

Abstract
Understanding immune tolerance mechanisms is a major goal of immunology research, but mechanistic studies have generally required the use of mouse models carrying untargeted or targeted antigen receptor transgenes, which distort lymphocyte development and therefore preclude analysis of a truly normal immune system. Here we demonstrate an advance in in vivo analysis of immune tolerance that overcomes these shortcomings. We show that custom superantigens generated by single chain antibody technology permit the study of tolerance in a normal, polyclonal immune system. In the present study we generated a membrane-tethered anti-Igκ–reactive single chain antibody chimeric gene and expressed it as a transgene in mice. B cell tolerance was directly characterized in the transgenic mice and in radiation bone marrow chimeras in which ligand-bearing mice served as recipients of nontransgenic cells. We find that the ubiquitously expressed, Igκ-reactive ligand induces efficient B cell tolerance primarily or exclusively by receptor editing. We also demonstrate the unique advantages of our model in the genetic and cellular analysis of immune tolerance.
Immunological tolerance is important in preventing autoimmunity and in promoting the efficiency of immune responses. Tolerance has been assessed experimentally in two general ways. First, tolerance in normal polyclonal individuals has been measured by evaluating responses revealed by subsequent immunization in vivo or in vitro (for review see references 1, 2). This first approach has the advantage of addressing a normal immune system, but because of the extreme heterogeneity of lymphocyte specificity and the low precursor frequency of antigen-specific lymphocytes, it makes analysis of tolerance mechanisms difficult or impossible. A second approach has involved the use of antigen receptor transgenic mice, which artificially increase the frequency of antigen-reactive cells by limiting or fixing expression of one or both receptor chains, permitting the visualization of cells with fully or partly defined specificities (3–6). Antigen receptor transgenics have become indispensable reagents in the study of lymphocyte biology, allowing analysis of development, selection, specificity, and memory. But transgenic models are designed to distort lymphocyte development, subverting the “allelic exclusion” mechanisms to generate quasi-monoclonal immune systems in defined lymphocyte subsets. Furthermore, antibody transgenics, or the more recent approach of targeted transgenics, have a number of inherent drawbacks, owing to nonphysiological changes, such as accelerated B cell development, a tendency to skewing in B cell subset, and nonphysiological DH-to-VDJH joining (7–10). Therefore, a variety of experimental conclusions based on the use of antigen receptor transgenic mice, in particular the relative contributions of different tolerance mechanisms to central and peripheral immune tolerance, require verification in normal, polyclonal immune systems.
One mechanism by which tolerance can occur in B cells is receptor editing, in which self-reactivity directly or indirectly induces ongoing recombinase activator gene (RAG) expression and secondary Ig light chain rearrangements that alter receptor specificity (6, 11–13). Receptor editing may be a major self-tolerance mechanism in immature B cells (14–20). Many studies indicate that the precursor frequency of autoreactive B cells is high and that autoreactivity may be corrected by light chain exchange (11, 12, 14, 21–26). Recent studies by Wardemann et al. show that reverse genetic analysis of human B cell antibody expression can be a fruitful approach to study repertoire and tolerance (23, 26). However, to date, studies demonstrating in vivo receptor editing have primarily relied upon analysis of receptor gene transgenic mice, the limitations of which have been discussed.
On the other hand, immune responses can sometimes also be analyzed in polyclonal models of the immune system, and with appropriate experimental tools this analysis can provide additional advantages over receptor gene transgenics because lymphocyte development is not radically altered. For example, lymphocyte responses can be modeled by stimulation with anti-Ig and anti-TCR reagents (27–34), or through the use of TCRβ-reactive superantigens, which among other things provided early evidence for clonal deletion and T cell anergy in lymphocytes (35, 36). B cell–reactive superantigens also exist, notably HIV gp120, Staphylococcal toxin protein A, and Peptostreptococcus protein L (37–39).
The premise of this study is that custom superantigens, generated by single chain Fv antibody engineering technology, can be expressed as transgenes to regulate or tolerize a fully normal, polyclonal immune system. To test this idea, we generated mice expressing a single chain antibody combining site reactive to the constant portion of mouse Igκ L chain, which was engineered to be expressed as a membrane protein. We then used these mice to assess the mechanisms of B cell tolerance in vivo in a polyclonal immune system. This approach provides the advantages of a ligand that reacts identically with the antigen receptors of a high frequency of precursor cells, but does so without prior skewing of lymphocyte repertoire or subset, and in fact can be used with entirely normal cells. We call our synthetic superantigens “macroself” antigens to distinguish them from natural superantigens. We show here that a ubiquitously expressed Igκ-macroself antigen can promote central tolerance and receptor editing in a polyclonal immune system in vivo. Furthermore, we show that transgenic mice expressing κ-macroself antigen can facilitate the analysis of mutations that affect tolerance processes.
Results
Generation of κ-macroself antigen constructs and transgenic mice
To express an Igκ-specific macroself antigen of the predicted structure indicated schematically in Fig. 1 A, we assembled the gene construct depicted in Fig. 1 B. The antigenic specificity was generated by forming a single chain Fv from the variable genes of a rat anti–mouse Cκ hybridoma. We also chose to include hinge regions and Fc portion from rat IgG1 to promote protein stability, bivalency, and flexibility, and to project the binding sites away from the plasma membrane. The transmembrane and intracytoplasmic regions of the protein were derived from the H-2Kb gene, which we assumed were compatible with ubiquitous cell surface expression. A construct carrying the chimeric gene driven by a human cytomegalovirus promoter was transfected into L cells and stable clones were selected. Flow cytometry analysis revealed that the transfectants expressed the predicted chimeric protein on the cell surface as they reacted with a monoclonal antibody to the Fc portion of rat IgG1 (Fig. 1 C, left). Most importantly, the transfectants bound a mouse IgG2b monoclonal antibody carrying κ L chain, but not to a IgG2b/λ antibody (Fig. 1 C, right), indicating that the synthetic gene encoded a cell surface protein with the desired specificity for mouse κ chain.
Figure 1.
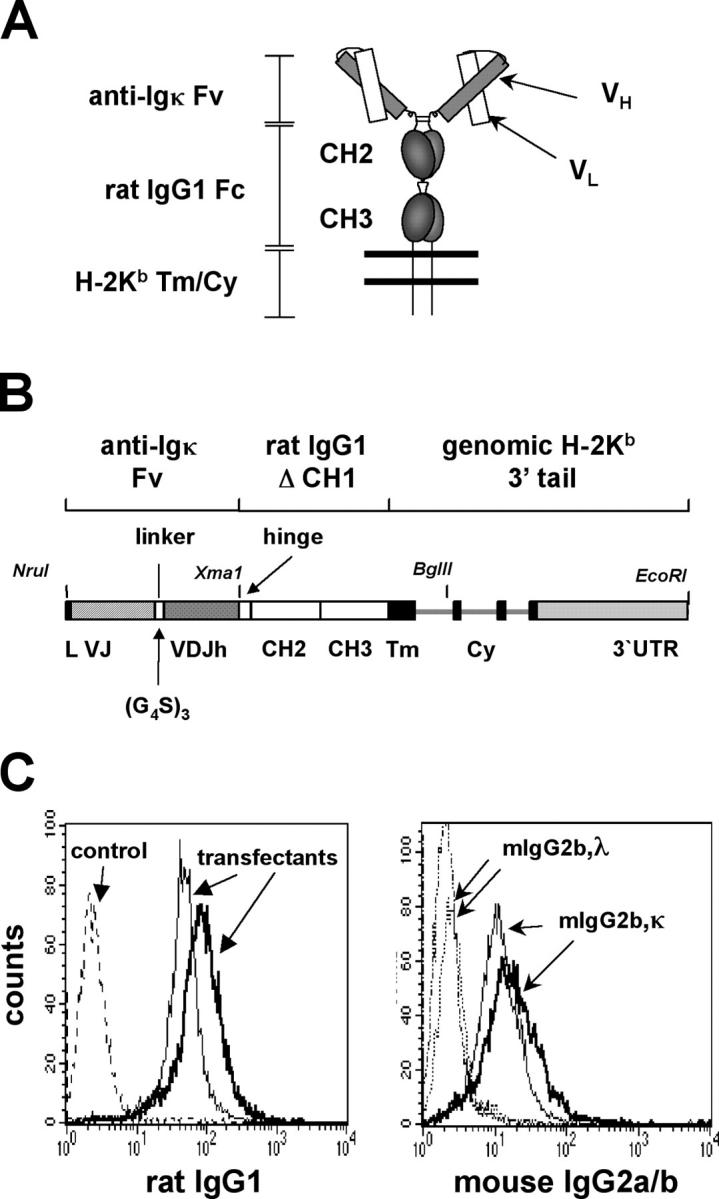
Design and in vitro testing of a synthetic B cell superantigen. (A) Schematic representation of the predicted protein structure of membrane bound anti–mouse Igκ-macroself Ag. A single chain Fv generated from the anti-κ hybridoma 187 is linked to the hinge and membrane proximal domains of rat IgG1 followed by transmembrane and cytoplasmic tail regions (Tm/Cy) of H-2Kb. (B) Gene construct encoding κ-macroself antigen showing intron/exon structure and selected features. Introns are depicted as thin lines. (G4S)3 refers to linker codons in one letter amino acid code: GGGGSGGGGSGGGGS. For stable transfection analysis, the gene shown was inserted into an expression vector providing a human cytomegalovirus promoter and zeocin resistance gene, generating plasmid pmSCA187ΔCH1. (C) Flow cytometry analysis of stably transfected cell lines. Two clones were analyzed for surface expression of the macroself Ag. (Left) Staining with an anti–rat IgG1 monoclonal antibody compared with empty vector-transfected control. (Right) Testing of binding specificity of the macroself antigen to mouse Ig light chain isotype. Two transfectant cell lines were incubated with soluble mouse IgG2b,κ or IgG2b,λ and binding revealed with a secondary rat anti–mouse IgG2a/b reagent.
