

Abstract
A recombinant prodrug, single-chain urokinase-type plasminogen activator (scuPA) fused to an anti–PECAM-1 antibody single-chain variable fragment (anti–PECAM scFv/scuPA) targets endothelium and augments thrombolysis in the pulmonary vasculature.1 To avoid premature activation and inactivation and to limit systemic toxicity, we replaced the native plasmin activation site in scFv/low-molecular-weight (lmw)–scuPA with a thrombin activation site, generating anti–PECAM scFv/uPA-T that (1) is latent and activated by thrombin instead of plasmin; (2) binds to PECAM-1; (3) does not consume plasma fibrinogen; (4) accumulates in mouse lungs after intravenous injection; and (5) resists PA inhibitor PAI-1 until activated by thrombin. In mouse models of pulmonary thrombosis caused by thromboplastin and ischemia-reperfusion (I/R), scFv/uPA-T provided more potent thromboprophylaxis and greater lung protection than plasmin-sensitive scFv/uPA. Endothelium-targeted thromboprophylaxis triggered by a prothrombotic enzyme illustrates a novel approach to time- and site-specific regulation of proteolytic reactions that can be modulated for therapeutic benefit.
Introduction
Plasminogen activators (PAs) used in the treatment of thrombosis convert the zymogen plasminogen into plasmin, which lyses fibrin and restores perfusion.2–4 Unfortunately, the utility of PAs is limited by inadequate delivery (rapid clearance, inactivation, and ineffective penetration of occlusive clots) and side effects, including hemorrhage and extravascular toxicity.5–7 Further, postevent thrombolytic therapy is marred by inevitable delays (time needed for diagnosis, transportation, injection, and lysis) causing ischemia-reperfusion (I/R) injury that perpetuates thrombosis and worsens outcome.8,9 Attempts to improve the effectiveness of PAs by increasing clot affinity have further diminished permeation into occluding thrombi due to retention on the clot surface5 and have yet to provide decisively better clinical outcomes.10–14
Thrombi often recur due to underlying procoagulation states, vascular damage, disturbance in blood flow, inflammation, or/and patients' immobility.15 Rethromboses often occur within hours to days after acute myocardial infarction, transient ischemic attack, ischemic stroke, and pulmonary embolism. The effectiveness of secondary prevention using existing anticoagulants and antiplatelet agents is limited and dosing is constrained by the risk of bleeding.16
In theory, prophylactic delivery of a PA into nascent thrombi, which are more susceptible to dissolution than mature occlusive clots, would expedite thrombolysis and minimize ischemia-reperfusion injury in patients at high risk of imminent thrombosis. Unfortunately, PAs are not suitable for thromboprophylaxis due to unfavorable pharmacokinetics and toxicity.
Conceivably, the failure to exploit PAs for thromboprophylaxis could be overcome by optimization of drug and targeting strategies. Single-chain urokinase-type plasminogen activator (scuPA) seems suitable for this purpose. It has minimal intrinsic enzymatic activity, but cleavage by plasmin at Lys158-Ile159 yields fully active 2-chain uPA (tcuPA). Low-molecular-weight scuPA (lmw-scuPA) is preferable for this purpose because it lacks the growth factor domain that mediates binding to uPA's cognate receptor (uPAR, CD87) on vascular and hematopoietic cells that may cause potentially harmful side effects.17 However, all forms of uPA pose the danger of causing hemorrhage by lysing hemostatic clots when injected at the high concentrations needed to overcome their rapid clearance and lack of affinity to vascular determinants. These limitations might be circumvented through the use of more targeted approaches to thromboprophylaxis.
We have shown that therapeutic enzymes can be targeted to the vascular surface of the endothelium1,18,19 (eg, using antibody to platelet-endothelial cell adhesion molecule-1 ([PECAM-1]), which is stably expressed at high density on the luminal surface of endothelium20 and does not mediate internalization of PECAM antibodies.21,22 We hypothesized that injection of an anti–PECAM single-chain variable fragment (scFv, avoiding PECAM cross-linking and Fc-fragment–mediated side effects) fused with a PA zymogen would lead to a high concentration of active enzyme formed at sites of clotting. In support of this concept, we recently synthesized an anti–PECAM scFv/lmw-scuPA (scFv/uPA) protein that was activated by plasmin and found that it accumulated preferentially in the lungs after intravenous injection in mice and augmented fibrinolytic potency in the pulmonary vasculature.1
However, both the full-length and lmw-scuPA generate plasmin over time, which in turn rapidly converts it to fully active tcuPA with the potential to cause indiscriminate activation of plasminogen and fibrinogen consumption in the circulation, predisposing to hemorrhage. Further, both forms of tcuPA are rapidly and irreversibly inhibited by plasminogen activator inhibitor-1 (PAI-1)23 and inactivated by thrombin cleavage at Arg156-Phe157, compromising activity at sites of thrombosis, the intended target area.24
To circumvent these problems and to design a targeted fibrinolytic for durable, safe, and locally controlled thromboprophylaxis, we generated a novel fusion protein composed of a plasmin-resistant lmw-scuPA that can be activated by thrombin (lmw-scuPA-T)25 linked to anti–PECAM scFv (ie, anti–PECAM scFv/lmw-scuPA-T, designated scFv/uPA-T for simplicity). We hypothesized that scFv/uPA-T would not be activated by plasmin, minimizing systemic plasminogen activation and premature inactivation, but would be tethered to endothelium and be activated by thrombin generated at the sites of thrombosis. Further, the scFv moiety contains an intrinsic thrombin-sensitive cleavage site, providing a built-in mechanism for local drug release into nascent clots.
