing HA-tagged KLF5, FLAG-PIAS1, or both. Lysates from the cells were subjected to ChIP assays with rabbit KLF5 (α KLF5), HA (α HA), or FLAG antibody (α FLAG) as indicated, or with normal rabbit serum (IgG) as negative control. In addition, the corresponding promoter input and GAPDH were amplified. C = control; K = KLF5; P = PIAS1; KP = KLF5 and PIAS1.
PIAS1 Enhances the Ability of KLF5 to Stimulate DNA Synthesis
Previous studies indicate that KLF5 stimulates proliferation of fibroblasts and intestinal epithelial cells by promoting G1/S transition and entry into mitosis (11–13). This is confirmed by BrdUrd incorporation in COS-7 cells transiently transfected with an expression vector containing KLF5 (Fig. 10A). Because the transfection did not significantly change the proportion of cells in S phase as analyzed by green fluorescent protein transfection (results not shown), we used cells transfected with vector alone as control. As shown in Fig. 10B, transfection with KLF5 significantly increased the percentage of cells incorporating BrdUrd to an average of 51% from 25% in the control. The presence of PIAS1 in KLF5-transfected cells further enhanced the ability of KLF5 to stimulate BrdUrd incorporation, to an average of 68% (Fig. 10B). These results indicate that PIAS1 enhances the ability of KLF5 to stimulate cellular proliferation.
FIGURE 10. PIAS1 enhances the ability of KLF5 to stimulate DNA synthesis.
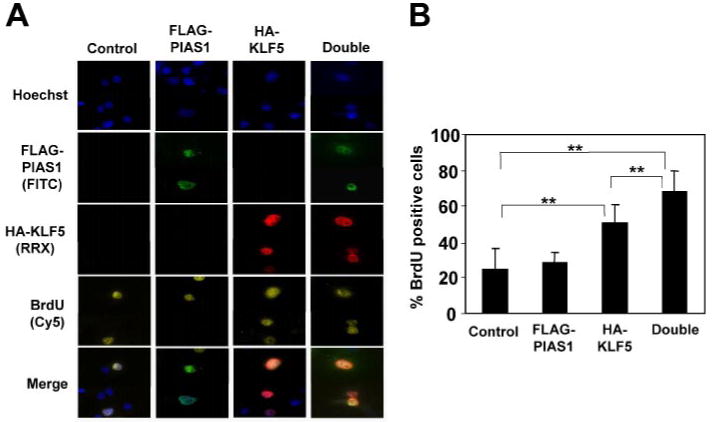
COS-7 cells were transfected with vector control, FLAG-PIAS1, HA-KLF5, or both. Sixteen hours after transfection, cells were pulse-labeled with BrdUrd for 30 min after which the DNA was denatured with HCl. Cells were then stained with rabbit HA, mouse BrdUrd, and FITC-conjugated chicken FLAG antibodies followed by RRX-conjugated goat anti-rabbit and Cy5-conjugated goat anti-mouse secondary antibodies. Several representative cells are shown in A. Between 100 and 300 cells were analyzed for the respective construct(s), and the percentages of KLF5-positive and PIAS1-positive cells, or both, that were also positive for BrdUrd are shown in B. n = 7; **, p < 0.01 by two-tailed t test.
Discussion
Proliferation of mammalian cells is stringently regulated. Abnormalities in the regulatory process cause diseases such as cancer. KLF5 is a transcription factor that regulates proliferation of multiple cell types, including the intestinal epithelial cells (7). In this study, we identified a novel partner for KLF5, PIAS1, which interacts with and positively regulates KLF5 at multiple levels. Evidence for a physical association between KLF5 and PIAS1 was documented by both yeast two-hybrid and co-immunoprecipitation. Consistent with these findings, indirect immunofluorescence microscopy demonstrated co-localization of the two in the nucleus and their enrichment in TBP-containing pol II nuclear foci. Transient transfection, ChIP, and reporter assays showed that PIAS1 markedly stimulates KLF5 promoter activity as well as increases KLF5 protein levels. In addition, PIAS1 significantly increases KLF5-regulated cyclin D1 and Cdc2 promoter activities, suggesting that PIAS1 is an activator of KLF5 and influences the ability of KLF5 to regulate cell cycle-related genes. Indeed, PIAS1 enhances the ability of KLF5 to promote DNA synthesis. Our results support a physiologic role for PIAS1 as a positive regulator of KLF5.