To study the effect of a ubiquitously expressed κ-macroself antigen on B cells in vivo, transgenic mice were generated using the construct shown in Fig. 2 A. A leader intron was included in this construct because it improved expression in transient transfection by ∼10-fold (unpublished data) and the ubiquitin C promoter was chosen because it provided a more uniform and ubiquitous expression than a mouse MHC class I H-2Kb promoter tested (unpublished data). To avoid potential toxicity caused by maternal Igκ antibodies, microinjected zygotes were implanted in foster mothers that were Igκ deficient (40). Several transgenic lines were generated, four of which were selected for further study. As shown in Fig. 2 B, all lines expressed macroself antigen on virtually all tested cells: lines #2, #37, and #20 expressed at relatively high, uniform levels in lymphoid tissues, whereas line #26 was expressed more weakly, particularly in bone marrow.
Figure 2.

Generation of κ-macroself antigen transgenics and in vivo transgene expression. (A) Schematic representation of DNA construct used for microinjection to generate transgenic mice. Elements shown are approximately to scale. The pUliκ construct is a derivative of the gene shown in Fig. 1 B. It contains the κ-macroself Ag under the control of the human ubiquitin C promoter. The construct contains a Ig light chain gene leader exon and first intron. (B) Flow cytometry analysis of κ-macroself antigen expression in bone marrow, spleen, and lymph nodes of pUliκ transgenic mice as detected using an anti–rat IgG1Fc antibody (dotted lines, nontransgenic cells; solid line, pUliκ transgenic cells). Results from four different pUliκ transgenic lines are shown.
Flow cytometry analyses were performed to determine if the κ-macroself antigen influenced the development of κ-expressing B cells and to see if there was an effect on λ1 B cells. All four transgenic lines had a similar, striking phenotype: in the κ-macroself transgenic mice, virtually all of the B220+ cells expressed λ L chains and failed to express κ L chains (shown in Fig. 3 for line #2 and #26 splenocytes, and summarized for all lines in Fig. 4, D and E). As shown in the lower right panels of Fig. 3, ∼75% of B cells in the spleens of κ-macroself antigen mice were stained with a monoclonal antibody to λ1–3. The remaining cells likely expressed Vλx because these cells expressed sIgM, but failed to react to anti-κ or anti-λ1–3 antibodies; moreover, κ-deficient mice (κ2/−) had a similar population of IgM+,λ1–3 − cells (Fig. 3). Similar losses of κ1 cells and increases in λ1 cells were observed in lymph nodes of the transgenic mice (Fig. 4, D and E). Peripheral B cell numbers were reduced ∼50% in all κ-macroself antigen transgenic lines (total splenic cells and the B220+ fraction were reduced to, respectively, 80 and 60% of control levels; Fig. 4, A and C); in contrast, bone marrow B220+ cell numbers were unchanged in transgenic mice (Fig. 4 C). The percentages of λ1 cells in peripheral lymphoid tissues of κ-macroself transgenic mice were increased relative to nontransgenics by over sevenfold (Fig. 4 E), representing an overall numerical increase of three- to fourfold. These results indicate that in all κ-macroself transgenic lines κ1 cells were absent from the peripheral lymphoid organs and λ1 cell numbers were substantially increased.
Figure 3.

Flow cytometry analysis of κ-macroself transgenic lymphoid tissues showing reduction in frequency of κ1 B cells and increases in λ1 B cells. Cells from the indicated organs of 8-wk-old normal littermate, κ2/−, and transgenic lines #2 and #26 were analyzed. Lymphocytes were gated on forward scatter versus side scatter to eliminate myeloid cells, dead cells, and cell debris from the analysis (lymphocyte gate). The results shown are representative of at least four experiments. (Top) Analysis of expression of B220 and Igκ. Middle row of panels, costaining for Igλ1−3 and Igκ. (Bottom) B220+ gated cells were analyzed for expression of IgM and Igλ1−3. The percentage in each quadrant, rounded to the nearest 1% is indicated in the upper right corner of each plot.
Figure 4.
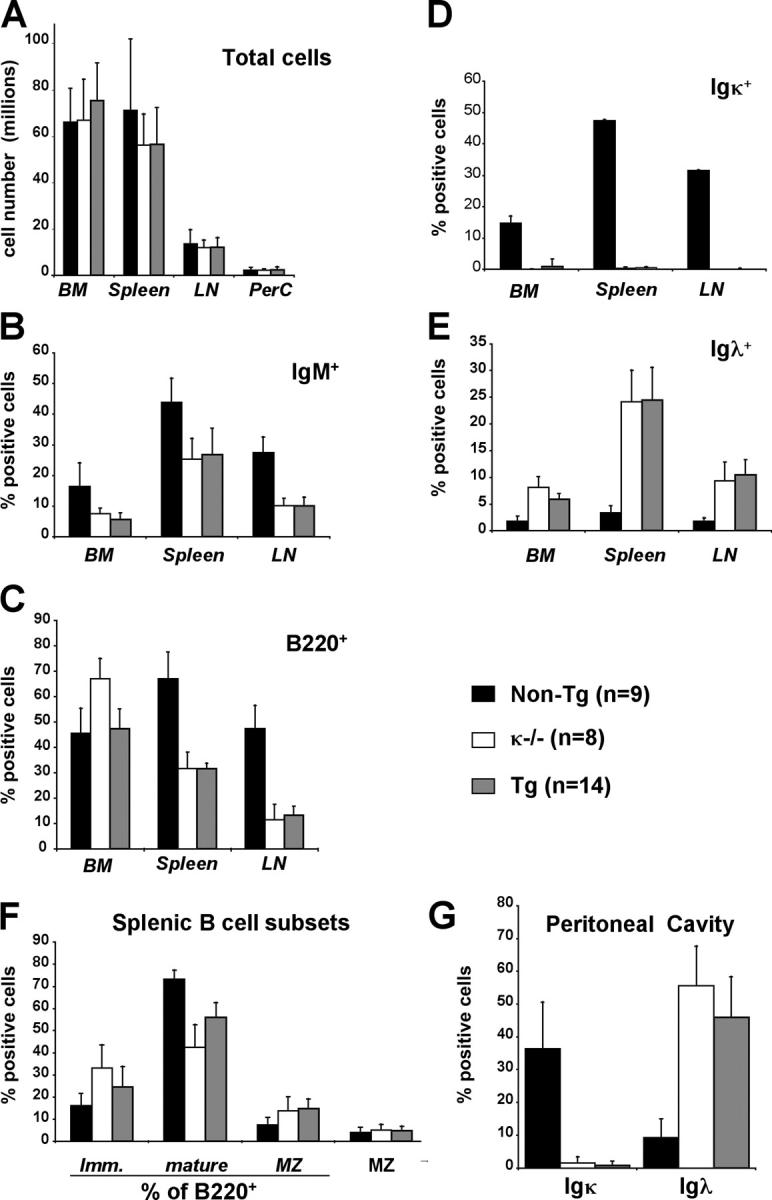
Elimination of Igκ B cell and increase of Igλ cell number in κ-macroself transgenic mice. (A–G) Histograms show mean of total cell numbers (A) and the percentages (±SEM) of (B) IgM+, (C) B220+, (D) Igκ and (E) Igλ in bone marrow, spleen, and lymph nodes of the indicated mouse genetic types. All data except A and G involved use of a lymphocyte gate. (F) Mean percentages of immature, mature, and marginal zone splenic B cells. (G) Shown are mean percentages of Igλ+ and Igκ+ B lymphocytes in the peritoneal cavity, gated on large, CD5+ lymphocytes to focus on the B-1 subset. These cells were B220intermediate, IgMhigh, and IgDlow. In the inset the number of experimental animals analyzed per group is noted in parentheses next to the symbol of mouse type.
Flow cytometry was also used to look for differences between normal and κ-macroself antigen-expressing mice in peripheral B cell subsets (Fig. 4, F and G). The κ-macroself antigen has the unique advantages of possessing comparable reactivity to receptors carried by most B cells present in all target populations. Among B220+ splenic cells there appeared to be similar or somewhat increased percentages of newly formed splenic B cells in κ-macroself transgenic lines compared with littermate controls, whereas the percentages of mature, follicular B cells were somewhat reduced and those of marginal zone B cells increased (Fig. 4 F). Taking into account the reduction of B220+ cells in the κ-macroself antigen-expressing mice, we estimate that marginal zone B cell numbers were not significantly reduced in the κ-macroself antigen transgenic mice, whereas follicular and newly formed B cells were reduced by ∼50 and 20%, respectively. Peritoneal B-1 cells in the κ-macroself antigen transgenic mice were similar in number to control littermates, but, like other peripheral B cells in these mice, expressed λ, rather than κ L chains (Fig. 4 G). Consistent with these observations, analysis of serum IgM and IgG2a/b revealed a loss of Igκ and an increase in λ-carrying Igs in κ-macroself transgenic mice (Table I). Thus, the immune system appears to readily adapt to the presence of an autoantigen reactive to 94% of normal B cell receptors.
