

Abstract
Lyme arthritis results from colonization of joints by Borrelia burgdorferi and the ensuing host response. Using gene array–based differential analysis of B. burgdorferi gene expression and quantitative reverse trancription-polymerase chain reaction, we identified two paralogous spirochete genes, bmpA and bmpB, that are preferentially up-regulated in mouse joints compared with other organs. Transfer of affinity-purified antibodies against either BmpA or BmpB into B. burgdorferi–infected mice selectively reduced spirochete numbers and inflammation in the joints. B. burgdorferi lacking bmpA/B were therefore generated to further explore the role of these proteins in the pathogenesis of Lyme disease. B. burgdorferi lacking bmpA/B were infectious in mice, but unable to persist in the joints, and they failed to induce severe arthritis. Complementation of the mutant spirochetes with a wild-type copy of the bmpA and bmpB genes partially restored the original phenotype. These data delineate a role for differentially produced B. burgdorferi antigens in spirochete colonization of mouse joints, and suggest new strategies for the treatment of Lyme arthritis.
Lyme arthritis results from infection with a tick-borne spirochete, Borrelia burgdorferi (1). Spirochetes are deposited into the skin when Ixodes scapularis feed on mice or humans. B. burgdorferi replicate in the dermis, and then disseminate to distant cutaneous sites and other organs, including the joints (2). Clinical manifestations of Lyme disease may involve a characteristic skin rash called erythema migrans, arthritis, carditis, or neurological symptoms (3, 4). Lyme arthritis occurs in a substantial number of untreated patients (1), often several weeks to months after the tick bite, and is associated with spirochete invasion of the joints (3). In most cases, antibiotic therapy is curative; however, some patients develop a form of antibiotic-resistant arthritis that is thought to be unrelated to persistent infection (5).
C3H/HeN mice infected with B. burgdorferi develop joint swelling that partially mimics human disease and has been helpful in understanding the pathogenesis of Lyme arthritis (6–9). Intradermally inoculated spirochetes reside in the mouse skin for ∼1 wk, after which they disseminate to many tissues. Arthritis begins to develop at ∼10 d and is prominent at 2 or 3 wk (2, 10). At 4 wk, the disease may begin to regress, and by 8 wk the arthritis has resolved (2, 6, 7, 11–13). The presence of B. burgdorferi in mouse joints, and the innate and adaptive host responses to the pathogen, contributes to the development of inflammation (1). Spirochete genes are also implicated in mouse Lyme arthritis, as B. burgdorferi lacking certain plasmids are less arthritogenic (14). Indeed, B. burgdorferi Arp is expressed in many tissues during infection, and antisera against Arp can attenuate mouse Lyme arthritis (15, 16). Specific spirochete antigens that are selectively induced in the joints and causally associated with the genesis of arthritis have, however, not yet been identified.
The preferential up-regulation of specific B. burgdorferi genes throughout the spirochete life cycle, both in the vector and mammalian host, plays an important role in pathogen survival (17–19). It is likely that the diverse metabolic or immune microenvironments within mammalian tissues may influence the ability of B. burgdorferi to persist in different organs (20–22). We determined, therefore, whether particular B. burgdorferi genes are selectively expressed in mouse joints, and if the gene products contribute to spirochete colonization of the joints and the development of arthritis. Characterization of microbial ligands that are expressed in a tissue-specific manner is critical for understanding the pathogenesis of complex infectious diseases.
RESULTS
Identification of B. burgdorferi genes up-regulated in mouse joints
We identified a subset of B. burgdorferi genes that are preferentially up-regulated in mouse joints using a microarray-based analysis to compare spirochete transcriptomes from different tissues. The differential expression analysis using a custom-amplified library (DECAL) technique (22, 23) specifically amplified spirochete mRNAs from infected skin, joints, heart, and bladder. To accomplish this, C3H/HeN mice were challenged with B. burgdorferi. At day 15, when arthritis was evident (14, 24) and was reflected by swelling of the tibiotarsal joints, total RNA was isolated from the skin, joints, heart, and bladder tissue. B. burgdorferi–specific mRNAs were selected and amplified using the DECAL technique and hybridized onto a B. burgdorferi genomic array, as detailed in the Materials and methods. Analysis of array data revealed that the expression of B. burgdorferi genes varied in different host tissues, and that the B. burgdorferi bmpA/B gene operon displayed the most significant up-regulation in joints.
The temporal expression of bmpA/B coincides with B. burgdorferi infection of mouse joints and the induction of Lyme arthritis
We used the gene array data as a guide to perform more detailed quantitative RT-PCR (qRT-PCR) analyses on the genes of interest. The B. burgdorferi bmp gene family has four paralogous members, bmpA, bmpB, bmpC, and bmpD (25), where bmpA and bmpB are on a bicistronic operon and may be controlled by a common promoter (26, 27). We assessed the independent expression of each bmp gene throughout the first few weeks of B. burgdorferi infection in multiple tissues using gene-specific qRT-PCR analysis. Groups of C3H/HeN mice were infected with B. burgdorferi, and tissues were isolated at 6, 9, 12, and 15 d. Isolated mRNA, without the DECAL enrichment, was converted to cDNA and subjected to qRT-PCR to measure the individual B. burgdorferi bmp transcripts. Expression of bmpA and bmpB in joints was dramatically and selectively up-regulated at 12 and 15 d compared with the skin, heart, and bladder (Fig. 1).bmpC and bmpD expression showed no differences between joint or other tissues and remained unchanged in the joints at 12 and 15 d; at earlier time points, spirochetes were not readily detectable in the joints, heart, or bladder (unpublished data). Furthermore, expression of both bmpA and bmpB was also minimal in spirochetes isolated from infected ticks; their expression levels were ∼8–10-fold lower than the corresponding levels in spirochetes isolated from joints at day 15. As bmpA and bmpB showed the greatest degree of selective up-regulation in joints, these gene products were further examined to explore the paradigm that tissue-specific gene expression contributes to arthritis.
Figure 1.