PIAS1 and KLF5 co-localize with the vital transcription factor TBP, and all three are significantly enriched in pol II nuclear foci (Figs. 2–4). The speckled pattern of PIAS1 localization is similar to those of SAF-A (scaffold attachment factor-A)/heterogeneous nuclear ribonucleoprotein U and SAF-B (43, 44). SAF-B binds to pol II and serine/arginine-rich RNA processing factors and may serve as an assembly point for transcriptosomal formation (44). A Drosophila melanogaster homolog of PIAS1, suppressor of position-effect variegation SU (var)2-10, is co-localized with lamin as spots in interphase nuclei (45). SAF-A, SAF-B, acinus, and a protein highly homologous to SAF-B (similar to KIAA0138 product) were all identified in purified ICGCs (46, 47). Although PIAS1 is localized to speckles, these speckles have not been characterized. In this study, we demonstrate that TBP is enriched in such speckles. Furthermore, results of the ChIP assays showed that PIAS1 binds to a number of promoters, providing critical evidence in addition to the previous finding that PIAS1 directly binds to A/T-rich DNA segments in vitro. As TBP is a prerequisite for initiation of transcription, co-localization of TBP and PIAS1, their physical interaction, binding of PIAS1 to several promoters and to KLF5, enrichment of PIAS1 and KLF5 to pol II nuclear foci, and enhancement of KLF5 transcriptional activity by PIAS1 prove beyond doubt that PIAS1 is a transcriptional co-regulator that directly regulates transcription by protein-protein interaction.
Results from our yeast two-hybrid screen lend further support to the observation that KLF5 and PIAS1 are enriched in pol II foci. It is well known that pol II foci contain pre-mRNA splicing factors and other proteins involved in coupled transcription and pre-mRNA splicing (33, 34, 36, 48). Multiple molecular chaperones, such as DnaJ protein homolog 2 and Hsp70 as well as a subset of ribosomal subunits, including 40s ribosomal protein S2 (RPS2), were identified in purified ICGCs (46, 47). RPS2, identified in this screen as an interacting partner of KLF5, is among the few ribosomal proteins localized to nucleoplasm but not to nucleoli (49). In our two-hybrid screen, two pre-mRNA splicing factors, SFRS15 and HNRPF, were among the preys. HNRPF is a pre-mRNA splicing factor that interacts with both pol II and TBP (50, 51). SFRS15 is an arginine/serine-rich pre-mRNA splicing factor and pol II carboxyl-terminal domain-binding protein (52). The carboxyl-terminal domain of pol II plays a major role in targeting splicing factors to transcription sites (53). We also isolated from the screen DnaJ homolog, subfamily C member 4, a predicted chaperone (54) and ANKRD17 (55). ANKRD17 contains a transcriptional regulator FadR domain, a DNA mismatch repair protein MutS carboxyl-terminal domain, and a K homology domain. It also contains multiple ankyrin repeats spanning half of the protein. Ankyrin repeat is a common protein-protein interaction motif found in transcriptional initiators, cell cycle regulators, and signal transducers (56). K homology domain, first identified in heterogeneous nuclear ribonucleoprotein K, is an RNA-binding domain present in a wide variety of nucleic acid-binding proteins (57, 58). An ANKRD17-related protein, multiple ankyrin repeats, single K homology domain (also called ANKRD1), is a known signaling protein (59). Our results pinpoint PIAS1 and highlight the other preys as possible molecular elements that coordinate the gene expression regulated by KLF5.
The preys also include four known or predicted enzymes involved in metabolism, DCK, FXNa, PCCA, and PMM2. Three of these, DCK, PCCA, and PMM2, have key clinical links. DCK maintains a dCTP pool for DNA replication but is also essential to the effectiveness of various anti-cancer and antiviral drugs (60). A minor fraction of DCK is located in the nucleus, and it is predominantly targeted to the nucleus when expressed at high levels (61). PCCA deficiency is the underlying cause of propionic acidemia, and nuclear localization of PCCA has been detected in neoplastic lesions as well as pregnancy-related endometrium (62). PMM2 is necessary for the synthesis of GDP-mannose, and its mutations are major causes of congenital disorder of glycosylation type Ia (63). Congenital disorder of glycosylation Ia also has gastrointestinal manifestations such as protein-losing enteropathy and hepatic fibrosis (64). Although cytoplasmic, PMM2 displays perinuclear localization in the cytoplasm, although the possibility that it might target to the nucleus under certain conditions cannot be completely ruled out (65). Interestingly, among various tissues, the most abundant PMM2 mRNA levels and enzymatic activity were found in intestinal mucosa (66). Recently, we reported that KLF4, which often plays a role opposite to KLF5, significantly inhibits the expression of several groups of genes involved in macromolecular synthesis, including protein biosynthesis, transcription, and cholesterol biosynthesis (67). These results imply that KLF5 may also be involved in regulating metabolism.
