

Abstract
Phosphodiesterases (PDEs) catalyze the hydrolysis of the second messengers cAMP and cGMP. However, little is known about how PDE activity regulates cyclic nucleotide signals in vivo because, outside of specialized cells, there are few methods with the appropriate spatial and temporal resolution to measure cyclic nucleotide concentrations. We have previously demonstrated that adenovirus-expressed, olfactory cyclic nucleotide–gated channels provide real-time sensors for cAMP produced in subcellular compartments of restricted diffusion near the plasma membrane (Rich, T.C., K.A. Fagan, H. Nakata, J. Schaack, D.M.F. Cooper, and J.W. Karpen. 2000. J. Gen. Physiol. 116:147–161). To increase the utility of this method, we have modified the channel, increasing both its cAMP sensitivity and specificity, as well as removing regulation by Ca2+-calmodulin. We verified the increased sensitivity of these constructs in excised membrane patches, and in vivo by monitoring cAMP-induced Ca2+ influx through the channels in cell populations. The improved cAMP sensors were used to monitor changes in local cAMP concentration induced by adenylyl cyclase activators in the presence and absence of PDE inhibitors. This approach allowed us to identify localized PDE types in both nonexcitable HEK-293 and excitable GH4C1 cells. We have also developed a quantitative framework for estimating the KI of PDE inhibitors in vivo. The results indicate that PDE type IV regulates local cAMP levels in HEK-293 cells. In GH4C1 cells, inhibitors specific to PDE types I and IV increased local cAMP levels. The results suggest that in these cells PDE type IV has a high K m for cAMP, whereas PDE type I has a low K m for cAMP. Furthermore, in GH4C1 cells, basal adenylyl cyclase activity was readily observable after application of PDE type I inhibitors, indicating that there is a constant synthesis and hydrolysis of cAMP in subcellular compartments near the plasma membrane. Modulation of constitutively active adenylyl cyclase and PDE would allow for rapid control of cAMP-regulated processes such as cellular excitability.
Keywords: adenylyl cyclase, G-protein signaling, calcium influx, GH4 pituitary cells, biosensors
INTRODUCTION
Cyclic nucleotide phosphodiesterases (PDEs), the crucial terminators of cAMP and cGMP signals, were discovered almost 40 yr ago (Drummond and Perrot-Yee 1961). Since then, PDEs have been classified into eleven families according to substrate specificity, regulation, pharmacology, and, more recently, amino acid homology (Beavo 1988, Beavo 1995; Conti et al. 1995). Studies have confirmed differential regulation of PDE families by Ca2+-calmodulin (CaM), G-proteins, phosphorylation, and cyclic nucleotides. The diversity of PDE families has led to the realization that PDE activity is a central element in the control of second messenger signaling, as important as adenylyl and guanylyl cyclase activity in shaping cyclic nucleotide signals. Yet, little is known about how PDE regulates cyclic nucleotide signals in vivo or how cyclic nucleotide signals differentially regulate hundreds of cellular targets because, outside of specialized cells, there are few convenient real-time measures of cyclic nucleotide concentration.
Retinal rod outer segments are the most closely examined second messenger signaling system, in part, because the endogenous CNG channels have been used as real-time detectors of cGMP concentration (Baylor et al. 1979). Information from biochemical measurements of guanylyl cyclase and cGMP-specific PDE have been combined with real-time measurements of cGMP concentration to characterize the relationship between these enzymes in regulating cGMP levels (Fung et al. 1981; Koch and Stryer 1988; Gorczyca et al. 1994; Koutalos et al. 1995a,Koutalos et al. 1995b; He, W., et al., 1998; Tsang et al. 1998; Chen et al. 2000; Leskov et al. 2000; Nikonov et al. 2000). The convergence of different approaches has led to an unprecedented understanding of feedback signaling within this model system (for review see Stryer 1991; Lagnado and Baylor 1992; Pugh and Lamb 1993; Yarfitz and Hurley 1994; Yau 1994; Polans et al. 1996; Pugh et al. 1997; Molday 1998).
We recently reported that cAMP concentration can be measured accurately with the rat wild-type (WT) olfactory CNG channel, encoded by an adenovirus vector (Rich et al. 2000). An important finding in multiple cell types was that cAMP is produced in subcellular compartments near the plasma membrane. Diffusion between these compartments and the bulk cytosol is severely restricted. These regions likely allow for rapid and energetically efficient activation of cAMP-dependent processes, and may help to explain how cAMP can differentially regulate large numbers of cellular targets. In this study, we have modified the WT CNG channel to increase its sensitivity and specificity for cAMP and to remove regulation by Ca2+-CaM. We also present a convenient, optical method for detecting changes in cAMP, taking advantage of the Ca2+ permeability of the channel. Using the modified CNG channels, this assay is sensitive to cAMP in the physiologic range (0.1–50 μM). We have used this approach to probe the interactions between adenylyl cyclase (AC) and PDE in regulating local cAMP concentration in two cell types, nonexcitable HEK-293 and excitable GH4C1 pituitary cells.
MATERIALS AND METHODS
Construction of CNG Channel–encoding Adenoviruses
Point mutations were introduced into the α subunit of the WT rat olfactory CNG channel (CNG2, CNCα3) using the QuikChange site-directed mutagenesis kit (Stratagene). Overlap extension PCR was used to delete the Ca2+-CaM binding site (amino acids 61–90). A replication-defective adenovirus, in which the channel coding sequence containing the E583M mutation replaced the E1 region, was constructed as described previously (Fagan et al. 1999). Briefly, the cDNA encoding the mutant channel was ligated into the plasmid pACCMV (Gomez-Foix et al. 1992) under the control of the cytomegalovirus major immediate early (CMV) promoter between the BamHI and SalI sites. The plasmid was then digested with SalI and ligated with a BstBI adaptor. The resultant plasmid was then digested with BstBI and XmnI and ligated with BstBI-digested Ad5dl327Bstβ-gal-terminal-protein complex, that had been isolated by banding purified Ad5dl327Bstβ-gal (Schaack et al. 1995) virions in 2.8 M CsCl, and 4 M guanidine-HCl. The ligation products were used to transfect HEK-293 cells using Ca3(PO4)2 precipitation (Jordan et al. 1996). The transfected cells were incubated for 7 d. The cells were frozen and thawed to release the virus, and dilutions were used to infect HEK-293 plates for plaque purification. The infected HEK-293 plates were overlaid with medium in Noble agar, fed after 4 d, and stained with X-gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and neutral red. Clear plaques, which were derived from viral chromosomes lacking the LacZ gene of the parental virus, were amplified and analyzed by PCR and restriction digestion for the presence of the mutated CNG channel cDNA.
Adenovirus transducing vectors encoding C460W/E583M and Δ61–90/C460W/E583M channels were constructed using the AdEasy system (He, T.-C., et al., 1998). The cDNA encoding the mutant channel was ligated into pShuttle-CMV between the KpnI and XbaI sites. The resultant plasmid was linearized by digestion with PmeI and used to transform Escherichia coli strain BJ5183 that had been transformed with pAdEasy-1. A plasmid containing the adenovirus chromosome encoding the mutated CNG channel was digested with PacI to release the adenovirus chromosome, and this DNA was used to transfect HEK-293 cells. After incubation for 7 d, the virus was released by freezing and thawing, and plaque purified. The purified virus was tested for the presence of the CNG channel cDNA by PCR. A virus containing the channel cDNA was grown in large scale in HEK-293 cells and purified by banding using CsCl step and isopycnic gradients.
Cell Culture and Channel Expression
HEK-293 cells were maintained in culture and infected with adenovirus as described previously (Rich et al. 2000). Briefly, HEK-293 cells were maintained in MEM (Life Technologies Inc.) supplemented with 26.2 mM NaHCO3, 10% (vol/vol) FBS (Gemini), penicillin (50 μg/ml), and streptomycin (50 μg/ml), pH 7.0, at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were plated at ∼60% confluence in 100-mm culture dishes 24 h before infection with the CNG channel–encoding adenovirus constructs (multiplicity of infection = 10 plaque forming units per cell). 2 h after infection, hydroxyurea was added to the cell media at 2 mM final concentration to partially inhibit viral replication. 24 h after infection cells were detached with PBS containing 0.03% EDTA, resuspended in serum-containing medium, and assayed within 12 h.
GH4C1 rat pituitary cells (American Type Culture Collection) were maintained in 13 ml Ham's F-10 medium (Life Technologies Inc.) supplemented with 14.3 mM NaHCO3, 15% donor horse serum (Gemini), and 2.5% FBS, pH 6.8, in 75-cm2 flasks at 37°C in a humidified atmosphere of 95% air and 5% CO2. Cells were split weekly (1:4) and washed with fresh medium twice weekly. Cells were plated at ∼60% confluence in 100-mm culture dishes 24 h before infection with the CNG channel–encoding adenovirus constructs (multiplicity of infection = 50 plaque forming units per cell). 48 h after infection cells were detached, resuspended in serum-containing medium, and assayed within 12 h.
Electrical Recording
To assess the cyclic nucleotide sensitivity of different CNG channel constructs, excised, inside-out patch recordings were made at room temperature (20–21°C) using an Axopatch-200A patch-clamp amplifier (Axon Instruments Inc.). Pipettes were pulled from borosilicate glass and heat polished. Pipettes were lowered onto the cells and gigaohm seals were formed. Patches were excised by shearing cells from the pipette with a jet of liquid. Ionic currents were elicited by 250-ms pulses to membrane potentials of +50 and −50 mV from a holding potential of 0 mV. Current records were sampled at five times the filter setting and stored on an IBM compatible computer. Records were corrected for errors due to series resistance (pipette resistance was 4.1 ± 0.1 MΩ). Both the pipette and bath solutions contained the following (in mM): 130 NaCl, 2 HEPES, 0.02 EDTA, and 1 EGTA, pH 7.6. Cyclic nucleotide–induced currents were obtained from the difference between currents in the presence and absence of cyclic nucleotides. Dose–response curves for cAMP and cGMP were obtained at +50 and −50 mV in the same patch. The effects of the modifications were assessed using the Hill equation, I/Imax = [cNMP]N/([cNMP]N+ K 1/2 N), where I/Imax is the fraction of maximal current, cNMP represents cyclic nucleotide, K 1/2 is the concentration that gives half-maximal current, and N is the Hill coefficient, an index of cooperativity.
Detection of cAMP in Populations of Cells
Homomultimeric CNG channels comprised of the rat olfactory channel α subunit are permeable to Ca2+ (Frings et al. 1995). We have taken advantage of this to detect changes in local cAMP concentration. In this assay, an increase in local cAMP concentration causes activation of CNG channels and a subsequent increase in Ca2+ entry (Rich et al. 2000). Thus, a change in the rate of Ca2+ influx in response to a stimulus reflects a change in the cAMP level. In this study, cAMP synthesis was stimulated using either forskolin or agonists of G-protein–coupled receptors. PDE activity was assessed using a variety of type-specific PDE inhibitors. Certain ones inhibit multiple PDE types; the efficacies and specificities of the inhibitors used in this study are listed in Table .
Table 1.
