

Abstract
The mammalian genome is highly organized within the cell nucleus. The nuclear position of many genes and genomic regions changes during physiological processes such as proliferation, differentiation, and disease. It is unclear whether disease-associated positioning changes occur specifically or are part of more global genome reorganization events. Here, we have analyzed the spatial position of a defined set of cancer-associated genes in an established mammary epithelial three-dimensional cell culture model of the early stages of breast cancer. We find that the genome is globally reorganized during normal and tumorigenic epithelial differentiation. Systematic mapping of changes in spatial positioning of cancer-associated genes reveals gene-specific positioning behavior and we identify several genes that are specifically repositioned during tumorigenesis. Alterations of spatial positioning patterns during differentiation and tumorigenesis were unrelated to gene activity. Our results demonstrate the existence of activity-independent genome repositioning events in the early stages of tumor formation.
Introduction
Chromosomes, subchromosomal domains, and individual gene loci occupy preferred positions within the 3D space of the cell nucleus (Cremer et al., 2006; Meaburn and Misteli, 2007). The high degree of spatial organization inside the nucleus is rapidly emerging as an important factor in nuclear function (Fraser and Bickmore, 2007; Lanctot et al., 2007; Misteli, 2007). The spatial organization of the genome varies between cell types (Boyle et al., 2001; Parada et al., 2004; Mayer et al., 2005; Neusser et al., 2007), upon differentiation (Kosak et al., 2002; Chambeyron and Bickmore, 2004; Kim et al., 2004; Foster et al., 2005), and with exit from the cell cycle (Bridger et al., 2000; Solovei et al., 2004; Meaburn et al., 2007a). Moreover, some, although not all, genes change their nuclear location upon changes in activity (Lanctot et al., 2007; Meaburn et al., 2007b) and in some cases repositioning occurs before the start of expression (Ragoczy et al., 2003, 2006).
The spatial organization of the genome is frequently altered in disease. For example, human chromosome (HSA) X becomes more internally located in neurons after epileptic seizures (Borden and Manuelidis, 1988). HSA 13 and 18 are relocated away from the nuclear periphery in fibroblasts of patients with laminopathies, a group of diseases caused by mutations in the nuclear envelope proteins lamin A/C (Meaburn et al., 2007a). In cancer, distinctive changes in number and morphology of nucleoli have long been used as indicators of cancerous transformation (Zink et al., 2004). Furthermore, distribution patterns of centromeres and telomeres can be altered in cancer cells (Chuang et al., 2004; Sarkar et al., 2007). More specifically, chromosomes carrying cancerous translocations or fusion genes generated by translocations are sometimes found in different positions than their normal counterparts (Cremer et al., 2003; Taslerova et al., 2003, 2006; Murmann et al., 2005). Further evidence for a reorganization of the genome in tumorigenesis comes from the observation that HSA 19 is commonly more peripherally located than HSA 18 in multiple cancers (Croft et al., 1999; Cremer et al., 2003). In pancreatic cancer, HSA 8 is more peripheral compared with normal tissue and the centromere of HSA 17 becomes more internally positioned in breast cancer tissues (Wiech et al., 2005). Interestingly, gains in copy numbers of whole chromosomes do not appear to alter the spatial organization of the genome (Croft et al., 1999 Sullivan et al., 2001; Sengupta et al., 2007).
All studies on genome reorganization to date have been performed on late-stage tumor cells or tissues. It is not known whether the changes in spatial positioning are an early or late event in the disease and whether repositioning events are specific or occur by default as part of global genome reorganization. Here, we have used a 3D mammary epithelial differentiation system to characterize the positioning of individual gene loci during early tumorigenesis. 3D mammary epithelial systems are accurate models of early tumorigenesis and are widely used to study the earliest events in tumor formation (Debnath and Brugge, 2005; Nelson and Bissell, 2005). In these systems, mammary epithelial cells (MECs) grown in contact with reconstituted basement membrane form growth-arrested spheroid structures termed acini. Mature acini consist of a single layer of polarized cells surrounding a hollow lumen; they closely resemble in vivo mammary gland morphology and recapitulate some glandular functions including milk protein secretion (Petersen et al., 1992; Muthuswamy et al., 2001; Nelson and Bissell, 2005). Overexpression of an active form of epidermal growth factor receptor ErbB2 in acini triggers tumorigenesis, as indicated by an increase in proliferation and a decrease in apoptosis leading to lumen filling and enlargement of the structures, which frequently contain multi-acinar units. This phenotype closely mimics events of early breast cancer, particularly ductal carcinoma in situ (Muthuswamy et al., 2001; Debnath et al., 2002). This system is highly physiologically relevant because ERBB2 is amplified and/or overexpressed in ∼25% of breast cancers and 60–90% of ductal carcinoma in situ (Hynes and Stern, 1994; Harari and Yarden, 2000). Using this system, we found changes in spatial positioning of a set of cancer-associated genes both during normal breast epithelial differentiation and early tumorigenesis. We found activity-independent specific repositioning events and identified several genes that exhibit cancer-specific spatial repositioning.
Results
Global genome reorganization during normal and tumorigenic early mammary epithelial differentiation
To assess the spatial organization of the genome during early tumorigenesis, we used a well-characterized 3D cell culture model system for early breast cancer (Muthuswamy et al., 1999, 2001). MCF-10A.B2 cells are nontransformed, near-diploid, immortalized MECs that express a synthetic ligand-inducible active ErbB2 variant (Soule et al., 1990; Muthuswamy et al., 2001). When grown in the presence of recombinant basement membrane extract, these cells undergo normal differentiation into acini within 20 d (Fig. 1 A; Muthuswamy et al., 2001; Debnath et al., 2003). Tumorigenesis can be triggered by the addition of the small molecule ligand AP1510, which induces homodimerization of the ErbB2 variant but not endogenous ErbB2 (Fig. 1 A; Muthuswamy et al., 1999). MCF-10A.B2 cells grown under these conditions replicate many features of early breast cancer and have been widely used to study mechanisms of early breast cancer formation (Muthuswamy et al., 1999, 2001; Seton-Rogers et al., 2004; Aranda et al., 2006; Witt et al., 2006).
Figure 1.

The nucleolus during mammary epithelial differentiation and early mammary tumorigenesis. (A) MCF-10.B2 cells were stained with ANA-N antibody to detect nucleoli (green) and counterstained with DAPI to delineate nuclei (blue). Projected image stacks are shown. For acini structures, projected stacks are approximately one nucleus thick and taken from the midsection of the acini. Bar, 10 μm. (B) Quantitation of nucleoli in proliferating 2D culture (Day 0), quiescent 2D cultures (Quiescent), and cells grown for 20 d under 3D growth conditions with 10 d of constitutive activation of ErbB2 (Day 20, ErbB2) or without activation (Day 20 control). Values represent means ± SEM from a minimum of three independent experiments. n = 270–1,000.
We first assessed global genome rearrangements during differentiation and early tumorigenesis by analyzing nucleolar distribution patterns (Fig. 1). Nucleoli form around nucleolar organizing regions (NORs), which in humans are located on the five acrocentric chromosomes. Before differentiation (day 0), MCF10A.B2 cells predominantly contained multiple nucleoli (median 3 and maximum 15) and only 3.4 ± 0.2% had a single nucleolus (Fig. 1). In contrast, the majority (79.8 ± 2.9%; P < 0.001 using Yates correlated χ2 analysis) of fully differentiated MCF-10A.B2 cells in acini contained a single nucleolus and no cells were found with more than six nucleoli. Because differentiation of MCF10A.B2 cells is accompanied by a transition from a highly proliferative state of precursors (98.5 ± 1.3% pKi-67 positive) to quiescence in acini, it was important to rule out that the observed reduction in nucleoli was not merely a consequence of quiescence. In undifferentiated MCF10A.B2 cells grown in low serum for 5 d (5.9 ± 0.03% pKi-67 positive), only 15.7 ± 2.3% of cells contained a single nucleolus (median 2 and maximum 8; Fig. 1). This was significantly different from differentiated day-20 acini (P < 0.001), which indicates that the reduction in nucleoli was caused by differentiation. The reduced number of nucleoli in acini implies large-scale spatial genome rearrangements during differentiation. Analysis of day-20 tumor acini cultures induced by activation of ErbB2 at day 10 in 3D culture indicated a similar nucleolar staining pattern to that of normal day-20 cultures (74.5 ± 4.26% with a single nucleolus, maximum 6; Fig. 1). Furthermore, no significant difference in the distribution of centromeres was detected between normal and tumor acini (unpublished data). We conclude that global and similar genome reorganization occurs both during normal and tumorigenic MEC differentiation.