Methods
Materials
All chemicals were obtained from Sigma-Aldrich (St Louis, MO) unless specified otherwise. QuickChange Site-Directed Mutagenesis kit was from Stratagene (La Jolla, CA). Bovine thrombin and horseradish peroxidase–coupled anti–mouse IgG were from Amersham Biosciences (Piscataway, NJ). Human leukocyte elastase, tryptase, chymase, anti-FLAG M2 affinity gel, mouse fibrinogen, and hirudin were from Sigma-Aldrich. lmw-tcuPA, activated protein C (APC), Spectrozyme UK chromogenic substrate, human Glu-plasminogen, Spectrozyme PL chromogenic substrate, monoclonal antibodies 394 against human uPA B-chain, 350 anti–fibrin β-chain, and PAI-1 were from American Diagnostica (Stamford, CT). TMB substrate was from Pierce (Rockford, IL). Biotinylated and nonbiotinylated rat anti–mouse fibrinogen antibodies were from Pharmingen (San Diego, CA). Dade Thromboplastin (rabbit brain) Plus C was from Fisher Scientific (Hampton, NH). Zymogram developing buffer and Simplyblue Safestain were from Invitrogen (Carlsbad, CA).
Construction and expression of scFv/uPA-T
Mutagenesis was performed according to the manufacturer's protocol. The 2 oligomers used were UKTsen: 5′-GTG GCC AAA AGA CTC TGA GGC CCC GCA TTA TTG GGG GAG AAT TCA CCA CCA TC-3′ and UKTrev, 5′-GAT GGT GGT GAA TTC TCC CCC AAT AAT GCG GGG CCT CAG AGT CTT TTG GCC AC-3′, which correspond to the DNA sequence encoding amino acids Cys148 to Ile167, except for the deletion of 6 nucleotides encoding amino acids Phe157 and Lys158. Production of the mutagenesis template scFv/uPA in the pMT/Bip vector, generation of the stable drosophila cell line expressing the fusion protein, and purification of the fusion protein scFv/uPA-T and lmw-scuPA were described previously.1
Biochemical characterization and enzymatic activity of fusion protein
The size and homogeneity of purified scFv/uPA-T was analyzed using 12% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) with or without addition of thrombin (150 nM). Conversion to its 2-chain derivative was determined in the presence of 50 mM dithiothreitol (DTT).
scFv/uPA-T or scuPA was incubated with various concentrations of thrombin, plasmin, or other indicated serine proteases, for 1 hour at 37°C. Thrombin was inactivated by heating at 100°C for 30 minutes to distinguish between the requirement for its catalytic and noncatalytic activities. The resultant amidolytic activity was assayed in activity buffer (50 mM Tris, pH 7.5, 30 KIU/mL aprotinin, and 33 U/mL hirudin) as described.1 To detect protease activity, 0.25 ng of nonreduced or reduced fusion protein or thrombin-treated protein was separated on 10% SDS-PAGE containing 1% nonfat milk and 20 μg/mL plasminogen and renatured by adding 2.5% Triton X-100 in PBS for 30 minutes. The gel was transferred to zymogram developing buffer and stained with Simplyblue Safestain. Fibrinolysis was measured using a plate assay as described previously.1
Enzyme inhibitor assay
Formation of SDS-stable complexes between scFv/uPA-T and PAI-1 was assessed by Western blot with an anti-uPA monoclonal antibody. Native or thrombin-treated scFv/uPA-T (10 ng) was incubated with PAI-1 at different molar ratios for 30 minutes at 37°C and the proteins were allowed to migrate on a 4% to 12% gradient SDS-PAGE. The proteins were then transferred to a nitrocellulose membrane subsequently blocked with 5% nonfat milk. Immunoblotting with anti-uPA was performed as described previously.1
PECAM-binding ability of scFv/uPA-T
cDNA encoding the extracellular domain of mouse PECAM-1 (amino acids Glu18-Lys590) was obtained by the reverse-transcription–polymerized chain reaction (RT-PCR) using mouse lung tissue total RNA. A FLAG affinity tag was fused to the N-terminus and subcloned into the BglII and NotI sites in the pMT/Bip vector. Generation of stable cell lines was performed as described.1 Soluble mouse PECAM was purified from secreted supernatant using M2 anti-FLAG affinity chromatography. Specific binding of scFv/uPA-T to soluble PECAM was measured by enzyme-linked immunosorbent assay (ELISA). Each well within a 96-well plate was coated with 5 μg/mL soluble mouse PECAM protein overnight, the unbound sites were blocked with PBS containing 5% bovine serum albumin (BSA) at 37°C for 1 hour, and various dilutions of scFv/uPA-T or wild-type lmw-scuPA were added for 1 hour. Unbound protein was removed by washing with PBS and 1 μg/mL anti-uPA monoclonal antibody in PBS containing 1% BSA was added. The wells were washed, and horseradish peroxidase–conjugated anti–mouse IgG was added. After the wells were washed again, TMB substrate was added and the optical density at 450 nm (OD450) was measured. A competition ELISA was used to demonstrate the specificity of binding. Purified scFv/uPA-T (10 μg/mL) mixed with various amounts of parental anti–PECAM IgG was incubated with the mouse PECAM-coated wells and the ELISA was performed as described in this paragraph.
Thrombin-mediated release of PECAM-bound fusion protein
Purified scFv/uPA-T (25 μg/mL) was added to each well of a mouse PECAM-coated 96-well plate for 2 hours at 37°C and washed with PBS. Thrombin was added for 2 hours at 37°C, the wells were washed, and bound fusion protein was measured by ELISA, as above.
Enzymatic activity of PECAM-associated fusion protein
To measure the amidolytic activity of PECAM-associated scFv/uPA-T, various concentrations of the fusion protein were added to mouse PECAM-coated wells for 1 hour at 37°C, 30 nM thrombin was added, and the generated amidolytic activity was measured as above. To further determine the plasminogen activator activity of PECAM-associated scFv/uPA-T, purified scFv/uPA-T (25 μg/mL) was incubated with a PECAM-coated plate, activated with 50 nM thrombin for 2 hours at 37°C, and incubated with 2 μM human Glu-plasminogen in activity assay buffer (50 mM Tris, pH 7.5, 33 U/mL hirudin). To determine the amount of plasmin activity generated, Spectrozyme PL was added, and optical density at 405 nm (OD405) was measured and compared with a standard curve generated by known amounts of plasmin.