Specificity of PDE Inhibitors for Different PDE Families
| Inhibitor | Reported specificity | I | II | III | IV | V | VI | VII | VIII | IX | X | XI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IBMX3,6,8,10–12,15,16,20–22 | nonselective | 2–3 | 20–50 | 2 | 5–15 | 4–7 | 10 | 2–8 | >200 | >200 | 2.6 | 49.8 |
| 8-methoxymethyl-IBMX2,4,14,15 | PDE I | 2–8 | 69 | >100 | ||||||||
| EHNA12,16,18–22 | PDE II | 0.5–1 | >500 | >100 | >100 | >100 | 69 | |||||
| Trequinsin23 | PDE III | 0.5–2 | 0.0003 | |||||||||
| Quazinone13 | PDE III | 0.6 | ||||||||||
| Etazolate1 | PDE IV | 2 | ||||||||||
| RO-20-17247,9,12,20,21,23 | PDE IV | 200 | >100 | 52 | 1–2 | 250 | >200 | >200 | ||||
| Rolipram5,8,9,15–17,20–23 | PDE IV | >150 | >100 | 0.05–2 | >100 | >100 | >200 | >200 | 47 | >100 | ||
| Zaprinast6,8,9,15,16,20–22 | PDE V–VI | 3–10 | 70 | 10 | 22 | 0.2–0.5 | 0.15 | >50 | >100 | 29–35 | 11 | 12 |
There are over 30 known forms of PDE that have been grouped into 11 families. Of these families, PDE types V, VI, and IX are cGMP-specific. The IC50 of PDE inhibitors given in μM. Recently, an isoform of PDE type IV, PDE4A, purified from U937 monocytic cells was shown to be inhibited by rolipram with an IC50 of 3 nM (MacKenzie and Houslay 2000). It should be noted that these efficacies may be modulated in vivo. For example, it has been shown that forms of PDE type IVA are ∼10-fold more sensitive to rolipram when they bind to the SRC family tyrosyl kinase LYN (McPhee et al. 1999). Data were obtained using heterologously expressed mouse (types VII, VIII, and X) and human (types I, II, IV, V, VII, IX, and XI) PDEs as well as endogenous PDEs from dog kidney (type IV), rat brain (type IV), bovine heart (types III and IV), human heart (type II), bovine aorta (types I, III, and V), rabbit aorta (types I, IV, and V), and bovine photoreceptor (type VI). Data compiled from: 1(Ahluwalia and Rhoads 1982), 2(Ahn et al. 1989), 3(Beavo 1988), 4(Bolger et al. 1993), 5(Bolger et al. 1997), 6(Coste and Grondin 1995), 7(Epstein et al. 1982), 8(Fawcett et al. 2000), 9(Fisher et al. 1998), 10(Gardner et al. 2000), 11(Harrison et al. 1988), 12(Hetman et al. 2000), 13(Holck et al. 1984), 14(Lorenz and Wells 1983), 15(Loughney et al. 1996), 16(Loughney et al. 1998), 17(Nemoz et al. 1985), 18(Podzuweit et al. 1995), 19(Rosman et al. 1997), 20(Soderling et al. 1998a), 21(Soderling et al. 1998b), 22(Soderling et al. 1999), 23(Whalin et al. 1991). Blank spaces indicate that data are not available.
We used the fluorescent indicator fura-2 to monitor Ca2+ influx in cell populations. Cells were loaded with 4 μM fura-2/AM (the membrane-permeant form) and 0.02% pluronic F-127 at room temperature for 30–40 min (HEK-293 cells) or 70 min (GH4C1 cells), in a buffer containing Ham's F-10 medium supplemented with 1 mg/ml BSA fraction V (BSA, Fisher) and 20 mM HEPES, pH 7.4. In some experiments, HEK-293 cells were loaded with a higher concentration of fura-2/AM (16 μM) for comparison (see below). Cells were washed twice, then resuspended in a solution containing the following (in mM): 145 NaCl, 11 d-Glucose, 10 HEPES, 4 KCl, 1 CaCl2, 1 MgCl2, and 1 mg/ml BSA, pH 7.4 (3–4 × 106 cells /3 ml buffer solution), and assayed using an LS-50B spectrofluorimeter (Perkin Elmer). Additions were made by pipetting stock solutions into a stirred cuvette. The mixing time was estimated to be on the order of 5 s. Fluorescence was measured at an excitation wavelength of 380 nm and an emission wavelength of 510 nm. Under these conditions, Ca2+ influx causes a decrease in fluorescence (ΔF), which was expressed relative to the pre-stimulus fluorescence (F0) to correct for variations in dye concentration, and to allow for comparison of results on different batches of cells. Data were sampled at 0.5 Hz and filtered at 0.1 Hz (see Fig. 3A and Fig. B) or sampled at 6 Hz and filtered at 1.2 Hz (see Fig. 3C and Fig. D).
Figure 3.
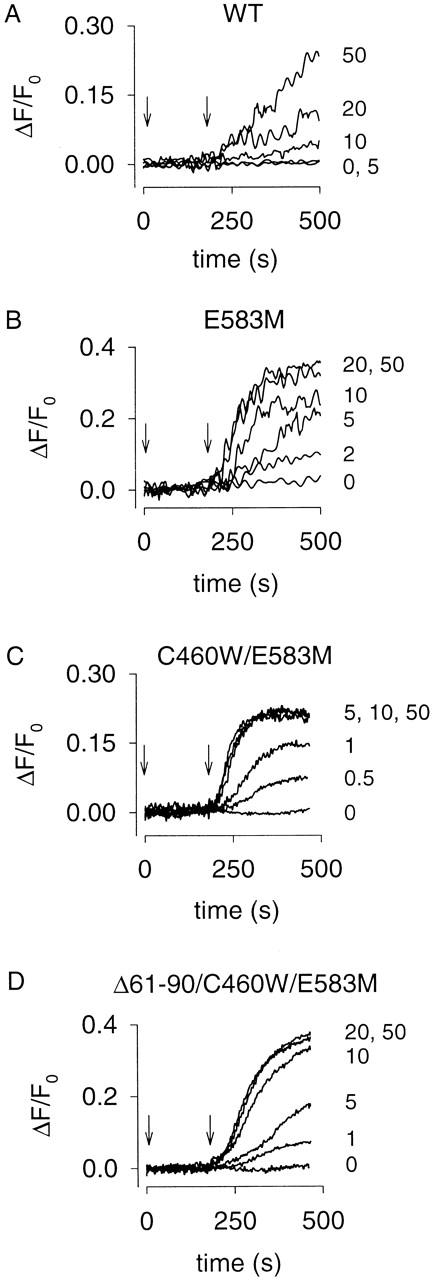
Forskolin-induced cAMP accumulation in HEK-293 cells monitored using Ca2+ influx through expressed CNG channels. 100 μM IBMX was added at time zero and different forskolin concentrations were added at 180 s (indicated by the arrows); concentrations (in micromolars) are indicated at the end of each trace. (A) Forskolin-induced Ca2+ influx was observed in cells expressing the WT channel; the cAMP response (slope) did not saturate in the forskolin range tested (0–50 μM). (B) Forskolin-induced Ca2+ influx was larger in cells expressing the E583M channel. More importantly, the responses saturated at 20 μM. (C) Forskolin-induced responses in cells expressing the C460W/E583M channel saturated at a lower forskolin concentration, 5 μM. (D) Forskolin-induced responses in cells expressing the Δ61–90/C460W/E583M channel were similar to those in cells expressing the E583M channel, saturating at 20 μM. For each construct, the variability in the response between batches of cells was <25% (n = 4).
The measurement of absolute Ca2+ influx rates using fluorescence requires that fura-2 overwhelm endogenous Ca2+ buffers, and that the relation between fluorescence changes and Ca2+ entry be known (Schneggenburger et al. 1993; Frings et al. 1995). However, in this study, we are concerned with relative Ca2+ influx rates, which report changes in cAMP levels. This requires only that fura-2 detect a fixed proportion of the entering Ca2+ in a given experiment. For this to be true, the concentration of unbound fura-2 should not change appreciably upon Ca2+ binding, and cellular Ca2+ buffers that are not overwhelmed by fura-2 should also not be significantly depleted by Ca2+ binding. Under these conditions, the equilibrium concentration of Ca2+-bound fura-2 (CaF), monitored by fluorescence at 380 nm, is given by:
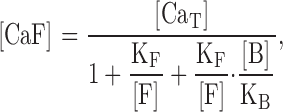 |
1 |
where F represents fura-2, KF the dissociation constant for Ca2+ binding to fura-2, B the endogenous buffer, KB the dissociation constant for Ca2+ binding to the endogenous buffer, and CaT the total Ca2+ concentration that freely exchanges between fura-2 and the endogenous buffer. The equilibrium assumption is reasonable given the time scale of the experiments presented here (tens to hundreds of seconds) and the time scale of binding and unbinding of Ca2+ from fura-2 and endogenous buffers (milliseconds). indicates that with [F] and [B] unchanged by the binding of Ca2+, [CaF] will be a constant fraction of [CaT], and therefore directly proportional to the amount of entering Ca2+. In experiments in which quantitative information was extracted (e.g., the in vivo estimate of KI for PDE inhibitors), we purposely worked at low levels of Ca2+ influx. A typical experiment is shown in Fig. 1, in which addition of 0.5 μM forskolin (an adenylyl cyclase activator) and 50 nM rolipram (a PDE inhibitor) caused a rise in cAMP and an increase in Ca2+ influx. There are several lines of evidence that free fura-2 levels did not change significantly at these low influx rates, and that depletion of endogenous buffers did not distort the measurements. First, after a brief delay (during which cAMP rose to steady level), the Ca2+ influx rate was constant for a substantial period (linear fit in Fig. 1). Second, the changes in fluorescence over which Ca2+ influx rates were measured were always a very small fraction of the total change in fluorescence measured when saturating Ca2+ was admitted into the cells (by adding 30 μl of 10% Triton to the cuvette). The fluorescence changes used in the linear fits were generally <10% of the saturated fura-2 response, indicating that unbound fura-2 was predominant. At high levels of Ca2+ influx, nonlinearities in the traces are likely due to depletion of fura-2 and Ca2+ pumping mechanisms. Third, increasing the external fura-2/AM concentration from 4 to 16 μM significantly altered the intracellular fura-2 concentration but had no effect on the measurement of relative Ca2+ influx rates or estimates of PDE inhibitor KI. The intracellular concentrations of fura-2 were not determined, but a previous study of neuroblastoma cells and isolated pulmonary artery endothelial cells found that 60 min loading with 10 μM fura-2/AM yielded intracellular fura-2 concentrations of ∼130 μM (Oakes et al. 1988). If HEK-293 cells behave similarly, we would expect intracellular concentrations of 35 and 140 μM under the two loading conditions.
Figure 1.

Measurement of Ca2+ influx through CNG channels in response to a rise in cAMP. Ca2+ influx causes a decrease in fluorescence (ΔF), which was expressed relative to the prestimulus fluorescence (F0). In this experiment, HEK-293 cells expressing C460W/E583M CNG channels responded to increased cAMP levels caused by exposure to 50 nM rolipram (a PDE type-IV–specific inhibitor) added at 0 s (first arrow) and 0.5 μM forskolin added at 180 s (second arrow). After a brief lag, ΔF changed in a linear fashion (slope = 6.2 × 10−4 s−1). Slopes were used to assess relative Ca2+ influx rates, for estimating the KI of PDE inhibitors (see and ).
