

Abstract
Aging is associated with a progressive loss of skeletal muscle mass and strength and the mechanisms mediating these effects likely involve mitochondrial DNA (mtDNA) mutations, mitochondrial dysfunction and the activation of mitochondrial mediated apoptosis. Because the mitochondrial genome is densely packed and close to the main generator of reactive oxygen species (ROS) in the cell, the electron transport chain (ETC), an important role for mtDNA mutations in aging has been proposed. Point mutations and deletions in mtDNA accumulate with age in a wide variety of tissues in mammals, including humans, and often coincide with significant tissue dysfunction. Here, we examine the evidence supporting a causative role for mtDNA mutations in aging and sarcopenia. We review experimental outcomes showing that mtDNA mutations, leading to mitochondrial dysfunction and possibly apoptosis, are causal to the process of sarcopenia. Moreover, we critically discuss and dispute an important part of the mitochondrial ‘vicious cycle’ theory of aging which proposes that accumulation of mtDNA mutations may lead to an enhanced mitochondrial ROS production and ever increasing oxidative stress which ultimately leads to tissue deterioration and aging. Potential mechanism(s) by which mtDNA mutations may mediate their pathological consequences in skeletal muscle are also discussed.
Keywords: skeletal, muscle, mitochondria, DNA, mutations, ROS, deletions, vicious, cycle, electron, transport, chain
Introduction
With aging a progressive loss of skeletal muscle mass and strength is observed, a condition termed sarcopenia. On average, aging individuals lose muscle mass at a rate of 1-2% per year past the age of 50 (1, 2). This age-related muscle atrophy is associated with muscle weakness and can have significant effects on physical function and quality of life as aging commences. At the cellular level, the aging process can activate stress-associated signal transduction pathways that may eventually result in the collapse of mitochondrial function, causing apoptotic cell death. Because the mitochondrion contains its own intron-less and histone-less ∼16-kilobase circular DNA that is located closely to the main ROS source in the cell, the mitochondrial ETC, a central role for mtDNA mutations in aging has been postulated (3-5). In fact, mtDNA mutations have been shown to accumulate with aging in several tissues of various species (6-11), including skeletal muscle.
Over the past two decades, increasing evidence suggests that mitochondrial dysfunction may play a causal role in the aging process. The essential role of mitochondria in cellular ATP production, the generation of reactive oxygen species (ROS), and the induction of apoptosis suggest a number of mechanisms for mitochondrial pathology. There is strong evidence that age induces alterations in the mitochondrial genome that lead to defects in mitochondrial function, especially in post-mitotic tissues with high energy requirements such as the heart, brain and skeletal muscle (12-14).
It was proposed that during an individual's life span, ROS, by-products of oxidative metabolism, accumulate and alter cell components (15). Mitochondria, one of the primary sources of ROS, are particularly affected, leading to changes in their structure as well as in the genetic information of mtDNA. The observed alterations of mtDNA include oxidative damage to DNA bases, point mutations and large scale deletions or duplications (6, 11, 16-23). MtDNA mutations are known to have deleterious effects on oxidative phosphorylation, especially in patients with mitochondrial diseases (24, 25), and tissues that rely heavily on oxidative phosphorylation are expected to be more affected.
Special focus on skeletal muscle is given in this review because this tissue is highly dependent on oxidative phosphorylation and suffers marked age-related degeneration. The information presented here focuses on the changes that are induced in mitochondrial ETC function as a result of aging, and the cellular impact of mtDNA mutations, by providing evidence for a causal role of mtDNA mutations in mammalian aging and sarcopenia. The plausibility of the mitochondrial ‘vicious cycle’ theory and recent controversial findings on the role of mtDNA mutations in mammalian aging are also discussed.
Mitochondrial DNA Mutations, Deletions and Electron Transport Chain Abnormalities in Aging
There is now growing evidence that the accumulation of mtDNA mutations and deletions, associated with aging, result in tissue dysfunction; impaired respiratory function and oxidative phosphorylation in muscle fibers is becoming increasingly evident (26, 27), and point-mutations and deletions in mtDNA have been found to correlate with this reduced capacity (19, 28, 29). Biochemical analyses of ETC complex activities performed in tissue homogenates from humans, rhesus monkeys and rodents have, in general, identified age-associated decreases in the activities of several ETC complexes. Previous cytochemical-immunocytochemical studies of oxidative phosphorylation enzymes in monkeys (10-25 years of age) showed complex III, complex IV and complex V defects in skeletal muscles, diaphragm, myocardium and extraocular muscles of 25-year-old animals. Decreased activity of complex I with age was also reported in gastrocnemius muscle of mice (30). These defects were randomly distributed and not associated with a loss of complex II, which is all nuclear encoded (30, 31).
Commonly used markers for mitochondrial ETC abnormalities include the loss of cytochrome c oxidase (COX) activity and the concomitant increase in succinate dehydrogenase (SDH) activity (COX-/SDH++) (COX-/SDH++ regions, also known as ragged red fibers (RRF)). Aiken's group has repeatedly demonstrated loss of COX (complex VI) staining combined with hyperactive SDH staining in aged rat, primate, and human skeletal muscle cross-sections (6, 28, 32-36). Interestingly, these segmental ETC abnormalities co-localized with clonal intracellular expansions of unique somatically derived mtDNA deletion mutations. Specifically, in rats, they showed that the proportion of ETC abnormal fibers that displayed the RRF phenotype increased significantly with age, and there were no ETC abnormal fibers with the RRF phenotype observed in the 5-month-old muscles, whereas 42% of the total ETS abnormal fibers in the 38-month-old animals displayed the RRF phenotype. They further detected shorter than wild type genomes in all of the RRFs, while mtDNA deletion mutations were not detected in ETS normal fibers from the same sections. Multiple microdissections along the same RRF amplified identically sized products, supporting the clonal nature mtDNA deletion mutations (6). Deletions occurred at the major arc of the mtDNA spanning the origin of replication, and different deletions were observed between the fibers (28). In humans similar results were obtained; the number of vastus lateralis muscle fibers exhibiting mitochondrial ETC abnormalities increased from an estimated 6% at age 49 years to 31% at age 92 years (36). Furthermore, mtDNA-deletion mutations were detected in all ETC-abnormal fibers (RRF) (36). Deletion mutations were also clonal within a fiber and concomitant to the COX-/SDH++ region. Quantitative PCR analysis of wild-type and deletion-containing mtDNA genomes within ETC-abnormal regions of single fibers demonstrated that these deletion mutations accumulate to detrimental levels (>90% of the total mtDNA) (36). Importantly, in the areas of the fiber where the mutation abundance surpassed 90% of the total mitochondrial genomes, the fibers lost COX activity and displayed abnormal morphology, such as, a striking decrease in cross sectional area indicative of fiber atrophy, fiber splitting and breakage, while morphologically normal areas of the same fiber contained only wild type mitochondrial genomes and did not exhibit de-regulation of ETC complex activities (6, 33). The same group also showed that the vastus lateralis muscle, which undergoes a high degree of sarcopenia, exhibited more ETS abnormalities and associated fiber loss than the soleus and adductor longus muscles, which are more resistant to sarcopenia, suggesting a direct association between ETS abnormalities and fiber loss (35). These findings strongly suggest a causal role for age-associated mitochondrial DNA deletion mutations and mitochondrial dysfunction in sarcopenia.