Table I.
Serum immunoglobulins of unimmunized κ-macroself transgenic mice
| Mouse strain (n) | Total IgM | IgM, κ | IgM, λ | IgG2a/b, κ | IgG2a/b, λ |
|---|---|---|---|---|---|
| μg/ml | μg/ml | units/ml | μg/ml | μg/ml | |
| Nontransgenic (4) | 113 ± 78 | 75 ± 21 | 6 ± 4 | 211.0 ± 91.0 | 0.6 ± 0.1 |
| κ2/− (4) | 163 ± 60 | < 0.6 | 79 ± 39 | 0.6 ± 0.1 | 42.0 ± 8.0 |
| Transgenic #2 (4) | 188 ± 81 | < 0.6 | 76 ± 33 | 0.7 ± 0.2 | 101.0 ± 29.0 |
Analysis of receptor editing
To determine if the increase in λ1 B cells in κ-macroself antigen-expressing mice was the result of receptor editing, rather than proliferation of preexisting λ1 cells, we first quantitated in the bone marrow the frequency of newly formed, B220intermediate/λ1 cells, a measurement that has been used as an indicator of the rate of new B cell production (41). As shown in Fig. 5, λ B cell production is increased three- to fourfold in κ-macroself antigen-expressing mice compared with nontransgenic littermates. This increase was comparable to the increase seen in κ2/− mice, in which bone marrow output of λ1 cells is known to be elevated (41). To independently measure the rate of new λ B cell production, we quantitated Igλ gene excision products (Fig. 6, A and C). These episomal DNAs generated by gene assembly fail to replicate upon cell division, and hence are a sensitive indicator of receptor editing (11). As shown in Fig. 6 C, the levels of Vλ1-to-Jλ1 excision products were elevated approximately threefold in isolated B220+ bone marrow cells of κ-macroself antigen-expressing mice, relative to littermate controls, consistent with the estimated increased bone marrow output of λ1 cells. An even greater increase in the relative levels of λ excision product was seen in splenic B220+ cells of transgenic mice (Fig. 6 C), suggesting that most splenic cells had undergone little or no proliferation since their generation. Once again, κ-macroself antigen-expressing mice revealed a phenotype similar to κ2/− mice. We conclude that κ-macroself transgenic mice have an increased rate of λ gene rearrangement and λ1 B cell production in the bone marrow.
Figure 5.

Increased output of Igλ1 cells in κ-macroself antigen mice. (A) Bone marrow cells from κ2/−, κ-macroself transgenic, or nontransgenic littermate mice were stained with B220 and Igλ1−3 and analyzed by flow cytometry. The B220intermediate/λ1 population is found in the lower box. Data shown was analyzed using a lymphocyte gate. (B) Summary of quantitation of newly formed λ1 cells as illustrated in A. Newly formed λ1 cell numbers (obtained from the bone marrow of two legs) were 0.3 (±0.1), 0.7 (±0.2), and 1 (±0.3) million cells in the nontransgenic littermate, the κ2/− and the κ-macroself transgenic mice, respectively.
Figure 6.

Evaluation of molecular parameters of receptor editing in κ-macroself transgenic, littermate control, and κ2/− mice. (A, top rows) PCR detection of Vλ1–Jλ1 rearrangement excision products in genomic DNA extracted from anti-B220 magnetic bead purified B cells of bone marrow and spleen. Fourfold DNA serial template dilutions were tested. Tail DNA was used as a negative control (lane 17). PCR products were quantitated by Southern blot using specific probes. (Middle rows) Detection of RS-to-IRS1 and RS-to-IRS2 joins. (Bottom rows) PCR product located 3′ to the 3′-RS Igκ sequence is used as a DNA loading control (control). (B) Quantitation of RAG1 and RAG2 mRNA levels in B220+ bone marrow cells by quantitative Northern blot. (C) Quantitation of Ig Vλ1-to-Jλ1 excision product rearrangements detected by PCR in DNA of B220+ cells isolated from bone marrow or spleens of the indicated mouse types.
Independent indices of receptor editing were measured to verify the additional predictions that recombinase expression and destructive Igκ rearrangements should be increased in κ-macroself transgenic mice. RAG1 and RAG2 mRNA expression levels in κ-macroself transgenic bone marrow B220+ cells were significantly elevated by 80 to 90% over nontransgenic controls (Fig. 6 B). Recombining sequence (RS; 42, 43) recombination to the acceptor sites in the J-Cκ intron was also evaluated as a measure of destructive V(D)J recombination on the κ locus of B cells (Fig. 6 A). RS, which is found ∼25-kb downstream of the Cκ exon, undergoes RAG-mediated recombination to Vκs and to two acceptor sites in the J-Cκ intron (IRS1 and IRS2) that inactivate the κ locus (44). RS recombinations are common in λ1 cells and are also seen in ∼10% of normal κ1 cells (16, 45, 46). RS recombination to IRS1 and IRS2 was elevated in bone marrow and spleens of κ-macroself transgenic mice (Fig. 6 A, RS to κ intron rows). Taken together with data showing similar increases in RAG expression and λ1 B cell production in κ-deficient and κ-macroself transgenic mice, the data suggested that κ-macroself antigen induces efficient central B cell tolerance in an otherwise normal immune system by a mechanism of developmental arrest and receptor editing.
Analysis of bone marrow B cell turnover and intracellular Ig expression
To further quantitate the relative roles of receptor editing and clonal deletion in the tolerance induced by κ-macroself antigen, the intracellular Igκ expression and BrdU uptake of newly formed bone marrow B cells was assessed. Despite the data indicating excess RAG expression, RS recombination and λ gene excision product DNA in the bone marrow of κ-macroself mice, it remained possible that clonal deletion depleted developing κ1 cells, allowing excess survival and a nonproliferative buildup of λ1 cells. Such a hypothesis predicts an increased turnover of B cells in κ-macroself bone marrow and a drastic reduction of developing B cells carrying κ chains, whereas editing should not increase B cell turnover. Turnover was assessed by daily injection of BrdU and assessment of uptake in bone marrow B cells over time (47). As shown in Fig. 7 A (left), BrdU uptake in the B220intermediate bone marrow B cell population was similar or slightly slower in κ-macroself transgenic mice, compared with wild-type littermates, indicating that the κ-macroself antigen does not accelerate bone marrow B cell turnover. BrdU uptake was delayed in the sIgM fraction of newly formed κ-macroself transgenic B cells compared with wild type, suggesting a longer average transit time (Fig. 7 A, right). These results were further supported by intracellular immunoglobulin staining experiments as κ-macroself antigen mice had significant numbers of intracellular κ1 B cells in bone marrow, but not in the spleen (Fig. 7 B, lower rows). The frequency of cytoplasmic Igκ1 cells was only marginally reduced (by ∼13%) among the newly formed, B220intermediate B cells in the presence of κ-macroself antigen (Fig. 7 D). Moreover, cytoplasmic κ1 cells costained with an IgM antibody (Fig. 7 C). Because both the anti-κ and anti-IgM antibodies used see only assembled immunoglobulins (48, 49), we infer that the bone marrow B cells of κ-macroself antigen mice that carry sIgκ undergo receptor down modulation followed by receptor editing.
Figure 7.
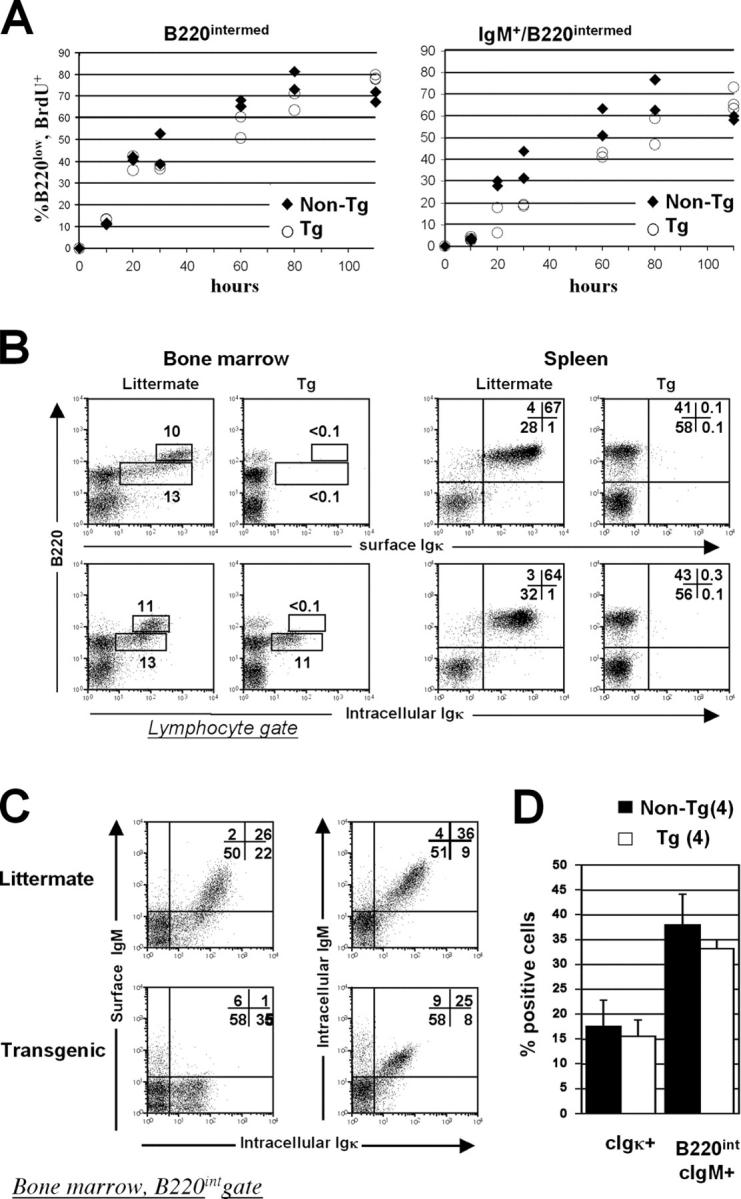
Intracellular immunofluorescence analysis of BrdU uptake and immunoglobulin expression in κ-macroself transgenic mice and littermates. (A) BrdU uptake with time of labeling in bone marrow B220intermediate cells of transgenic mice (open circles) or littermates (diamonds). (Left) BrdU incorporation in total B220intermediate cells; (right) BrdU incorporation in sIgM+/B220intermediate cells. (B) Comparison of surface Igκ staining (top), with intracellular Igκ staining (bottom). (C) Costaining for intracellular Igκ and either sIgM alone (left) or both surface and cytoplasmic IgM (right). (D) Statistical analysis of experiments shown in b and c. Left pair of bars shows the percentages of cells found in lower analysis boxes of B220/intracellular Igκ stain (i.e., B, bottom). The right pair of bars shows percentages of cells in upper two quadrants of intracellular IgM/intracellular Igκ stain with B220low gate (C, right). Means and standard deviations are indicated. Filled bars, transgenic; open bars, nontransgenic littermates.