Fibrinogen depletion studies
Male C57BL/B6 mice, 6 to 10 weeks of age, were used throughout. All in vivo experiments were performed in accordance with National Institutes of Health guidelines and with the written approval of the University of Pennsylvania Animal Use committee. Various amounts of lmw-scuPA, scFv/uPA, and scFv/uPA-T were incubated in 0.25 mL citrated-pooled mouse plasma at 37°C for 3 hours, and the concentration of residual fibrinogen was measured.26 Briefly, each well in 96-well plates was coated with 2 μg/mL rat anti–mouse fibrinogen antibody, nonreactive sites were blocked with 5% PBS-BSA, and serially diluted mouse plasma was added for 1 hour at 37°C. Wells were incubated sequentially with 1 μg/mL biotinylated rat anti–mouse fibrinogen antibody, streptavidin-peroxidase, and TMB substrate, and the OD450 was measured. Fibrinogen concentration was determined by comparison with a standard curve based on purified mouse fibrinogen. The plasma concentration of fibrinogen in mice given an equal volume of saline was used as the baseline. To measure protein stability in vivo, 2 nmol lmw-scuPA, scFv/uPA, or scFv/uPA-T, or an equal volume (150 μL) of saline was injected intravenously. After 1 hour, 300 μL of whole blood was extracted from the jugular vein in citrate solution (0.32% final concentration).
In vivo biodistribution of scFv/uPA-T and lmw-scuPA
Biodistribution studies of 125I-labeled scFv/uPA-T and nontargeted scuPA were carried out in mice as previously described.1 Briefly, labeled proteins were injected via the jugular vein at the indicated time points, blood was drawn, and the mice were killed. Radioactivity in the organs was measured in a gamma-counter and data were calculated as the percentage of injected dose per gram of tissue (%ID/g).
Mouse model of pulmonary thrombosis induced by thromboplastin
Thrombin-mediated thrombosis was induced in mice by thromboplastin as described.27 To quantify fibrinolysis, fibrin/fibrinogen deposition in lungs was measured as described.28 Briefly, scFv/uPA-T (300 μg), an equal molar amount of lmw-scuPA or scFv/uPA were injected intravenously. Thirty minutes or 3 hours later, reconstituted thromboplastin (85 μL/kg) premixed with buffer containing 125I-fibrinogen (150 000 cpm) was injected through the jugular vein. Ninety minutes later, the mice were given heparin (400 U/mouse) and killed immediately, the lungs were harvested and washed, and tissue radioactivity was measured. Radioactivity in the lungs was corrected for the background level of 125I-fibrinogen within the pulmonary circulation at the time of killing. The amount of 125I-fibrinogen within the pulmonary circulation (cpmFg) was obtained from mice injected with the identical fibrinogen solution in the absence of thromboplastin. The residual lung radioactivity was normalized to tissue weight and the percentage of fibrinolysis was calculated using the formula: fibrinolysis (%) = (residual radioactivityPBS − residual radioactivitydrug)/(residual radioactivityPBS − residual radioactivityFg) × 100. Therefore, lower levels of residual radioactive fibrin(ogen) in the lungs correspond to more extensive fibrinolysis (Figure 6).
Figure 6.

Vascular-targeted scFv/uPA-T provides “on-demand” prophylactic fibrinolysis triggered by thrombin. Lysis of pulmonary clots initiated by injection of thromboplastin in mice 0.5 hour or 3 hours after a bolus injection of equal molar amounts (5 nmol) of scFv/uPA-T, scFv/uPA, and lmw-scuPA (n = 3–4; *P < .05, scFv/uPA-T vs lmw-scuPA; #P < .05, scFv/uPA-T vs scFv/uPA). Error bars represent SEM.
Mouse model of unilateral in situ lung ischemia-reperfusion
The protective effect of scFv/uPA-T was tested in a mouse model of lung ischemia-reperfusion. Thirty minutes prior to causing ischemia-reperfusion, 75 μg scFv/uPA-T or scFv/uPA or the same volume of PBS was injected intravenously. Unilateral left lung ischemia-reperfusion was created using a protocol previously described in detail.8,9 Briefly, after performing a thoracotomy in ventilated anesthetized mice, the hilum of the left lung was cross-clamped for 120 minutes followed by 150 minutes of reperfusion, while being ventilated with 95% O2. Five minutes prior to killing, 400 U heparin was injected intravenously to prevent postmortem clotting and the left lobe of the lung was excised. Sham-operated mice underwent the same procedure except that clamping of the hilum was omitted. Lung specimens were weighed and homogenized in PBS (pH 7.2) containing protease inhibitor cocktail (Sigma), aprotinin (30 KIU/mL), heparin 15 U/mL, and 100 mM 6-aminohexanoic acid (EACA). Aliquots of tissue homogenates were prepared for analysis. Lung fibrin was extracted from one aliquot, detected by Western blot using fibrin β-chain–specific antibody 350 (American Diagnostica), as described previously.29 Total lung proteins were extracted with 1% SDS and 1% Triton X-100 and subjected to Western blot using anti–β-actin polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Lung fibrin was quantified by densitometric analysis and normalized to β-actin. Antithrombotic effect was calculated by comparing lung fibrin deposition in the PA-treated and PBS-treated groups. Oxygen tension in the arterial blood was measured as previously described using I-Stat (Abbott Laboratories, Abbott Park, IL).22
Data analysis
All data are presented as the means plus or minus standard error of the mean of at least 3 separate experiments. Differences between groups were tested for statistical significance using Student t test or analysis of variance (ANOVA). Statistical significance was set at P less than .05.