Neher and Augustine 1992 have shown in adrenal chromaffin cells that 98–99% of entering Ca2+ binds to endogenous buffers that are present at high concentration (375–750 μM) but have a low affinity for Ca2+ (KB ∼5–10 μM). Thus, the ratio [B]/KB in was estimated to be 75. KF for fura-2 was estimated to be 150 nM. Thus, the [F]/KF values under our two loading conditions are expected to be 233 and 933. At the higher loading condition, fura-2 may be overwhelming the low affinity cellular buffers. However, as pointed out above, this is not necessary as long as the buffers are not being depleted. It is very likely that low affinity buffers are present at a high enough concentration that they are not being depleted by the low amounts of entering Ca2+. Two of the lines of evidence cited above indicate that buffers (including any high affinity buffers that may be present) are not affecting the proportion of Ca2+ detected by fura-2: the linearity of the influx traces (Fig. 1) and the observation that increasing fura-2 by a factor of four does not alter the measurement of relative Ca2+ influx rates. These results strongly suggest that any high affinity buffers are being overwhelmed by the fura-2 concentrations used.
Unless otherwise stated, data are representative of four experiments, and quantified results are given as mean ± standard error. Forskolin, VIP, nimodipine, and PDE inhibitors were obtained from Calbiochem. Fura-2/AM and pluronic F-127 were purchased from Molecular Probes. Unless otherwise noted, all other chemicals were obtained from Sigma-Aldrich.
RESULTS
Improving the cAMP-sensing Capabilities of CNG Channels
We have previously shown that adenovirus-expressed rat olfactory CNG channels have several properties that make them excellent cAMP sensors (Rich et al. 2000). These properties include their location at the plasma membrane, rapid gating kinetics, and lack of desensitization. Furthermore, these channels appear to colocalize with AC in discrete regions of the membrane, which allows the measurement of localized cAMP signals (Rich et al. 2000). However, there are several limitations to the use of WT CNG channels to detect changes in cAMP. First, WT channels have a low apparent affinity for cAMP (Table and Fig. 2 A), which makes it difficult to detect the low cAMP concentrations that activate PKA. Second, these channels are activated more readily by cGMP than cAMP (Fig. 2 A; see also Dhallan et al. 1990). Third, they can also be activated directly by nitric oxide (NO; Broillet 2000). Fourth, the binding of the Ca2+–CaM complex to these channels strongly inhibits channel opening (Liu et al. 1994). To overcome these limitations, we have modified the properties of the WT channel.
Table 2.
Characteristics of CNG Channels Used as cAMP Sensors
| Membrane potential | K1/2 cAMP | NcAMP | K1/2 cGMP | NcGMP | Imax cGMP/Imax cAMP | |
|---|---|---|---|---|---|---|
| mV | μM | μM | ||||
| WT | +50 | 36 ± 5 | 2.2 ± 0.1 | 1.6 ± 0.1 | 2.3 ± 0.1 | 1.0 |
| E583M | 10.5 ± 0.2 | 2.4 ± 0.2 | 28 ± 2 | 2.4 ± 0.2 | 0.40 ± 0.10 | |
| C460W/E583M | 1.2 ± 0.3 | 2.7 ± 0.2 | 12 ± 2 | 2.8 ± 0.3 | 0.84 ± 0.10 | |
| Δ61-90/C460W/E583M | 14.5 ± 1.8 | 2.1 ± 0.1 | 36 ± 4 | 1.8 ± 0.2 | 0.16 ± 0.08 | |
| WT | −50 | 36 ± 5 | 2.2 ± 0.1 | 1.3 ± 0.4 | 2.5 ± 0.4 | 1.0 |
| E583M | 9 ± 2 | 2.2 ± 0.1 | 32 ± 4 | 1.9 ± 0.2 | 0.35 ± 0.16 | |
| C460W/E583M | 0.89 ± 0.23 | 2.2 ± 0.3 | 6.2 ± 1 | 2.7 ± 0.1 | 0.50 ± 0.10 | |
| Δ61-90/C460W/E583M | 10.5 ± 0.1 | 2.2 ± 0.1 | 16 ± 1 | 2.6 ± 0.2 | 0.09 ± 0.06 |
Data are presented as mean ± SD of three experiments. Hill equation parameters are defined in materials and methods. Imax cGMP/Imax cAMP is the current induced by saturating cGMP divided by the current induced by saturating cAMP.
Figure 2.
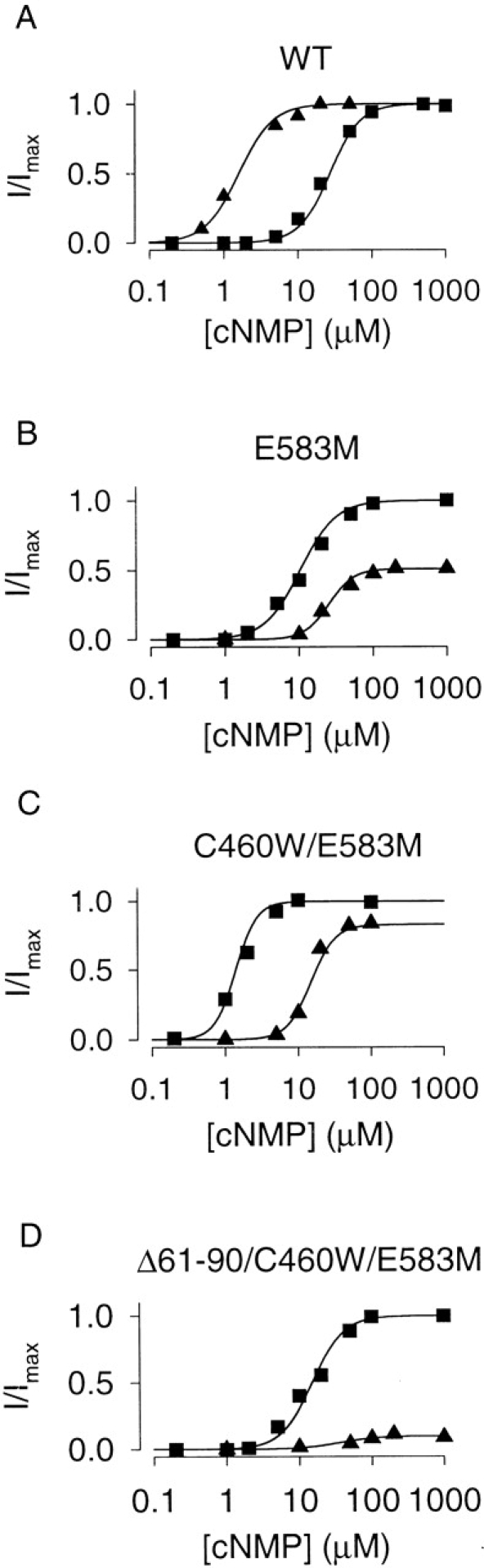
Comparison of CNG channel constructs. Dose–response relations of α subunit homomultimers of WT (A), E583M (B), C460W/E583M (C), and Δ61–90/C460W/E583M (D) rat olfactory CNG channels for cAMP (squares) and cGMP (triangles), evaluated at a membrane potential of +50 mV. The modified CNG channels are more sensitive to cAMP than WT channels, and less sensitive to cGMP. Solid lines are fits to the Hill equation. For the experiments shown, K1/2 cAMP = 30, 10.3, 1.4, and 15 μM; K1/2 cGMP = 1.6, 25, 11, and 36 μM, for WT, E583M, C460W/E583M, and Δ61–90/C460W/E583M channels, respectively. Other fit parameters are given in Table .
We first introduced the mutation E583M into the rat olfactory CNG channel α subunit, guided by the work of Varnum et al. 1995. They showed that mutation of the corresponding residue in the α subunit of the bovine retinal rod CNG channel (D604M) increased the sensitivity to cAMP and decreased the sensitivity to cGMP. The olfactory channel has a higher overall sensitivity to cyclic nucleotides than the rod channel, and, in excised patches, the E583M channel displayed increased sensitivity for cAMP and decreased sensitivity for cGMP (Table II; compare Fig. 2, panel A with B). Furthermore, cGMP was only a partial agonist of the E583M channel. The current elicited by saturating cGMP was ∼40% of the current elicited by saturating cAMP (Table and Fig. 2 B).
To further increase the sensitivity to cAMP, we constructed a second mutant, C460W/E583M, guided by studies showing that modifications to C460 (or C481 in the retinal channel) increased the sensitivity of CNG channels to cyclic nucleotides (Finn et al. 1995; Gordon et al. 1997; Brown et al. 1998; Zong et al. 1998) and eliminated sensitivity to NO (Broillet 2000). The double mutant was considerably more sensitive to cAMP than was the E583M channel (∼10-fold lower K 1/2, Table and Fig. 2 C).
To remove regulation of the channel by Ca2+-CaM binding, we deleted residues 61–90, described previously by Liu et al. 1994. This channel, Δ61–90/C460W/E583M, is almost as sensitive to cAMP as the E583M channel (Table and Fig. 2 D), yet it is virtually insensitive to cGMP. In fact, the current elicited by saturating cGMP was <20% of the current elicited by saturating cAMP (Table and Fig. 2 D). Thus, we have generated two very useful channel constructs for the measurement of cAMP: the Δ61–90/C460W/E583M channel that is sensitive to cAMP at the upper end of the physiological range (∼1–50 μM), and the C460W/E583M channel that is sensitive to cAMP at the lower end of the physiological range (∼0.1–5 μM).
Assessment of the Relative cAMP Sensitivity of Channel Constructs In Vivo
We next wished to assess the ability of each channel construct to detect increases in local cAMP concentration. Changes in cAMP concentration were detected using fura-2 to monitor Ca2+ influx through CNG channels (see materials and methods for details). We measured Ca2+ influx induced by different concentrations of forskolin, an AC activator, in the presence of 100 μM IBMX, a nonselective PDE inhibitor. This approach allowed us to compare the cAMP sensitivities of the different channels to changes in cAMP, regardless of variations in expression levels between experiments. In HEK-293 cells expressing the WT channel, addition of forskolin was followed by a brief delay and an increase in Ca2+ influx (Fig. 3 A). The delay decreased and the rate of Ca2+ influx increased in a dose-dependent manner. Neither effect was saturated at 50 μM forskolin. In cells expressing the E583M channel, addition of forskolin was also followed by a brief delay and an increase in Ca2+ influx (Fig. 3 B). As with the WT channel, the delay and the initial rate of Ca2+ influx were dose-dependent. However, both effects saturated at 20 μM forskolin (the 20- and 50-μM forskolin traces overlap; Fig. 3 B). In cells expressing the C460W/E583M channel, both the delay and the rate of Ca2+ influx were saturated at 5 μM forskolin (Fig. 3 C). In cells expressing the Δ61–90/C460W/E583M channel, addition of forskolin caused similar increases in Ca2+ influx as the E583M channel, including saturation of the delay and rate of influx at 20 μM (Fig. 3, compare B and D). The increased response of the modified channels to forskolin treatment is consistent with the apparent cAMP affinities measured in excised patches in the absence of Ca2+-CaM (C460W/E583M > E583M ≈ Δ61–90/C460W/E583M > WT channels). There are several factors that may have contributed to the apparent lack of a Ca2+-CaM effect on the channels. As pointed out in materials and methods, the concentrations of fura-2 being used are likely overwhelming high affinity cellular Ca2+ buffers, in terms of the fraction of incoming Ca2+ that is bound and, therefore, fura-2 is probably significantly reducing the effects of Ca2+-CaM. It is also possible that HEK-293 cells do not produce enough CaM to regulate heterologously expressed CNG channels, or that the CaM concentration within microdomains is too low to significantly regulate the channels. Very small forskolin-induced changes in Ca2+ influx were observed in HEK-293 cells not expressing CNG channel constructs (Fig. 4).