Transplantation of mutant bone marrow in κ-macroself antigen-expressing hosts
Because the κ-macroself transgenics express antigen on all cells tested, including B cells, we assessed B cell tolerance in radiation bone marrow chimeras in which κ-macroself transgenic line #2 mice served as hosts and donor bone marrow cells lacked macroself transgenes (Fig. 8 and Table II). Three types of bone marrow donors were used in these experiments: nonmutant controls (wt), mice carrying a hemizygous deficiency in the RAG1 gene (RAG+/−; 50), and mice carrying a Bcl2 transgene expressed in the B cell compartment (51). Use of wild-type donors assessed the ability of κ-macroself antigen to promote tolerance in a population of cells that was completely normal and lacking in any genetic modifications. The results observed in line #2 hosts carrying wild-type donor cells were generally similar to those obtained in unmanipulated line #2 mice, except that cells in the bone marrow carrying low levels of Igκ were more apparent (Fig. 8 A, compare lower left pair of panels). Splenic B cells in transgenic #2 recipients were largely devoid of κ expression, but instead expressed λ chains (Fig. 8 C, right panels under “wt”). In addition, transgenic #2 recipients manifested an increased frequency of B220intermediate, λ1 cells in the bone marrow (Fig. 8 A). Thus, nontransgenic B cells developing in κ-macroself line #2 appeared to undergo central tolerance and receptor editing.
Figure 8.
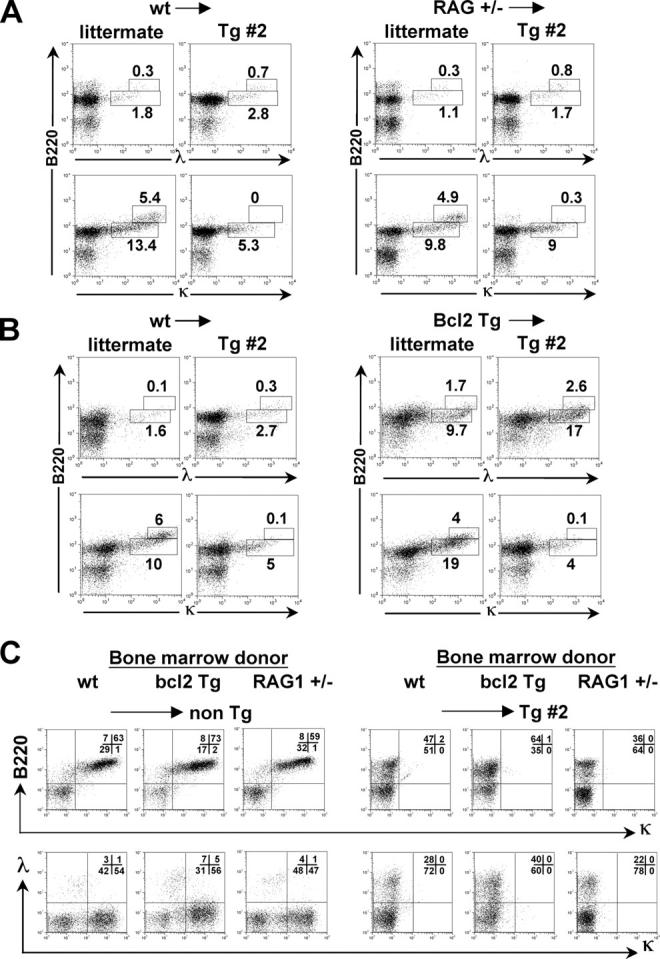
Flow cytometry analysis of tolerance induction in radiation chimeras using κ-macroself transgenic hosts. Mice were analyzed at 6 wk post reconstitution. (A and B) Analysis of bone marrow lymphocytes using anti-L chain and B220 antibodies. Bone marrow donors are indicated to the left of the arrows, the recipient mouse genotypes are shown just below. Newly formed lymphocytes carrying Ig-κ or -λ, were identified as falling in the lower analysis boxes, as indicated. (A) B6 (wt) or RAG+/− bone marrow was used to reconstitute lethally irradiated CD45.1+/κ-macroself transgenic #2 (Tg #2) or littermate recipients. (B) Comparison of chimeras generated with bone marrow from Bcl-2 transgenic or littermate (wt) donors, using as irradiated recipients either transgenic #2 or littermate. (C) Analysis of spleen cells from the indicated radiation chimeras. Recipient mouse genotype is shown to the right of arrows above dot plots. Cells were stained with B220 and anti-κ antibodies (top) or anti-κ and anti-λ antibodies (bottom).
Table II.
Effect of Bcl2 overexpression and RAG1+/− mutation in donor cells on receptor editing and tolerance induction in κ-macroself radiation chimeras
| Percent positive mean ± SD |
||||||||
|---|---|---|---|---|---|---|---|---|
| Donor → Host (n)
|
Cells /106
mean ± SD |
B220
|
IgM
|
κ
|
λ
|
κ+B220int
|
λ+B220int
|
|
| Bone marrow | ||||||||
| Wild type | → wt (5) | 68 ± 6.6 | 78 ± 13 | 15 ± 2 | 21.0 ± 2.0 | 2.7 ± 0.5 | 12.8 ± 2.0 | 1.7 ± 0.1 |
| → Tg#2 (4) | 50 ± 13 | 74 ± 9 | 8 ± 3a
|
5.2 ± 1.8a | 4.9 ± 1.4a | 4.4 ± 1.3a | 2.7 ± 0.3a | |
| Bcl2 | → wt (4) | 56 ± 13 | 84 ± 5 | 35 ± 2 | 33.0 ± 7.0 | 8.6 ± 1.2 | 22.5 ± 3.0 | 9.3 ± 0.6 |
| → Tg#2 (3) | 61 ± 17 | 74 ± 8 | 22 ± 2a | 5.7 ± 0.4a | 15.0 ± 2.5a | 4.5 ± 0.4a | 15.0 ± 3.0a | |
| RAG1+/− | → wt (4) | 52 ± 7 | 67 ± 11 | 16 ± 2 | 21.0 ± 3.0 | 2.3 ± 0.7 | 9.7 ± 0.4 | 1.5 ± 0.4 |
| → Tg#2 (3) | 42 ± 2 | 60 ± 10 | 6 ± 2a | 8.3 ± 2.0a | 2.5 ± 0.7 | 7.4 ± 0.5a | 1.3 ± 0.2 | |
| Spleen | ||||||||
| Wild type | → wt (5) | 67 ± 23 | 66 ± 3 | 50 ± 2 | 5.0 ± 6.0 | 4.0 ± 1.0 | ||
| → Tg#2 (4) | 43 ± 7a | 36 ± 7a | 28 ± 5a | 0.6 ± 0.3a | 27.0 ± 7.0a | |||
| Bcl2 | → wt (4) | 146 ± 33 | 84 ± 3 | 63 ± 14 | 65.0 ± 8.0 | 10.0 ± 2.0 | ||
| → Tg#2 (3) | 83 ± 20a | 73 ± 15 | 49 ± 10 | 1.0 ± 0.6a | 41.0 ± 5.0a | |||
| RAG1+/− | → wt (4) | 64 ± 6 | 68 ± 4 | 50 ± 3 | 61.0 ± 4.0 | 4.7 ± 0.5 | ||
| → Tg#2 (3) | 39 ± 18a | 29 ± 6a | 26 ± 3a | 0.2 ± 0.1a | 19.0 ± 7.0a | |||
Bone marrow sample data shows total cell counts, but percentages of B cell subsets are based on a lymphocyte gate. B220int refers to cells with intermediate levels of B220 and the indicated Ig light chain.