Results
Generation of thrombin-activatable anti–PECAM scFv/uPA
We used cDNA encoding lmw-scuPA fused to anti–PECAM scFv1 as a template to generate scFv/uPA-T by deleting the plasmin cleavage site Phe157-Lys158, which creates the thrombin-cleavage site Pro155-Arg156-Ile157-Ile158 (Figure 1A). Purified scFv/uPA-T migrated on SDS-PAGE as a single band at the predicted molecular weight (∼ 59 kDa) under reduced conditions, excluding activation in vitro (Figure 1B). Analysis of the cDNA sequence revealed that anti–PECAM scFv contains a potential thrombin cleavage site, Pro232-Arg233-Ala234, predicted to yield protein fragments of approximately 30 kDa. Thus, thrombin cleaved the fusion protein within scFv, generating 2 proteins with distinct migrations under nonreduced conditions (Figure 1B). Under reduced conditions, the B-chain dissociated from thrombin-cleaved scFv/uPA-T leading to faster migration (Figure 1B).
Figure 1.

Molecular design and biochemical characterization of the scFv/uPA-T fusion protein. (A) Single-chain variable fragment (scFv) fused with thrombin-inducible lmw-scuPA (scFv/uPA-T) was generated by deleting Phe157 and Lys158 from the previously described construct, scFv/uPA. This converts the plasmin activation site Pro155-Arg156-Phe157-Lys158-Ile159-Ile160 (PRFKII) into the sequence Pro155-Arg156-Ile157-Ile158 (PRII), which is cleaved by thrombin after Arg156. (B) Migration of the purified fusion protein in the absence or presence of thrombin was analyzed using SDS-PAGE under nonreduced or reduced conditions.
Figure S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article) demonstrates that thrombin cleavage of scFv/scuPA evidently generates 2 bands with distinct molecular sizes using nonreducing SDS-PAGE. In our original paper describing scFv/scuPA, we had shown that plasmin cleavage of scFv/scuPA does not cause a band shift using nonreducing SDS-PAGE, but generates 2 bands that migrate with similar sizes under reduced conditions.1 This result shows that the thrombin-sensitive cleavage site is present in the scFv moiety of the prototype scFv/scuPA as well as in the scFv/scuPA-T described in this paper.
Enzymatic properties of scFv/uPA-T
Cleavage of scFv/uPA-T by thrombin, but not other serine proteases, including plasmin, activated protein C (APC), or heat-inactivated thrombin, generated amidolytic activity (Figure 2A). Conversely, wild-type scuPA was activated by plasmin but not by thrombin (data not shown). Native scFv/uPA-T does not have plasminogen activator activity on zymography, whereas its cleavage by thrombin generated an approximately 30-kDa fragment that activated plasminogen and lost this activity under reduced conditions, consistent with the expected requirement for disulfide bonding (Figure 2B). scFv/uPA-T did not lyse fibrin clots containing trace amounts of plasminogen, whereas thrombin-activated scFv/uPA-T expressed fibrinolytic activity (Figure 2C left columns) comparable with plasmin-generated lmw-tcuPA used as a positive control (Figure 2C middle column). The low intrinsic PA activity of lmw-scuPA was eliminated by thrombin, as expected (Figure 2C right columns). Native scFv/uPA-T did not bind PAI-1 even at 5-fold molar excess inhibitor (Figure 2D left lanes). However, thrombin generated an approximately 30-kDa 2-chain uPA fragment of scFv/uPA-T that expressed enzymatic activity (Figure 2B) and formed SDS-resistant complexes30 with the inhibitor detectable by a shift in the molecular weight of the lmw-tcuPA fragment to approximately 80 kDa (Figure 2D right lanes).
Figure 2.

Activation by thrombin and PAI-1 resistance of the scFv/uPA-T. (A) Specific activation of scFv/uPA-T by thrombin. scFv/uPA-T was incubated with various serine proteases for 1 hour. Thrombin (5 nM) generated amidolytic activity from scFv/uPA-T, whereas the other serine proteases or heat-inactivated thrombin did not, even when 40 nM enzyme was added. APC indicates activated protein C. Error bars represent SEM. (B) Activity of scFv/uPA-T measured by zymography before or after incubation with thrombin under reduced or nonreduced conditions. (C) Fibrinolytic activity. The indicated amounts of scFv/uPA-T, thrombin-treated scFv/uPA-T, lmw-tcuPA, lmw-scuPA, and thrombin-treated lmw-scuPA were incubated on a fibrin-coated plate at 37°C. Lytic zones were counterstained using impregnation of fibrin by trypan blue. (D) Susceptibility of scFv/uPA-T to PAI-1. Native scFv/uPA-T does not bind PAI-1. After addition of thrombin, a dose-dependent increase in lmw-tcuPA-PAI-1 complexes is evident.
Interaction of scFv/uPA-T with mouse PECAM-1
scFv/uPA-T, but not nontargeted lmw-scuPA, bound to the immobilized mouse PECAM (Figure 3A). Binding was inhibited by a PECAM monoclonal antibody (Figure 3B). Thrombin added to PECAM-coated plastic wells preincubated with scFv/uPA-T released uPA-T from PECAM-bound scFv/uPA-T (Figure 3C). Adding thrombin, to PECAM-coated plastic wells preincubated with scFv/uPA-T but not lmw-scuPA, generated amidolytic activity (Figure 3D), indicating that antigen-bound scFv/uPA-T maintains its susceptibility to activation by thrombin. Moreover, upon addition of thrombin, PECAM-associated scFv/uPA-T retained the ability to activate its in vivo substrate plasminogen to plasmin (Figure 3E). These results imply that the prothrombotic enzyme thrombin might both activate and liberate lmw-uPA at sites of thrombosis in vivo and initiate “on-demand” thrombolysis.
Figure 3.