Gene-specific repositioning during normal mammary epithelial differentiation
We next sought to determine how specific genes behaved during normal and cancerous mammary differentiation. To this end, we first mapped the radial position of 11 cancer-associated genes during normal acinar differentiation using interphase FISH (Figs. 2 and Figs.3; Tables I and II; and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200708204/DC1). These genes map to a range of chromosomes and are involved in various aspects of ErbB2-dependent and -independent tumor biology (Table I). To ask whether any of these genes had undergone repositioning within the nuclear space during differentiation, their radial distribution, normalized to the size of the nucleus, was determined. Typically, ∼200 nuclei were analyzed per experiment and only nuclei with two signals per gene were incorporated in the analysis (see Materials and methods; Fig. S1). Statistical differences between samples were probed using the 1D Kolmogorov-Smirnov test (KS test) on cumulative distribution graphs (Fig. 3). 7 of the 11 genes analyzed were found to be repositioned during differentiation, with only BCL2, ERBB2IP, FGFR1, and TP53 not altering their location (Fig. 3). Six of the seven genes became more internally localized during differentiation (Figs. 2 and Figs.3). One gene, TGFB3, shifted from two subpopulations at day 0 to a single population at the end of differentiation ( Figs. 3 J and S1 J). Because mature acini consist of nonproliferative cells, it was important to rule out the possibility that the differentiation-associated repositioning events merely reflected the quiescent nature of acini. To this end, we mapped gene positions in quiescent undifferentiated cultures. 9 out of 11 analyzed genes were positioned significantly differently between quiescent undifferentiated cultures and day-20 acini (Fig. 3 L, Table II), which demonstrates that quiescence alone cannot account for the observed repositioning during differentiation. These data demonstrate that the transition from growth in a monolayer to 3D tissue architecture alters the position of some but not all genes.
Figure 2.

Gene positioning during normal differentiation and early mammary tumorigenesis. Indicated gene loci (red) were detected in PFA-fixed, undifferentiated, proliferating 2D culture cells (Day 0) and cells grown for 20 d under 3D growth conditions with 10 d of constitutive activation of ErbB2 (Day 20, ErbB2) or without activation (Day 20 control). Projected stacks of nuclei are shown. Note the difference in scale for Day 0 compared with 3D cultured cells. Bars, 5 μm.
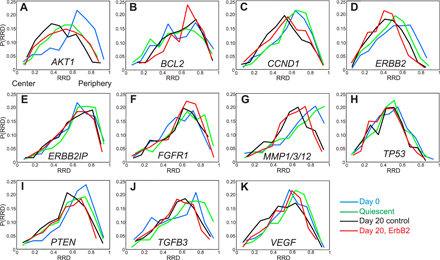
Figure 3.

Radial distribution of genes during differentiation of MECs. Gene loci were detected in PFA-fixed cells using specific BAC FISH probes (Table I). (A–K) Cumulative frequency graphs to quantify the radial distribution (RRD) for each gene in MCF-10.B2 cells, in proliferating 2D culture nuclei (Day 0), after growth for 20 d under 3D culture conditions (Day 20 control), and in quiescent 2D cultures (Quiescent). (L) Pairwise comparisons of cumulative radial distributions using a 1D KS test. n = 195–220 per BAC per growth condition.
Table I. Test set of cancer-associated genes.
| Gene symbol | Full gene name | Chromosomal location | Cancer related | BAC |
|---|---|---|---|---|
| AKT1 | V-akt murine thymoma viral oncogene homologue 1 |
14q32.3 | Oncogene; proliferation, survival, and mobility |
RP11-477I4 |
| BCL2 | B-cell CLL/lymphoma 2 | 18q21.3 | Oncogene; survival | RP11-299P2D |
| CCND1 | Cyclin D1 | 11q13 | Oncogene; cell cycle | RP11-300I6 |
| ERBB2 | V-erb-b2 avian erythroblastic leukemia viral oncogene homologue 2 |
17q21.1 | Oncogene; proliferation, survival, and mobility |
RP11-62N23 |
| ERBB2IP | ErbB2 interacting protein | 5q12 | Regulates ErbB2 function and localization |
RP11-720M2 |
| FGFR1 | Fibroblast growth factor receptor 1 | 8p11.2-p12 | Oncogene; proliferation, survival, and invasion |
RP11-100B16 |
| MMP1/3/12 | Matrix metallopeptidase 1, 3, or 12 | 11q22-q23 | Oncogene; metastasis | RP11-686G6 |
| TP53 | Tumor protein p53 | 17p13 | Tumor suppressor genea | RP11-89D11 |
| PTEN | Phosphatase and tensin homologue | 10q23.3 | Tumor suppressor genea | RP11-383D9 |
| TGFB3 | Transforming growth factor β3 | 14q24 | In early cancer, tumor suppressor gene |
RP11-270M14 |
| VEGF | Vascular endothelial growth factor | 6p12 | Oncogene; angiogenesis, migration, and survival |
RP11-1152J4 |
Official gene names and symbols were taken from the OMIM/Entrez gene.
Negatively regulated ErbB2/AKT1 signaling pathway.
Table II. Summary of gene expression and gene positioning data.
| Day 0→day 20
|
Day 0→Q
|
Q→day 20
|
Day 0→day 20E
|
Day 20→day 20E
|
Q→day 20E
|
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Posn | Exp | Posn | Exp | Posn | Exp | Posn | Exp | Posn | Exp, | Posn | Exp | |
| AKT1 | In | NSD | In | ↓ | In | ↑ | In | NSD | P | NSD | NSD | NSD |
| BCL2 | NSD | ↓ | NSD | ↓ | P | ↑ | P | ↓ | P | NSD | P | NSD |
| CCND1 | In | ↓ | NSD | ↓ | In | ↑ | In | ↓ | NSD | NSD | In | ↑ |
| ERBB2 | In | NSD | In | ↑ | In | NSD | In | ↑ | In | NSD | In | ↑ |
| ERBB2IP | NSD | ↑ | NSD | ↑ | In | NSD | NSD | ↑ | NSD | NSD | In | NSD |
| FGFR1 | NSD | NSD | P | NSD | In | ↓ | P | NSD | NSD | ↑ | In | NSD |
| MMP1 | In | ↑ | NSD | NSD | In | ↑ | In | ↑ | NSD | ↑ | In | ↑ |
| TP53 | NSD | ↑ | NSD | NSD | NSD | ↑ | In | NSD | NSD | ↓ | In | NSD |
| PTEN | In | ↑ | In | ↑ | In | NSD | In | ↑ | NSD | ↓ | In | ↓ |
| TGFB3 | Altereda | NSD | Altereda | NSD | NSD | NSD | Altereda | NSD | NSD | NSD | NSD | NSD |
| VEGF | In | ↑ | P | NSD | In | NSD | NSD | ↑ | P | NSD | In | ↑ |
Heading row shows growth conditions being compared. Changes in gene positions (Posn) and in gene expression (Exp) between growth conditions are shown.
Day 0, proliferating 2D cultures; Day 20, control cultures grown in 3D culture for 20 d; Day 20E, ErbB2-activated 3D cultures; In, internal movement; NSD, no significant difference between the growth conditions; P, peripheral movement; Q, quiescent 2D cultures; ↑, up-regulation; ↓, down-regulation.
TGFB3 shifts from two subpopulations at day 0 to a single population distribution pattern in all other growth conditions.