Items marked represent significant (P < 0.05) differences between cells maturing in transgenic #2 compared to wild-type littermate hosts. Items in bold represent significant (P < 0.05) differences associated with the mutant donor type, compared to wild-type donor type in the comparable host.
The RAG+/− mutant and Bcl2 transgenic donors were tested to determine the feasibility of using κ-macroself transgenics to rapidly screen mouse mutant strains for defects in immune tolerance. We reasoned that enforced Bcl2 expression should suppress apoptosis, whereas heterozygous deficiency in RAG1 might hinder receptor editing. Bone marrow chimeras carrying RAG+/− or Bcl2 transgenic bone marrow manifested subtle, but reproducible, differences from chimeras reconstituted with wild-type bone marrow (shown in bold in Table II). Chimeras reconstituted with Bcl2 transgenic marrow had elevated frequencies of λ1 cells in the absence of κ-macroself antigen, and an even further enhanced λ frequency in κ-macroself recipients (Fig. 8 C); furthermore, these increases were paralleled by a rise in newly formed B220intermediate/λ1 cells in the bone marrow (Fig. 8 B). We conclude that the survival-enhancing effects of enforced Bcl2 expression do not block central B cell tolerance to κ-macroself antigen, but instead promote increased generation of λ1 cells. This increase in λ, along with the further-reduced frequency of κ1 cells in the bone marrows of κ-macroself recipients reconstituted with Bcl2 transgenic cells, likely occurs because of an extended lifetime for editing. In contrast, chimeras reconstituted with RAG+/− bone marrow had a subnormal peripheral B cell frequency that was exacerbated by the presence of κ-macroself antigen. Furthermore, RAG+/− cells had a significantly smaller κ-macroself antigen-induced increase in the rate of λ1 B cell production relative to wild type (Fig. 8 A). An additional anomaly observed in RAG+/− chimeras was a relative retention in the bone marrow of κ-macroself antigen recipients of cells carrying high levels of Igκ (Fig. 8 A, lower right). Thus, loss of surface Igκ expression in bone marrow B220+ cells of κ-macroself antigen transgenic mice is partly related to editing and is not wholly a result of receptor protein down modulation. Similar results were obtained in RAG+/− mice carrying the line #26 transgene (unpublished data). These results suggest that reduced RAG1 gene dosage suppresses or, more likely, slows tolerance-induced receptor editing, reducing the κ to λ isotype switch in developing B cells. This finding in turn may suggest that RAG1 levels are limiting for receptor editing.
Discussion
The κ-macroself antigen approach that we introduce here is a strategy to study immunological tolerance in a large polyclonal cohort of cells. In the present study, we have used these mice to study immune tolerance in an otherwise unmanipulated immune system. We were able to revisit the question of whether or not B cell tolerance to a ubiquitous, membrane-tethered self antigen occurs by receptor editing or deletion (or if it occurs at all in normal cells). Importantly, our approach allowed us to assess the relative contributions of editing and deletion in a normal immune system. The data indicate that both cell death and receptor editing contribute to tolerance because substantial increases in λ light chain gene recombination and new λ1 B cell production occur in κ-macroself antigen mice, but their overall B cell numbers are reduced to ∼50%. It is known that λ loci almost always recombine after κ loci (45, 52), typically ∼24 h later (53, 54). However, we believe that apoptosis induced rapidly by autoreactivity (clonal deletion) appears to play a minimal role in our model, whereas the cellular time limit for editing to eliminate an autoreactive receptor exerts a more severe restriction. Because ∼94% of B cells in nontransgenic mice express κ, the reduction in B cell numbers is less than predicted if generated κ1 B cells are rapidly eliminated by cell death and not replaced by other cells. Indeed, B cells in κ-macroself transgenic mice resembled those of κ2/− mice, which cannot be subject to negative selection of sIgκ (Fig. 3). Tolerance purely by rapid clonal deletion of developing B cells would be predicted to reduce κ1 B cell output while leaving λ1 B cell generation unchanged. Results of BrdU uptake studies and Igκ cytoplasmic staining were incompatible with significant rapid clonal deletion. Indeed, in the presence of κ-macroself antigen the turnover of immature B cells was actually slowed, while the fraction of newly formed λ1 cells increased substantially. Rather, the results are most easily explained by a tolerance-induced developmental arrest, followed by significant, but incomplete, rescue by editing.
If editing occurs by developmental arrest without induced deletion in κ-macroself transgenic mice these transgenics should resemble κ2/− mice. This is in fact the case: their percentages of B cells in various lymphoid compartments are quite similar. We have assessed mice carrying κ-macroself antigen on a background in which one of the κ constant alleles carried the human sequence (17) and found minimal B cell attrition compared with wild type (unpublished data). This would be consistent with findings indicating that the efficiency of rescue of autoreactive cells by editing is visibly affected only by severely reducing the number of L chain genes available for editing, such as by knocking out the κ locus, or limiting the available repertoire of Jκs (20, 25, 55–57). In contrast to these studies, however, our present results were obtained in a context in which B cell development was unaltered.
Anti-Ig suppression of B cell development in vivo and in vitro has been studied for many years (27–32, 34, 58–60). Work first done in chickens, then later in mice and rats, showing that antibodies introduced into developing embryos suppressed B cell development represented the first evidence for clonal abortion (for review see reference 61). In some studies, evidence of developmental arrest was seen, including “irreversible” receptor down-regulation (28, 59). Typically, mature B cells were absent from the periphery while B cell progenitors were retained in the bone marrow. However, in other studies, anergy was induced, rather than deletion (30, 32). Often, competitive advantage was had by B cells that did not react with antibody (31, 58, and for review see reference 61). Our results are similar in several ways to a study of anti-κ suppressed mice by Weiss et al., in which the loss of κ1 B cells and κ immunoglobulin in serum was compensated for by an increase of peripheral λ1 B cells and serum immunoglobulin λ (31). In any case, the prior studies were often consistent with our conclusions in that the immature B cells receiving anti-Ig constant region stimulus did not die immediately, though none of these studies proposed an editing type escape mechanism.
Our ability to probe immune tolerance using macroself transgenic mice as adoptive hosts for normal or mutant bone marrow is important for several reasons. First, it establishes that the macroself antigen need not be expressed by B cells to induce tolerance in wild-type B cells. Second, we establish that wild-type B cells are tolerance susceptible by editing in vivo, a finding that to our knowledge has never before been directly demonstrated in a non-Ig transgenic mouse. Third, we demonstrate that macroself transgenic mice allow one to screen mutant bone marrow for defects in immunological tolerance. We confirmed earlier results, obtained in 3–83 antibody transgenic mice carrying autoreactive receptors, indicating that enforced B cell expression of Bcl2 facilitated receptor editing, but did not rescue autoreactive B cells from central tolerance (62). This last is consistent with the notion that the incomplete editing to λ in κ-macroself antigen transgenic mice is limited by the life span of editing competent cells, which can be artificially prolonged by enforced Bcl2 expression. In other words, we believe that autoreactive cells arrested in development and undergoing light chain rearrangements are subject to death by neglect, which can be slowed by Bcl2. Furthermore, we have made the novel additional finding that reducing RAG1 gene dosage impairs B cell production and receptor editing. (This result was independently confirmed using conventional Ig transgenic mice; unpublished data).
Although antigen receptor transgenic mice are a valuable resource for many studies, by design they perturb lymphocyte development, resulting in a number of anomalies. For example, targeted or randomly integrated transgenes are typically prematurely expressed, and are generally in an unusual antigen receptor gene context, such as in conventional IgH transgenics, which cannot undergo H chain class switch, or in targeted VDJh transgenics, where the introduced gene is downstream of germline D elements and can be eliminated by DJ joining. Furthermore, antigen receptor transgenes usually skew lymphocyte subset distributions, such as CD4/CD8 ratios in T cells or B-1/follicular ratios in B cells. Macroself antigen mice overcome many of these drawbacks. Importantly, the macroself approach can, in theory, be easily used for any antigen receptor for which there is a monoclonal antibody.
We note that B cells have the striking propensity to alter their Ig constant region usage during development, a feature that makes their analysis by challenge with macroself Ags potentially fruitful. Early in development, when bone marrow B cells express exclusively sIgM, receptor editing can cause successive Ig-κ allele usage, and can lead to a progression from usage of κ to λ L chain. Later in development, B cells up-regulate sIgD, marking the beginning of a phase that includes migration to the spleen and final preimmune maturation. After encounter with antigen, reactive B cells often undergo a heavy chain class switch, leading to the loss of IgM and IgD, and their replacement by downstream H chain C regions. Hence, macroself Ags reactive to Ig-κ, IgD, and IgG C-regions would be expected to promote immune tolerance in a polyclonal immune system at different developmental stages, and quite possibly by different mechanisms. As macroself antigens with specificity to other antigen receptors, such as TCRs, can be easily generated, they may provide useful tools for studies in other cell types. Future studies will focus on such possibilities.