Mouse PECAM-1 binding properties of scFv/uPA-T. (A) ELISA: binding of anti–PECAMscFv/uPA-T and free lmw-scuPA to immobilized soluble mouse PECAM. (B) Inhibition of binding of fusion protein to soluble mouse PECAM by parental anti–PECAM IgG. (C) Thrombin-mediated release of the lmw-uPA-T moiety from PECAM-bound scFv/uPA-T. scFv/uPA-T was incubated with PECAM-immobilized wells and treated with thrombin as indicated in “Thrombin-mediated release of PECAM-bound fusion protein.” Error bars indicate standard error of the mean (SEM). (D) Amidolytic activity of scFv/uPA-T and lmw-scuPA bound to immobilized mouse PECAM after addition of thrombin. (E) Activation of plasminogen to plasmin by PECAM-associated scFv/uPA-T upon addition of thrombin. Error bars represent SEM.
scFv/uPA-T is a latent prodrug that does not cause consumption of fibrinogen in vitro and in vivo
We then asked whether scFv/uPA-T caused less indiscriminate systemic plasminogen activation than wild-type lmw-uPA. To exclude pharmacokinetic factors, we first assessed fibrinogen consumption in vitro. Both lmw-scuPA and the prototype plasmin-sensitive scFv/uPA fusion construct (0.3 and 1 μM) depleted fibrinogen levels in mouse plasma to approximately 50% and 20% of normal, respectively. In contrast, fibrinogen levels remained more than 80% of normal after incubation of plasma with scFv/uPA-T (Figure 4A). Further, 1 hour after injection of 120 μg (2 nmol) scFv/uPA-T in vivo, the concentration of fibrinogen in mouse blood was unaffected, while the same molar amounts of lmw-scuPA and scFv/uPA caused approximately 15% depletion, indicating activation of circulating plasminogen (P < .05; Figure 4B).
Figure 4.

Plasmin-sensitive lmw-scuPA and scFv/uPA, but not scFv/uPA-T, deplete fibrinogen from mouse plasma. (A) Concentration of fibrinogen in pooled mouse plasma treated with lmw-scuPA, scFv/uPA or scFv/uPA-T for 3 hours. (B) Concentration of fibrinogen in plasma of mice injected with scFv/uPA-T (120 μg) or the same dose of lmw-scuPA and scFv/uPA. scFv/uPA-T–injected mice showed intact plasma fibrinogen level, in contrast to lmw-scuPA– or scFv/uPA-treated mice (*P < .05, vs lmw-scuPA cohort; #P < .05, vs scFv/uPA cohort). Error bars represent SEM.
In vivo biodistribution of scFv/uPA-T
scFv/uPA-T accumulated in the lungs after intravenous injection in mice, whereas lmw-scuPA did not (Figure 5). This result is consistent with previous data showing that PECAM-targeted drugs accumulate in the pulmonary vasculature as a result of binding to endothelial PECAM in this highly vascularized organ.1,21,22 Three hours after injection, the amount of scFv/uPA-T in the lungs was approximately 5-fold higher than of nontargeted uPA (Figure 5B). Both scFv/uPA-T and wild-type lmw-scuPA disappeared rapidly from the circulation (4% and 6% of the injected dose/gram blood at 1 hour after injection, respectively, Figure 5A). The amount of scFv/uPA-T in the blood was lower than that of lmw-scuPA throughout, consistent with more rapid depletion due to endothelial binding.
Figure 5.

Endothelial targeting of scFv/uPA-T. In vivo biodistribution of scFv/uPA-T versus wild-type lmw-scuPA 1 hour (A) or 3 hours (B) after intravenous injection in mice. The data are shown as the percentage (±SEM) of the injected dose per gram of tissue (%ID/g); n = 3.
Thromboprophylaxis against thromboplastin-induced pulmonary thrombosis in mice: the effect of scFv/uPA-T is more durable than scFv/uPA
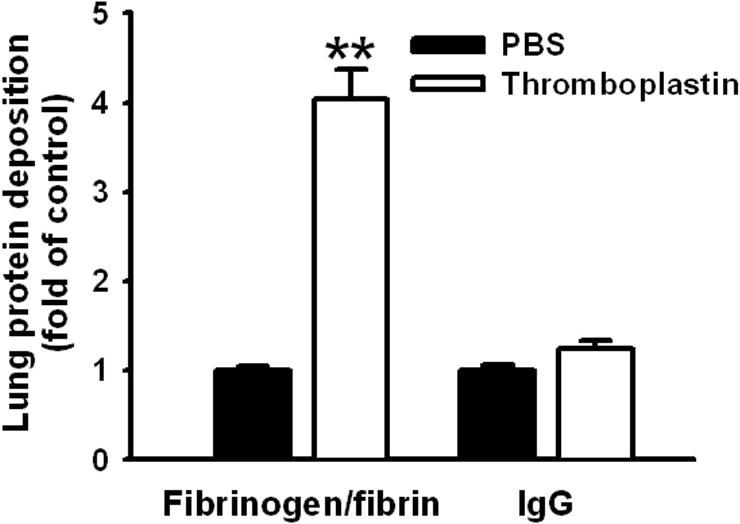
We first tested thromboprophylaxis in vivo in a mouse model of thrombosis caused by intravenous injection of thromboplastin. To detect formation, deposition, and lysis of thrombi in the pulmonary vasculature, we injected a 125I-fibrinogen/thromboplastin mixture intravenously. In agreement with previous reports, thromboplastin markedly increased the pulmonary radioactivity in control animals by generating thrombin.27,31 Figure S2 shows that injection of thromboplastin did not increase IgG deposition in mouse lungs, whereas pulmonary deposition of radiolabeled fibrinogen was increased approximately 4-fold (from 5% of injected radioactivity/gram tissue to 22% of injected radioactivity/gram tissue, P < .01). These results are consistent with another tissue factor–mediated thrombotic model.28 Since the molecular size of fibrinogen (340 kDa) is larger than IgG (160 kDa), these results indicate that extravasation of plasma proteins contributes very little to the increased lung fibrinogen deposition. Rather, the accumulation of radiolabeled fibrinogen results from thrombosis.