Human studies conducted by other groups also provide evidence for an increase in mtDNA mutations with aging, and a correlation between mtDNA mutations and the occurrence of skeletal muscle abnormalities with advancing age (19, 29, 37, 38). Muscle biopsies from aged humans revealed that randomly deleted mtDNA appeared mainly in the oldest subjects (beyond 80 years old), affecting up to 70% of mtDNA molecules, and coincides with a decrease in the activities of complexes III and IV of the ETC, which contain subunits encoded by mtDNA (38).
Furthermore, Wang et al., showed that muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication; specifically, they demonstrated that most of 26 individuals 53 to 92 years old, without a known history of neuromuscular disease, exhibited an accumulation of two new point mutations, i.e., A189G and T408A, at mtDNA replication control sites in muscle which were absent or marginally present in 19 young individuals. These two mutations were not found in fibroblasts from 22 subjects 64 to 101 years of age (T408A), or were present only in three subjects in very low amounts (A189G) (11). The investigators suggested that the striking tissue specificity of the muscle mtDNA mutations detected, and their mapping at critical sites for mtDNA replication, strongly point to the involvement of a specific mutagenic machinery and to the functional relevance of these mutations.
Similar to Aiken's experimental findings regarding mtDNA deletions, Fayet et al. identified high levels of clonally expanded mtDNA point mutations in cytochrome c oxidase deficient muscle fibers, from old individuals without muscle disease, while no point mutations were detected in any of the normal fibers (39). Immunohistochemical experiments showed that the majority of the cytochrome c oxidase deficient muscle fibers expressed reduced levels of subunit II of cytochrome c oxidase, which is encoded by mitochondrial DNA, whereas there was normal or increased expression of subunit IV of cytochrome c oxidase, which is encoded by nuclear DNA (39). The authors concluded that mtDNA point mutations are associated with cytochrome c oxidase deficient muscle fiber segments in aging, the focal accumulation of which may cause significant impairment of mitochondrial function in individual cells in spite of low overall levels of mitochondrial DNA mutations in muscle (39). Indeed, although only a few cells develop COX deficiency, the resultant cellular dysfunction might have substantial effects, especially if the cell is part of a complex network (40). It is therefore, likely that, in skeletal muscle of aged individuals, normal mtDNA devoid of deletions or point mutations may represent a minority of the total mtDNA pool. There is also evidence that the rate of mitochondrial mutagenesis is faster in mice than humans per unit time (41), a necessary condition if mitochondrial mutations are causally linked to aging. When taken as a whole, these studies provide compelling evidence for an important role of mitochondrial DNA mutations in aging.
Evidence for a Causal Role of mtDNA Mutations in Aging
As discussed above, there is an ever growing body of research that supports an important role for mtDNA mutations in aging by providing experimental support for an association between mtDNA mutations, ETC abnormalities and tissue dysfunction, particularly in long-lived post mitotic cells. However, most of the studies to date are correlative in nature. In particular, until recently, it has been unclear whether mtDNA mutations are simply associated with aging in various tissues, or if they actually cause alterations in tissue function.
In 2004, Larsson and colleagues published the first experimental evidence providing a causative link between mtDNA mutations and mammalian aging (42). In order to test the in vivo effects of somatic mtDNA mutation accumulation, they constructed knock-in mice that expressed a proof-reading-deficient version of PolgA, the nucleus-encoded catalytic subunit of the mitochondrial DNA polymerase γ (POLG mice). The specific mutation in the exonuclease domain of Polg resulted in increased spontaneous mutation rates in mtDNA in mice homozygous for the mutation. Specifically, mice with mtDNA mutator phenotype exhibited a threefold to fivefold increase in the levels of point mutations, as well as increased amounts of deleted mtDNA (42). This increase in somatic mtDNA mutations was associated with severely reduced lifespan and premature onset of aging-related phenotypes such as alopecia, kyphosis, osteoporosis, heart enlargement and sarcopenia. Importantly, they demonstrated that mtDNA mutations and deletions are responsible for a progressive decline in respiratory function of mitochondrially encoded complexes, that was evident as early as 12 weeks, resulting in decreased oxygen consumption and ATP production (42, 43).
A year later we corroborated Trifunovic's findings regarding the impact of mtDNA mutations in aging using mice with the same mutation in PolgA (POLG mice)(44). Specifically, in skeletal muscle of mutant mice, we found profound decreases in mitochondrial O2 consumption during state 3, and significantly reduced ATP content compared to wild type mice (WT) at ∼11-mo of age, a time point when the sarcopenic phenotype is also evident in mutator mice (A. Hiona unpublished data). Interestingly, we showed that the accumulation of mtDNA mutations was not associated with increased levels of oxidative stress or a defect in cellular proliferation, but was correlated with the induction of apoptotic markers, suggesting that accumulation of mtDNA mutations that promote apoptosis may be a central mechanism driving mammalian aging (44) and age-related phenotypes, such as skeletal muscle loss.
Moreover, Zassenhaus and colleagues studied mice that express a proofreading-deficient Polg specifically in the heart, and develop cardiac mtDNA mutations, in order to determine whether low frequency mtDNA mutations are pathogenic. They found that sporadic myocytic death occurred in all regions of the heart, due to apoptosis as assessed by histological analysis and terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling (TUNEL) staining (45). They also pointed out that cytochrome c was released from mitochondria and concluded that mtDNA mutations are pathogenic, and seem to trigger apoptosis through the mitochondrial pathway (45).