With the advent of germline mouse knockout technology, autoimmune-prone congenic strains, and genome-wide ethylnitrosourea mutagenesis studies, there is an increasing need to devise methods to rapidly screen mutant mice for immune tolerance phenotypes. At the present time, this is typically approached by crossing of the knockout in question to antigen receptor transgenic mice and then introducing the cognate (auto)antigen. Because it requires a minimum of two generations of breeding, and extensive mouse genotyping, this approach is time consuming and expensive, and therefore not ideal for the screening of large numbers of mutants. Generation of well-conceived macroself Ag mice could provide a means to greatly speed up this screening process. Our results show that mutant mouse bone marrow or fetal liver precursor cells could be used to reconstitute lethally irradiated macroself Ag-expressing mice, and the development and function of the transferred cells could be directly monitored. Because the macroself Ag mice carry specific ligands that react with a subset of normal lymphocytes, no special breeding is required, particularly if the macroself Ag transgene is maintained on the same genetic background. Macroself Ag transgenic mice with specificity for defined lymphocyte receptor elements can facilitate the analysis of mouse mutants, particularly those with known signaling defects.
Materials and Methods
Generation of κ-macroself gene constructs
RNA was isolated from the rat anti-κ hybridoma 187(49) (American Type Culture Collection), and expressed antibody genes were cloned by 5′ rapid amplification and cloning of ends (5′-RACE) using the RLM-RACE PCR kit (Ambion) according to the manufacturer's instructions. The variable genes were amplified using sense primer specific to the 5′ adaptor either with a rat Cκ-specific anti-sense primer (5′-CTAACTGTTCCGTGGATGGTGGG) to amplify the light chain variable region, or a rat IgG1 anti-sense specific primer (5′-GGCTCCAGAGTTCCAGGTCACGG) to amplify the heavy chain variable region, together with their respective leader sequences. PCR products were then cloned in a plasmid vector using the TOPO TA cloning kit (Invitrogen) and several clones sequenced to obtain consensus sequences and to facilitate the identification of clones lacking mutations introduced by the amplification and cloning. Independently, rat IgG1 H chain constant regions were amplified from cDNA. To generate a single chain antibody gene and linked elements a PCR sewing approach was taken using the following oligonucleotide primers: primer 1 (5′VL) 5′-TCGCGAATCGCCGACAGGTGCGATGGACATGAGGGCCCATGCTC-3′; primer 2 (3′VL) 5′-CCTCCCGAGCCACCGCCTCCGCTGCCTCCGCCTCCCCGTTTTATTTCCAACTTCGTCCCG-3′; primer 3 (5′VH) 5′-GCAGCGGAG-GCGGTGGCTCGGGAGGCGGAGGCTCGCAGGTACAGCTGAAAGAGTCAGG-3′; primer 4 (3′VH) 5′-CCCGGGTTTCTGGGGGCTG-TTGTTTCAGCTGAGGAGAC-5′; primer 5 (5′hinge) 5′-CAGAAACC-CGGGAGGTGATTGCAAGCC; primer 6 (3′CH3) GGGAGTGGGAG-AGACTCTTCTCAGTATGGTGG; 7 (5′Tm) 5′-GTCTCTCCCACTC CCCCGGTAAAGAGCCTCCTCCATCCAC; 8 (3′Tm) 5′-CCAACTTCACTCCAATGTCCCC. The “overlapping” sequences are underlined; those recognized by restriction endonucleases are in italics. PCR products corresponding to the LVJL and the VDJH region were initially amplified with the primers 1 and 2, and 3 and 4, respectively. Rat Cγ1 cDNA lacking the CH1 domain (Cγ1ΔCH1) was amplified using primers 5 and 6. A segment of the genomic H-2Kb locus encoding the transmembrane (Tm) exon 5 was amplified using primers 7 and 8. The single chain Fv gene, including the intervening flexible peptide codons (Gly4-Ser)3, was assembled by a second PCR step with the outer primers 1 and 4, using as templates the LVL and VH PCR products at a 1:1 molar ratio (total of 50 ng of DNA). Similarly, the hinge, Fc, and Tm coding regions were assembled together by recombinant PCR with the primers 5 and 8 using as DNA templates PCR products of primers 5 + 6 and 7 + 8. The Fv and the Cγ1ΔCH1 coding regions were then successively cloned upstream of the genomic cytoplasmic tail (exon 6–8) and the 3′ UTR of the H-2Kb gene in pBluescript II SK as NruI/XmaI and XmaI/BglII restriction fragments, respectively. The resulting chimeric gene was then cloned downstream the cytomegalovirus gene promoter (pCMV) as a NruI /EcoRI restriction fragment into the pBudCE4.1 expression vector (Invitrogen), generating plasmid pBudSCA187ΔCH1, which was used for transfection analysis. The construct used for the production of transgenic mice (pUIik) was obtained as follows: the leader and the first intron of the RF IgL chain (a gift from M. Shlomchik, Yale University, New Haven, CT; reference 63) were added upstream of the 187.1 Fv by recombinant PCR using the following primers: primer 9 (5′-leader) 5′-TCGCGAATCGCCGACAGGTGCGATGGAATCACAGACCCAGGTCC; primer 10 (3′-intron) 5′-GGTCATCTGAATGTCTGCACAGGCACCTGTAATAATTAATAGGC; primer 11 (5′-scFv) 5′-TACAGGTG CCTGTGCAGACATTCAGATGACCC; and primer 4 (see above). The leader intron and the scFv 187.1 were first amplified by PCR using the primers 9 and 10, and 11 and 4, respectively. The leader intron and the scFv 187.1 were fused together by recombinant PCR using the outer primers 9 and 4, and then cloned as a NruI/XmaI restriction fragment upstream of the mCγ1ΔCHI coding sequence. Finally, the 1.2-kb human ubiquitin C gene promoter (64) was cloned upstream of the coding sequences as a HindIII/NruI restriction fragment in pBluescript II SK.
Stable transfection of L929 cells
L929 cells were transfected with pBudSCA187ΔCH1 constructs using Lipofectamine/Plus reagent (Invitrogen) on six-well plates according the manufacturer's recommendations. Stable transfectants were selected after 3 wk of growth in complete IMDM medium containing 100 μg/ml of Zeocin.
Mice
All mice were bred and maintained in the TSRI Animal Resources facility according to The Scripps Research Institute Institutional Animal Care and Use guidelines. C57BL6/J (B6), B6.RAG1−/− (50) and B6.CD45.1 mice were obtained from Jackson ImmunoResearch Laboratories. Germline Ig Jκ-Cκ–deleted mice (40) were provided by D. Huzsar (GenPharm Intl., San Jose, CA). EmuBcl-2-22 transgenic (Bcl2 Tg) mice (51), were provided by Drs. Strasser and Harris (The Walter and Eliza Hall Institute of Medical Research, Melbourne, Australia).
Production of transgenic mice
The 4-kb pUliκ transgene construct was separated from bacterial vector sequences by a digestion with HindIII/EcoRI and agarose gel electrophoresis. The fragment was isolated and purified on Elutip-d columns (Schleicher & Schuell) according the manufacturer's recommendations and dialyzed overnight against zygote injection grade Tris-HCl-EDTA. Transgenic mice were produced by classical microinjection techniques at the TSRI Mouse Genetics Core Facility. (B6 x DBA/2)F1 zygotes were microinjected with the pUliκ transgene and reimplanted into the oviducts of pseudo pregnant κ2/− foster mothers. Mice analyzed had been backcrossed three times to the B6.CD45.1 background.
Bone marrow chimeras
Recipient mice were κ-macroself transgenics or littermate controls; all carried the CD45.1 allele. Recipients received 950 rads γ radiation from a Cs source on the day of transfer. Bone marrow donors were all of the CD45.2 allotype. Bone marrow cell suspensions were depleted of T cells by treatment with biotinylated anti-Thy1.2 antibodies and antibiotin magnetic beads (Miltenyi Biotec) according to the suggested protocol. Five million cells were transferred i.v. per recipient. After 6 wk, recipients were killed and their lymphoid tissues analyzed by flow cytometry. Only chimeras in which ≥98% of cells in bone marrow and spleen were donor derived were included in the analysis.
Flow cytometry analysis.
For the analysis of mouse cells ex vivo, nucleated cell suspension were prepared from bone marrow, spleen, mesenteric lymph nodes, and peritoneal cavity. Erythrocytes were eliminated from the spleen and bone marrow preparations by ammonium chloride treatment. Cells were stained in staining buffer containing appropriately diluted combinations of the following monoclonal antibodies: biotin-coupled mouse anti–rat IgG1 (BD Biosciences) developed with streptavidin-PE; PE and biotin rat anti–mouse Igκ (187.1; BD Biosciences); biotin–anti-Igλ 1-3 (BD Biosciences) followed by PE- or allophycocyanin-streptavidin; PerCP- or FITC-coupled anti-CD45R/B220 (RA3-6B2; BD Biosciences); Cy5-coupled anti-IgM (331.12), PE-coupled anti-CD45.1 (eBioscience); FITC-coupled anti-CD45.2 (eBioscience). L929-transfected cells were harvested using PBS, 0.5 mM EDTA, washed twice, and resuspended in staining buffer (PBS, 1% BSA, 0.01% NaN3). Cells were incubated with biotin mouse anti–rat IgG1 (BD Biosciences), which reacts with the linker region of the macroself antigen, or mouse IgG2b monoclonal antibody carrying κ or λ L chains, followed by biotin-coupled rat anti–mouse IgG2a (BD Biosciences) to assess Fv binding site specificity. Biotin-coupled antibodies were revealed with streptavidin-phycoerythrin (PE; BD Biosciences). Cells were gated on the basis of forward and side scatter criteria to avoid contamination by dead cells or debris. For intracellular staining and BrdU uptake studies, surface stained cells were fixed and permeabilized using a kit (Cytofix/Cytoperm™; BD Biosciences) and stained according to the manufacturer's instructions. FITC anti-BrdU antibody was used (BD Biosciences). Stained cells were analyzed on a FACSCaliber flow cytometer (Becton Dickinson) using the FlowJo software package.