Notably, four proteins involved in intracellular trafficking, ARFGAP3, α-taxilin, SNAP23, and SNX3, were identified as KLF5-interacting proteins in the screen. In addition, among the preys was p85α, the main regulatory subunit of PI 3-kinase, which is a major kinase in regulating intracellular trafficking and a vast array of other important events (68). It would be interesting to determine whether KLF5 is phosphorylated by PI 3-kinase including Akt or modulates PI 3-kinase signaling. Strong connections exist regarding biochemical and biological functions of these proteins. For instance, all five proteins are either structural components of intracellular vesicles or major regulators of vesicle transport. SNX3 possesses a PX domain that directly interacts with phosphatidylinositol 3-phosphate, the product of PI 3-kinase (69). α-Taxilin interacts with almost all syntaxin family members that are part of a target-soluble N-ethylmaleimide-sensitive factor attachment protein receptor (t-SNARE) complex essential to vesicle docking and donor/acceptor membrane fusion (70). Although α-taxilin is primarily cytoplasmic, a closely related isoform, γ-taxilin, is localized to the nucleus in osteoblasts and inhibits activating transcription factor 4 (71). Biological correlations to the digestive system have also been described. SNAP23 is induced by various cytokines such as interleukin-2, -3, -5, and -10, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, and erythropoietin (72). Remarkably, the gastrointestinal tract has the highest expression of SNAP23, with the highest expression in the small intestine and very high levels of expression in stomach and liver as well (72). Mice lacking p85α of PI 3-kinase were severely deficient in gastrointestinal mast cells, whereas dermal mast cells were present (73). Intracellular sorting and vesicle transport have fundamental roles in signaling and maintaining polarity of epithelial cells. In intestinal cells apical membrane participates in nutrient uptake, whereas basolateral membrane mediates cell adhesion and active signaling. KLF5 may play important, yet unidentified, roles in maintaining a polarized structure of epithelial cells. On the other hand, a polarized structure may be critical for KLF5 to regulate epithelial and adjacent cells. Although mainly cytoplasmic, SNAP23 and γ-taxilin have been shown to be localized to the nucleus (71, 72), and the distribution of ARFGAP3 and SNX3 is largely perinuclear (72, 74). We cannot completely rule out the possibility that the latter two may target to the nucleus upon certain stimuli. Similarly, we cannot rule out the possibility that KLF5 may be localized outside the nucleus under certain circumstances and interact with proteins in the cytoplasm or elsewhere. We hypothesize that this cluster of vesicle transport proteins reflects a novel aspect of KLF5 interaction and predicts either regulation of intracellular transport by KLF5 or, vice versa, regulation of KLF5 by intracellular transport.
Mice with heterozygous deletion of Klf5 (Klf5+/−) exhibited less arterial wall thickness and angiogenesis, distorted intestinal villi, and decreased number of mesenchymal cells in the gastrointestinal tract (75). Activation and proliferation of smooth muscle cells and fibroblasts were impaired (75). The gastrointestinal phenotypes of Klf5+/− and Pdgfa−/− mice were similar. PDGF-A expression was diminished in the gastrointestinal tract and injured arteries of Klf5+/− mice (75). Consistent with these findings, KLF5 and PIAS1 up-regulate the PDGF-A promoter (Fig. 9A). To lend further support to the notion that KLF5 and PIAS1 jointly regulate physiologic processes, both activate transcription of genes specific for smooth muscle cells such as those encoding smooth muscle myosin heavy chain and smooth muscle α-actin (76–78). PIAS1 and KLF5 may therefore play broader roles in regulating cell growth and development.
Mice with homozygous deletion of Pias1 had partial lethality (79). The surviving Pias1−/− mice were smaller, despite that Pias1 knock-out had no effect on apoptosis mediated by p53 or global SUMOylation (79). The runt phenotype may be caused by abnormal regulation of transcription factors in addition to disrupted STAT1 signaling (79). Based on the positive roles for KLF5 in promoting cell proliferation and blood vessel development (9, 11–13, 75) and activation of KLF5 by PIAS1, it would be interesting to examine whether KLF5 dysregulation could account for the phenotype in Pias1-deficient mice.