Figure 4.

Typical forskolin-induced Ca2+ influx in control HEK-293 cells. HEK-293 cells not expressing CNG channel constructs were treated in the same manner as those in Fig. 3. Forskolin (0, 0.5, 5, or 50 μM) was added 180 s after treatment with 100 μM IBMX. As expected, little or no Ca2+ influx was observed in control HEK-293 cells under any of the experimental conditions described in this paper.
PDE Activity in HEK-293 Cells
As described earlier, CNG channels monitor cAMP produced in subcellular compartments near the plasma membrane. To assess the extent to which PDE activity affects cAMP levels, we measured forskolin-induced Ca2+ influx in the presence and absence of PDE inhibitors. Initially we examined the effects of the nonselective PDE inhibitor, IBMX, using each CNG channel construct as a cAMP sensor. In cells expressing the WT channel, there was little or no change in Ca2+ influx in response to an intermediate forskolin concentration (10 μM; Fig. 5 A). After a 3-min pretreatment with 100 μM IBMX only a modest forskolin-induced increase in Ca2+ influx was observed (Fig. 5 A), arising from Ca2+ entry through WT channels. In cells expressing the E583M channel, a moderate forskolin-induced increase in Ca2+ influx was observed in the absence of PDE inhibitors (Fig. 5 B). After pretreatment with IBMX, forskolin caused a robust increase in Ca2+ influx. In cells expressing the C460W/E583M channel, forskolin-induced increases in Ca2+ influx were readily observable, even in the absence of PDE inhibitors (Fig. 5 C). Thus, this channel is capable of detecting cAMP in cells with low AC activity. Using the Δ61–90/C460W/E583M channel as a sensor gave similar forskolin-induced changes in Ca2+ influx as the E583M channel, both in the absence and presence of IBMX (Fig. 5, compare B and D). These results demonstrate that, in HEK-293 cells, basal PDE activity limits the accumulation of cAMP after a moderate stimulus. However, even in the absence of PDE inhibitors, after stimulation of AC with high concentrations of forskolin (50–100 μM), cAMP reaches levels high enough to activate WT CNG channels (not shown).
Figure 5.
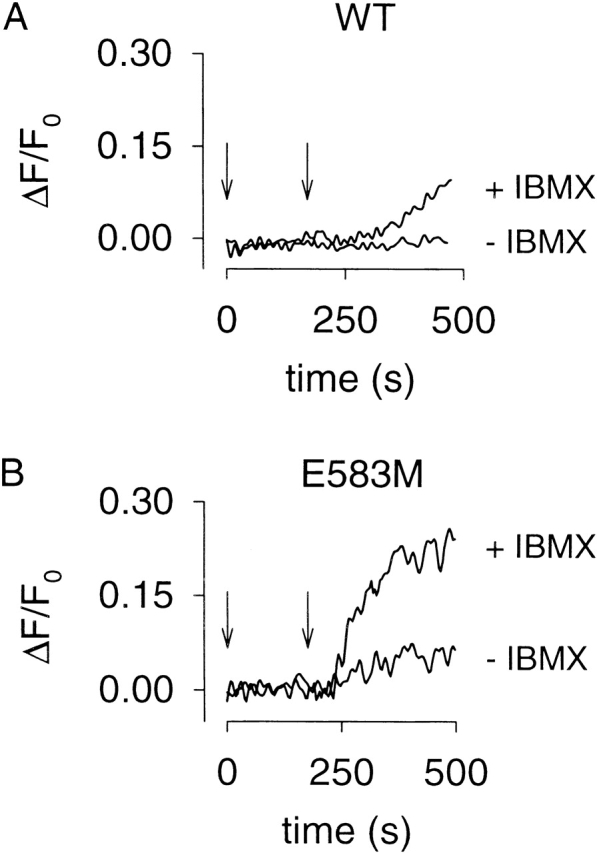
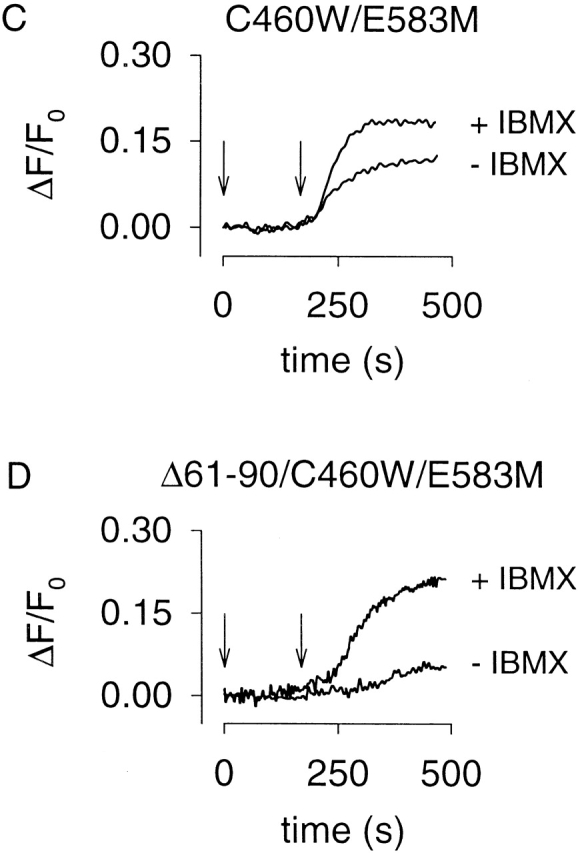
PDE activity reduces forskolin-induced Ca2+ influx through CNG channels in HEK-293 cells. Ca2+ influx through WT (A), E583M (B), C460W/E583M (C), and Δ61–90/C460W/E583M (D) CNG channels in response to 10 μM forskolin (180 s, indicated by arrow), after a 3-min pretreatment with either vehicle (−IBMX) or 100 μM IBMX. Note that a robust forskolin-induced response was easily monitored in the absence of IBMX using C460W/E583M channels as cAMP sensors (C).
We next sought to identify pharmacologically the PDE type(s) that regulates cAMP levels near CNG channels. The C460W/E583M channels were used to monitor forskolin-stimulated cAMP accumulation in the presence and absence of a series of PDE inhibitors. The reported IC50 values of inhibitors used in this study for each PDE type are given in Table . The PDE inhibitor concentrations used were typically at least fivefold higher than the most potent IC50. Either vehicle or PDE inhibitors were added at 0 s and 1 μM forskolin was added at 180 s (Fig. 6). In the absence of PDE inhibitors, little or no forskolin-induced Ca2+ influx was observed; whereas, in the presence of IBMX (10 or 100 μM), or the PDE type-IV–specific inhibitor RO-20-1724 (10 μM) significant forskolin-induced Ca2+ influx was observed (Fig. 6A, Fig. B, and Fig. G). Two other PDE type-IV inhibitors significantly increased the forskolin-induced Ca2+ influx, rolipram (see Fig. 7 C) and etazolate (not shown). Inhibitors specific to other PDE families did not affect forskolin-induced Ca2+ influx. Similar results were obtained in HEK-293 cells expressing either the WT or Δ61–90/C460W/E583M channels by stimulating cAMP production with higher forskolin concentrations (10 μM, not shown), or when prostaglandin E1 (1 μM) was used to stimulate AC activity (not shown). In these experiments, we used inhibitor concentrations at which specific PDE types should be inhibited; however, many PDE inhibitors are not completely specific (Table ). Also, PDE types VIII and IX are insensitive to IBMX as well as most PDE inhibitors (see references in Table ). Unfortunately, dipyridamole, the inhibitor to which they are most sensitive, fluoresces and, as such, cannot be used in this assay.
Figure 6.
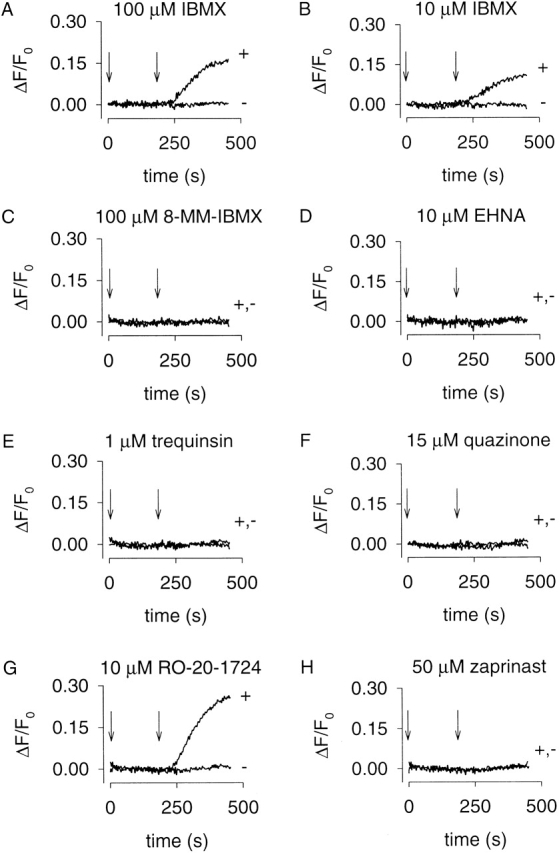
Effects of different PDE inhibitors on forskolin-induced Ca2+ influx in HEK-293 cells. C460W/E583M channels were used to monitor cAMP accumulation in the presence and absence of PDE inhibitors. PDE inhibitors were added at time zero, and 1 μM forskolin was added at 180 s (indicated by arrows). The IC50 for each PDE inhibitor is given in Table . Only the nonspecific PDE inhibitor IBMX (A and B) and the PDE type-IV–specific inhibitor RO-20-1724 (G) influenced the time course of forskolin-induced Ca2+ influx. In general, there was little variability in responses within a single batch of cells.
Figure 7.