Mitochondrial DNA Damage, Mutagenesis, and the Mitochondrial ‘Vicious Cycle’ Theory of Aging
By far, the predominant kind of insult, to which mtDNA is exposed, is oxidative damage. It is also well demonstrated that oxidative damage to mtDNA is much greater compared to nuclear DNA in various species and tissues examined (22, 23). This susceptibility of the mitochondrial genome to oxidative DNA damage may be due to a number of factors including: 1) greater exposure of mtDNA to reactants: mtDNA is in close proximity to the ETC, whose complexes I and III are believed to be the predominant sites of ROS production inside the cell, 2) the lack of protective histones, and 3) the lack of introns and the compactness of its genetic information, so that damage at any point in the genome will likely occur in a gene. ROS - induced damage to mtDNA is believed to be the primary source of mutagenesis in mitochondria (46), giving rise to both mtDNA mutations and deletions. Furthermore, the greater occurrence of point-mutations and deletions observed in mtDNA compared to nuclear DNA could be due to a less advanced DNA repair system (47, 48). In fact, mitochondria are believed to entirely lack nucleotide excision repair (NER), which constitutes a major nuclear defense system acting on various nuclear DNA lesions including pyrimidine dimmers (47, 48). Mitochondria also have other discrepancies and may also have less sophisticated mismatch repair (MMR) system (48). Like crosslinks between DNA bases (such as thymine dimmers), DNA–protein crosslinks, or bulky DNA-adducts can cause a stall during mtDNA replication which can induce DNA double-strand brakes (49, 50), contributing to the occurrence of mtDNA deletions with aging, and as previous research suggests, it is likely for mtDNA containing deletions, to acquire a replicative advantage over longer wild type mtDNA (28).
Despite the fact that in animal cells mtDNA comprises only 1–3% of genetic material, its contribution to cellular physiology could be much greater than would be expected from its size alone (51). For instance, as previously mentioned, (a) it mutates at higher rates than nuclear DNA; (b) it encodes either polypeptides of ETC or components required for their synthesis, such as tRNAs and rRNAs, and therefore, any coding mutations in mtDNA will affect the ETC as a whole; this could affect both the assembly and function of the products of numerous nuclear genes in ETC complexes; (c) defects in the ETC can have pleiotropic effects because they affect cellular energetics as a whole (51). The phenotypic implications of mtDNA mutations are dependent on which gene product is disrupted and one might predict that damage may occur in those complexes to which the mitochondrial genome makes the greatest contributions.
The mitochondrial ‘vicious cycle’ theory of aging (Figure 1) can be considered as an extension and refinement of the free radical theory which was first put forward by Harman (4, 52). Its major premise is that mtDNA mutations accumulate progressively during life, as a side effect of respiration, and are directly responsible for a measurable deficiency in cellular oxidative phosphorylation activity, leading to an enhanced ROS production (51). In turn, increased ROS production results in an increased rate of mtDNA damage and mutagenesis, thus causing a ‘vicious cycle’ of exponentially increasing oxidative damage and dysfunction, which ultimately culminates in death (Figure 1) (51, 53).
Figure 1. The mitochondrial ‘Vicious Cycle’ theory.
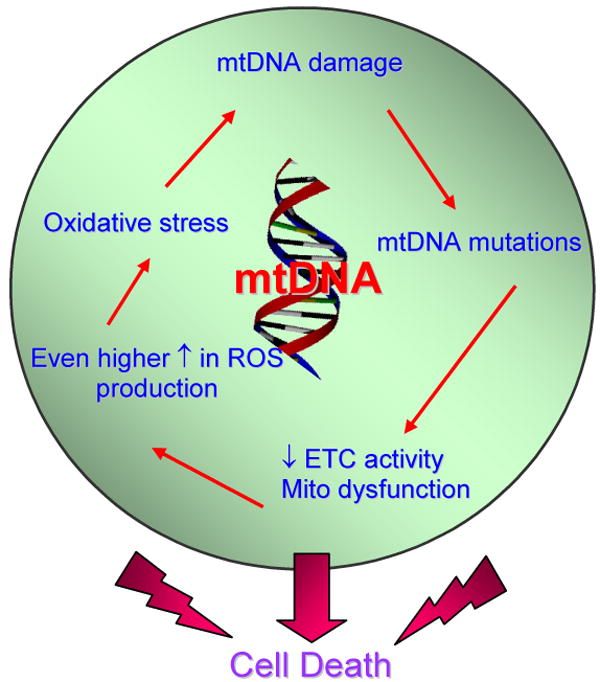
The theory proposes that chronic ROS generation and increases in oxidative stress can be damaging to mtDNA. Much of this damage can be mutagenic giving rise to mtDNA mutations that may accumulate progressively during life. MtDNA mutations in turn, can be directly responsible for a measurable deficiency in cellular oxidative phosphorylation activity, leading to an enhanced mitochondrial ROS production, according to the theory. Increased ROS generation results in further increases in oxidative stress and an increased rate of mtDNA damage and mutagenesis, thus causing a ‘vicious cycle’ of exponentially increasing oxidative damage and dysfunction, which ultimately culminates in cell death.
Challenging the Mitochondrial ‘Vicious Cycle’ Theory of Aging
Bandy and Davison were the first to put forward a mechanistic elaboration of what later became known as the mitochondrial ‘vicious cycle’ theory (54). While they showed that mtDNA mutations may have the same effect on the respiratory chain as small-molecule inhibitors of respiration, that is, to stimulate the one-electron reduction of molecular oxygen to superoxide (therefore, increasing ROS production), they also carefully noted that not all mutations stimulate superoxide production. Specifically, they pointed out that a mutation preventing the synthesis of cytochrome b would actually abolish any superoxide production at complex III that normal mitochondria might exhibit, because without cytochrome b in place, complex III cannot be assembled (54).