Pretreatment of the animals with free lmw-scuPA, either 30 minutes or 3 hours prior to injecting the 125fibrinogen/thromboplastin mixture, did not have a significant effect on the deposition of radiolabeled fibrin(ogen) in the lungs (P > .3; Figure 6). Pretreatment with the prototype plasmin-sensitive scFv/uPA 30 minutes prior to challenge led to a significant thrombolysis, but when the interval was extended to 3 hours, only 50% of the initial fibrinolytic effect was retained. By comparison, scFv/uPA-T injected 30 minutes prior to 125fibrinogen/thromboplastin augmented pulmonary thrombolysis relative to scFv/uPA. Further, even when the interval between pretreatment and challenge was extended to 3 hours, approximately 80% of the fibrinolytic activity expressed at 30 minutes was retained (Figure 6).
scFv/uPA-T protects against ischemia-reperfusion–induced lung thrombosis and injury
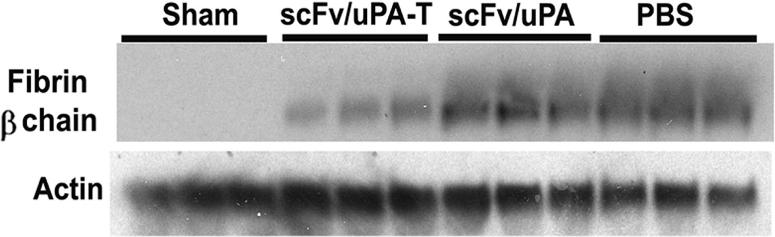
Next, we compared the effects of scFv/uPA and scFv/uPA-T in a mouse model of in situ unilateral pulmonary I/R. In agreement with previous reports,8,9 we observed marked accumulation of fibrin in the ipsilateral lung after I/R, which was significantly and more profoundly attenuated by pretreatment with scFv/uPA-T than with scFv/uPA (Figure 7A; Figure S4). Further, scFv/uPA-T, but not scFv/uPA, caused a statistically significant improvement of arterial blood oxygenation after lung I/R, reaching levels close to normal (Figure 7B).
Figure 7.

Thrombin-mediated thrombolysis prevents ischemia-reperfusion (I/R)–induced lung injury. (A) Effects of scFv/uPA-T and scFv/uPA on lung fibrin deposition induced by I/R. Thirty minutes before inducing lung I/R, mice were given an intravenous injection of 1.25 nmol scFv/uPA or scFv/uPA-T, or the same volume of PBS. Antifibrin β-chain and anti–β-actin Western blots were performed using lung fibrin extracts and total protein extracts from the same animal, respectively. The amount of fibrin in the lung was measured by densitometric scan and normalized to the amount of β-actin. The bar graph shows quantification of fibrin in each group. Fibrin deposition in PBS-treated animals was designated as 100% (n = 5–6). *P < .05; **P < .01. (B) scFv/uPA-T improves lung gas exchange. Arterial blood oxygen tension in the various cohorts was compared (n = 3–6). The dashed line represents the paO2 in sham-operated animals. *P < .05. Error bars represent SEM.
Discussion
Local generation of thrombin plays a key role in initiating and propagating thrombosis.32 Prophylactic targeting of releasable fibrinolytic agents to the endothelial lumen may inhibit clot growth and permit lysis before irreversible vascular occlusion impedes delivery. Indeed, uPA targeting to PECAM-1 prevents clot accretion in a model of acute pulmonary embolism1 and cerebrovascular thrombosis.33 However, for use as thromboprophylaxis, endothelium-targeted PAs must resist premature inactivation by inhibitors and be activated preferentially at sites of incipient thrombosis.
To meet these criteria, we fused an anti–PECAM scFv to a thrombin-activated uPA mutant (lmw-scuPA-T).25 We reasoned that therapeutic lysis by a circulating thrombin-inducible PA would still be limited by blood clearance and would require activation by the limited amount of thrombin in the circulation, where both enzymes are rapidly inactivated, or within the clot itself, which is difficult to permeate.5 In contrast, PECAM-targeted thrombin-inducible PA seemed especially well suited for use as thromboprophylaxis. scFv/uPA-T prebound to the vascular lumen is positioned to be activated locally when and where thrombin is generated and then released from PECAM-1 into the porous nascent fibrin mesh, which remains susceptible to lysis.
The results of this study indicate that scFv/uPA-T (1) is resistant to plasmin, but is activated by thrombin (Figure 2); (2) is targeted to PECAM-1 and released from the antigen-binding portion of the protein by thrombin (Figure 3); (3) does not consume plasma fibrinogen (Figure 4); (4) accumulates in the lung in vivo (Figure 5); and (5) facilitates dissolution of nascent clots in vivo for hours after delivery resulting in improvement of lung function (Figures 6,7). The latent character of scFv/uPA-T translates into enhanced resistance to PAI-1 (Figure 2) and less systemic fibrin(ogen) lysis (Figure 4), which helps prevent premature inactivation and provides more durable prophylaxis than plasmin-sensitive scFv/uPA (Figures 6,7). Subsequent inactivation of thrombin-generated tcuPA by PAI-1 provides a mechanism for eventual down-regulation once thrombin generation has abated. The fact that scFv/uPA-T is not sensitive to plasmin, but depends on thrombin for activation, also suggests this prodrug is less likely to attack stable mural hemostatic clots, which generate much less thrombin than newly formed clots that cause tissue ischemia.