The repositioning events were gene specific, as indicated by the fact that AKT1 and TGFB3 both map to HSA 14 yet reposition differently from each other after exit from the cell cycle and during differentiation (Fig. 3, A, J, and L). Furthermore, endogenous ERBB2 and TP53 both map to HSA 17 and, although the position of endogenous ERBB2 was affected by differentiation and growth arrest, TP53 position was not (Fig. 3, D, H, and L). To further probe if the gene repositioning events were related to underlying chromosome movements, the positions of selected whole chromosomes were evaluated in proliferating and quiescent 2D cultures (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200708204/DC1). The positions of neither HSA 11 nor the genes that map to this chromosome (CCND1 and MMP1) were affected by exit from the cell cycle (Figs. 3 and S2). The more internal position of HSA 17 in quiescent cells matched the movement of ERBB2, however, and TP53 did not significantly alter position. HSA 8 and 14 were not repositioned after exit from the cell cycle, yet the genes that map to them (FGFR1 [HSA 8], AKT1, and TGFB3) did reposition. We conclude that the repositioning of single gene loci did not simply reflect changes in the position of their chromosomes. Taken together, these observations suggest the existence of gene-specific, proliferation-independent gene repositioning events during early mammary differentiation.
Gene-specific repositioning during tumorigenic differentiation
We next asked whether repositioning occurs in a similar manner during tumorigenic differentiation. To this end, we compared the positioning patterns of our panel of genes between undifferentiated cells and day-20 ErbB2-activated tumor acini (Fig. 4 and Fig. S1, and Table II). Analysis of radial distributions showed that 9 out of 11 genes were in statistically different positions in day-20 ErbB2-activated 3D cultured acini compared with undifferentiated cells (Fig. 4). All genes that underwent repositioning during normal differentiation also changed their location during tumorigenic differentiation with the exception of VEGF. In addition to this, BCL2, FGFR1, and TP53, which did not undergo relocalization during normal differentiation, repositioned in tumorigenic differentiation. Six of the genes became more internally localized between day-0 and -20 ErbB2-activated nuclei. In contrast, BCL2 and FGFR1 became more peripherally localized. The radial positions of ERBB2IP and VEGF were not significantly affected by tumorigenic differentiation (Figs. 2 and Figs.4). These results were not confounded by the formation of global genome rearrangements in response to ErbB2 activation because no large-scale cytogenetic rearrangements were detected in metaphase spreads from ErbB2-activated cells compared with normal cells (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200708204/DC1). The absence of large-scale chromosome rearrangements upon ErbB2 expression is further indicated by the fact that no amplifications or losses of any of the 11 genes were found upon expression of ErbB2 (Fig. 2 and not depicted).
Figure 4.

Quantitation of the radial distribution of specific loci during tumorigenic differentiation. (A–K) FISH was performed on PFA-fixed cells grown in 3D culture for 20 d with constitutive activation of ErbB2 for the final 10 d (Day 20, ErbB2). The radial distributions (RRD) for each gene in proliferating 2D cultures are shown for comparison (Day 0). Pairwise comparisons of cumulative radial distributions were performed using a 1D KS test. n = 195–220 per BAC per growth condition.
Identification of tumor-specific repositioning events
Having determined the repositioning patterns during normal and tumorigenic acini formation, we sought to identify tumorigenesis-specific gene repositioning events. To this end, we directly compared the positions of the entire set of genes in normal and tumor acini (Fig. 5 and Table II). 4 of the 11 tested cancer-associated genes showed significantly distinct positions. AKT1, BCL2, and VEGF loci shifted to a more peripheral position during oncogenic activation compared with normal acini, whereas endogenous ERBB2 became more internally localized than in normal acini (Figs. 2 and Figs.5). These events were specific because several genes on the same chromosome did not behave in the same manner. Among a pair of loci on HSA 14 (AKT1 and TGFB3) and HSA 17 (ERBB2 and TP53), the spatial position of one was affected by the overactivation of ErbB2, whereas the other was not. Both gene loci on the q arm of HSA 11 (CCND1 and MMP1), however, did not alter location after ErbB2 activation. This early tumor-specific reorganization was not a consequence of changes in cell cycle status because BCL2 and ERBB2 did not change positions in proliferating compared with quiescent 2D cultures (Fig. 3). Moreover, oncogenic activation induced a peripheral movement of VEGF (Fig. 5 K), whereas the locus was more internally positioned in proliferating undifferentiated cells compared with quiescent undifferentiated nuclei (Fig. 3, K and L; and Table II). Furthermore, although AKT1 was more internally positioned in quiescent cells compared with proliferating cells, in both 2D and 3D cultures, cell cycle differences alone do not appear to determine the position of AKT1 because the gene occupies distinct nuclear positions in quiescent undifferentiated cells compared with quiescent normal acini (Fig. 3, A and L; and Fig. S1 A). Additionally, AKT1 was found in a similar position in proliferating tumor acini cultures as in quiescent 2D cells (P > 0.05; Table II, Fig. S1 A, and not depicted). We conclude that we have identified four genes that exhibit tumor-specific nuclear repositioning.
Figure 5.

Loci-specific gene repositioning during early breast tumorigenesis. (A–K) Quantitation of the radial distribution (RRD) of 11 cancer-associated genes grown in 3D culture for 20 d with 10 d of constitutive activation of ErbB2 (Day 20, ErbB2) or without activation (Day 20 control). Pairwise comparisons of cumulative radial distributions were performed using a 1D KS test. n = 195–220 per BAC per growth condition.
Spatial gene repositioning is independent of gene activity
To ask whether repositioning of gene loci during differentiation and tumorigenic events is related to changes in their activity, the expression levels of all genes during normal and tumor differentiation as well as during their transition into quiescence was measured by quantitative RT-PCR and compared with their positioning behavior (Fig. 6 and Table II). We find that repositioning behavior is independent of changes in gene activity. Overall, among 66 comparisons of gene activity under various conditions, only 22 correlated with changes in position. For more than half of the comparisons (35/66), a change in either expression or position, but not both, was detected (Figs. 3–6 and Table II). Furthermore, even in cases when a change in activity was accompanied by repositioning, the direction of the gene movement often differed among conditions and did not correlate with the direction of the change in activity. For example, peripheral repositioning of BCL2 occurred with an increase in expression between quiescent 2D and control acini but also with a decrease during tumorigenesis. Similarly, internal repositioning of some genes, e.g., CCND1, occurred in situations with up-regulation, such as when quiescent 2D cultures were compared with either normal or ErbB2-activated acini, but the same internal repositioning was also observed upon down-regulation of the same gene such as during normal or tumorigenic differentiation (Table II).
Figure 6.

Quantitative RT-PCR analysis during differentiation of MECs. (A) Total RNA was harvested from cells grown as standard 2D cultures (Day 0), quiescent 2D cultures (Quiescent), and cultures grown for 20 d under 3D growth conditions with 10 d of constitutive activation of ErbB2 (Day 20, ErbB2) or without activation (Day 20 control). Values represent means ± SEM from three independent experiments. Expression levels are normalized to cyclophilin. (B) Pairwise comparisons for expression using a t test.
The absence of a correlation between gene expression and positioning applied to all conditions. During normal differentiation, the expression of seven genes was altered (Fig. 6). Of these, three genes (BCL2, ERBB2IP, and TP53) were not repositioned at all. Of the other four, MMP1, PTEN, and VEGF were more internally positioned and up-regulated, but CCND1, which was also more internally positioned, was down-regulated. However, of the four genes that did not alter expression during differentiation, three (AKT1, ERBB2, and TGFB3) underwent repositioning. Similarly, no correlation was detected between repositioning during exit from the cell cycle in 2D culture and gene activity (Figs. 3 and Figs.6 and Table II). Withdrawal from proliferation decreased the expression of three genes (AKT1, BCL2, and CCND1), none of which were repositioned. The expression of three genes was unchanged in quiescent cells, two of which were more internally positioned (ERBB2 and PTEN), whereas ERBB2IP was not repositioned. Of the five genes that did not alter expression, three were repositioned (FGFR1, TGFB3, and VEGF; Figs. 3 and Figs.6 and Table II).
During tumorigenic differentiation, four genes (AKT1, FGFR1, TP53, and TGFB3) that were significantly repositioned did not undergo a detectable change in expression (Fig. 6). In contrast, two genes (ERBB2IP and VEGF) that were significantly up-regulated in tumorigenic differentiation were not repositioned. For some genes (ERBB2, MMP1, and PTEN), internal positioning correlated with an increase in expression, whereas for CCND1, it correlated with a decrease in expression. BCL2, which was also down-regulated, was more peripherally positioned. Finally, of the four genes that altered location after constitutive activation of ErbB2 (AKT1, BCL2, ERBB2, and VEGF) compared with normal acini, no change in expression was detected (Fig. 6). Moreover, ErbB2 activation significantly modulated the expression of four genes (FGFR1, MMP1, TP53, and PTEN) but did not alter their location upon ErbB2 activation (Fig. 6). Taken together, these results demonstrate that there is no general correlation between gene activity and radial nuclear position.