Later studies reported that respiration-deficient cells of several tissues, including skeletal muscle, possessed mutations that would indisputably preclude assembly of both the enzyme complexes known to be responsible for mitochondrial ROS production, complexes I and III (55-57). These mutations were large deletions, which eliminated the genes for at least a couple of respiratory chain subunits, but also removed at least one tRNA gene. There is no redundancy of tRNA genes in the mtDNA, so the loss of any such gene abolishes the synthesis of all 13 mtDNA-encoded proteins. These findings are highly relevant to the plausibility of the ‘vicious cycle’ theory in normal aging.
Recently, there is an increasing body of evidence challenging the ‘vicious cycle’ theory of aging (43, 44, 58). Kujoth et al. have shown that in ∼9 month old POLG mice, when accelerated aging has commenced and aging phenotypes are evident, there were no differences in mitochondrial hydrogen peroxide production, in mitochondrial and cytosolic protein carbonylation, or in total DNA oxidation in heart and liver tissues, compared to age-matched WT animals (44). In fact, RNA oxidation measured by the levels of 7,8-dihydro-8-oxoguanine (8-oxoG) was actually lower in liver RNA of mutant mice (44). Interestingly, in most tissues examined, including skeletal muscle, apoptosis was evident either by TUNEL staining or by activation of the effector caspase-3 (44), suggesting that apoptosis may be a central mechanism of tissue dysfunction associated with the accumulation of mtDNA mutations. It is therefore possible that the accumulation of mtDNA mutations may be associated with, or instigate apoptosis, irrespective of elevations in ROS production and oxidative stress in mitochondria with age. Similarly, Trifunovic et al., further substantiated the oxidative stress findings in mitochondrial mutator mice, by showing that mouse embryonic fibroblasts (MEFs) from POLG mice produced normal levels of superoxide and hydrogen peroxide, compared to WT (43). Furthermore, they also found normal aconitase activity, no differences in protein oxidation and no up-regulation of manganese superoxide dismutase (MnSOD) and glutathione peroxidase 1 (GPX1) in the heart and liver of POLG mice compared to age-matched WT mice (43). Importantly, these experimental observations do not support the idea that mtDNA mutations contribute to increased ROS production and oxidative stress in mitochondria with age, placing the mitochondrial “vicious cycle” theory of aging in question.
Zassenhaus and colleagues also tested the hypothesis that the production of reactive oxygen species causes the pathogenic effects of mtDNA mutations by using transgenic mice that develop cardiomyopathy due to the accumulation of mitochondrial DNA mutations specifically in the heart (58). In agreement with the above findings, they have demonstrated no elevations in protein carbonyls, no differences in mtDNA oxidative damage measured by 8-oxo-7,8-dihydro-2′-deoxyguanine (8-oxodG) levels, no up-regulation of antioxidant defense systems, normal ratios of reduced to oxidized glutathione (GSH/GSSG), normal ubiquitination levels and intact (not oxidatively damaged) iron-sulfur centers in aconitase enzyme in the hearts of transgenic mice (58). These outcomes provide further support to the notion that the mechanism of pathogenesis does not involve increased oxidative stress.
In skeletal muscle of ∼11-mo old POLG mice our lab detected mitochondrial dysfunction, evident by compromised mitochondrial oxygen consumption, significantly lower membrane potential during both state 3 (phosphorylative state) and state 4 (resting state), and lower ATP content. However, this dysfunction was not accompanied by an increase in mitochondrial ROS production or oxidative damage. In fact, we detected a decrease in the rate of hydrogen peroxide production by intact POLG mitochondria and no difference in mtDNA oxidative modification measured by 8-oxodG, compared to WT (A. Hiona unpublished data). This is also in contrast to the mitochondrial “vicious cycle” theory of aging which suggests that mtDNA mutations may lead to mitochondrial dysfunction via further increases in mitochondrial ROS production. Interestingly, we found that POLG mice had reduced mitochondrial protein yield and decreased protein content of complexes I, III and IV, all of which contain subunits encoded by mtDNA (A. Hiona unpublished data). This abolishment of ETC complexes in POLG skeletal muscle appears to be the primary mechanism of the mitochondrial dysfunction associated with the accumulation of mtDNA mutations in the mutant mice. Furthermore, we detected DNA laddering and an increase in the amount of cytosolic mono- and oligo-nucleosomes in POLG mice compared to WT, indicative of apoptosis. The activities of both the initiator caspase-9, and the final effector caspase-3, as well as, the protein content of caspase-3 were all elevated in POLG mice suggesting that: a) apoptosis in mutant mice is mitochondrial-mediated, and is conferred upon mitochondrial dysfunction and b) mutations in mtDNA play a causal role in sarcopenia, through enhancing apoptosis induced by mitochondrial dysfunction. Based on these recent experimental data in POLG skeletal muscle we proposed a hypothetical mechanism for the skeletal muscle loss induced by high load of somatic mtDNA mutations, that is likely to be operating during normal aging (Figure 2). According to the mechanism, abrogation of ETC complexes in POLG mice suggests that possibly fewer fully functional electron transport chains exist per skeletal muscle mitochondrion in POLG mice. If fewer ETCs assemble per mitochondrion this would leave the mitochondrion at energy deficit, leading to mitochondrial dysfunction, and ultimately mitochondrial-mediated apoptosis, which would be responsible for the loss of skeletal muscle (Figure 2).
Figure 2. Proposed mechanism for the skeletal muscle loss induced by accumulation of somatic mtDNA mutations.