We have shown by electron and confocal fluorescent microscopy that PECAM antibodies and conjugates injected intravascularly in animals bind to endothelium, not other cell types in the pulmonary vasculature.34 However, PECAM-1 targeting is not lung specific; in fact, local arterial infusion of anti-PECAM conjugates and fusion proteins via the coronary or carotid artery permits enhanced drug delivery to their respective organs.33,34 These observations extend the potential utility of the proposed approach to diverse vascular beds. Nevertheless, the pulmonary vasculature collects the approximately 30% of injected dose after systemic intra-arterial or intravenous injection, consistent with its contribution to the total endothelial surface of the body; the lungs receive 50% of the total cardiac output and the slow rate of perfusion to the high-capacity, low-resistance pulmonary vasculature further promotes binding of PECAM targeted compounds.21,22,35
This study shows that it is possible to convert the prothrombotic enzyme thrombin into a functioning prothrombolytic agent in vivo. This approach is more likely to have a role in the management and prevention of ischemia-reperfusion and recurrent thrombosis rather than in postevent therapy of acute thrombosis. Clinical settings potentially amenable to thromboprophylaxis include postsurgical (re)thromboses in immobilized patients, pulmonary thrombosis in acute lung injury, ischemia-reperfusion, and posttransplantation thrombosis,9 among others. Additional studies involving models simulating recurrent arterial and venous thrombosis will be needed to assess the benefits and limitations of increased PA localization and activation suggested by this approach. Conceivably, this approach can be generalized to target other classes of therapeutic molecules (eg, antiadhesives, antiproteases) to the surface of diverse vascular or hematopoietic cells (eg, leukocytes), where they will be activated by enzymes released in high concentrations by those specific cell types (eg, elastase). Thus, this study provides a paradigm for local drug delivery/activation while lowering systemic toxicity that might prove applicable in the treatment of diverse disorders in which the local enzymatic mediators of pathological process have been identified.
Supplementary Material
Acknowledgments
The authors are grateful to Ms Alice Kuo and Dr Bdeir for advice in experimental precedures, Dr Shuraev for radiolabeling proteins, and Ms Evguenia Arguiri for technical help with animal experiments.
This work was supported by HL71175, HL071174, HL079063, DOD PR 012262, and a grant from the University of Pennsylvania Research Foundation (V.R.M.), and HL076406, HL076206, CA83121, and a grant from the Research Management Group (D.B.C.). B.-S.D. is a recipient of a Predoctoral Fellowship from the American Heart Association. J.-C.M. is a recipient of Ramón y Cajal Foundation Award and grant from Fondo de Investigaciones Sanitarias PI040961. CNIC is supported by the Spanish Ministry of Health and Consumer Affairs and the Pro-CNIC Foundation.
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: B.-S.D., D.B.C., and V.R.M. designed the study; B.-S.D., N.H., J.-C.M., and K.G. performed the experiments; B.-S.D., N.H., J.-C.M., K.G., C.G., M.C.-S., A.B.F., S.M.A., D.B.C., and V.R.M analyzed the data; and B.-S.D., D.B.C., and V.R.M. wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Vladimir R. Muzykantov, IFEM, 1 John Morgan Bldg, University of Pennsylvania Medical Center, 3620 Hamilton Walk, Philadelphia, PA, 19104-6068; e-mail: muzykant@mail.med.upenn.edu.
References
- 1.Ding BS, Gottstein C, Grunow A, et al. Endothelial targeting of a recombinant construct fusing a PECAM-1 single-chain variable antibody fragment (scFv) with prourokinase facilitates prophylactic thrombolysis in the pulmonary vasculature. Blood. 2005;106:4191–4198. doi: 10.1182/blood-2005-05-2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topol EJ, Morris DC, Smalling RW, et al. A multicenter, randomized, placebo-controlled trial of a new form of intravenous recombinant tissue-type plasminogen activator (activase) in acute myocardial infarction. J Am Coll Cardiol. 1987;9:1205–1213. doi: 10.1016/s0735-1097(87)80457-6. [DOI] [PubMed] [Google Scholar]
- 3.Benedict CR, Refino CJ, Keyt BA, et al. New variant of human tissue plasminogen activator (TPA) with enhanced efficacy and lower incidence of bleeding compared with recombinant human TPA. Circulation. 1995;92:3032–3040. doi: 10.1161/01.cir.92.10.3032. [DOI] [PubMed] [Google Scholar]
- 4.Marler JR, Goldstein LB. Medicine: stroke–tPA and the clinic. Science. 2003;301:1677. doi: 10.1126/science.1090270. [DOI] [PubMed] [Google Scholar]
- 5.Sakharov DV, Rijken DC. Superficial accumulation of plasminogen during plasma clot lysis. Circulation. 1995;92:1883–1890. doi: 10.1161/01.cir.92.7.1883. [DOI] [PubMed] [Google Scholar]
- 6.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 7.Zhu Y, Carmeliet P, Fay WP. Plasminogen activator inhibitor-1 is a major determinant of arterial thrombolysis resistance. Circulation. 1999;99:3050–3055. doi: 10.1161/01.cir.99.23.3050. [DOI] [PubMed] [Google Scholar]
- 8.Okada K, Fujita T, Minamoto K, Liao H, Naka Y, Pinsky DJ. Potentiation of endogenous fibrinolysis and rescue from lung ischemia/reperfusion injury in interleukin (IL)-10-reconstituted IL-10 null mice. J Biol Chem. 2000;275:21468–21476. doi: 10.1074/jbc.M002682200. [DOI] [PubMed] [Google Scholar]
- 9.Fujita T, Toda K, Karimova A, et al. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med. 2001;7:598–604. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- 10.Fujise K, Revelle BM, Stacy L, et al. A tissue plasminogen activator/P-selectin fusion protein is an effective thrombolytic agent. Circulation. 1997;95:715–722. doi: 10.1161/01.cir.95.3.715. [DOI] [PubMed] [Google Scholar]