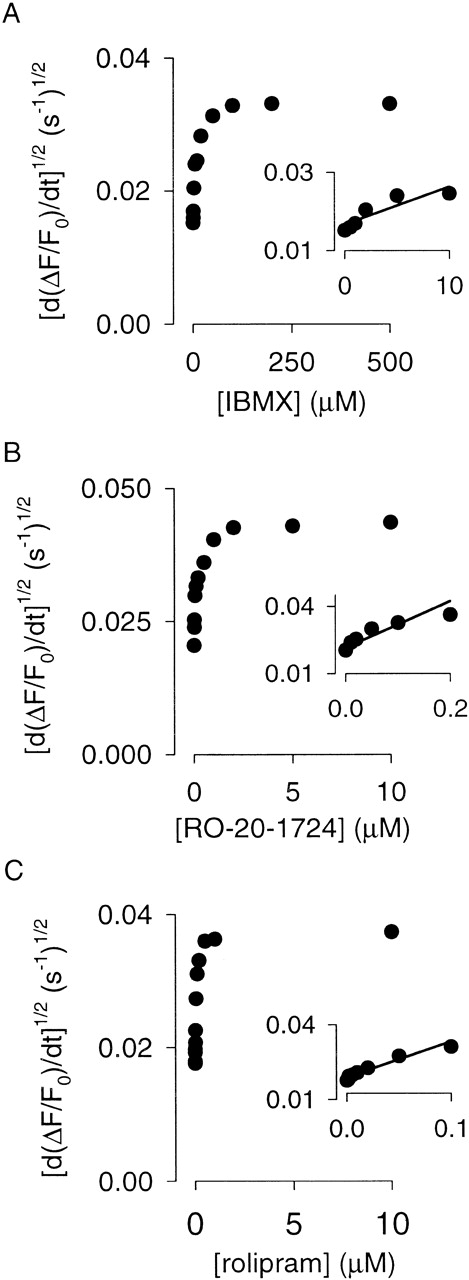
Estimates of KI for three different PDE inhibitors reveal that PDE type IV regulates local cAMP levels in HEK-293 cells. C460W/E583M channels were used to monitor cAMP accumulation triggered by 0.5 μM forskolin (180 s) in the presence and absence of PDE inhibitors (0 s). d(ΔF/F0)/dt was determined as in Fig. 1. The linear fits used to estimate KI values are shown in the insets (see results for explanation). The KI's of the PDE inhibitors were 10 μM (IBMX), 0.14 μM (RO-20-1724), and 0.09 μM (rolipram) for the experiments shown.
In Vivo Estimates of PDE Inhibitor KI
To further establish that PDE type IV is responsible for the observed IBMX-sensitive PDE activity, we estimated the KI's (inhibition constants) of IBMX and two PDE type-IV inhibitors (RO-20-1724 and rolipram) in vivo. This required developing a quantitative framework to assess the relationship between cAMP synthesis, hydrolysis, and redistribution throughout the cell. Thus, we adopted the following formalism:
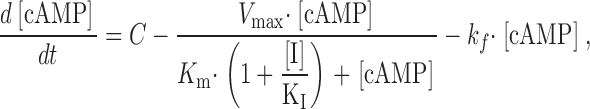 |
2 |
where C is the steady-state rate of cAMP synthesis by AC, V max is the maximal rate of cAMP hydrolysis, K m is the Michaelis constant for PDE, and k f is the rate constant of cAMP flux out of the microdomain. To estimate KI for the PDE inhibitors, we made two assumptions. First, we assumed that at low levels of AC stimulation and PDE inhibition, the concentration of local cAMP is low and diffusion of cAMP out of the microdomain is negligible. Second, we assumed that cAMP levels reach steady-state shortly after AC stimulation (i.e., equal rates of synthesis and hydrolysis). With these assumptions, can be simplified to:
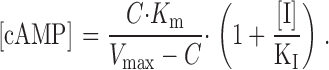 |
3 |
Interestingly, this equation reveals that when the inhibitor concentration is equal to KI the cAMP concentration is twice that in the absence of inhibitor. At low cAMP concentrations (<K1/2 for the channel), the cAMP concentration is proportional to the square root of the Ca2+ influx rate (the Hill coefficient for channel activation is ∼2). Thus, KI can be estimated using a linear fit to the square root of the slopes of the fluorescence traces as a function of inhibitor concentration (Fig. 7).
The high cAMP affinity C460W/E583M channels were used to detect changes in cAMP concentration after pretreatment with PDE inhibitors (added at 0 s) and modest forskolin stimulation (0.5 μM added at 180 s). IBMX, RO-20-1724, and rolipram were completely equilibrated across the plasma membrane of HEK-293 cells in <180 s (data not shown). The assumption that cAMP levels reach steady state is supported by the long-lasting linear rise in Ca2+ concentration after forskolin stimulation (a steady Ca2+ influx rate reflects a constant cAMP level; Fig. 1). Dose–response relations for the three PDE inhibitors are shown in Fig. 7. Based upon fits to this data with (Fig. 7, A–C, insets), the KI values were estimated to be 11 ± 2 μM (IBMX), 0.13 ± 0.02 μM (RO-20-1724), and 0.07 ± 0.02 μM (rolipram), n = 4, which are consistent with published IC50 values from in vitro experiments (Table ). To ensure that the results were independent of channel construct, we also estimated the KI for RO-20-1724 using the lower cAMP affinity Δ61–90/C460W/E583M channels. The KI (0.15 ± 0.02 μM, n = 3) was indistinguishable from that estimated using C460W/E583M channels. These data strongly suggest that PDE type IV is primarily responsible for the IBMX-sensitive component of PDE activity monitored using CNG channels and, thus, for regulating localized cAMP concentration in HEK-293 cells.
At higher PDE inhibitor concentrations, the square root of Ca2+ influx rate, which is proportional to cAMP concentration, deviates from linearity (Fig. 7). It is likely that at higher inhibitor concentrations cAMP may reach levels ≥K 1/2 of the channel. At these concentrations, the relationship between Ca2+ influx rate and cAMP concentration deviates from a simple square law. Importantly, this does not explain why at high levels of PDE inhibition the Ca2+ influx rate reaches a plateau. When PDE activity is completely inhibited, forskolin-stimulated cAMP accumulation would continually increase in a confined region of free diffusion. This, in turn, would lead to higher Ca2+ influx rates. To ensure that the plateau was not primarily due to channel saturation or Ca2+ homeostatic mechanisms, we measured Ca2+ influx rates induced by 50 μM forskolin and maximal PDE inhibition in the same experiment. Under these conditions, the Ca2+ influx rate greatly exceeded the influx rates induced by 0.5 μM forskolin with PDE inhibition. At increased cAMP concentrations, cAMP efflux from the microdomain is expected to increase; this increased efflux could create the observed plateau. This would be consistent with the diffusionally restricted microdomain model (Rich et al. 2000).
PDE Activity in GH4C1 Cells
To further test the utility of this approach, we have examined PDE activity in excitable GH4C1 pituitary cells. In all the experiments shown (Fig. 8 Fig. 9 and Table ), 1 μM nimodipine was added at time zero to block Ca2+ influx though voltage-gated Ca2+ channels (triggered by membrane depolarization due to Ca2+ and Na+ influx through CNG channels). This concentration of nimodipine was sufficient to block Ca2+ influx through voltage-gated Ca2+ channels activated by membrane depolarization in 24 mM external KCl, and did not alter forskolin or pCPT-cGMP induced Ca2+ influx through CNG channels expressed in HEK-293 cells (data not shown). Fig. 8 Fig. 9 each depict experiments done on a single batch of cells. As before, similar results were obtained on three other batches.
Figure 8.

Detection of AC and PDE activity in GH4C1 cells. (A) GH4C1 cells expressing WT CNG channels. Either 100 μM IBMX or vehicle (−IBMX) was added at time zero. 50 μM forskolin was added at 180 s. (B) Control cells (not expressing CNG channels). No forskolin-induced Ca2+ influx was observed under these conditions, or any of the experimental conditions presented. (C and D) GH4C1 cells expressing C460W/E583M channels. IBMX or vehicle was added at 60 s (first arrow). Either 10 μM forskolin (C) or 100 nM VIP (D) was added at 240 s (second arrow). In the absence of IBMX, large forskolin- or VIP-induced increases in Ca2+ influx were observed. When the local PDE activity was inhibited by 100 μM IBMX, substantial basal AC activity was revealed. This level of basal AC activity was quite different from that observed in HEK-293 cells (see Fig. 5 and Fig. 6). 1 μM nimodipine was added at time 0 to block endogenous voltage-gated Ca2+ channels.
Figure 9.

Evidence that PDE type IV is responsible for localized, high K m PDE activity in GH4C1 cells. Δ61–90/C460W/E583M channels were used to detect changes in cAMP accumulation in the presence and absence of PDE inhibitors. Inhibitors were added at 60 s and 10 μM forskolin was added at 240 s. 1 μM nimodipine was added at time zero to block voltage-gated Ca2+ channels. The nonspecific PDE inhibitor IBMX (A), the PDE type-I–specific inhibitor 8-methoxymethyl- IBMX (8-MM-IBMX, B), and the PDE type-IV–specific inhibitors RO-20-1724 (C) and rolipram (D) increased forskolin-induced Ca2+ influx through CNG channels.
Table 3.
Effect of PDE Inhibitors on Basal cAMP Levels in GH4C1 Cells
| Inhibitor | d(ΔF/F0)/dt | No. of experiments |
|---|---|---|
| μM | s−1 | |
| 100 IBMX | 0.0025 ± 0.0009 | 5 |
| 100 8-methoxymethyl-IBMX | 0. 0029 ± 0.0011 | 4 |
| 10 EHNA | 0 | 3 |
| 1 Trequinsin | 0 | 3 |
| 15 Quazinone | 0 | 3 |
| 100 RO-20-1724 | 0 | 4 |
| 100 Rolipram | 0 | 4 |
| 50 Zaprinast | 0 | 3 |
cAMP concentration was monitored using Ca2+ influx through C460W/E583M channels. The experimental protocol is described in the legend of Fig. 8 C. d(ΔF/F0)/dt, which is proportional to the Ca2+ influx rate, was estimated as described in materials and methods. Data are given as mean ± standard error.
Fig. 8 A shows the forskolin-induced responses of cells expressing WT channels. Either vehicle or 100 μM IBMX was added at 0 s and 50 μM forskolin was added at 180 s. After the addition of forskolin, there was a short delay followed by an increased Ca2+ influx. The slope of the Ca2+ influx was greater in the presence of IBMX, indicating higher cAMP levels. In control cells (cells not expressing CNG channel constructs), no forskolin-induced Ca2+ influx was observed in either the presence or absence of IBMX (Fig. 8 B). This was true of controls done for all experimental protocols.
We next examined forskolin-induced responses and the effects of IBMX in cells expressing the high cAMP affinity construct, C460W/E583M. In these experiments, either vehicle or 100 μM IBMX was added at 60 s (Fig. 8C and Fig. D). Interestingly, the addition of IBMX triggered Ca2+ influx that was not observed in either control cells or cells expressing the WT channel. Subsequent addition of 10 μM forskolin (Fig. 8 C) or 100 nM vasoactive intestinal peptide (VIP; Fig. 8D) caused additional cAMP accumulation and Ca2+ influx. A comparison of responses measured with the WT and C460W/E583M channels indicates that the IBMX-induced response was not due to an increase in local cGMP concentration. Moreover, 100 μM IBMX did not alter CNG channel activity monitored in excised patches (data not shown). Thus, these data indicate that the IBMX-induced Ca2+ influx was due primarily to an increase in cAMP arising from basal AC activity.