PIAS1 regulates G1/S transition (80). Its overexpression reduces the proportion of cells in the G1 phase and causes cell cycle reentry (80). In contrast, RNA interference of PIAS1 reduces the proportion of cells in the S phase and induces G2 arrest (80). Although these effects are at least mediated through p73 (80), the exact mechanism by which PIAS1 regulates the cell cycle is largely undefined. Co-transfection of COS-7 cells with PIAS1 and KLF5 resulted in a synergistic increase in BrdUrd incorporation above and beyond those transfected with KLF5 alone (Fig. 10). As KLF5 is known to enhance G1/S and G2/M transitions (11, 12), our results suggest that PIAS1 cooperates with KLF5 to accelerate the cell cycle at critical checkpoints.
PIAS1 interacts with both the amino terminus and the carboxyl-terminal zinc finger DNA-binding domain of KLF5, although only the acidic domain of PIAS1 is required for the interaction (Fig. 6). The strong positive charges from zinc ions may provide an electrostatic bridge for the highly acidic domain of PIAS1 to bind to the DNA-binding domain of KLF5. The pattern of KLF5-PIAS1 interaction is reminiscent of the interaction between PIAS1 and STAT1, a major target of PIAS1, as a region near the carboxyl terminus of PIAS1 (amino acids 392–541) interacts with the amino terminus of STAT1 (81). The association between KLF5 and PIAS1 is negatively regulated by the amino terminus of PIAS1. Interestingly, the amino terminus of PIAS1 also inhibits PIAS1-STAT1 interaction, preventing the binding of the carboxyl terminus of PIAS1 to STAT1 (81). As a result, partial but not full-length PIAS1 interacted with STAT1 (16, 81). KLF5 and STAT1 could exist in the same complex or compete with each other for PIAS1 binding.
Although PIAS1 acts as a SUMO E3 ligase, we have not detected SUMOylation of KLF5 to date. This could be due to the low abundance of SUMOylated KLF5. However, PIAS1 and SUMO can regulate cellular processes without SUMOylation (82). Whether KLF5 is regulated by SUMOylation in addition to PIAS1 binding is being investigated.
Both KLF5 and PIAS1 are downstream targets of mitogen-activated protein kinase (MAPK). The Ras-activated MAPK pathway activates KLF5 and stimulates cell proliferation (11, 12, 83). Interestingly, a PIAS1-related protein, PIASxβ, is also activated by MAPK (84). Activation of MAPK by transforming growth factor-β (TGF-β) not only enhances PIASxβ protein stability but also up-regulates the expression of PIASxβ mRNA (84). However, the underlying mechanisms were not identified. In this study, we show that KLF5 increases the protein abundance of co-expressed PIAS1, and PIAS1 activates its own promoter. Because TGF-β is a known target of KLF5 and activates p38 MAPK (75, 84), KLF5 may stabilize PIAS through TGF-β. It is possible that in some cell types, growth factors such as epidermal growth factor and TGF-β, which activate Ras/MAPK and p38 MAPK, respectively, may coordinately activate KLF5 and PIAS and promote cell proliferation stimulated by mitogens.
Acknowledgments
We are grateful to Dr. J. Lingrel for providing the mouse KLF5 cDNA; Dr. K. Morohashi for 3xFLAG-PIAS1 and -3; and Drs. D. Kaetzel, R. Pestell, and C. Glass for PDGF-A, cyclin D1, and Cdc2 reporter plasmids.
Footnotes
The abbreviations used are: KLF, Krüppel-like factor; ANKRD17, ankyrin-repeat domain 17; ARFGAP3, ADP-ribosylation factor GTPase-activating protein 3; BrdUrd, bromodeoxyuridine; BSA, bovine serum albumin; ChIP, chromatin immunoprecipitation; DCK, deoxycytidine kinase; FITC, fluorescein isothiocyanate; FXNa, putative amino peptidase FXNa; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HA, hemagglutinin; HNRPF, heterogeneous ribonucleoprotein F; ICGC, interchromatin granule cluster; MAPK, mitogen-activated protein kinase; NEM, N-ethylmaleimide; PCCA, propionyl coenzyme A carboxylase, α; PIAS1, protein inhibitor of activated STAT1; PI 3-kinase, phosphatidylinositol 3-kinase; PMM2, phosphomannomutase 2; pol II, RNA polymerase II; RRX, Rhodamine Red-X; SAF-A, scaffold attachment factor A; SAF-B, scaffold attachment factor B; SFRS15, splicing factor, arginine/serine-rich 15; SNAP23, synaptosomal associated protein 23; SNX3, sorting nexin 3; STAT1, signal transducer and activator of transcription 1; SUMO, small ubiquitin-like modifier; TBP, TATA-binding protein; TGF-β, transforming growth factor-β; X-gal, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside.