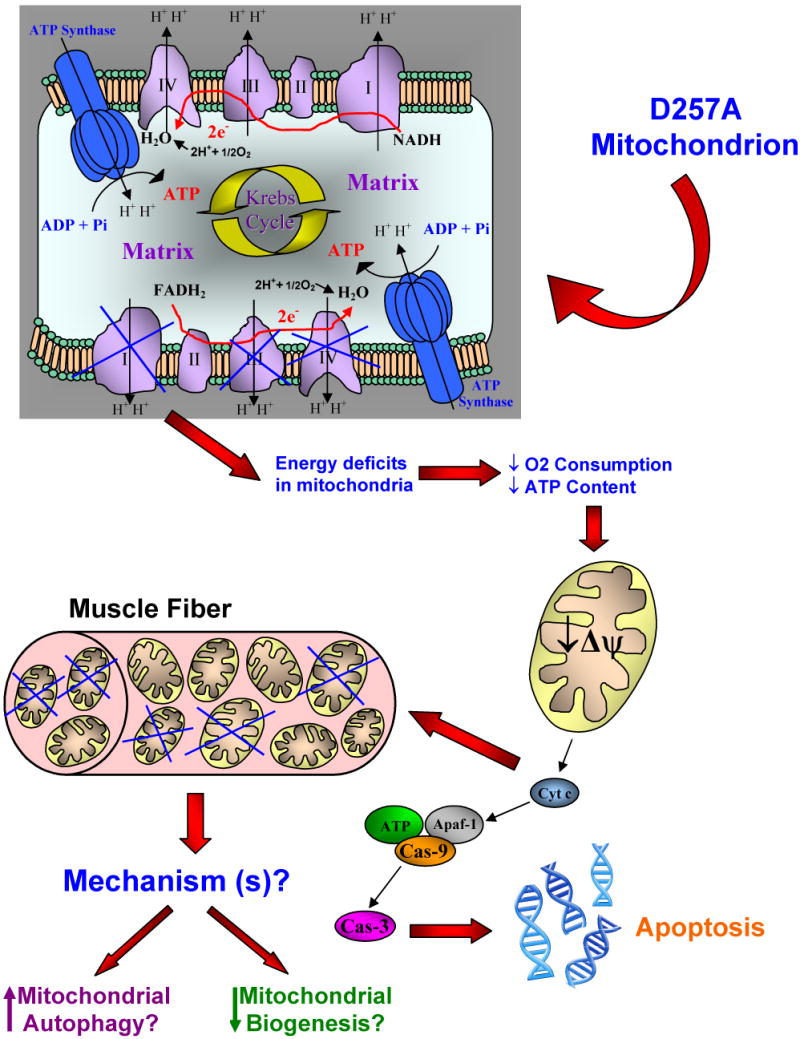
Abolishment of ETC complexes in POLG mice (POLG mice with a D257A mutation express a proof-reading-deficient version of PolgA, the nucleus-encoded catalytic subunit of the mitochondrial DNA polymerase γ, resulting in the accumulation of somatic mtDNA mutations) leads to assembly of less functional electron transport chains per mutant mitochondrion. This can create energy deficits leading to mitochondrial dysfunction, evident by severely compromised mitochondrial respiration and reduced ATP content in POLG muscle. Ultimately, this dysfunction results in significant drop in mitochondrial membrane potential and release of cytochrome c from the intermembrane space into the cytosol. Cytochrome c in the cytosol results in apoptosome formation, activation of caspase-9 and downstream activation of caspase-3 which ultimately results in apoptotic DNA fragmentation. Apoptosis appears to be a central mechanism of skeletal muscle loss in POLG mice. Moreover, the observation of reduced mitochondrial yield in POLG skeletal muscle suggests that mitochondria are eliminated. The mechanism for their elimination still remains to be determined although up-regulation of autophagy, down-regulation of mitochondrial biogenesis or both are likely mechanisms.
Similar to the oxidative stress findings in transgenic animal models, Hutter et al. recently demonstrated that normally aged human skeletal muscle produced less ROS, as assessed by in situ staining with dihydroethidium, and did not show any increases in protein oxidative modification, compared to young skeletal muscle (59). Interestingly, in this study the authors found that mitochondrial respiration was fully preserved with aging in skeletal muscle, a finding which is not similar to the POLG mice (42, 43) (A. Hiona unpublished data). Furthermore, the extent/accumulation of mtDNA mutations were not assessed in this report but the investigators identified the accumulation of lipofuscin as a robust marker of human muscle aging (59). As lipofuscin formation itself may be symptomatic of mitochondrial damage, further investigation is warranted in order to gain more insight as to the relationship between lipofuscin deposits and mitochondrial function. The oxidative stress outcomes in this study have further implications for the ‘vicious cycle’ theory, supporting a model where ROS-inflicted molecular damage may be continuously removed with age (59).
Taken all together, although chronic accumulation of ROS production and oxidative stress are undoubtedly important factors contributing to mtDNA damage and mutagenesis over the course of a lifetime, leading to aging and age-related phenotypes, recent experimental findings by our group as well as other groups, using transgenic mouse models, provide concrete evidence against an important part of the mitochondrial ‘vicious cycle’ theory of aging; the hypothesis that mutagenesis, partly due to chronic ROS insults to mtDNA, leads to further increases in ROS production and oxidative stress. Hence, it appears that oxidative stress may not be an obligate mediator of tissue pathology and demise provoked my mtDNA mutations. In the case of skeletal muscle, it is proposed that respiratory chain dysfunction per se may be the primary inducer of the sarcopenic phenotype associated with the accumulation of mtDNA mutations.
Recent controversy on the role of mtDNA mutations in mammalian aging
Very recently, Loeb's group published experimental work suggesting that mitochondrial point mutations do not limit the natural lifespan of mice (60). They used a new, highly sensitive assay to redefine the relationship between mtDNA mutations and age in WT and POLG mice. In their experiment, they used an adaptation of the random mutation capture (RMC) assay, a quantitative PCR-based approach that relies on PCR amplification of single molecules for mutation detection, but is not limited by polymerase fidelity, like other methods. This methodology allowed for exact determination of mutation frequencies in high-throughput screens that interrogated millions of base pairs simultaneously (60). The authors found that the mutational burden in mitochondria of WT mice was more than ten times lower than previously reported (44), and suggested that this discrepancy was most likely the result of mutations introduced ex vivo on damaged DNA templates during PCR before cloning steps in conventional assays, as PCR amplification, prior to application of the RMC assay, increased the mutation frequency at least 32-fold (60). Moreover, the previously reported mtDNA mutational rate of three- to eightfold higher in homozygous mutant POLG mice compared with WT mice, as measured by a standard DNA sequencing approach, was corrected to be ∼2,500-fold. Since measurements in young POLG mice were equivalent to what was previously reported (42, 44) with conventional assays, the authors attributed this difference solely to the increased sensitivity of the RMC assay, which allowed for correctly determining a very low mutation frequency in wild-type mice (60). Importantly, heterozygous POLG mice, with corrected mtDNA mutational burden up to 500-fold higher compared to age-matched WT mice, did not show an accelerated aging phenotype, a significant reduction in mean lifespan or a significant increase in age related pathology, consistent with previous reports (42, 44). Furthermore, because heterozygous mice are born with a 30-fold higher mutation burden than the oldest WT animals without suffering a phenotype that resembles premature aging, the authors concluded that the threshold at which mitochondrial mutations become limiting for lifespan is unlikely to be reached in wild-type mice (60).