To identify the PDE family or families that regulate the local cAMP concentration in GH4C1 cells, cAMP accumulation was monitored in the presence and absence of PDE inhibitors (Table ). Again, the high cAMP affinity C460W/E583M channels were used. PDE inhibitors were added at 60 s. Although the addition of IBMX alone triggered Ca2+ influx, we added 10 μM forskolin at 240 s as a positive control for CNG channel activity. Only the nonspecific PDE inhibitor, IBMX, and the PDE type-I–specific inhibitor, 8-methoxymethyl-IBMX, induced Ca2+ influx through CNG channels (Table ). This PDE is likely to have a low K m for cAMP because it is capable of regulating cAMP at concentrations too low for the WT or Δ61–90/C460W/E583M channels to detect. Thus, it is likely that a Ca2+-CaM–stimulated PDE (type I) is primarily responsible for controlling basal cAMP signals in GH4C1 cells.
We also monitored PDE activity using Δ61–90/C460W/E583M channels to detect changes in cAMP concentration (Fig. 9). PDE inhibitors and forskolin were added as described above. With this construct, we were unable to observe changes in cAMP concentration due to inhibition of PDE activity alone. However, PDE inhibitors caused an increase in forskolin-induced Ca2+ influx. As observed using the high cAMP affinity C460W/E583M channels, both IBMX and the PDE type-I–specific inhibitor 8-methoxymethyl-IBMX significantly inhibited PDE activity (Fig. 9A and Fig. B). Interestingly, the PDE type-IV–specific inhibitors, RO-20-1724 and rolipram, also inhibited PDE activity (Fig. 9C and Fig. D). Inhibitors specific to other PDE types had no effect on the forskolin-induced Ca2+ influx (data not shown). No RO-20-1724- or rolipram-sensitive PDE was observed using the high cAMP affinity channel (Table ). It is likely that Ca2+ influx through the C460W/E583M channels was saturated at cAMP concentrations below the K m for this PDE. Thus, there are likely two different PDE types that regulate local cAMP signals in GH4C1 cells: a low K m, 8-methoxymethyl-IBMX–sensitive PDE (type I) and a high K m, RO-20-1724-rolipram–sensitive PDE (type IV).
DISCUSSION
We have made several modifications to the WT rat olfactory CNG channel that significantly enhance its utility as a cAMP sensor. Three novel constructs were tested. The E583M channel is more sensitive to cAMP and less sensitive to cGMP. Two additional mutations produced channels that make even better cAMP sensors. The C460W/E583M channel has a high affinity for cAMP, allowing it to detect cAMP at levels which activate PKA. The Δ61–90/C460W/E583M channel is almost as sensitive to cAMP as E583M. However, the Δ61–90/C460W/E583M channel has two important advantages: it is not inhibited by Ca2+-CaM, and it is barely activated by cGMP.
Using both WT and modified CNG channels, we have developed a convenient assay for detecting changes in local cAMP concentration, and used this assay to examine PDE activity in two cell types. There are several advantages of measuring Ca2+ influx through CNG channels as a way to monitor changes in cAMP. First, this assay is very simple to implement in either cell populations or single cells. Second, the measurement has a high signal-to-noise ratio. Third, it is sensitive to small changes in AC activity. This is due, in part, to the fact that CNG channels detect local rather than total cellular cAMP. With traditional assays, high agonist and PDE inhibitor concentrations are often required to detect cAMP accumulation. In contrast, the channels described here readily detect changes in cAMP caused by subsaturating agonist concentration in the absence of PDE inhibitors. Overall, the assay presented here can be used as an initial screen for changes in cAMP in response to physiological stimuli or pharmacological agents.
There are a few shortcomings to this approach that should be mentioned. The temporal resolution is somewhat limited. It is also difficult to extract the precise cAMP concentration from this assay because the responses cannot be calibrated (with known intracellular cAMP), and because Ca2+ handling and depletion of free fura-2 become issues at high Ca2+ influx rates. When more precise measurements of local cAMP are required, cAMP concentration can be determined using electrophysiological methods (Rich et al. 2000). Although such experiments are considerably more difficult, the increased temporal resolution, higher dynamic range, and more accurate measurement of cAMP concentration will be required for a detailed understanding of cAMP signals.
We have previously demonstrated in HEK-293 and C6–2B glioma cells that CNG channels measure cAMP produced in subcellular compartments or microdomains near the plasma membrane, and that diffusion of cAMP between the microdomains and the bulk cytosol is severely hindered (Rich et al. 2000). This conclusion was based on several lines of evidence. First, forskolin-induced increases in cAMP concentration measured using CNG channels (>25 μM) were much greater than the increases in cAMP concentration averaged throughout the accessible cell volume (1–2 μM). Second, forskolin-induced cAMP accumulation was easily detected by CNG channels in the rapidly dialyzed, whole-cell patch-clamp configuration. Third, the wash-in of cAMP from the patch pipette to the CNG channels was much slower than would be expected based upon the rapid exchange of the bulk cytosol. All of these results were described by a three-compartment model (microdomain, cytosol, and whole-cell pipette) in which the transfer rates between compartments were determined using data from the wash-in experiments.
Much has been learned about the 2-D localization of proteins involved in cellular signaling. For example, certain isoforms of PDE are differentially distributed between particulate (membrane-localized) and supernatant (cytosolic) cellular fractions; PDE kinetics may vary with cellular localization (Bolger et al. 1997). Elements of G-protein signaling pathways have been shown to preferentially localize within caveolae (distinct regions of the plasma membrane; Rybin et al. 2000). In addition, a recent report has described the localization of different voltage-gated K+ channels to distinct populations of lipid rafts (Martens et al. 2001).
However, 2-D localization within the plasma membrane does not account for the 3-D compartmentalization of cAMP signals. We have shown that proximity to AC does not provide sufficient cAMP concentrations for activation of effector proteins because, in essence, each cAMP molecule diffuses away more rapidly than the next molecule is synthesized (Rich et al. 2000). Thus, PDE activity alone is unlikely to be responsible for compartmentalized cAMP signals because PDE activity only lowers cAMP levels. It is more likely that PDEs regulating cAMP levels detected by CNG channels are localized within the same 3-D compartment as AC and CNG channels. Importantly, by regulating cAMP concentration within this compartment, PDE activity affects the rate of cAMP flux between compartments. This PDE is likely to be a component of the previously described particulate PDE fractions. Cytosolic PDE is likely to regulate cAMP in other cellular compartments.
In the current study, we have used Ca2+ influx through CNG channels to detect changes in cAMP concentration in both nonexcitable HEK-293 cells and excitable pituitary GH4C1 cells. The specific goal was to assess the PDE subtypes responsible for shaping cAMP signals near the plasma membrane. To study the effects of PDEs, it is important to be able to first detect cAMP changes in the absence of PDE inhibitors. We had no difficulty with this in either cell type, despite the modest levels of cAMP accumulation in response to forskolin or VIP stimulation reported previously (Mollard et al. 1992; Fagan et al. 1996, Fagan et al. 2000). In HEK-293 cells, only nonselective and PDE type-IV–selective inhibitors decreased PDE activity. The in vivo estimates of KI for three PDE inhibitors (IBMX, RO-20-1724, and rolipram) presented here are consistent with IC50 values estimated in vitro for PDE type IV. These data are also consistent with a previous report that identified two isoforms of PDE type IV (PDE4D3 and PDE4D5) in HEK-293 cells based on Western blot analysis (Hoffmann et al. 1999). Inhibition of PDE activity did not reveal basal AC activity in HEK-293 cells. In GH4C1 cells, inhibitors selective to PDE types I and IV decreased PDE activity. Using the high cAMP affinity CNG channels, inhibition of PDE type I activity led to a rise in local cAMP concentration in the absence of AC stimulation. This increase revealed substantial basal AC activity in GH4C1 cells. Under the same conditions, no change in cAMP level was observed when PDE type-IV inhibitors were added. However, using the lower cAMP affinity, Δ61–90/C460W/E583M channels, inhibition of either PDE type I or type IV increased forskolin-induced cAMP accumulation. These observations point to the presence of two different PDE types: a low K m, PDE type I and a higher K m, PDE type IV. Based on the apparent cAMP affinities of the two channel constructs, we estimate the two K m values to be <1 μM and >5 μM.
Interestingly, the data in GH4C1 cells indicate the presence of a “futile cycle” of cAMP synthesis and hydrolysis. Obviously, restricting this phenomenon to subcellular compartments would be advantageous energetically, but why expend the additional energy at all? One possibility is that constant AC and PDE activity allow the system to respond rapidly to a stimulus. In essence, the enzymes are poised to respond to changes in active G-protein or changes in internal Ca2+, allowing for rapid increases or decreases in cAMP levels. A similar situation exists in light-adapted photoreceptor cells; for a detailed discussion see Nikonov et al. 2000.
The existence of PDE type I in GH4C1 cells is consistent with previous work showing that CaM antagonists inhibit the hydrolysis of cAMP (Sletholt et al. 1987). Furthermore, a high K m (28.6 μM) and a low K m (0.66 μM) PDE have been identified in both GH3 and GH4C1 cells, each PDE with a different IC50 for theophylline (Gautvik et al. 1982). These K m values are consistent with our in vivo estimates of the K m values for PDEs that regulate local cAMP levels. The presence of both high and low K m PDEs offers two possibilities for the shaping of cAMP signals: (1) that both PDE types regulate cAMP levels within a single compartment, or (2) that they differentially regulate cAMP levels in different cellular compartments.
If the two PDE types coexist in the same subcellular compartment then the low K m PDE would be likely to regulate cAMP levels under basal conditions. Upon further activation of AC, this PDE would be overwhelmed, and large concentrations of cAMP would accumulate locally. The high K m PDE would be able to efficiently hydrolyze cAMP at these elevated concentrations. Furthermore, these PDE types could be differentially regulated, allowing different feedback mechanisms to control cAMP levels within this compartment. If the activity of the two PDE types is compartmentalized, then distinct cyclic nucleotide signals could occur simultaneously in different regions of the cell. Although this has not yet been demonstrated, it seems likely because cAMP can be excluded from areas of the cell by PDE activity (Jurevicius and Fischmeister 1996), cAMP is produced in microdomains with restricted diffusional access to the bulk cytosol (Rich et al. 2000), and different pools of cGMP have different functional effects in ECV304 epithelial cells (Zolle et al. 2000).
In the future, it should be possible with the improved sensors to measure local cAMP signals in response to a variety of extracellular stimuli. The ultimate goal is to understand how cAMP can differentially regulate >200 cellular targets. This requires a quantitative knowledge of how cAMP signals are initiated and terminated. Different PDE subtypes are likely to play crucial roles in shaping the signals in different cellular compartments. Within these compartments, cAMP levels are likely to vary dynamically (Brooker 1973; Cooper et al. 1995, Cooper et al. 1998), particularly in excitable cells where Ca2+ levels oscillate during a train of action potentials. For example, modulation of PDE type I by Ca2+-CaM in excitable GH4C1 cells could contribute to transient or oscillating cAMP signals. In general, dynamic cAMP signals would escape detection with conventional techniques.
Acknowledgments
We would like to thank Dr. R.R. Reed for providing the cDNA encoding the olfactory CNG channel, Dr. K.-W. Yau for helpful advice concerning the Ca2+-CaM deletion, Dr. M.M. Tamkun for providing HEK-293 cells, Dr. Y. He and X. Wang for technical assistance, and Dr. D.M.F. Cooper for helpful discussions and for use of spectrofluorimeter and tissue culture facilities.