This work was supported in part by National Institutes of Health Grants DK52230, DK64399, and CA84197.
References
- 1.Bieker JJ. J Biol Chem. 2001;276:34355–34358. doi: 10.1074/jbc.R100043200. [DOI] [PubMed] [Google Scholar]
- 2.Black AR, Black JD, Azizkhan-Clifford J. J Cell Physiol. 2001;188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 3.Dang DT, Pevsner J, Yang VW. Int J Biochem Cell Biol. 2000;32:1103–1121. doi: 10.1016/s1357-2725(00)00059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaczynski J, Cook T, Urrutia R. Genome Biol. 2003;4:206.1–206.8. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Conkright MD, Wani MA, Anderson KP, Lingrel JB. Nucleic Acids Res. 1999;27:1263–1270. doi: 10.1093/nar/27.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi H, Zhang Z, Wang X, Liu S, Teng CT. Nucleic Acids Res. 1999;27:4807–4815. doi: 10.1093/nar/27.24.4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ghaleb AM, Nandan MO, Chanchevalap S, Dalton WB, Hisamuddin IM, Yang VW. Cell Res. 2005;15:92–96. doi: 10.1038/sj.cr.7290271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohnishi S, Laub F, Matsumoto N, Asaka M, Ramirez F, Yoshida T, Terada M. Dev Dyn. 2000;217:421–429. doi: 10.1002/(SICI)1097-0177(200004)217:4<421::AID-DVDY9>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 9.Bateman NW, Tan D, Pestell RG, Black JD, Black AR. J Biol Chem. 2004;279:12093–12101. doi: 10.1074/jbc.M311532200. [DOI] [PubMed] [Google Scholar]
- 10.Chanchevalap S, Nandan MO, Merlin D, Yang VW. FEBS Lett. 2004;578:99–105. doi: 10.1016/j.febslet.2004.10.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nandan MO, Chanchevalap S, Dalton WB, Yang VW. FEBS Lett. 2005;579:4757–4762. doi: 10.1016/j.febslet.2005.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nandan MO, Yoon HS, Zhao W, Ouko LA, Chanchevalap S, Yang VW. Oncogene. 2004;23:3404–3413. doi: 10.1038/sj.onc.1207397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun R, Chen X, Yang VW. J Biol Chem. 2001;276:6897–6900. doi: 10.1074/jbc.C000870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shuai K, Liu B. Nat Rev Immunol. 2005;5:593–605. doi: 10.1038/nri1667. [DOI] [PubMed] [Google Scholar]
- 15.Chung CD, Liao J, Liu B, Rao X, Jay P, Berta P, Shuai K. Science. 1997;278:1803–1805. doi: 10.1126/science.278.5344.1803. [DOI] [PubMed] [Google Scholar]
- 16.Liu B, Liao J, Rao X, Kushner SA, Chung CD, Chang DD, Shuai K. Proc Natl Acad Sci U S A. 1998;95:10626–10631. doi: 10.1073/pnas.95.18.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jackson PK. Genes Dev. 2001;15:3053–3058. doi: 10.1101/gad.955501. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt D, Muller S. Cell Mol Life Sci. 2003;60:2561–2574. doi: 10.1007/s00018-003-3129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dohmen RJ. Biochim Biophys Acta. 2004;1695:113–131. doi: 10.1016/j.bbamcr.2004.09.021. [DOI] [PubMed] [Google Scholar]
- 20.Gill G. Curr Opin Genet Dev. 2003;13:108–113. doi: 10.1016/s0959-437x(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 21.Gill G. Genes Dev. 2004;18:2046–2059. doi: 10.1101/gad.1214604. [DOI] [PubMed] [Google Scholar]
- 22.Girdwood DW, Tatham MH, Hay RT. Semin Cell Dev Biol. 2004;15:201–210. doi: 10.1016/j.semcdb.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 23.Hay RT. Mol Cell. 2005;18:1–12. doi: 10.1016/j.molcel.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Johnson ES. Annu Rev Biochem. 2004;73:355–382. doi: 10.1146/annurev.biochem.73.011303.074118. [DOI] [PubMed] [Google Scholar]