- 11.Hagemeyer CE, Tomic I, Weirich U, et al. Construction and characterization of a recombinant plasminogen activator composed of an anti-fibrin single-chain antibody and low-molecular-weight urokinase. J Thromb Haemost. 2004;2:797–803. doi: 10.1111/j.1538-7836.2004.00697.x. [DOI] [PubMed] [Google Scholar]
- 12.Holvoet P, Laroche Y, Lijnen HR, et al. Characterization of a chimeric plasminogen activator consisting of a single-chain Fv fragment derived from a fibrin fragment D-dimer-specific antibody and a truncated single-chain urokinase. J Biol Chem. 1991;266:19717–19724. [PubMed] [Google Scholar]
- 13.Runge MS, Quertermous T, Zavodny PJ, et al. A recombinant chimeric plasminogen activator with high affinity for fibrin has increased thrombolytic potency in vitro and in vivo. Proc Natl Acad Sci U S A. 1991;88:10337–10341. doi: 10.1073/pnas.88.22.10337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnee JM, Runge MS, Matsueda GR, et al. Construction and expression of a recombinant antibody-targeted plasminogen activator. Proc Natl Acad Sci U S A. 1987;84:6904–6908. doi: 10.1073/pnas.84.19.6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenberg RD, Aird WC. Vascular-bed–specific hemostasis and hypercoagulable states. N Engl J Med. 1999;340:1555–1564. doi: 10.1056/NEJM199905203402007. [DOI] [PubMed] [Google Scholar]
- 16.Topol EJ, Byzova TV, Plow EF. Platelet GPIIb-IIIa blockers. Lancet. 1999;353:227–231. doi: 10.1016/S0140-6736(98)11086-3. [DOI] [PubMed] [Google Scholar]
- 17.Blasi F, Carmeliet P. uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol. 2002;3:932–943. doi: 10.1038/nrm977. [DOI] [PubMed] [Google Scholar]
- 18.Muzykantov VR, Barnathan ES, Atochina EN, Kuo A, Danilov SM, Fisher AB. Targeting of antibody-conjugated plasminogen activators to the pulmonary vasculature. J Pharmacol Exp Ther. 1996;279:1026–1034. [PubMed] [Google Scholar]
- 19.Murciano JC, Muro S, Koniaris L, et al. ICAM-directed vascular immunotargeting of antithrombotic agents to the endothelial luminal surface. Blood. 2003;101:3977–3984. doi: 10.1182/blood-2002-09-2853. [DOI] [PubMed] [Google Scholar]
- 20.Newman PJ. The biology of PECAM-1. J Clin Invest. 1997;100:S25–S29. [PubMed] [Google Scholar]
- 21.Muzykantov VR, Christofidou-Solomidou M, Balyasnikova I, et al. Streptavidin facilitates internalization and pulmonary targeting of an anti-endothelial cell antibody (platelet-endothelial cell adhesion molecule 1): a strategy for vascular immunotargeting of drugs. Proc Natl Acad Sci U S A. 1999;96:2379–2384. doi: 10.1073/pnas.96.5.2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozower BD, Christofidou-Solomidou M, Sweitzer TD, et al. Immunotargeting of catalase to the pulmonary endothelium alleviates oxidative stress and reduces acute lung transplantation injury. Nat Biotechnol. 2003;21:392–398. doi: 10.1038/nbt806. [DOI] [PubMed] [Google Scholar]
- 23.Eitzman DT, McCoy RD, Zheng X, et al. Bleomycin-induced pulmonary fibrosis in transgenic mice that either lack or overexpress the murine plasminogen activator inhibitor-1 gene. J Clin Invest. 1996;97:232–237. doi: 10.1172/JCI118396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichinose A, Fujikawa K, Suyama T. The activation of pro-urokinase by plasma kallikrein and its inactivation by thrombin. J Biol Chem. 1986;261:3486–3489. [PubMed] [Google Scholar]
- 25.Yang WP, Goldstein J, Procyk R, Matsueda GR, Shaw SY. Design and evaluation of a thrombin-activable plasminogen activator. Biochemistry. 1994;33:606–612. doi: 10.1021/bi00174a043. [DOI] [PubMed] [Google Scholar]
- 26.Gulledge AA, Rezaee F, Verheijen JH, Lord ST. A novel transgenic mouse model of hyperfibrinogenemia. Thromb Haemost. 2001;86:511–516. [PubMed] [Google Scholar]
- 27.Leon C, Freund M, Ravanat C, Baurand A, Cazenave JP, Gachet C. Key role of the P2Y(1) receptor in tissue factor-induced thrombin-dependent acute thromboembolism: studies in P2Y(1)-knockout mice and mice treated with a P2Y(1) antagonist. Circulation. 2001;103:718–723. doi: 10.1161/01.cir.103.5.718. [DOI] [PubMed] [Google Scholar]
- 28.Lawson CA, Yan SD, Yan SF, et al. Monocytes and tissue factor promote thrombosis in a murine model of oxygen deprivation. J Clin Invest. 1997;99:1729–1738. doi: 10.1172/JCI119337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson LL, Berggren KN, Szaba FM, Chen W, Smiley ST. Fibrin-mediated protection against infection-stimulated immunopathology. J Exp Med. 2003;197:801–806. doi: 10.1084/jem.20021493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Manchanda N, Schwartz BS. Interaction of single-chain urokinase and plasminogen activator inhibitor type 1. J Biol Chem. 1995;270:20032–20035. doi: 10.1074/jbc.270.34.20032. [DOI] [PubMed] [Google Scholar]
- 31.Smyth SS, Reis ED, Vaananen H, Zhang W, Coller BS. Variable protection of beta 3-integrin–deficient mice from thrombosis initiated by different mechanisms. Blood. 2001;98:1055–1062. doi: 10.1182/blood.v98.4.1055. [DOI] [PubMed] [Google Scholar]
- 32.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8:1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 33.Danielyan K, Ding BS, Gottstein C, Cines DB, Muzykantov VR. Delivery of anti-platelet-endothelial cell adhesion molecule single-chain variable fragment-urokinase fusion protein to the cerebral vasculature lyses arterial clots and attenuates postischemic brain edema. J Pharmacol Exp Ther. 2007;321:947–952. doi: 10.1124/jpet.107.120535. [DOI] [PubMed] [Google Scholar]
- 34.Scherpereel A, Rome JJ, Wiewrodt R, et al. Platelet-endothelial cell adhesion molecule-1-directed immunotargeting to cardiopulmonary vasculature. J Pharmacol Exp Ther. 2002;300:777–786. doi: 10.1124/jpet.300.3.777. [DOI] [PubMed] [Google Scholar]
- 35.Muzykantov VR. Biomedical aspects of targeted delivery of drugs to pulmonary endothelium. Expert Opin Drug Deliv. 2005;2:909–926. doi: 10.1517/17425247.2.5.909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}