Discussion
We present here a systematic analysis of genome reorganization events during early tumorigenesis. We have characterized several genes that change position during normal and tumor mammary differentiation and identified genes that are differentially positioned in normal and tumor acini.
The transition from undifferentiated MECs to mature acini involves differential expression of ∼500 genes, based on microarray analysis (Fournier et al., 2006). Consistent with these extensive changes in the genome expression program associated with differentiation, our results demonstrate that global reorganization of the genome occurs during both normal and tumorigenic mammary differentiation. Large-scale reorganization is indicated by the formation of a single nucleolus from multiple nucleoli upon differentiation. This observation is in agreement with a previous study on the parental MCF-10A cell line and normal adult breast tissue (Underwood et al., 2006). Furthermore, large-scale genome reorganization is supported by our observation that 7 of 11 tested genes occupied distinct radial positions in acini compared with undifferentiated MECs. Importantly, five of the genes that repositioned were not on NOR-containing chromosomes, which indicates that genome reorganization is not limited to NOR-containing chromosomes. Our observations extend earlier indirect evidence for global genome reorganization during normal mammary differentiation based on redistribution of nuclear proteins, including heterochromatin protein HP1, splicing factor SRm160, nuclear matrix protein NuMA, telomere associated protein TIN2, and histone modifications on histones 3 and 4 (Lelievre et al., 1998; Plachot and Lelievre, 2004; Kaminker et al., 2005; Le Beyec et al., 2007). We cannot entirely rule out the possibility that some of the observed genome reorganization is a consequence of global changes in nuclear shape and size because MEC nuclei in acini are rounder with a smaller diameter than the flat ellipsoid nuclei of their 2D cultured counterparts (Le Beyec et al., 2007). We think this is unlikely, however, because not all genes were repositioned in the same way during the differentiation process. In addition, no systematic correlation between nuclear shape and chromosome positioning has been observed in several cell types from different tissues in both human and mouse (Croft et al., 1999; Bridger et al., 2000; Boyle et al., 2001; Cremer et al., 2003; Mayer et al., 2005; Meaburn et al., 2007a). Finally, the positional changes of several genes were distinct during normal differentiation and when comparing quiescent 2D cells to mature acini, both of which are situations where cells undergo similar shape changes.
During the 3D culture differentiation process, MECs become quiescent (Petersen et al., 1992; Muthuswamy et al., 2001; Fournier et al., 2006). Although the spatial organization of the genome can change with cell cycle status (Figs. 3 and S2; Bridger et al., 2000; Solovei et al., 2004; Meaburn et al., 2007a), withdrawal from the cell cycle did not account for the observed repositioning during mammary differentiation. The percentage of quiescent monolayer cells with a single nucleolus was dramatically lower than in acini and 9 out of the 11 genes were positioned differently in 2D quiescent cultures compared with quiescent acini. Taken together, these data suggest that the global genome reorganization observed during acinar formation is not caused by withdrawal from the cell cycle but is rather a consequence of the differentiation process itself.
A key goal of our study was to determine whether malignant mammary differentiation is distinct from normal mammary differentiation. Several global aspects of nuclear architecture have previously been demonstrated to be affected during MEC tumorigenesis, including patterns of epigenetic modifications, promyelocytic leukemia bodies, nuclear speckles, and the nuclear lamins (Chandramouly et al., 2007). Conversely, we did not find any major differences in the global reorganization of the genome between normal and early mammary tumor formation. We found, however, cancer-specific repositioning of several gene loci. AKT1, BCL2, endogenous ERBB2, and VEGF loci were specifically repositioned in tumor acini compared with normal acini. Once again, changes in cell cycle status alone could not account for the repositioning of these genes. Thus, these specific repositioning events appear to be a consequence of tumorigenic progression itself. Our results differ from earlier observations in a HER2-negative breast cancer sample, where ERBB2 occupied a similar nuclear position to that of normal breast tissue (Wiech et al., 2005). These differences are likely caused by the fact that, unlike the model system we used, HER2 negative cancers, by definition, do not have amplifications or overactivation of ErbB2, and they suggest that reorganization events may differ depending on the nature of the oncogene that triggers tumorigenesis.
The observation in several experimental systems of differential positioning of active and inactive genes has lead to the suggestion that radial positioning is functionally important and that positioning is dependent on gene activity. This hypothesis was based on observation of a few selected, mostly loci-specific or developmentally regulated genes, particularly β-globin, IgK, and IgH during B cell differentiation and Mash1 in neuronal differentiation, but does not apply to others including CD4 and CD8 during T cell differentiation (Skok et al., 2001; Kosak et al., 2002; Kim et al., 2004; Ragoczy et al., 2006; Williams et al., 2006). Our analysis of 11 genes represents the largest unbiased test to date of a link between gene activity and positioning. In this set of genes, we found no general correlation between radial locus positioning and gene activity level in multiple cross comparisons between different physiological conditions. Our finding of a lack of correlation between activity level and nuclear radial positioning is consistent with a growing number of studies indicating that there is no strong and direct link between the activity of a gene and its radial position (Kim et al., 2004; Scheuermann et al., 2004) or its position within the chromosome territory (Kurz et al., 1996; Bartova et al., 2002; Mahy et al., 2002; Clemson et al., 2006). The lack of correlation between expression and radial position does not seem to be restricted to individual genes and applies to larger subchromosomal regions because their radial positioning is influenced by local gene density but not gene expression (Kupper et al., 2007). It is important to point out that our results cannot be interpreted to mean that nuclear positioning per se (e.g., relative to other genes or chromatin domains) is not of functional importance for gene expression (Misteli, 2007; Osborne et al., 2007). Most genes for which strong correlations between activity and positioning have been reported undergo a transition from a silenced to an active state or vice versa, whereas most genes in our analysis merely undergo changes in expression levels. It thus seems that the radial position of a gene is not directly determined by the level of its expression but whether it is permanently silenced or activated, e.g., in a tissue-specific manner. One likely possibility is that the higher-order organization of the chromatin fiber around a gene locus plays a key role in its nuclear location (Misteli, 2007). Although transition from a repressed to an active state, or vice versa, dramatically alters higher-order chromatin structure, modulation of the level of transcriptional output, even transient inhibition, likely does not. One can envision that the higher-order organization of the chromatin fiber directly affects a locus's ability to explore its nuclear environment and interact with other gene loci or structural elements of the nucleus, thus determining its position with the nucleus (Ragoczy et al., 2006; Fraser and Bickmore, 2007; Soutoglou and Misteli, 2007). Regardless of the precise functional relevance of gene positioning, the identification of a set of genes that are specifically repositioned in tumor acini compared with normal acini found here might pave the way for the application of spatial genome positioning patterns as a novel strategy for early cancer diagnosis.