- 25.Ferrier V. Nat Cell Biol. 2002;4:E57. doi: 10.1038/ncb0302-e57. [DOI] [PubMed] [Google Scholar]
- 26.Kahyo T, Nishida T, Yasuda H. Mol Cell. 2001;8:713–718. doi: 10.1016/s1097-2765(01)00349-5. [DOI] [PubMed] [Google Scholar]
- 27.Liang M, Melchior F, Feng XH, Lin X. J Biol Chem. 2004;279:22857–22865. doi: 10.1074/jbc.M401554200. [DOI] [PubMed] [Google Scholar]
- 28.Megidish T, Xu JH, Xu CW. J Biol Chem. 2002;277:8255–8259. doi: 10.1074/jbc.C200001200. [DOI] [PubMed] [Google Scholar]
- 29.Schmidt D, Muller S. Proc Natl Acad Sci U S A. 2002;99:2872–2877. doi: 10.1073/pnas.052559499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okubo S, Hara F, Tsuchida Y, Shimotakahara S, Suzuki S, Hatanaka H, Yokoyama S, Tanaka H, Yasuda H, Shindo H. J Biol Chem. 2004;279:31455–31461. doi: 10.1074/jbc.M403561200. [DOI] [PubMed] [Google Scholar]
- 31.Prigge JR, Schmidt EE. J Biol Chem. 2006;281:12260–12269. doi: 10.1074/jbc.M510835200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timmers HT, Meyers RE, Sharp PA. Proc Natl Acad Sci U S A. 1992;89:8140–8144. doi: 10.1073/pnas.89.17.8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iborra FJ, Pombo A, Jackson DA, Cook PR. J Cell Sci. 1996;109:1427–1436. doi: 10.1242/jcs.109.6.1427. [DOI] [PubMed] [Google Scholar]
- 34.Jackson DA, Hassan AB, Errington RJ, Cook PR. EMBO J. 1993;12:1059–1065. doi: 10.1002/j.1460-2075.1993.tb05747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dundr M, Misteli T. Biochem J. 2001;356:297–310. doi: 10.1042/0264-6021:3560297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mintz PJ, Spector DL. J Struct Biol. 2000;129:241–251. doi: 10.1006/jsbi.2000.4213. [DOI] [PubMed] [Google Scholar]
- 37.Kim JL, Nikolov DB, Burley SK. Nature. 1993;365:520–527. doi: 10.1038/365520a0. [DOI] [PubMed] [Google Scholar]
- 38.Handwerger KE, Murphy C, Gall JG. J Cell Biol. 2003;160:495–504. doi: 10.1083/jcb.200212024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komatsu T, Mizusaki H, Mukai T, Ogawa H, Baba D, Shirakawa M, Hatakeyama S, Nakayama KI, Yamamoto H, Kikuchi A, Morohashi K. Mol Endocrinol. 2004;18:2451–2462. doi: 10.1210/me.2004-0173. [DOI] [PubMed] [Google Scholar]
- 40.Sapetschnig A, Rischitor G, Braun H, Doll A, Schergaut M, Melchior F, Suske G. EMBO J. 2002;21:5206–5215. doi: 10.1093/emboj/cdf510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Q, Pedigo N, Shenoy S, Khalili K, Kaetzel DM. Gene (Amst) 2005;348:25–32. doi: 10.1016/j.gene.2004.12.050. [DOI] [PubMed] [Google Scholar]
- 42.Aizawa K, Suzuki T, Kada N, Ishihara A, Kawai-Kowase K, Matsumura T, Sasaki K, Munemasa Y, Manabe I, Kurabayashi M, Collins T, Nagai R. J Biol Chem. 2004;279:70–76. doi: 10.1074/jbc.M306621200. [DOI] [PubMed] [Google Scholar]
- 43.Aravind L, Koonin EV. Trends Biochem Sci. 2000;25:112–114. doi: 10.1016/s0968-0004(99)01537-6. [DOI] [PubMed] [Google Scholar]