Although these data argue against a causal role for mitochondrial mutations in aging, it should be noted that despite the ability of RMC assay to detect point mutations, large mtDNA deletions cannot be detected with this method. As mtDNA deletions have been correlated with the demise of certain specialized tissues such as the substantia nigra (21, 61), skeletal muscle dysfunction, and mitochondrial ETC abnormalities (6, 28, 33, 34, 36), these data fail to address the relevance of large scale mtDNA deletion mutations in the aging phenotype. Furthermore, in the above study (60), the authors did not find any evidence for clonal expansions during spectrum analysis, although clonal expansion, especially of deletion mutations has been repeatedly and very thoroughly demonstrated by other groups in the past (6, 33-36). It is also important to point out that age-induced skeletal muscle dysfunction is associated with a pattern of focal accumulation of mtDNA mutations within a muscle fiber and a mosaic distribution of mutations between different fibers (6, 28, 33, 34), a phenomenon that is likely not present in POLG mutant mice. In light of the above recent findings, the question of whether mitochondrial mutations cause mammalian aging, or are merely correlated with it, remains unanswered, and it is still an area of intense debate.
Conclusions
In summary, our review of the literature suggests that: a) aging is associated with an increase in mtDNA mutations and deletions, coinciding with an increase in mitochondrial abnormalities in skeletal muscle of several species, including humans, and b) the age-associated accumulation of mtDNA mutations, leading to mitochondrial dysfunction, may be an important contributor to sarcopenia.
It is important to note that aging and aging-associated phenotypes are complex processes that are likely to have multi-factorial causes. Mitochondrial DNA mutations can arise directly from sporadic errors during mitochondrial DNA replication (62) or from mtDNA replication errors resulting from oxidative damage. Besides mtDNA mutations, oxidative stress may also generate damaged proteins that might be able to directly signal apoptosis through a misfolded protein response (62, 63). Respiratory deficiency could contribute to apoptotic signaling or be directly responsible for some aspects of tissue dysfunction (62). The limited and sometimes contradictory evidence available concerning the capacity of pathogenic mtDNA mutations to start and support the development of tissue dysfunction and demise and the role of ROS production and subsequent oxidative stress in this phenomenon makes it difficult to reach general conclusions.
Last, because cells may have hundreds of mitochondria, with each carrying multiple copies of mtDNA, the contribution of mtDNA mutations and deletions to normal aging and aging phenotypes, remains a controversial issue. It is clear, however, that progress in this area will lead to a better understanding of the cause and role of mtDNA mutations in aging, and the resources available to the cell for compensating and possibly reversing the process leading to cell death, with potential implications for the therapy of sarcopenia, as well as degenerative diseases associated with mtDNA mutations.
Acknowledgments
We would like to thank Drs Hofer and Servais for their critical input into this manuscript. This research was supported by a grant to CL from the National Institute on Aging (AG17994 and AG21042) and an American Heart Association Predoctoral Fellowship to AH (0415166B) and this work was supported by the University of Florida Institute on Aging and the Claude D. Pepper Older Americans Independence Center NIH Grant # 1 P30 AG028740.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
List of References
- 1.Marcell TJ. Sarcopenia: causes, consequences, and preventions. J Gerontol A Biol Sci Med Sci. 2003;58:M911–916. doi: 10.1093/gerona/58.10.m911. [DOI] [PubMed] [Google Scholar]
- 2.Roubenoff R. Sarcopenia and its implications for the elderly. Eur J Clin Nutr. 2000;54 3:S40–47. doi: 10.1038/sj.ejcn.1601024. [DOI] [PubMed] [Google Scholar]
- 3.Harman D. Free radical theory of aging: dietary implications. Am J Clin Nutr. 1972;25:839–843. doi: 10.1093/ajcn/25.8.839. [DOI] [PubMed] [Google Scholar]
- 4.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 5.Fleming JE, Miquel J, Cottrell SF, Yengoyan LS, Economos AC. Is cell aging caused by respiration-dependent injury to the mitochondrial genome? Gerontology. 1982;28:44–53. doi: 10.1159/000212510. [DOI] [PubMed] [Google Scholar]
- 6.Wanagat J, Cao Z, Pathare P, Aiken JM. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. Faseb J. 2001;15:322–332. doi: 10.1096/fj.00-0320com. [DOI] [PubMed] [Google Scholar]
- 7.Khaidakov M, Heflich RH, Manjanatha MG, Myers MB, Aidoo A. Accumulation of point mutations in mitochondrial DNA of aging mice. Mutat Res. 2003;526:1–7. doi: 10.1016/s0027-5107(03)00010-1. [DOI] [PubMed] [Google Scholar]
- 8.Corral-Debrinski M, Shoffner JM, Lott MT, Wallace DC. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat Res. 1992;275:169–180. doi: 10.1016/0921-8734(92)90021-g. [DOI] [PubMed] [Google Scholar]
- 9.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet. 1992;2:324–329. doi: 10.1038/ng1292-324. [DOI] [PubMed] [Google Scholar]