Materials and methods
Cell culture
MCF-10A.B2 cells are a stable MCF-10A cell line containing p75.B2, a chimeric ErbB2 receptor that is activated by the synthetic ligand AP1510 (ARIAD Pharmaceuticals, Inc.; Muthuswamy et al., 1999, 2001). MCF10.B2 cells were provided by S. Muthuswamy (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). MCF-10A is a nontumorigenic, immortalized MEC line derived from the breast tissue of a 36-yr-old woman with fibrocystic changes (Soule et al., 1990). MCF-10A have a near diploid karyotype with a t(3:9)(p13;p22) and few minor additional aberrations in several subclones (Soule et al., 1990; Cowell et al., 2005; Worsham et al., 2006). We used the same subclone at similar passage numbers throughout this study, reducing the possible variation in karyotype. Monolayer (2D) cultures were maintained in DME/F12 (Invitrogen) supplemented with 5% horse serum (Invitrogen), 20 ng/ml EGF (PeproTech), 10 μg/ml insulin (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 0.5 μg/ml hydrocortisone (Sigma-Aldrich), 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen) in standard tissue culture plastic dishes. To enrich the culture for quiescent cells, 3 d after passaging, the cells were rinsed in serum-free DME/F12 medium and cultured for an additional 5 d in reduced serum medium (0.5% horse serum). Proliferating 2D cultured cells were grown on 12-mm coverslips for 2 or 3 d before fixation, whereas quiescent cultures were grown on 12-mm coverslips for 8 d. For 3D culture, 5,000 cells were seeded per well into a Laboratory-Tek 8-chambered glass slide (Thermo Fisher Scientific) that had been coated with thin layer of reduced growth factor reconstituted basement membrane extract (rBME; Trevigen), and were maintained in assay media (DME/F12 supplement with 2% reduced-growth rBME, 2% horse serum, 10 μg/ml insulin, 100 ng/ml cholera toxin, 0.5 μg/ml hydrocortisone, 50 U/ml penicillin, and 50 μg/ml streptomycin) containing 5 ng/ml EGF. For quantitative RT-PCR, 3D cultures were grown in 24-well plates. The media was changed 4, 8, 10, 14, and 18 d after seeding and cells were fixed or harvested at day 20. To activate the ErbB2 variant, EGF-containing assay media was replaced with assay media containing 1 μM AP1510 at day 10 and all subsequent changes of medium. All cells were maintained in a humidified atmosphere at 37°C at 5% CO2.
Indirect immunofluorescence
Indirect immunofluorescence was performed on 2D culture cells as described previously (Meaburn et al., 2007a) after a 10-min fixation in 4% (wt/vol) PFA/PBS and 10 min permeabilization in 1% Triton X-100/PBS. Cell fixation and indirect immunofluorescence were performed on 3D culture cells as described previously (Debnath et al., 2003) with the following modifications: sodium azide was not used in the IF buffer and goat serum was replaced with fetal bovine serum (Invitrogen), the second block was omitted, consequently antibodies were diluted in primary blocking solution, and cells were counterstained and mounted using DAPI-containing mounting medium (Vectashield; Vector Laboratories). Antibodies used were human autoimmune sera ANA-N (anti-nucleolar; Sigma-Aldrich), pKi-67 (clone 35; BD Biosciences), anti-centromere proteins (Antibodies Incorporated), goat anti–human FITC (Vector Laboratories), and Alexa Fluor 488 or 568 goat anti–mouse (Invitrogen). Cells were observed on a confocal microscope (LSM 510 META; Carl Zeiss, Inc.) and images were acquired using a 63× 1.4 NA oil objective lens (Carl Zeiss, Inc.) using an optical step size of 0.3 or 1 μm. A minimum of 200 cells for a specific staining pattern were scored, up to 1,000 cells in total for three independent experiments. With the exception of pKi-67 staining, mitotic cells were not counted. Statistical significance between growth conditions was determined by Yates correlated χ2 analysis, where a p-value of <0.05 was considered significant.
FISH
To produce probes for gene FISH, bacterial artificial chromosomes (BACs; BACPAC Resources Center) were labeled by nick translations with dUTP conjugated with biotin (Roche) or digoxigenin (Roche) using the human BAC clones detailed in Table I. The ERBB2 probe detected only the endogenous alleles and not the chimera construct; only two signals were detected for this probe per nuclei in MCF-10A.B2. Single or dual probe FISH experiments were performed. For 2D cultured cells, probes consisted of the following: 150–300 ng of digoxigenin- and/or biotin-labeled probe DNA combined with 3 μg human COT1 DNA (Roche) and 20 μg tRNA (Sigma-Aldrich) resuspended in 7 μl of hybridization mix (10% dextran sulfate, 50% formamide/2× SSC, and 1% Tween 20). For each well of 3D culture, ∼1 μg of digoxigenin- and/or biotin-labeled probe DNA, 15 μg human COT1, and 71 μg tRNA were resuspended in 50 μl of hybridization mix.
For 3D FISH, 2D cultured cells were fixed in 4% PFA/PBS for 10 min, permeabilized for 20 min in 0.5% (wt/vol) saponin (Sigma-Aldrich)/0.5% (vol/vol) Triton X-100/PBS, and incubated in 0.1 N HCl for 15 min with PBS washes between each step. After a 2× SSC wash, cells were equilibrated in 50% formamide/2× SSC. Probes were predenatured at 95°C for 5 min just before use. Nuclei and probes were then denatured together at 75°C for 5 min and left to hybridize at 37°C overnight in a humidified chamber. FISH on whole acini was performed using a modified version of the protocol used for 2D cultured cells: incubation in 0.5% saponin/0.5% Triton X-100/PBS was increased to 40 min, the 0.1 N HCl incubation was increased to 30 min, and the probe and nuclei were co-denatured at 85°C for 10 min.
The next day, 2D cultured cells were washed three times with 50% formamide/2× SSC at 45°C for 5 min each and thrice with 1× SSC at 60°C for 5 min each. Next, the cells were placed in 0.05% Tween 20/4× SSC at room temperature to cool and blocked for 20 min in 3% BSA/0.05% Tween 20/4× SSC. Detection antibodies (anti–digoxigenin-rhodamine [Roche] and fluorescein avidin DN [Vector Laboratories]) were diluted 1:200 in blocking solution and incubated with cells for 30 min at 37°C. Coverslips were mounted in DAPI-containing Vectashield mounting medium after three 5-min washes in 0.05% Tween 20/4× SSC at 42°C. Acini cultures were washed as described for 2D cultured cells but with the following exceptions: for the 1× SSC wash, the solution was prewarmed at 60°C but the washes were performed at 45°C and acini were incubated for 2 h with detection antibodies at 37°C.
For whole chromosome FISH, 2D cultured cells were fixed in 4% PFA/PBS for 10 min and permeabilized for 20 min in 0.5% (wt/vol) saponin/0.5% (vol/vol) Triton X-100/PBS before a 1-h incubation in 20% glycerol/PBS. Subsequently, the coverslips were subjected to seven freeze–thaw cycles and incubated in PBS for 30–45 min. The cells were then incubated for 30 min at 37°C in 100 μg/ml RNase A (Sigma-Aldrich)/2× SSC and for 15 min 0.1 N HCl. Preceding each step, the cells were washed three times in PBS. After a 2× SSC wash, cells were equilibrated in 50% formamide/2× SSC. Nuclei were co-denatured at 75°C for 5 min with whole chromosome probes (L. Christensen and T. Ried, National Cancer Institute, National Institutes of Health, Bethesda, MD) labeled in either biotin or digoxigenin. The nuclei were left to hybridize with the probes over two nights at 37°C in a humidified chamber. Posthybridization washes and probe detection were performed as for the gene FISH, with the exceptions that 0.1× SSC was used in place of 1× SSC and 0.1% Tween 20/4× SSC replaced 0.05% Tween 20/4× SSC.
Image acquisition
For gene FISH, 3D z stacks were acquired using a 63× 1.4 NA oil objective lens on an LSM 510 META confocal microscope at zoom level 3 with an optical step size of 0.3 μm with LSM 510 acquisition software V3.2 SP2 (Carl Zeiss, Inc.). In some cases, 2D cultured cells were imaged with an IX70 microscope (Olympus) controlled by a Deltavision System (Applied Precision) with SoftWoRx 3.5.1 (Applied Precision) and fitted with a charge-coupled device camera (CoolSnap; Photometrics) using a 60× 1.4 oil objective lens (Olympus) and an auxiliary magnification of 1.5 using an optical step size of 0.2 μm. Identical results were obtained on the two microscope systems. For chromosome territory FISH, all images were acquired using the IX70 Deltavision microscope fitted with a charge-coupled device camera using a 60× 1.4 oil objective lens with an optical step size of 0.2 μm. In all cases, for 2D cultured cells, the focal planes covered the entire nucleus (∼3–6 μm for proliferating and ∼3–5 μm for quiescent cultures). Typically, 15–20 sections for 2D cultures were acquired when imaged on the LSM 510 and ∼25 sections were aquired when imaging on the IX70. For the acini, whole acini were not imaged to reduce bleaching and increase the number of acini analyzed. Instead, the optical sections imaged totaled ∼15–20 μm in thickness. 2D maximal projections were generated and analyzed for radial distribution of FISH signals as described in the following section. For acini, projections for analysis were generated from subsections of the focal stack approximately one nucleus thick (∼4–6 μm for day 20 and ∼5–7 μm for day 20 ErbB2 activated) to generate multiple projections containing nonoverlapping whole nuclei. 195–220 nuclei were analyzed per BAC per growth condition or 100 nuclei for each chromosome territory analysis. Only nuclei with two signals per probe were chosen for analysis. Because cells in each acinus are derived from a single cell (Muthuswamy et al., 2001), nuclei were analyzed from a minimum of 10 different acini to reduce any possible clonal affect.