- 44.Nayler O, Stratling W, Bourquin JP, Stagljar I, Lindemann L, Jasper H, Hartmann AM, Fackelmayer FO, Ullrich A, Stamm S. Nucleic Acids Res. 1998;26:3542–3549. doi: 10.1093/nar/26.15.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hari KL, Cook KR, Karpen GH. Genes Dev. 2001;15:1334–1348. doi: 10.1101/gad.877901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saitoh N, Spahr CS, Patterson SD, Bubulya P, Neuwald AF, Spector DL. Mol Biol Cell. 2004;15:3876–3890. doi: 10.1091/mbc.E04-03-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mintz PJ, Patterson SD, Neuwald AF, Spahr CS, Spector DL. EMBO J. 1999;18:4308–4320. doi: 10.1093/emboj/18.15.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bregman DB, Du L, van der Zee S, Warren SL. J Cell Biol. 1995;129:287–298. doi: 10.1083/jcb.129.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antoine M, Reimers K, Wirz W, Gressner AM, Muller R, Kiefer P. Biochem Biophys Res Commun. 2005;335:146–153. doi: 10.1016/j.bbrc.2005.07.069. [DOI] [PubMed] [Google Scholar]
- 50.Yoshida T, Kokura K, Makino Y, Ossipow V, Tamura T. Genes Cells. 1999;4:707–719. doi: 10.1046/j.1365-2443.1999.00295.x. [DOI] [PubMed] [Google Scholar]
- 51.Yoshida T, Makino Y, Tamura T. FEBS Lett. 1999;457:251–254. doi: 10.1016/s0014-5793(99)01048-0. [DOI] [PubMed] [Google Scholar]
- 52.Yuryev A, Patturajan M, Litingtung Y, Joshi RV, Gentile C, Gebara M, Corden JL. Proc Natl Acad Sci U S A. 1996;93:6975–6980. doi: 10.1073/pnas.93.14.6975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Misteli T, Spector DL. Mol Cell. 1999;3:697–705. doi: 10.1016/s1097-2765(01)80002-2. [DOI] [PubMed] [Google Scholar]
- 54.Silins G, Grimmond S, Hayward N. Biochem Biophys Res Commun. 1998;243:273–276. doi: 10.1006/bbrc.1997.8091. [DOI] [PubMed] [Google Scholar]
- 55.Watt AJ, Jones EA, Ure JM, Peddie D, Wilson DI, Forrester LM. Mech Dev. 2001;100:205–215. doi: 10.1016/s0925-4773(00)00530-x. [DOI] [PubMed] [Google Scholar]
- 56.Sedgwick SG, Smerdon SJ. Trends Biochem Sci. 1999;24:311–316. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- 57.Nagai K. Curr Opin Struct Biol. 1996;6:53–61. doi: 10.1016/s0959-440x(96)80095-9. [DOI] [PubMed] [Google Scholar]
- 58.Grishin NV. Nucleic Acids Res. 2001;29:638–643. doi: 10.1093/nar/29.3.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith RK, Carroll PM, Allard JD, Simon MA. Development (Camb) 2002;129:71–82. doi: 10.1242/dev.129.1.71. [DOI] [PubMed] [Google Scholar]
- 60.Eriksson S, Cederlund E, Bergman T, Jornvall H, Bohman C. FEBS Lett. 1991;280:363–366. doi: 10.1016/0014-5793(91)80332-w. [DOI] [PubMed] [Google Scholar]
- 61.Hatzis P, Al-Madhoon AS, Jullig M, Petrakis TG, Eriksson S, Talianidis I. J Biol Chem. 1998;273:30239–30243. doi: 10.1074/jbc.273.46.30239. [DOI] [PubMed] [Google Scholar]
- 62.Gamachi A, Kashima K, Daa T, Nakatani Y, Tsujimoto M, Yokoyama S. Mod Pathol. 2003;16:1124–1131. doi: 10.1097/01.MP.0000092953.20717.48. [DOI] [PubMed] [Google Scholar]
- 63.Van Schaftingen E, Jaeken J. FEBS Lett. 1995;377:318–320. doi: 10.1016/0014-5793(95)01357-1. [DOI] [PubMed] [Google Scholar]
- 64.Damen G, de Klerk H, Huijmans J, den Hollander J, Sinaasappel M. J Pediatr Gastroenterol Nutr. 2004;38:282–287. doi: 10.1097/00005176-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 65.Cromphout K, Keldermans L, Snellinx A, Collet JF, Grunewald S, De Geest N, Sciot R, Vanschaftingen E, Jaeken J, Matthijs G, Hartmann D. Eur J Neurosci. 2005;22:991–996. doi: 10.1111/j.1460-9568.2005.04266.x. [DOI] [PubMed] [Google Scholar]
- 66.Pirard M, Achouri Y, Collet JF, Schollen E, Matthijs G, Van Schaftingen E. Biochem J. 1999;339:201–207. [PMC free article] [PubMed] [Google Scholar]