This work was supported by the National Institutes of Health grants EY09275, NS28389, HL58344, and DC00385, and American Heart Association grant DM0020554Z.
Footnotes
Abbreviations used in this paper: AC, adenylyl cyclase; AKAP, A-kinase anchoring protein; CaM, calmodulin; HEK-293, human embryonic kidney cell line; IBMX, 3-isobutyl-1-methylxanthine; NO, nitric oxide; pCPT-cGMP, 8-p-chlorophenylthio-cGMP; PDE, phosphodiesterase; RO-20-1724, 4-(3-butoxy-4-methoxybenzyl)-2-imidazolidinone; VIP, vasoactive intestinal peptide; WT, wild-type.
References
- Ahluwalia G.S., Rhoads A.R. Selective inhibition of cyclic AMP and cyclic GMP phosphodiesterases of cardiac nuclear fraction. Biochem. Pharmacol. 1982;31:665–669. doi: 10.1016/0006-2952(82)90447-6. [DOI] [PubMed] [Google Scholar]
- Ahn H.S., Crim W., Romano M., Sybertz E., Pitts B. Effects of selective inhibitors on cyclic nucleotide phosphodiesterases of rabbit aorta. Biochem. Pharmacol. 1989;38:3331–3339. doi: 10.1016/0006-2952(89)90631-x. [DOI] [PubMed] [Google Scholar]
- Baylor D.A., Lamb T.D., Yau K.-W. The membrane current of single rod outer segments. J. Physiol. 1979;288:589–611. [PMC free article] [PubMed] [Google Scholar]
- Beavo J.A. Multiple isozymes of cyclic nucleotide phosphodiesterase. Adv. Second Messenger Phosphoprot. Res. 1988;22:1–38. [PubMed] [Google Scholar]
- Beavo J.A. Cyclic nucleotide phosphodiesterasesfunctional implications of multiple isoforms. Physiol. Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- Bolger G.B., Erdogan S., Jones R.E., Loughney K., Scotland G., Hoffmann R., Wilkinson I., Farrell C., Houslay M.D. Characterization of five different proteins produced by alternatively spliced mRNAs from the human cAMP-specific phosphodiesterase PDE4D gene. Biochem. J. 1997;328:539–548. doi: 10.1042/bj3280539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger G., Michaeli T., Martins T., St. John T., Steiner B., Rodgers L., Riggs M., Wigler M., Ferguson K. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol. Cell. Biol. 1993;13:6558–6571. doi: 10.1128/mcb.13.10.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broillet M.-C. A single intracellular cysteine residue is responsible for the activation of the olfactory cyclic nucleotide-gated channel by NO. J. Biol. Chem. 2000;275:15135–15141. doi: 10.1074/jbc.275.20.15135. [DOI] [PubMed] [Google Scholar]
- Brooker G. Oscillation of cyclic adenosine monophosphate concentration during the myocardial contraction cycle. Science. 1973;182:933–934. doi: 10.1126/science.182.4115.933. [DOI] [PubMed] [Google Scholar]
- Brown R.L., Snow S.D., Haley T.L. Movement of gating machinery during the activation of rod cyclic nucleotide-gated channels. Biophys. J. 1998;75:825–833. doi: 10.1016/s0006-3495(98)77571-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.K., Burns M.E., He W., Wensel T.G., Baylor D.A., Simon M.I. Slowed recovery of rod photoresponse in mice lacking the GTPase accelerating protein RGS9-1. Nature. 2000;403:557–560. doi: 10.1038/35000601. [DOI] [PubMed] [Google Scholar]
- Conti M., Nemoz G., Sette C., Vicini E. Recent progress in understanding the hormonal regulation of phosphodiesterases. Endocrine Rev. 1995;16:370–389. doi: 10.1210/edrv-16-3-370. [DOI] [PubMed] [Google Scholar]
- Cooper D.M.F., Karpen J.W., Fagan K.A., Mons N.E. Ca2+-sensitive adenylyl cyclases. Adv. Second Messenger Phosphoprot. Res. 1998;32:23–51. [PubMed] [Google Scholar]
- Cooper D.M.F., Mons N., Karpen J.W. Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature. 1995;374:421–424. doi: 10.1038/374421a0. [DOI] [PubMed] [Google Scholar]
- Coste H., Grondin P. Characterization of a novel potent and specific inhibitor of type V phosphodiesterase. Biochem. Pharmacol. 1995;50:1577–1585. doi: 10.1016/0006-2952(95)02031-4. [DOI] [PubMed] [Google Scholar]
- Dhallan R.S., Yau K.-W., Schrader K.A., Reed R.R. Primary structure and functional expression of a cyclic nucleotide-activated channel from olfactory neurons. Nature. 1990;347:184–187. doi: 10.1038/347184a0. [DOI] [PubMed] [Google Scholar]
- Drummond G.I., Perrot-Yee S. Enzymatic hydrolysis of adenosine 3′,5′-phosphoric acid. J. Biol. Chem. 1961;236:1126–1129. [PubMed] [Google Scholar]
- Epstein P.M., Strada S.J., Sarada K., Thompson W.J. Catalytic and kinetic properties of purified high-affinity cyclic AMP phosphodiesterase from dog kidney. Arch. Biochem. Biophys. 1982;218:119–133. doi: 10.1016/0003-9861(82)90327-7. [DOI] [PubMed] [Google Scholar]
- Fagan K.A., Graf R.A., Tolman S., Schaack J., Cooper D.M.F. Regulation of Ca2+-sensitive adenylyl cyclase in an excitable cell. J. Biol. Chem. 2000;275:40187–40194. doi: 10.1074/jbc.M006606200. [DOI] [PubMed] [Google Scholar]
- Fagan K.A., Mahey R., Cooper D.M.F. Functional co-localization of transfected Ca2+-stimulated adenylyl cyclases with capacitative Ca2+ entry sites. J. Biol. Chem. 1996;271:12438–12444. doi: 10.1074/jbc.271.21.12438. [DOI] [PubMed] [Google Scholar]
- Fagan K.A., Rich T.C., Tolman S., Schaack J., Karpen J.W., Cooper D.M.F. Adenovirus-mediated expression of an olfactory cyclic nucleotide-gated channel regulates the endogenous Ca2+-inhibitable adenylyl cyclase in C6-2B glioma cells. J. Biol. Chem. 1999;274:12445–12453. doi: 10.1074/jbc.274.18.12445. [DOI] [PubMed] [Google Scholar]
- Fawcett L., Baxendale R., Stacey P., McGrouther C., Harrow I., Soderling S., Hetman J., Beavo J.A., Phillips S.C. Molecular cloning and characterization of a distinct human phosphodiesterase gene familyPDE11A. Proc. Natl. Acad. Sci. USA. 2000;97:3702–3707. doi: 10.1073/pnas.050585197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn J.T., Li J., Yau K.-W. C-terminus involvement in the gating of cyclic nucleotide-activated channels as revealed by Ni2+ and NEM Biophys. J. 68 1995. A253(Abstr [Google Scholar]
- Fisher D.A., Smith J.F., Pillar J.S., St. Denis S.H., Cheng J.B. Isolation and characterization of PDE9A, a novel human cGMP-specific phosphodiesterase. J. Biol. Chem. 1998;273:15559–15564. doi: 10.1074/jbc.273.25.15559. [DOI] [PubMed] [Google Scholar]
- Frings S., Seifert R., Godde M., Kaupp U.B. Profoundly different calcium permeation and blockage determine the specific function of distinct cyclic nucleotide-gated channels. Neuron. 1995;15:169–179. doi: 10.1016/0896-6273(95)90074-8. [DOI] [PubMed] [Google Scholar]
- Fung B.K., Hurley J.B., Stryer L. Flow of information in the light-triggered cyclic nucleotide cascade of vision. Proc. Natl. Acad. Sci. USA. 1981;78:152–156. doi: 10.1073/pnas.78.1.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner C., Robas N., Cawkill D., Fidock M. Cloning and characterization of the human and mouse PDE7B, a novel cAMP-specific cyclic nucleotide phosphodiesterase. Biochem. Biophys. Res. Commun. 2000;272:186–192. doi: 10.1006/bbrc.2000.2743. [DOI] [PubMed] [Google Scholar]
- Gautvik K.M., Kriz M., Jahnsen T., Haug E., Hansson V. Relationship between stimulated prolactin release from GH cells and cyclic AMP degradation and formation. Mol. Cell. Endocrinology. 1982;26:295–308. doi: 10.1016/0303-7207(82)90118-6. [DOI] [PubMed] [Google Scholar]
- Gomez-Foix A.M., Coats W.S., Baque S., Alam T., Gerard R.D., Newgard C.B. Adenovirus-mediated transfer of the muscle glycogen phosphorylase gene into hepatocytes confers altered regulation of glycogen metabolism. J. Biol. Chem. 1992;267:25129–25134. [PubMed] [Google Scholar]
- Gorczyca W.A., Gray-Keller M.P., Detwiler P.B., Palczewski K. Purification and physiological evaluation of a guanylate cyclase activating protein from retinal rods. Proc. Natl. Acad. Sci. USA. 1994;91:4014–4018. doi: 10.1073/pnas.91.9.4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S.E., Varnum M.D., Zagotta W.N. Direct interaction between amino- and carboxyl-terminal domains of cyclic nucleotide-gated channels. Neuron. 1997;19:431–441. doi: 10.1016/s0896-6273(00)80951-4. [DOI] [PubMed] [Google Scholar]
- Harrison S.A., Beier N., Martins T.J., Beavo J.A. Isolation and comparison of bovine heart cGMP-inhibited and cGMP-stimulated phosphodiesterases. Methods Enzymol. 1988;159:685–702. doi: 10.1016/0076-6879(88)59064-x. [DOI] [PubMed] [Google Scholar]
- He T.-C., Zhou S., DaCosta L.T., Yu J., Kinzler K.W., Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He W., Cowan C.W., Wensel T.G. RGS9, a GTPase accelerator for phototransduction. Neuron. 1998;20:95–102. doi: 10.1016/s0896-6273(00)80437-7. [DOI] [PubMed] [Google Scholar]
- Hetman J.M., Soderling S.H., Glavas N.A., Beavo J.A. Cloning and characterization of PDE7B, a cAMP-specific phosphodiesterase. Proc. Natl. Acad. Sci. USA. 2000;97:472–476. doi: 10.1073/pnas.97.1.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R., Baillie G.S., MacKenzie S.J., Yarwood S.J., Houslay M.D. The MAP kinase ERK2 inhibits the cyclic AMP-specific phosphodiesterase HSPDE4D3 by phosphorylating it at Ser579. EMBO J. 1999;18:893–903. doi: 10.1093/emboj/18.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holck M., Sebastien T., Muggli R., Eigenmann R. Studies on the mechanism of positive inotropic activity of Ro 13-6438, a structurally novel cardiotonic agent with vasodilating properties. J. Cardiovasc. Pharm. 1984;6:520–530. doi: 10.1097/00005344-198405000-00022. [DOI] [PubMed] [Google Scholar]