Quantitative analysis of FISH signal distributions
For the quantitative analyses of FISH signal distributions, software generated by S. Lockett and P. Gudla (National Cancer Institute, Bethesda, MD) was used. For automated nucleus segmentation and detection of FISH signals, a three-stage process was used that involved (1) noise reduction, (2) segmentation, and (3) postprocessing. For FISH analysis, background noise was removed in each channel by applying an adaptive nonlinear noise-reduction technique (Smith and Brady, 1997). A fuzzy C-means clustering algorithm was applied (Castleman, 1996; Duda et al., 2000) on the noise-reduced gray channels to delineate objects within them. Fuzzy images from the segmentation process were converted into binary images (hard segmentation) in order to obtain a collection of potential segmented objects (both FISH signals and nucleus). Gray-weighted thresholds were used to select only high-intensity FISH signals and we used a sequence of 2D morphological operations (opening and closing) for filling holes and boundary smoothing of the segmented nucleus. Manual comparison demonstrated successful identification of >99% of FISH signals and a false positive rate of <1%.
For quantitation of signal distributions, we computed intensity gravity center, area of nucleus, theoretical ellipse fitted to the nucleus boundary (Fitzgibbon et al., 1999), orientation of principal axis, and the ratio of the exclusive or area of segmented nucleus boundary and theoretical ellipse to the segmented nucleus. For each FISH signal, its intensity gravity center and the nearest pixels on the actual segmented nucleus boundary and the theoretical ellipse were determined. These values were used to determine the radial position of each signal. MCF-10.B2 cells have fairly regularly shaped nuclei in all growth conditions and the vast majority of nuclei fitted the theoretical template ellipse well. To ensure that nuclei that did not fit the template ellipse did not bias the results, a verification step was performed and nuclei that did not fit the template ellipse well were manually rejected from the analysis.
For statistical analysis to compare the distances of FISH signals from the intensity gravity center across multiple nuclei, each FISH signal was transformed into a template ellipse by applying a two-stage affine transformation on each segmented nucleus. The principal axis was horizontally oriented and each FISH signal was projected into a user-defined, common template ellipse via homographic projection between the template ellipse and the ellipse fitted to the segmented nucleus. Statistical differences (P < 0.05) between the distributions of a gene in different growth conditions were determined using 1D KS test. Pilot experiments demonstrated high reproducibility between repeat experiments, with variation of <4% of each data value. P-values between repeat experiments were never significant. All analysis tools were implemented using custom software written in MATLAB (The Mathworks, Inc.) with the DIPImage toolbox.
RNA extraction and quantitative RT-PCR
RNA from MCF-10A.B2 cells was isolated using the RNeasy mini kit (QIAGEN) according to the manufacturer's instructions. For each sample, 2 μg RNA was retrotranscribed using oligo dT or random primers and a High Capacity cDNA archive kit (Applied Biosystems) for 2 h at 42°C after a 5-min denaturation at 65°C. To stop the reaction, the cDNA was heated at 85°C for 10 min. Primers were generated to span two exons of the gene. The primer pairs generated an amplicon product of between 75 and 318 bp in length. The primers for ERBB2 were checked to ensure homology with only the endogenous ERBB2 and not the variant ERBB2. The following primer pairs were used for RT-PCR: AKT1 forward (F; 5′-CACCATCACACCACCTGACC-3′) and reverse (R; 5′-CCCTCCAAGCTATCGCCAG-3′), BCL2 F (5′-GATGACTGAGTACCTGAACCG-3′) and R (5′-GCCAGGAGAAATCAAACAGAG-3′), CCND1 F (5′-TCCTCTCCAAAATGCCAGAG-3′) and R (5′-GGCGGATTGGAAATGAACTT-3′), ERBB2 F (5′-TGTGGACATGCACAAAAGTG-3′) and R (5′-GCAGGTAGGTGAGTTCCAGG-3′), ERBB2IP F (5′-CAACCTGAAGGACCAGCATC-3′) and R (5′-GACACTGCTTGTCCATGTTC-3′), FGFR1 F (5′-GTCACAGCCACACTCTGCAC-3′) and R (5′-GGGACCAGGAAGGACTCCACT-3′), MMP1 F (5′-CCAAATGGGCTTGAAGCTGC-3′) and R (5′-GTCCCTGAACAGCCCAGTAC-3′), TP53 F (5′-GCTTTCCACGACGGTGAC-3′) and R (5′-GCTCGACGCTAGGATCTGAC-3′), PTEN F (5′-GAGCGTGCAGATAATGACAAGG-3′) and R (5′-GGATTTGACGGCTCCTCTACTG-3′), TGFB3 F (5′-CTCCGCAGTGCAGACACAAC-3′) and R (5′-CATAGTACAGGATGGTCAGG-3′), and VEGF F (5′-AGCCTTGCCTTGCTGCTCTA-3′) and R (5′-GTGCTGGCCTTGGTGAGG-3′). Cyclophilin was used as the housekeeping control gene (F, 5′-GTCAACCCCACCGTGTTCTT-3′; R, 5′-CTGCTGTCTTTGGGACCTTGT-3′). For each primer pair, 1 μl of each cDNA sample was analyzed by real-time RT-PCR in a 25-μl reaction using iQ SYBR green supermix (Bio-Rad Laboratories) in an iCycler (Bio-Rad Laboratories) under the following reaction conditions: 3 min at 95°C followed by 41 amplification cycles (20 s at 95°C and 30 s at 60°C). Melting curves of the amplified products were obtained to verify that a single amplicon was generated. Samples were analyzed using RNA extracted from each growth condition in three independent experiments and each RT-PCR reaction was run in duplicate. Statistical significance between the normalized expression levels of a gene between growth conditions was assessed using a t test, in Excel (Microsoft). A p-value of <0.05 was considered significant.
Metaphase spreads
Proliferating monocultures were harvested and swollen in hypotonic buffer (75 mM KCl) for 15 min at RT before fixation in ice-cold 3:1 (vol/vol) methanol/acetic acid. Cells were dropped onto humid slides and air dried before incubation at 70°C for 1 h. Slides were mounted in DAPI-containing Vectashield mounting medium. Metaphases were imaged with an IX70 microscope fitted with a charge-coupled device camera using a 40× 1.35 oil objective lens. 24 metaphase spreads were analyzed each for control and ErbB2-activated cultures. The colors of the grayscale images were inverted using Photoshop (Adobe).
Online supplemental materials
Fig. S1 presents the radial distribution of genes during differentiation of MECs and early tumorigenesis as histograms. Fig. S2 presents quantification of the radial distribution of chromosome territories after exit from the cell cycle in 2D cultures. Fig. S3 shows the maintenance of karyotype upon activation of ErbB2. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200708204/DC1.
Supplemental Material
Acknowledgments
We wish to thank Dr. T. Karpova for help with microscopy; Drs. T. Takizawa and P. Scaffidi and all members of the Misteli laboratory for discussions; and Dr. M. Takizawa for help with metaphase spreads. For providing reagents we are grateful to Dr S. Muthuswamy, ARIAD Pharmaceuticals, L. Christensen, and Dr. T. Ried. We also wish to thank Drs. S. Lockett and P. Gudla for FISH positioning analysis software. Fluorescence imaging was performed at the National Cancer Institute Fluorescence Imaging Facility.
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Abbreviations used in this paper: BAC, bacterial artificial chromosome; HSA, human chromosome; KS test, Kolmogorov-Smirnov test; MEC, mammary epithelial cell; NOR, nucleolar organizing region.