- 67.Whitney EM, Ghaleb AM, Chen X, Yang VW. Gene Expr. 2006;13:85–96. doi: 10.3727/000000006783991908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Otsu M, Hiles I, Gout I, Fry MJ, Ruiz-Larrea F, Panayotou G, Thompson A, Dhand R, Hsuan J, Totty N, Smith AD, Morgan SJ, Courtneidge SA, Parker PJ, Waterfield MD. Cell. 1991;65:91–104. doi: 10.1016/0092-8674(91)90411-q. [DOI] [PubMed] [Google Scholar]
- 69.Xu Y, Hortsman H, Seet L, Wong SH, Hong W. Nat Cell Biol. 2001;3:658–666. doi: 10.1038/35083051. [DOI] [PubMed] [Google Scholar]
- 70.Nogami S, Satoh S, Nakano M, Shimizu H, Fukushima H, Maruyama A, Terano A, Shirataki H. Genes Cells. 2003;8:17–28. doi: 10.1046/j.1365-2443.2003.00612.x. [DOI] [PubMed] [Google Scholar]
- 71.Yu VW, Gauthier C, St-Arnaud R. Ann N Y Acad Sci. 2006;1068:131–142. doi: 10.1196/annals.1346.027. [DOI] [PubMed] [Google Scholar]
- 72.Morikawa Y, Nishida H, Misawa K, Nosaka T, Miyajima A, Senba E, Kitamura T. Blood. 1998;92:129–135. [PubMed] [Google Scholar]
- 73.Fukao T, Yamada T, Tanabe M, Terauchi Y, Ota T, Takayama T, Asano T, Takeuchi T, Kadowaki T, Hata Ji J, Koyasu S. Nat Immunol. 2002;3:295–304. doi: 10.1038/ni768. [DOI] [PubMed] [Google Scholar]
- 74.Liu X, Zhang C, Xing G, Chen Q, He F. FEBS Lett. 2001;490:79–83. doi: 10.1016/s0014-5793(01)02134-2. [DOI] [PubMed] [Google Scholar]
- 75.Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami H, Nishimatsu H, Ishikawa T, Suzuki T, Morita H, Maemura K, Sata M, Hirata Y, Komukai M, Kagechika H, Kadowaki T, Kurabayashi M, Nagai R. Nat Med. 2002;8:856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- 76.Watanabe N, Kurabayashi M, Shimomura Y, Kawai-Kowase K, Hoshino Y, Manabe I, Watanabe M, Aikawa M, Kuro-o M, Suzuki T, Yazaki Y, Nagai R. Circ Res. 1999;85:182–191. doi: 10.1161/01.res.85.2.182. [DOI] [PubMed] [Google Scholar]
- 77.Liu Y, Sinha S, Owens G. J Biol Chem. 2003;278:48004–48011. doi: 10.1074/jbc.M301902200. [DOI] [PubMed] [Google Scholar]
- 78.Kawai-Kowase K, Kumar MS, Hoofnagle MH, Yoshida T, Owens GK. Mol Cell Biol. 2005;25:8009–8023. doi: 10.1128/MCB.25.18.8009-8023.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu B, Mink S, Wong KA, Stein N, Getman C, Dempsey PW, Wu H, Shuai K. Nat Immun. 2004;5:891–898. doi: 10.1038/ni1104. [DOI] [PubMed] [Google Scholar]
- 80.Munarriz E, Barcaroli D, Stephanou A, Townsend PA, Maisse C, Terrinoni A, Neale MH, Martin SJ, Latchman DS, Knight RA, Melino G, De Laurenzi V. Mol Cell Biol. 2004;24:10593–10610. doi: 10.1128/MCB.24.24.10593-10610.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liao J, Fu Y, Shuai K. Proc Natl Acad Sci U S A. 2000;97:5267–5272. doi: 10.1073/pnas.97.10.5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee YS, Jang MS, Lee JS, Choi EJ, Kim E. EMBO Rep. 2005;6:949–955. doi: 10.1038/sj.embor.7400511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kawai-Kowase K, Kurabayashi M, Hoshino Y, Ohyama Y, Nagai R. Circ Res. 1999;85:787–795. doi: 10.1161/01.res.85.9.787. [DOI] [PubMed] [Google Scholar]
- 84.Ohshima T, Shimotohno K. J Biol Chem. 2003;278:50833–50842. doi: 10.1074/jbc.M307533200. [DOI] [PubMed] [Google Scholar]
- 85.Song J, Zhang Z, Hu W, Chen Y. J Biol Chem. 2005;280:40122–40129. doi: 10.1074/jbc.M507059200. [DOI] [PubMed] [Google Scholar]