- 10.Lee CM, Chung SS, Kaczkowski JM, Weindruch R, Aiken JM. Multiple mitochondrial DNA deletions associated with age in skeletal muscle of rhesus monkeys. J Gerontol. 1993;48:B201–205. doi: 10.1093/geronj/48.6.b201. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Michikawa Y, Mallidis C, Bai Y, Woodhouse L, Yarasheski KE, Miller CA, Askanas V, Engel WK, Bhasin S, Attardi G. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc Natl Acad Sci U S A. 2001;98:4022–4027. doi: 10.1073/pnas.061013598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ojaimi J, Masters CL, Opeskin K, Mckelvie P, Byrne E. Mitochondrial respiratory chain activity in the human brain as a function of age. Mech Ageing Dev. 1999;111:39–47. doi: 10.1016/s0047-6374(99)00071-8. [DOI] [PubMed] [Google Scholar]
- 13.Trounce I, Byrne E, Marzuki S. Decline in skeletal muscle mitochondrial respiratory chain function: possible factor in ageing. Lancet. 1989;1:637–639. doi: 10.1016/s0140-6736(89)92143-0. [DOI] [PubMed] [Google Scholar]
- 14.Cooper JM, Mann VM, Schapira AH. Analyses of mitochondrial respiratory chain function and mitochondrial DNA deletion in human skeletal muscle: effect of ageing. J Neurol Sci. 1992;113:91–98. doi: 10.1016/0022-510x(92)90270-u. [DOI] [PubMed] [Google Scholar]
- 15.Harman D. The aging process. Proc Natl Acad Sci U S A. 1981;78:7124–7128. doi: 10.1073/pnas.78.11.7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace DC, Shoffner JM, Trounce I, Brown MD, Ballinger SW, Corral-Debrinski M, Horton T, Jun AS, Lott MT. Mitochondrial DNA mutations in human degenerative diseases and aging. Biochim Biophys Acta. 1995;1271:141–151. doi: 10.1016/0925-4439(95)00021-u. [DOI] [PubMed] [Google Scholar]
- 17.Simonetti S, Chen X, Dimauro S, Schon EA. Accumulation of deletions in human mitochondrial DNA during normal aging: analysis by quantitative PCR. Biochim Biophys Acta. 1992;1180:113–122. doi: 10.1016/0925-4439(92)90059-v. [DOI] [PubMed] [Google Scholar]
- 18.Schwarze SR, Lee CM, Chung SS, Roecker EB, Weindruch R, Aiken JM. High levels of mitochondrial DNA deletions in skeletal muscle of old rhesus monkeys. Mech Ageing Dev. 1995;83:91–101. doi: 10.1016/0047-6374(95)01611-3. [DOI] [PubMed] [Google Scholar]
- 19.Pesce V, Cormio A, Fracasso F, Vecchiet J, Felzani G, Lezza AM, Cantatore P, Gadaleta MN. Age-related mitochondrial genotypic and phenotypic alterations in human skeletal muscle. Free Radic Biol Med. 2001;30:1223–1233. doi: 10.1016/s0891-5849(01)00517-2. [DOI] [PubMed] [Google Scholar]
- 20.Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood) 2007;232:592–606. [PubMed] [Google Scholar]
- 21.Kraytsberg Y, Kudryavtseva E, Mckee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006;38:518–520. doi: 10.1038/ng1778. [DOI] [PubMed] [Google Scholar]
- 22.Barja G, Herrero A. Oxidative damage to mitochondrial DNA is inversely related to maximum life span in the heart and brain of mammals. Faseb J. 2000;14:312–318. doi: 10.1096/fasebj.14.2.312. [DOI] [PubMed] [Google Scholar]
- 23.Herrero A, Barja G. 8-oxo-deoxyguanosine levels in heart and brain mitochondrial and nuclear DNA of two mammals and three birds in relation to their different rates of aging. Aging (Milano) 1999;11:294–300. doi: 10.1007/BF03339803. [DOI] [PubMed] [Google Scholar]
- 24.Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature. 1988;331:717–719. doi: 10.1038/331717a0. [DOI] [PubMed] [Google Scholar]
- 25.Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, 2nd, Nikoskelainen EK. Mitochondrial DNA mutation associated with leber's hereditary optic neuropathy. Science. 1988;242:1427–1430. doi: 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- 26.Drew B, Phaneuf S, Dirks A, Selman C, Gredilla R, Lezza A, Barja G, Leeuwenburgh C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am J Physiol Regul Integr Comp Physiol. 2003;284:R474–480. doi: 10.1152/ajpregu.00455.2002. [DOI] [PubMed] [Google Scholar]
- 27.Short KR, Bigelow ML, Kahl J, Singh R, Coenen-Schimke J, Raghavakaimal S, Nair KS. Decline in skeletal muscle mitochondrial function with aging in humans. Proc Natl Acad Sci U S A. 2005;102:5618–5623. doi: 10.1073/pnas.0501559102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cao Z, Wanagat J, Mckiernan SH, Aiken JM. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: analysis by laser-capture microdissection. Nucleic Acids Res. 2001;29:4502–4508. doi: 10.1093/nar/29.21.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lezza AM, Pesce V, Cormio A, Fracasso F, Vecchiet J, Felzani G, Cantatore P, Gadaleta MN. Increased expression of mitochondrial transcription factor A and nuclear respiratory factor-1 in skeletal muscle from aged human subjects. Febs Lett. 2001;501:74–78. doi: 10.1016/s0014-5793(01)02628-x. [DOI] [PubMed] [Google Scholar]
- 30.Desai VG, Weindruch R, Hart RW, Feuers RJ. Influences of age and dietary restriction on gastrocnemius electron transport system activities in mice. Arch Biochem Biophys. 1996;333:145–151. doi: 10.1006/abbi.1996.0375. [DOI] [PubMed] [Google Scholar]
- 31.Muller-Hocker J, Schafer S, Link TA, Possekel S, Hammer C. Defects of the respiratory chain in various tissues of old monkeys: a cytochemical-immunocytochemical study. Mech Ageing Dev. 1996;86:197–213. doi: 10.1016/0047-6374(95)01692-9. [DOI] [PubMed] [Google Scholar]
- 32.Gokey NG, Cao Z, Pak JW, Lee D, Mckiernan SH, Mckenzie D, Weindruch R, Aiken JM. Molecular analyses of mtDNA deletion mutations in microdissected skeletal muscle fibers from aged rhesus monkeys. Aging Cell. 2004;3:319–326. doi: 10.1111/j.1474-9728.2004.00122.x. [DOI] [PubMed] [Google Scholar]
- 33.Herbst A, Pak JW, Mckenzie D, Bua E, Bassiouni M, Aiken JM. Accumulation of mitochondrial DNA deletion mutations in aged muscle fibers: evidence for a causal role in muscle fiber loss. J Gerontol A Biol Sci Med Sci. 2007;62:235–245. doi: 10.1093/gerona/62.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mckenzie D, Bua E, Mckiernan S, Cao Z, Aiken JM. Mitochondrial DNA deletion mutations: a causal role in sarcopenia. Eur J Biochem. 2002;269:2010–2015. doi: 10.1046/j.1432-1033.2002.02867.x. [DOI] [PubMed] [Google Scholar]