References
- Aranda, V., T. Haire, M.E. Nolan, J.P. Calarco, A.Z. Rosenberg, J.P. Fawcett, T. Pawson, and S.K. Muthuswamy. 2006. Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat. Cell Biol. 8:1235–1245. [DOI] [PubMed] [Google Scholar]
- Bartova, E., S. Kozubek, P. Jirsova, M. Kozubek, H. Gajova, E. Lukasova, M. Skalnikova, A. Ganova, I. Koutna, and M. Hausmann. 2002. Nuclear structure and gene activity in human differentiated cells. J. Struct. Biol. 139:76–89. [DOI] [PubMed] [Google Scholar]
- Borden, J., and L. Manuelidis. 1988. Movement of the X chromosome in epilepsy. Science. 242:1687–1691. [DOI] [PubMed] [Google Scholar]
- Boyle, S., S. Gilchrist, J.M. Bridger, N.L. Mahy, J.A. Ellis, and W.A. Bickmore. 2001. The spatial organization of human chromosomes within the nuclei of normal and emerin-mutant cells. Hum. Mol. Genet. 10:211–219. [DOI] [PubMed] [Google Scholar]
- Bridger, J.M., S. Boyle, I.R. Kill, and W.A. Bickmore. 2000. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr. Biol. 10:149–152. [DOI] [PubMed] [Google Scholar]
- Castleman, K.R. 1996. Digital Image Processing. Prentice Hall, Englewood Cliffs, NJ. 667 pp.
- Chambeyron, S., and W.A. Bickmore. 2004. Chromatin decondensation and nuclear reorganization of the HoxB locus upon induction of transcription. Genes Dev. 18:1119–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandramouly, G., P.C. Abad, D.W. Knowles, and S.A. Lelievre. 2007. The control of tissue architecture over nuclear organization is crucial for epithelial cell fate. J. Cell Sci. 120:1596–1606. [DOI] [PubMed] [Google Scholar]
- Chuang, T.C., S. Moshir, Y. Garini, A.Y. Chuang, I.T. Young, B. Vermolen, R. van den Doel, V. Mougey, M. Perrin, M. Braun, et al. 2004. The three-dimensional organization of telomeres in the nucleus of mammalian cells. BMC Biol. 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson, C.M., L.L. Hall, M. Byron, J. McNeil, and J.B. Lawrence. 2006. The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc. Natl. Acad. Sci. USA. 103:7688–7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowell, J.K., J. LaDuca, M.R. Rossi, T. Burkhardt, N.J. Nowak, and S. Matsui. 2005. Molecular characterization of the t(3;9) associated with immortalization in the MCF10A cell line. Cancer Genet. Cytogenet. 163:23–29. [DOI] [PubMed] [Google Scholar]
- Cremer, M., K. Kupper, B. Wagler, L. Wizelman, J. Hase Jv, Y. Weiland, L. Kreja, J. Diebold, M.R. Speicher, and T. Cremer. 2003. Inheritance of gene density–related higher order chromatin arrangements in normal and tumor cell nuclei. J. Cell Biol. 162:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremer, T., M. Cremer, S. Dietzel, S. Muller, I. Solovei, and S. Fakan. 2006. Chromosome territories–a functional nuclear landscape. Curr. Opin. Cell Biol. 18:307–316. [DOI] [PubMed] [Google Scholar]
- Croft, J.A., J.M. Bridger, S. Boyle, P. Perry, P. Teague, and W.A. Bickmore. 1999. Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145:1119–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath, J., and J.S. Brugge. 2005. Modelling glandular epithelial cancers in three-dimensional cultures. Nat. Rev. Cancer. 5:675–688. [DOI] [PubMed] [Google Scholar]
- Debnath, J., K.R. Mills, N.L. Collins, M.J. Reginato, S.K. Muthuswamy, and J.S. Brugge. 2002. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell. 111:29–40. [DOI] [PubMed] [Google Scholar]
- Debnath, J., S.K. Muthuswamy, and J.S. Brugge. 2003. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 30:256–268. [DOI] [PubMed] [Google Scholar]
- Duda, R.O., P.E. Hart, and D.G. Stork. 2000. Pattern Classification. John Wiley & Sons Inc. New York. 654 pp.
- Fitzgibbon, A., M. Pilu, and R.B. Fisher. 1999. Direct least square fitting of ellipses. IEEE Trans. Pattern Anal. Mach. Intell. 21:476–480. [Google Scholar]
- Foster, H.A., L.R. Abeydeera, D.K. Griffin, and J.M. Bridger. 2005. Non-random chromosome positioning in mammalian sperm nuclei, with migration of the sex chromosomes during late spermatogenesis. J. Cell Sci. 118:1811–1820. [DOI] [PubMed] [Google Scholar]
- Fournier, M.V., K.J. Martin, P.A. Kenny, K. Xhaja, I. Bosch, P. Yaswen, and M.J. Bissell. 2006. Gene expression signature in organized and growth-arrested mammary acini predicts good outcome in breast cancer. Cancer Res. 66:7095–7102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, P., and W. Bickmore. 2007. Nuclear organization of the genome and the potential for gene regulation. Nature. 447:413–417. [DOI] [PubMed] [Google Scholar]
- Harari, D., and Y. Yarden. 2000. Molecular mechanisms underlying ErbB2/HER2 action in breast cancer. Oncogene. 19:6102–6114. [DOI] [PubMed] [Google Scholar]
- Hynes, N.E., and D.F. Stern. 1994. The biology of erbB-2/neu/HER-2 and its role in cancer. Biochim. Biophys. Acta. 1198:165–184. [DOI] [PubMed] [Google Scholar]
- Kaminker, P., C. Plachot, S.H. Kim, P. Chung, D. Crippen, O.W. Petersen, M.J. Bissell, J. Campisi, and S.A. Lelievre. 2005. Higher-order nuclear organization in growth arrest of human mammary epithelial cells: a novel role for telomere-associated protein TIN2. J. Cell Sci. 118:1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S.H., P.G. McQueen, M.K. Lichtman, E.M. Shevach, L.A. Parada, and T. Misteli. 2004. Spatial genome organization during T-cell differentiation. Cytogenet. Genome Res. 105:292–301. [DOI] [PubMed] [Google Scholar]
- Kosak, S.T., J.A. Skok, K.L. Medina, R. Riblet, M.M. Le Beau, A.G. Fisher, and H. Singh. 2002. Subnuclear compartmentalization of immunoglobulin loci during lymphocyte development. Science. 296:158–162. [DOI] [PubMed] [Google Scholar]
- Kupper, K., A. Kolbl, D. Biener, S. Dittrich, J. von Hase, T. Thormeyer, H. Fiegler, N.P. Carter, M.R. Speicher, T. Cremer, and M. Cremer. 2007. Radial chromatin positioning is shaped by local gene density, not by gene expression. Chromosoma. 116:285–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz, A., S. Lampel, J.E. Nickolenko, J. Bradl, A. Benner, R.M. Zirbel, T. Cremer, and P. Lichter. 1996. Active and inactive genes localize preferentially in the periphery of chromosome territories. J. Cell Biol. 135:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanctot, C., T. Cheutin, M. Cremer, G. Cavalli, and T. Cremer. 2007. Dynamic genome architecture in the nuclear space: regulation of gene expression in three dimensions. Nat. Rev. Genet. 8:104–115. [DOI] [PubMed] [Google Scholar]
- Le Beyec, J., R. Xu, S.Y. Lee, C.M. Nelson, A. Rizki, J. Alcaraz, and M.J. Bissell. 2007. Cell shape regulates global histone acetylation in human mammary epithelial cells. Exp. Cell Res. 313:3066–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelievre, S.A., V.M. Weaver, J.A. Nickerson, C.A. Larabell, A. Bhaumik, O.W. Petersen, and M.J. Bissell. 1998. Tissue phenotype depends on reciprocal interactions between the extracellular matrix and the structural organization of the nucleus. Proc. Natl. Acad. Sci. USA. 95:14711–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahy, N.L., P.E. Perry, S. Gilchrist, R.A. Baldock, and W.A. Bickmore. 2002. Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J. Cell Biol. 157:579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer, R., A. Brero, J. von Hase, T. Schroeder, T. Cremer, and S. Dietzel. 2005. Common themes and cell type specific variations of higher order chromatin arrangements in the mouse. BMC Cell Biol. 6:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaburn, K.J., and T. Misteli. 2007. Cell biology: chromosome territories. Nature. 445:379–781. [DOI] [PubMed] [Google Scholar]