Excision product and RS-to-IRS PCR assays
B cells were isolated from spleen and bone marrow using an anti-B220 magnetic bead cell purification system (Miltenyi Biotec). The purity of these preparations was >90% in all cases. Genomic DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN) according the manufacturer's recommendations. PCR reaction were done in a final volume of 50 μl containing 100, 25, 12, and 6 ng of B cell genomic DNA. Vλ1-to-Jλ1 excision product DNA rearrangements were detected using the oligonucleotides and PCR conditions described (11). RS-to-IRS PCR assay was performed using primers B and C (16). Samples were amplified for 25 cycles: 1 min at 94°C, 1 min at 60°C, and 1 min at 72°C. PCR products were electrophoresed in 1.5% agarose gels, blotted on nylon membranes, and probed with intervening sequence probe as described previously (16).
Serum Ig determinations
Polyvinylchloride plastic microplates (Falcon) were coated with a rat monoclonal antibodies specific for IgG2a,b (BD Biosciences), IgM (331.12), or Igλ1-3 (R26-46; BD Biosciences). After washing and blocking, sera (diluted in PBS supplemented with 1% BSA) were incubated 3 h at room temperature. Bound Ig was detected using biotinylated anti–mouse Igλ1-3, anti–mouse IgM (R6-60.2; BD Biosciences) or horseradish peroxidase–conjugated anti–mouse Igκ (187.1; BD Biosciences). Biotinylated antibodies were revealed using streptavidin-peroxidase (Sigma-Aldrich) followed by addition of the chromogenic substrate 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (Sigma-Aldrich) in McIlvain's buffer (84 mM Na2PO4/48 mM citrate, pH 4.6). Absorbance was measured on a Spectra MAX250 plate reader (Molecular Devices). Standard curves were obtained using a mouse IgM,κ (G 155–228; BD Biosciences) and a mouse IgM,λ (11E10; Southern Biotechnology Associates, Inc.) or a mouse IgG2b,κ (BD Biosciences).
Acknowledgments
We thank Klaus Karjaleinen for the suggestion to generate an anti-IgM mouse that inspired this project; Norman Klinman, Michael McHeyzer-Williams, and Argyrios Theofilopoulos for their critiques of the manuscript; Richard Hardy for cell lines; Mark Shlomchik for plasmid pBKS; Andreas Strasser for Bcl2 transgenic mice; Jo Davies for RAG1−/− mice; Jean da Silva Correia for technical advice; Julie Lang for permitting us to cite her unpublished data, and Glen Nemerow for use of the ELISA plate reader.
This work was supported by research and training grants from the National Institutes of Health (RO1AI59714, R21AI49940, T32 HL07195, and F31 AI52484) and the Arthritis Foundation.
The authors have no conflicting financial interests.
Abbreviations used: RAG, recombinase activator gene; RS, recombining sequence.
J. Peters' present address is University Medical Centre, St. Radboud, Nijmegen University, 6500 HB Nijmegen, Netherlands.
References
- 1.Klinman, N.R. 1996. The “clonal selection hypothesis” and current concepts of B cell tolerance. Immunity. 5:189–195. [DOI] [PubMed] [Google Scholar]
- 2.Nossal, G.J. 1983. Cellular mechanisms of immunologic tolerance. Annu. Rev. Immunol. 1:33–62. [DOI] [PubMed] [Google Scholar]
- 3.Storb, U. 1987. Transgenic mice with immunoglobulin genes. Annu. Rev. Immunol. 5:151–174. [DOI] [PubMed] [Google Scholar]
- 4.Goodnow, C.C. 1992. Transgenic mice and analysis of B-cell tolerance. Annu. Rev. Immunol. 10:489–518. [DOI] [PubMed] [Google Scholar]
- 5.Miller, J.F., and A. Basten. 1996. Mechanisms of tolerance to self. Curr. Opin. Immunol. 8:815–821. [DOI] [PubMed] [Google Scholar]
- 6.Nemazee, D. 2000. Receptor selection in B and T lymphocytes. Annu. Rev. Immunol. 18:19–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spanopoulou, E., C.A. Roman, L.M. Corcoran, M.S. Schlissel, D.P. Silver, D. Nemazee, M.C. Nussenzweig, S.A. Shinton, R.R. Hardy, and D. Baltimore. 1994. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. Genes Dev. 8:1030–1042. [DOI] [PubMed] [Google Scholar]
- 8.Arnold, L.W., C.A. Pennell, S.K. McCray, and S.H. Clarke. 1994. Development of B-1 cells: segregation of phosphatidyl choline-specific B cells to the B-1 population occurs after immunoglobulin gene expression. J. Exp. Med. 179:1585–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taki, S., F. Schwenk, and K. Rajewsky. 1995. Rearrangement of upstream DH and VH genes to a rearranged immunoglobulin variable region gene inserted into the DQ52-JH region of the immunoglobulin heavy chain locus. Eur. J. Immunol. 25:1888–1896. [DOI] [PubMed] [Google Scholar]
- 10.Chen, C., Z. Nagy, E.L. Prak, and M. Weigert. 1995. Immunoglobulin heavy chain gene replacement: a mechanism of receptor editing. Immunity. 3:747–755. [DOI] [PubMed] [Google Scholar]
- 11.Tiegs, S.L., D.M. Russell, and D. Nemazee. 1993. Receptor editing in self-reactive bone marrow B cells. J. Exp. Med. 177:1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gay, D., T. Saunders, S. Camper, and M. Weigert. 1993. Receptor editing: an approach by autoreactive B cells to escape tolerance. J. Exp. Med. 177:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen, C., E.L. Prak, and M. Weigert. 1997. Editing disease-associated autoantibodies. Immunity. 6:97–105. [DOI] [PubMed] [Google Scholar]
- 14.Radic, M.Z., J. Erikson, S. Litwin, and M. Weigert. 1993. B lymphocytes may escape tolerance by revising their antigen receptors. J. Exp. Med. 177:1165–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Melamed, D., and D. Nemazee. 1997. Self-antigen does not accelerate immature B cell apoptosis, but stimulates receptor editing as a consequence of developmental arrest. Proc. Natl. Acad. Sci. USA. 94:9267–9272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Retter, M.W., and D. Nemazee. 1998. Receptor editing occurs frequently during normal B cell development. J. Exp. Med. 188:1231–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Casellas, R., T.A. Shih, M. Kleinewietfeld, J. Rakonjac, D. Nemazee, K. Rajewsky, and M.C. Nussenzweig. 2001. Contribution of receptor editing to the antibody repertoire. Science. 291:1541–1544. [DOI] [PubMed] [Google Scholar]
- 18.Braun, U., K. Rajewsky, and R. Pelanda. 2000. Different sensitivity to receptor editing of B cells from mice hemizygous or homozygous for targeted Ig transgenes. Proc. Natl. Acad. Sci. USA. 97:7429–7434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kouskoff, V., G. Lacaud, K. Pape, M. Retter, and D. Nemazee. 2000. B cell receptor expression level determines the fate of developing B lymphocytes: receptor editing versus selection. Proc. Natl. Acad. Sci. USA. 97:7435–7439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halverson, R., R.M. Torres, and R. Pelanda. 2004. Receptor editing is the main mechanism of B cell tolerance toward membrane antigens. Nat. Immunol. 5:645–650. [DOI] [PubMed] [Google Scholar]
- 21.Rolink, A.G., T. Radaszkiewicz, and F. Melchers. 1987. The autoantigen-binding B cell repertoires of normal and of chronically graft-versus-host-diseased mice. J. Exp. Med. 165:1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Louzoun, Y., E. Luning Prak, T. Friedman, S. Litwin, and M. Weigert. 2002. Comment on Langman and Cohn. Semin. Immunol. 14:231–232. [DOI] [PubMed] [Google Scholar]
- 23.Wardemann, H., S. Yurasov, A. Schaefer, J.W. Young, E. Meffre, and M.C. Nussenzweig. 2003. Predominant autoantibody production by early human B cell precursors. Science. 301:1374–1377. [DOI] [PubMed] [Google Scholar]
- 24.Radic, M.Z., M.A. Mascelli, J. Erikson, H. Shan, and M. Weigert. 1991. Ig H and L chain contributions to autoimmune specificities. J. Immunol. 146:176–182. [PubMed] [Google Scholar]
- 25.Yachimovich, N., G. Mostoslavsky, Y. Yarkoni, I. Verbovetski, and D. Eilat. 2002. The efficiency of B cell receptor (BCR) editing is dependent on BCR light chain rearrangement status. Eur. J. Immunol. 32:1164–1174. [DOI] [PubMed] [Google Scholar]
- 26.Wardemann, H., J. Hammersen, and M.C. Nussenzweig. 2004. Human autoantibody silencing by immunoglobulin light chains. J. Exp. Med. 200:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawton, A.R., and M.D. Cooper. 1974. Modification of B lymphocyte differentiation by anti-immunoglobulins. Contemp. Top. Immunobiol. 3:193–225. [DOI] [PubMed] [Google Scholar]