- Jordan M., Schallhorn A., Wurm F.M. Transfecting mammalian cellsoptimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res. 1996;24:596–601. doi: 10.1093/nar/24.4.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurevicius J., Fischmeister R. cAMP compartmentation is responsible for a local activation of cardiac Ca2+ channels by β-adrenergic agonists. Proc. Natl. Acad. Sci. USA. 1996;93:295–299. doi: 10.1073/pnas.93.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch K.W., Stryer L. Highly cooperative feedback control of retinal rod guanylate cyclase by calcium ions. Nature. 1988;334:64–66. doi: 10.1038/334064a0. [DOI] [PubMed] [Google Scholar]
- Koutalos Y., Nakatani K., Yau K.-W. The cGMP-phosphodiesterase and its contribution to sensitivity regulation in retinal rods J. Gen. Physiol. 106 1995. 891 921a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutalos Y., Nakatani K., Yau K.-W. Characterization of guanylate cyclase activity in single retinal rod outer segments J. Gen. Physiol. 106 1995. 863 890b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagnado L., Baylor D.A. Signal flow in visual transduction. Neuron. 1992;8:995–1002. doi: 10.1016/0896-6273(92)90122-t. [DOI] [PubMed] [Google Scholar]
- Leskov I.B., Klenchin V.A., Handy J.W., Whitlock G.G., Govardovskii V.I., Bownds M.D., Lamb T.D., Pugh E.N., Jr., Arshavsky V.Y. The gain of rod phototransductionreconciliation of biochemical and electrophysiological measurements. Neuron. 2000;27:525–537. doi: 10.1016/s0896-6273(00)00063-5. [DOI] [PubMed] [Google Scholar]
- Liu M., Chen T.Y., Ahamed B., Li J., Yau K.-W. Calcium-calmodulin modulation of the olfactory cyclic nucleotide-gated cation channel. Science. 1994;266:1348–1354. doi: 10.1126/science.266.5189.1348. [DOI] [PubMed] [Google Scholar]
- Lorenz K.L., Wells J.N. Potentiation of the effects of sodium nitroprusside and of isoproterenol by selective phosphodiesterase inhibitors. Mol. Pharmacol. 1983;23:424–430. [PubMed] [Google Scholar]
- Loughney K., Hill T.R., Florio V.A., Uher L., Rosman G.J., Wolda S.L., Jones B.A., Howard M.L., McAllister-Lucas L.M., Sonnenburg W.K. Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3′,5′-cyclic nucleotide phosphodiesterase. Gene. 1998;216:139–147. doi: 10.1016/s0378-1119(98)00303-5. [DOI] [PubMed] [Google Scholar]
- Loughney K., Martins T.J., Harris E.A.S., Hicks J.B., Sonnenburg W.K., Beavo J.A., Ferguson K. Isolation and characterization of cDNAs corresponding to two human calcium, calmodulin-regulated, 3′-5′-cyclic nucleotide phosphodiesterases. J. Biol. Chem. 1996;271:796–806. doi: 10.1074/jbc.271.2.796. [DOI] [PubMed] [Google Scholar]
- MacKenzie S.J., Houslay M.D. Action of rolipram on specific PDE4 cAMP phosphodiesterase isoforms and on the phosphorylation of cAMP-response-element-binding protein (CREB) and p38 mitogen-activated protein (MAP) kinase in U937 monocytic cells. Biochem. J. 2000;347:571–578. doi: 10.1042/0264-6021:3470571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens J.R., Sakamoto N., Sullivan S.A., Grobaski T.D., Tamkun M.M. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. J. Biol. Chem. 2001;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- McPhee I., Yarwood S.J., Scotland G., Huston E., Beard M.B., Ross A.H., Houslay E.S., Houslay M.D. Association with the SRC family tyrosyl kinase LYN triggers a conformational change in the catalytic region of human cAMP-specific phosphodiesterase HSPDE4A4B. J. Biol. Chem. 1999;274:11796–11810. doi: 10.1074/jbc.274.17.11796. [DOI] [PubMed] [Google Scholar]
- Molday R.S. Photoreceptor membrane proteins, phototransduction, and retinal degenerative diseasesthe Friedenwald lecture. Invest. Opthalmol. Vis. Sci. 1998;39:2493–2513. [PubMed] [Google Scholar]
- Mollard P., Zhang Y., Rodman D., Cooper D.M.F. Limited accumulation of cyclic AMP underlies a modest vasoactive-intestinal-peptide-mediated increase in cytosolic [Ca2+] transients in GH3 pituitary cells. Biochem. J. 1992;284:637–640. doi: 10.1042/bj2840637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E., Augustine G.J. Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoz G., Prigent A.F., Moueqqit M., Fougier S., Macovschi O., Pacheco H. Selective inhibition of one of the cyclic AMP phosphodiesterases from rat brain by the neurotropic compound rolipram. Biochem. Pharmacol. 1985;15:2997–3000. doi: 10.1016/0006-2952(85)90029-2. [DOI] [PubMed] [Google Scholar]
- Nikonov S., Lamb T.D., Pugh E.N., Jr. The role of steady phosphodiesterase activity in the kinetics of the light-adapted salamander rod photoresponse. J. Gen. Physiol. 2000;116:795–824. doi: 10.1085/jgp.116.6.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes S.G., Martin W.J., Lisek C.A., Powis G. Incomplete hydrolysis of the calcium indicator precursor fura-2 pentaacetoxymethyl ester (fura-2 AM) by cells. Anal. Biochem. 1988;169:159–166. doi: 10.1016/0003-2697(88)90267-9. [DOI] [PubMed] [Google Scholar]
- Podzuweit T., Nennstiel P., Muller A. Isozyme selective inhibition of cGMP-stimulated cyclic nucleotide phosphodiesterases by erythro-9-(2-hydroxy-3-nonyl) adenine. Cell Signal. 1995;7:733–738. doi: 10.1016/0898-6568(95)00042-n. [DOI] [PubMed] [Google Scholar]
- Polans A., Baehr W., Palczewski K. Turned on by Ca2+! The physiology and pathology of Ca2+-binding proteins in the retina. Trends Neurosci. 1996;19:547–554. doi: 10.1016/s0166-2236(96)10059-x. [DOI] [PubMed] [Google Scholar]
- Pugh E.N., Jr., Duda T., Sitaramayya A., Sharma R.K. Photoreceptor guanylate cyclasesa review. Biosci. Rep. 1997;17:429–473. doi: 10.1023/a:1027365520442. [DOI] [PubMed] [Google Scholar]
- Pugh E.N., Jr., Lamb T.D. Amplification and kinetics of the activation steps in phototransduction. Biochim. Biophys. Acta. 1993;1141:111–149. doi: 10.1016/0005-2728(93)90038-h. [DOI] [PubMed] [Google Scholar]
- Rich T.C., Fagan K.A., Nakata H., Schaack J., Cooper D.M.F., Karpen J.W. Cyclic nucleotide–gated channels colocalize with adenylyl cyclase in regions of restricted cAMP diffusion. J. Gen. Physiol. 2000;116:147–161. doi: 10.1085/jgp.116.2.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosman G.J., Martins T.J., Sonnenburg W.K., Beavo J.A., Ferguson K., Loughney K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene. 1997;191:89–95. doi: 10.1016/s0378-1119(97)00046-2. [DOI] [PubMed] [Google Scholar]
- Rybin V.O., Xu X., Lisanti M.P., Steinberg S.F. Differential targeting of β-adrenergic receptor subtypes and adenylyl cyclase to cardiomyocyte caveolae. J. Biol. Chem. 2000;275:41447–41457. doi: 10.1074/jbc.M006951200. [DOI] [PubMed] [Google Scholar]
- Schaack J., Langer S., Guo X. Efficient selection of recombinant adenoviruses by vectors that express beta-galactosidase. J. Virol. 1995;69:3920–3923. doi: 10.1128/jvi.69.6.3920-3923.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R., Zhou Z., Konnerth A., Neher E. Fractional contribution of calcium to the cation current through glutamate receptor channels. Neuron. 1993;11:133–143. doi: 10.1016/0896-6273(93)90277-x. [DOI] [PubMed] [Google Scholar]
- Sletholt K., Haug E., Gordeladze J., Sand O., Gautvik K.M. Effects of calmodulin antagonists on hormone release and cAMP levels in GH3 pituitary cells. Acta. Physiol. Scand. 1987;130:333–343. doi: 10.1111/j.1748-1716.1987.tb08145.x. [DOI] [PubMed] [Google Scholar]
- Soderling S.H., Bayuga S.J., Beavo J.A. Cloning and characterization of a cAMP specific cyclic nucleotide phosphodiesterase Proc. Natl. Acad. Sci. USA. 95 1998. 8991 8996a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling S.H., Bayuga S.J., Beavo J.A. Identification and characterization of a novel family of cyclic nucleotide phosphodiesterases J. Biol. Chem. 273 1998. 15553 15558b [DOI] [PubMed] [Google Scholar]
- Soderling S.H., Bayuga S.J., Beavo J.A. Isolation and characterization of a dual-substrate phosphodiesterase gene familyPDE10. Proc. Natl. Acad. Sci. USA. 1999;96:7071–7076. doi: 10.1073/pnas.96.12.7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L. Visual excitation and recovery. J. Biol. Chem. 1991;266:10711–10714. [PubMed] [Google Scholar]
- Tsang S.H., Burns M.E., Calvert P.D., Gouras P., Baylor D.A., Goff S.P., Arshavsky V.Y. Role for the target enzyme in deactivation of photoreceptor G protein in vivo. Science. 1998;282:117–121. doi: 10.1126/science.282.5386.117. [DOI] [PubMed] [Google Scholar]
- Varnum M.D., Black K.D., Zagotta W.N. Molecular mechanism for ligand discrimination of cyclic nucleotide-gated channels. Neuron. 1995;15:619–625. doi: 10.1016/0896-6273(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Whalin M.E., Scammell J.G., Strada S.J., Thompson W.J. Phosphodiesterase II, the cGMP-activatable cyclic nucleotide phosphodiesterase, regulates cyclic AMP metabolism in PC12 cells. Mol. Pharmacol. 1991;39:711–717. [PubMed] [Google Scholar]
- Yarfitz S., Hurley J.B. Transduction mechanisms of vertebrate and invertebrate photoreceptors. J. Biol. Chem. 1994;269:14329–14332. [PubMed] [Google Scholar]
- Yau K.-W. Phototransduction mechanism in retinal rods and conesThe Friedenwald Lecture. Invest. Ophthalmol. Vis. Sci. 1994;35:9–32. [PubMed] [Google Scholar]
- Zolle O., Lawrie A.M., Simpson A.W.M. Activation of the particulate and not the soluble guanylate cyclase leads to the inhibition of Ca2+ extrusion through localized elevation of cGMP. J. Biol. Chem. 2000;275:25892–25899. doi: 10.1074/jbc.M000786200. [DOI] [PubMed] [Google Scholar]
- Zong X., Zucker H., Hofmann F., Biel M. Three amino acids in the C-linker are major determinants of gating in cyclic nucleotide-gated channels. EMBO J. 1998;17:353–362. doi: 10.1093/emboj/17.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]