- Meaburn, K.J., E. Cabuy, G. Bonne, N. Levy, G.E. Morris, G. Novelli, I.R. Kill, and J.M. Bridger. 2007. a. Primary laminopathy fibroblasts display altered genome organization and apoptosis. Aging Cell. 6:139–153. [DOI] [PubMed] [Google Scholar]
- Meaburn, K.J., T. Misteli, and E. Soutoglou. 2007. b. Spatial genome organization in the formation of chromosomal translocations. Semin. Cancer Biol. 17:80–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misteli, T. 2007. Beyond the sequence: cellular organization of genome function. Cell. 128:787–800. [DOI] [PubMed] [Google Scholar]
- Murmann, A.E., J. Gao, M. Encinosa, M. Gautier, M.E. Peter, R. Eils, P. Lichter, and J.D. Rowley. 2005. Local gene density predicts the spatial position of genetic loci in the interphase nucleus. Exp. Cell Res. 311:14–26. [DOI] [PubMed] [Google Scholar]
- Muthuswamy, S.K., M. Gilman, and J.S. Brugge. 1999. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol. Cell. Biol. 19:6845–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy, S.K., D. Li, S. Lelievre, M.J. Bissell, and J.S. Brugge. 2001. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat. Cell Biol. 3:785–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C.M., and M.J. Bissell. 2005. Modeling dynamic reciprocity: engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Semin. Cancer Biol. 15:342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusser, M., V. Schubel, A. Koch, T. Cremer, and S. Muller. 2007. Evolutionarily conserved, cell type and species-specific higher order chromatin arrangements in interphase nuclei of primates. Chromosoma. 116:307–320. [DOI] [PubMed] [Google Scholar]
- Osborne, C.S., L. Chakalova, J.A. Mitchell, A. Horton, A.L. Wood, D.J. Bolland, A.E. Corcoran, and P. Fraser. 2007. Myc dynamically and preferentially relocates to a transcription factory occupied by Igh. PLoS Biol. 5:e192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada, L.A., P.G. McQueen, and T. Misteli. 2004. Tissue-specific spatial organization of genomes. Genome Biol. 7:R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, O.W., L. Ronnov-Jessen, A.R. Howlett, and M.J. Bissell. 1992. Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc. Natl. Acad. Sci. USA. 89:9064–9068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plachot, C., and S.A. Lelievre. 2004. DNA methylation control of tissue polarity and cellular differentiation in the mammary epithelium. Exp. Cell Res. 298:122–132. [DOI] [PubMed] [Google Scholar]
- Ragoczy, T., A. Telling, T. Sawado, M. Groudine, and S.T. Kosak. 2003. A genetic analysis of chromosome territory looping: diverse roles for distal regulatory elements. Chromosome Res. 11:513–525. [DOI] [PubMed] [Google Scholar]
- Ragoczy, T., M.A. Bender, A. Telling, R. Byron, and M. Groudine. 2006. The locus control region is required for association of the murine beta-globin locus with engaged transcription factories during erythroid maturation. Genes Dev. 20:1447–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar, R., A. Guffei, B.J. Vermolen, Y. Garini, and S. Mai. 2007. Alterations of centromere positions in nuclei of immortalized and malignant mouse lymphocytes. Cytometry A. 71:386–392. [DOI] [PubMed] [Google Scholar]
- Scheuermann, M.O., J. Tajbakhsh, A. Kurz, K. Saracoglu, R. Eils, and P. Lichter. 2004. Topology of genes and nontranscribed sequences in human interphase nuclei. Exp. Cell Res. 301:266–279. [DOI] [PubMed] [Google Scholar]
- Sengupta, K., M.B. Upender, L. Barenboim-Stapleton, Q.T. Nguyen, S.M. Wincovitch Sr., S.H. Garfield, M.J. Difilippantonio, and T. Ried. 2007. Artificially introduced aneuploid chromosomes assume a conserved position in colon cancer cells. PLoS ONE. 2:e199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seton-Rogers, S.E., Y. Lu, L.M. Hines, M. Koundinya, J. LaBaer, S.K. Muthuswamy, and J.S. Brugge. 2004. Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc. Natl. Acad. Sci. USA. 101:1257–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skok, J.A., K.E. Brown, V. Azuara, M.L. Caparros, J. Baxter, K. Takacs, N. Dillon, D. Gray, R.P. Perry, M. Merkenschlager, and A.G. Fisher. 2001. Nonequivalent nuclear location of immunoglobulin alleles in B lymphocytes. Nat. Immunol. 2:848–854. [DOI] [PubMed] [Google Scholar]
- Smith, S.M., and J.M. Brady. 1997. SUSAN – a new approach to low level image processing. International Journal of Computer Vision. 23:45–78. [Google Scholar]
- Solovei, I., L. Schermelleh, K. During, A. Engelhardt, S. Stein, C. Cremer, and T. Cremer. 2004. Differences in centromere positioning of cycling and postmitotic human cell types. Chromosoma. 112:410–423. [DOI] [PubMed] [Google Scholar]
- Soule, H.D., T.M. Maloney, S.R. Wolman, W.D. Peterson Jr., R. Brenz, C.M. McGrath, J. Russo, R.J. Pauley, R.F. Jones, and S.C. Brooks. 1990. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50:6075–6086. [PubMed] [Google Scholar]
- Soutoglou, E., and T. Misteli. 2007. Mobility and immobility of chromatin in transcription and genome stability. Curr. Opin. Genet. Dev. 17:435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, G.J., J.M. Bridger, A.P. Cuthbert, R.F. Newbold, W.A. Bickmore, and B. McStay. 2001. Human acrocentric chromosomes with transcriptionally silent nucleolar organizer regions associate with nucleoli. EMBO J. 20:2867–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taslerova, R., S. Kozubek, E. Lukasova, P. Jirsova, E. Bartova, and M. Kozubek. 2003. Arrangement of chromosome 11 and 22 territories, EWSR1 and FLI1 genes, and other genetic elements of these chromosomes in human lymphocytes and Ewing sarcoma cells. Hum. Genet. 112:143–155. [DOI] [PubMed] [Google Scholar]
- Taslerova, R., S. Kozubek, E. Bartova, P. Gajduskova, R. Kodet, and M. Kozubek. 2006. Localization of genetic elements of intact and derivative chromosome 11 and 22 territories in nuclei of Ewing sarcoma cells. J. Struct. Biol. 155:493–504. [DOI] [PubMed] [Google Scholar]
- Underwood, J.M., K.M. Imbalzano, V.M. Weaver, A.H. Fischer, A.N. Imbalzano, and J.A. Nickerson. 2006. The ultrastructure of MCF-10A acini. J. Cell. Physiol. 208:141–148. [DOI] [PubMed] [Google Scholar]
- Wiech, T., S. Timme, F. Riede, S. Stein, M. Schuricke, C. Cremer, M. Werner, M. Hausmann, and A. Walch. 2005. Human archival tissues provide a valuable source for the analysis of spatial genome organization. Histochem. Cell Biol. 123:229–238. [DOI] [PubMed] [Google Scholar]
- Williams, R.R., V. Azuara, P. Perry, S. Sauer, M. Dvorkina, H. Jorgensen, J. Roix, P. McQueen, T. Misteli, M. Merkenschlager, and A.G. Fisher. 2006. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci. 119:132–140. [DOI] [PubMed] [Google Scholar]
- Witt, A.E., L.M. Hines, N.L. Collins, Y. Hu, R.N. Gunawardane, D. Moreira, J. Raphael, D. Jepson, M. Koundinya, A. Rolfs, et al. 2006. Functional proteomics approach to investigate the biological activities of cDNAs implicated in breast cancer. J. Proteome Res. 5:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worsham, M.J., G. Pals, J.P. Schouten, F. Miller, N. Tiwari, R. van Spaendonk, and S.R. Wolman. 2006. High-resolution mapping of molecular events associated with immortalization, transformation, and progression to breast cancer in the MCF10 model. Breast Cancer Res. Treat. 96:177–186. [DOI] [PubMed] [Google Scholar]
- Zink, D., A.H. Fische, and J.A. Nickerson. 2004. Nuclear structure in cancer cells. Nat. Rev. Cancer. 4:677–687. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}