- 28.Raff, M.C., J.J. Owen, M.D. Cooper, A.R. Lawton, M. Megson, and W.E. Gathings. 1975. Differences in susceptibility of mature and immature mouse B lymphocytes to anti-immunoglobulin-induced immunoglobulin suppression in vitro. Possible implications for B-cell tolerance to self. J. Exp. Med. 142:1052–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker, D.C., D.C. Wadsworth, and G.B. Schneider. 1980. Activation of murine B lymphocytes by anti-immunoglobulin is an inductive signal leading to immunoglobulin secretion. J. Exp. Med. 152:138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pike, B.L., A.W. Boyd, and G.J. Nossal. 1982. Clonal anergy: the universally anergic B lymphocyte. Proc. Natl. Acad. Sci. USA. 79:2013–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weiss, S., K. Lehmann, W.C. Raschke, and M. Cohn. 1984. Mice completely suppressed for the expression of immunoglobulin kappa light chain. Proc. Natl. Acad. Sci. USA. 81:211–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gause, A., N. Yoshida, C. Kappen, and K. Rajewsky. 1987. In vivo generation and function of B cells in the presence of a monoclonal anti-IgM antibody: implications for B cell tolerance. Eur. J. Immunol. 17:981–990. [DOI] [PubMed] [Google Scholar]
- 33.Shi, Y.F., R.P. Bissonnette, N. Parfrey, M. Szalay, R.T. Kubo, and D.R. Green. 1991. In vivo administration of monoclonal antibodies to the CD3 T cell receptor complex induces cell death (apoptosis) in immature thymocytes. J. Immunol. 146:3340–3346. [PubMed] [Google Scholar]
- 34.Parry, S.L., J. Hasbold, M. Holman, and G.G. Klaus. 1994. Hypercross-linking surface IgM or IgD receptors on mature B cells induces apoptosis that is reversed by costimulation with IL-4 and anti-CD40. J. Immunol. 152:2821–2829. [PubMed] [Google Scholar]
- 35.Kappler, J.W., N. Roehm, and P. Marrack. 1987. T cell tolerance by clonal elimination in the thymus. Cell. 49:273–280. [DOI] [PubMed] [Google Scholar]
- 36.Rammensee, H.G., R. Kroschewski, and B. Frangoulis. 1989. Clonal anergy induced in mature V beta 6+ T lymphocytes on immunizing Mls-1b mice with Mls-1a expressing cells. Nature. 339:541–544. [DOI] [PubMed] [Google Scholar]
- 37.Berberian, L., L. Goodglick, T.J. Kipps, and J. Braun. 1993. Immunoglobulin VH3 gene products: natural ligands for HIV gp120. Science. 261:1588–1591. [DOI] [PubMed] [Google Scholar]
- 38.Sasso, E.H., G.J. Silverman, and M. Mannik. 1991. Human IgA and IgG F(ab′)2 that bind to staphylococcal protein A belong to the VHIII subgroup. J. Immunol. 147:1877–1883. [PubMed] [Google Scholar]
- 39.Goodyear, C.S., and G.J. Silverman. 2004. Staphylococcal toxin induced preferential and prolonged in vivo deletion of innate-like B lymphocytes. Proc. Natl. Acad. Sci. USA. 101:11392-11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen, J., M. Trounstine, C. Kurahara, F. Young, C.C. Kuo, Y. Xu, J.F. Loring, F.W. Alt, and D. Huszar. 1993. B cell development in mice that lack one or both immunoglobulin kappa light chain genes. EMBO J. 12:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takeda, S., Y.R. Zou, H. Bluethmann, D. Kitamura, U. Muller, and K. Rajewsky. 1993. Deletion of the immunoglobulin kappa chain intron enhancer abolishes kappa chain gene rearrangement in cis but not lambda chain gene rearrangement in trans. EMBO J. 12:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Durdik, J., M.W. Moore, and E. Selsing. 1984. Novel kappa light-chain gene rearrangements in mouse lambda light chain-producing B lymphocytes. Nature. 307:749–752. [DOI] [PubMed] [Google Scholar]
- 43.Siminovitch, K.A., M.W. Moore, J. Durdik, and E. Selsing. 1987. The human kappa deleting element and the mouse recombining segment share DNA sequence homology. Nucleic Acids Res. 15:2699–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selsing, E., and L.E. Daitch. 1995. Immunoglobulin λ genes. Immunoglobulin Genes. T. Honjo and F. Alt, editors. Academic Press Limited, London. 194-203.
- 45.Nadel, B., P.A. Cazenave, and P. Sanchez. 1990. Murine lambda gene rearrangements: the stochastic model prevails over the ordered model. EMBO J. 9:435–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dunda, O., and D. Corcos. 1997. Recombining sequence recombination in normal kappa-chain-expressing B cells. J. Immunol. 159:4362–4366. [PubMed] [Google Scholar]
- 47.Förster, I., and K. Rajewsky. 1990. The bulk of the peripheral B-cell pool in mice is stable and not rapidly renewed from the bone marrow. Proc. Natl. Acad. Sci. USA. 87:4781–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Velardi, A., H. Kubagawa, and J.F. Kearney. 1984. Analysis of the reactivity of four anti-mouse IgM allotype antibodies with mu+ B lineage cells at various stages of differentiation. J. Immunol. 133:2098–2103. [PubMed] [Google Scholar]
- 49.Yelton, D.E., C. Desaymard, and M.D. Scharff. 1981. Use of monoclonal anti-mouse immunoglobulin to detect mouse antibodies. Hybridoma. 1:5–11. [DOI] [PubMed] [Google Scholar]
- 50.Mombaerts, P., J. Iacomini, R.S. Johnson, K. Herrup, S. Tonegawa, and V.E. Papaioannou. 1992. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 68:869–877. [DOI] [PubMed] [Google Scholar]
- 51.Strasser, A., S. Whittingham, D.L. Vaux, M.L. Bath, J.M. Adams, S. Cory, and A.W. Harris. 1991. Enforced BCL2 expression in B-lymphoid cells prolongs antibody responses and elicits autoimmune disease. Proc. Natl. Acad. Sci. USA. 88:8661–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hieter, P.A., S.J. Korsmeyer, T.A. Waldmann, and P. Leder. 1981. Human immunoglobulin kappa light-chain genes are deleted or rearranged in lambda-producing B cells. Nature. 290:368–372. [DOI] [PubMed] [Google Scholar]
- 53.Arakawa, H., T. Shimizu, and S. Takeda. 1996. Re-evaluation of the probabilities for productive arrangements on the kappa and lambda loci. Int. Immunol. 8:91–99. [DOI] [PubMed] [Google Scholar]
- 54.Engel, H., A. Rolink, and S. Weiss. 1999. B cells are programmed to activate kappa and lambda for rearrangement at consecutive developmental stages. Eur. J. Immunol. 29:2167–2176. [DOI] [PubMed] [Google Scholar]
- 55.Luning Prak, E., M. Trounstine, D. Huszar, and M. Weigert. 1994. Light chain editing in kappa-deficient animals: a potential mechanism of B cell tolerance. J. Exp. Med. 180:1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li, H., Y. Jiang, E.L. Prak, M. Radic, and M. Weigert. 2001. Editors and editing of anti-DNA receptors. Immunity. 15:947–957. [DOI] [PubMed] [Google Scholar]
- 57.Li, Y., H. Li, and M. Weigert. 2002. Autoreactive B cells in the marginal zone that express dual receptors. J. Exp. Med. 195:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mage, R., and S. Dray. 1965. Persistent altered phenotypic expression of allelic gamma-G-immunoglobulin allotypes in heterozygous rabbits exposed to isoantibodies in fetal and neonatal life. J. Immunol. 95:525–535. [PubMed] [Google Scholar]
- 59.Sidman, C.L., and E.R. Unanue. 1975. Receptor-mediated inactivation of early B lymphocytes. Nature. 257:149–151. [DOI] [PubMed] [Google Scholar]
- 60.Cerny, A., A.W. Hugin, S. Sutter, C.H. Heusser, N. Bos, S. Izui, H. Hengartner, and R.M. Zinkernagel. 1985. Suppression of B cell development and antibody responses in mice with polyclonal rabbit and monoclonal rat anti-IgM antibodies. I. Characterization of the suppressed state. Exp. Cell Biol. 53:301–313. [DOI] [PubMed] [Google Scholar]
- 61.Cooper, M.D., J.F. Kearney, W.E. Gathings, and A.R. Lawton. 1980. Effects of anti-Ig antibodies on the development and differentiation of B cells. Immunol. Rev. 52:29–53. [DOI] [PubMed] [Google Scholar]
- 62.Lang, J., B. Arnold, G. Hammerling, A.W. Harris, S. Korsmeyer, D. Russell, A. Strasser, and D. Nemazee. 1997. Enforced Bcl-2 expression inhibits antigen-mediated clonal elimination of peripheral B cells in an antigen dose-dependent manner and promotes receptor editing in autoreactive, immature B cells. J. Exp. Med. 186:1513–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang, H., and M.J. Shlomchik. 1997. High affinity rheumatoid factor transgenic B cells are eliminated in normal mice. J. Immunol. 159:1125–1134. [PubMed] [Google Scholar]
- 64.Schorpp, M., R. Jager, K. Schellander, J. Schenkel, E.F. Wagner, H. Weiher, and P. Angel. 1996. The human ubiquitin C promoter directs high ubiquitous expression of transgenes in mice. Nucleic Acids Res. 24:1787–1788. [DOI] [PMC free article] [PubMed] [Google Scholar]