- 35.Bua EA, Mckiernan SH, Wanagat J, Mckenzie D, Aiken JM. Mitochondrial abnormalities are more frequent in muscles undergoing sarcopenia. J Appl Physiol. 2002;92:2617–2624. doi: 10.1152/japplphysiol.01102.2001. [DOI] [PubMed] [Google Scholar]
- 36.Bua E, Johnson J, Herbst A, Delong B, Mckenzie D, Salamat S, Aiken JM. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cormio A, Milella F, Vecchiet J, Felzani G, Gadaleta MN, Cantatore P. Mitochondrial DNA mutations in RRF of healthy subjects of different age. Neurobiol Aging. 2005;26:655–664. doi: 10.1016/j.neurobiolaging.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 38.Chabi B, De Camaret BM, Chevrollier A, Boisgard S, Stepien G. Random mtDNA deletions and functional consequence in aged human skeletal muscle. Biochem Biophys Res Commun. 2005;332:542–549. doi: 10.1016/j.bbrc.2005.04.153. [DOI] [PubMed] [Google Scholar]
- 39.Fayet G, Jansson M, Sternberg D, Moslemi AR, Blondy P, Lombes A, Fardeau M, Oldfors A. Ageing muscle: clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul Disord. 2002;12:484–493. doi: 10.1016/s0960-8966(01)00332-7. [DOI] [PubMed] [Google Scholar]
- 40.Chinnery PF, Samuels DC, Elson J, Turnbull DM. Accumulation of mitochondrial DNA mutations in ageing, cancer, and mitochondrial disease: is there a common mechanism? Lancet. 2002;360:1323–1325. doi: 10.1016/S0140-6736(02)11310-9. [DOI] [PubMed] [Google Scholar]
- 41.Wang E, Wong A, Cortopassi G. The rate of mitochondrial mutagenesis is faster in mice than humans. Mutat Res. 1997;377:157–166. doi: 10.1016/s0027-5107(97)00091-2. [DOI] [PubMed] [Google Scholar]
- 42.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 43.Trifunovic A, Hansson A, Wredenberg A, Rovio AT, Dufour E, Khvorostov I, Spelbrink JN, Wibom R, Jacobs HT, Larsson NG. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc Natl Acad Sci U S A. 2005;102:17993–17998. doi: 10.1073/pnas.0508886102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 45.Zhang D, Mott JL, Farrar P, Ryerse JS, Chang SW, Stevens M, Denniger G, Zassenhaus HP. Mitochondrial DNA mutations activate the mitochondrial apoptotic pathway and cause dilated cardiomyopathy. Cardiovasc Res. 2003;57:147–157. doi: 10.1016/s0008-6363(02)00695-8. [DOI] [PubMed] [Google Scholar]
- 46.Hamilton ML, Van Remmen H, Drake JA, Yang H, Guo ZM, Kewitt K, Walter CA, Richardson A. Does oxidative damage to DNA increase with age? Proc Natl Acad Sci U S A. 2001;98:10469–10474. doi: 10.1073/pnas.171202698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bohr VA, Dianov GL. Oxidative DNA damage processing in nuclear and mitochondrial DNA. Biochimie. 1999;81:155–160. doi: 10.1016/s0300-9084(99)80048-0. [DOI] [PubMed] [Google Scholar]
- 48.Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005;5:89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 49.Lewis LK, Resnick MA. Tying up loose ends: nonhomologous end-joining in saccharomyces cerevisiae. Mutat Res. 2000;451:71–89. doi: 10.1016/s0027-5107(00)00041-5. [DOI] [PubMed] [Google Scholar]
- 50.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. Embo J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alexeyev MF, Ledoux SP, Wilson GL. Mitochondrial DNA and aging. Clin Sci (Lond) 2004;107:355–364. doi: 10.1042/CS20040148. [DOI] [PubMed] [Google Scholar]
- 52.Harman D. Free radical theory of aging. Triangle. 1973;12:153–158. [PubMed] [Google Scholar]
- 53.Miquel J, Economos AC, Fleming J, Johnson JE., Jr Mitochondrial role in cell aging. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 54.Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Radic Biol Med. 1990;8:523–539. doi: 10.1016/0891-5849(90)90152-9. [DOI] [PubMed] [Google Scholar]
- 55.Katayama M, Tanaka M, Yamamoto H, Ohbayashi T, Nimura Y, Ozawa T. Deleted mitochondrial DNA in the skeletal muscle of aged individuals. Biochem Int. 1991;25:47–56. [PubMed] [Google Scholar]
- 56.Muller-Hocker J, Seibel P, Schneiderbanger K, Kadenbach B. Different in situ hybridization patterns of mitochondrial DNA in cytochrome c oxidase-deficient extraocular muscle fibres in the elderly. Virchows Arch A Pathol Anat Histopathol. 1993;422:7–15. doi: 10.1007/BF01605127. [DOI] [PubMed] [Google Scholar]
- 57.Brierley EJ, Johnson MA, Lightowlers RN, James OF, Turnbull DM. Role of mitochondrial DNA mutations in human aging: implications for the central nervous system and muscle. Ann Neurol. 1998;43:217–223. doi: 10.1002/ana.410430212. [DOI] [PubMed] [Google Scholar]
- 58.Mott JL, Zhang D, Stevens M, Chang S, Denniger G, Zassenhaus HP. Oxidative stress is not an obligate mediator of disease provoked by mitochondrial DNA mutations. Mutat Res. 2001;474:35–45. doi: 10.1016/s0027-5107(00)00159-7. [DOI] [PubMed] [Google Scholar]
- 59.Hutter E, Skovbro M, Lener B, Prats C, Rabol R, Dela F, Jansen-Durr P. Oxidative stress and mitochondrial impairment can be separated from lipofuscin accumulation in aged human skeletal muscle. Aging Cell. 2007;6:245–256. doi: 10.1111/j.1474-9726.2007.00282.x. [DOI] [PubMed] [Google Scholar]
- 60.Vermulst M, Bielas JH, Kujoth GC, Ladiges WC, Rabinovitch PS, Prolla TA, Loeb LA. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 61.Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and parkinson disease. Nat Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 62.Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. Plos Genet. 2007;3:E24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mott JL, Zhang D, Zassenhaus HP. Mitochondrial DNA mutations, apoptosis, and the misfolded protein response. Rejuvenation Res. 2005;8:216–226. doi: 10.1089/rej.2005.8.216. [DOI] [PubMed] [Google Scholar]