Joint-specific up-regulation of B. burgdorferi bmpA and bmpB in infected mice. The relative expression levels of bmpA (black bar) and bmpB (gray bar) are represented as copies of bmp transcript per 1,000 copies of flaB transcript. Total RNA was isolated from multiple tissues of B. burgdorferi–infected mice (10 mice/group) and converted to cDNA for measuring bmp transcripts using qPCR 12 and 15 d after challenge. Bars represent the mean ± the SEM from three independent experiments. The transcript levels in joints were significantly higher than those in skin, heart, or bladder tissue. n = 3. P < 0.01–0.001.
Antibodies against BmpA or BmpB interfere with B. burgdorferi persistence most dramatically in joints
To explore the physiological relevance of the joint-specific up-regulation of bmpA and bmpB, we assessed whether specific antibodies were capable of interfering with B. burgdorferi persistence in the joints. To achieve this goal, we produced recombinant BmpA and BmpB in Escherichia coli and generated antisera against the individual proteins in rabbits. As antisera against BmpA may cross-react with BmpB, or vice versa (28), monospecific BmpA and BmpB antibodies were isolated by affinity purification, as detailed in the Materials and methods. We then characterized the specificity of the purified BmpA or BmpB antibodies and their capacity to bind to the spirochete surface. Immunoblots demonstrated substantial binding of purified BmpA or BmpB antibodies to their respective proteins, with minimal cross-reactivity (Fig. 2 A). In addition, immunofluorescence microscopy showed that both of the Bmp antibodies were able to bind to the surface of unfixed cultured B. burgdorferi (Fig. 2 B) and had partial borreliacidal activity in vitro (Fig. 2 C).
Figure 2.

Characterization of affinity-purified BmpA and BmpB antibodies. (A) 1 ng of recombinant Bmp proteins (rBmpA and rBmpB) was probed with either affinity-purified BmpA (Affi-BmpA) or BmpB (Affi-BmpB) antibodies. The arrowhead and arrow indicate the positions of BmpA and BmpB, respectively. (B) BmpA or BmpB antibodies bind the surface of intact unfixed B. burgdorferi (arrows). Spirochetes were immobilized on glass slides and probed with affinity-purified BmpA (Affi-BmpA), BmpB (Affi-BmpB) antibodies, or normal rabbit sera (NRS). Images were acquired using the 40× objective lens of an epifluorescence microscope (Axiovert 200M; Carl Zeiss, Inc.). Bars, 10 μm. (C) Borreliacidal activities of BmpA or BmpB antibodies in vitro. Spirochetes were incubated with normal rabbit sera (NRS), affinity-purified BmpA (Affi-BmpA), BmpB (Affi-BmpB), or rabbit OspA antibodies (OspA). The number of viable spirochetes was assessed by fluorescent microscopy after labeling the spirochete preparations with vital dyes at 48 h of incubation. Data represent the mean number of viable spirochetes/microscopic field after antibody incubation ± the SEM from three independent experiments. The number of viable spirochetes exposed to the affinity-purified BmpA or BmpB antibodies were significantly diminished compared with spirochetes that received NRS. n = 3. p < 0.01. As expected, OspA antisera resulted in the complete killing of B. burgdorferi.
We next performed passive immunization studies to assess whether purified BmpA or BmpB antibodies interfered with selective B. burgdorferi persistence in joints. To accomplish this, groups of C3H/HeN mice (15 animals/group) were infected with B. burgdorferi, and 3 d after spirochete challenge, mice were administered 100 μl of BmpA, BmpB, or control antibodies. Skin, bladder, heart, and joints were collected on day 12, 15, and 18. B. burgdorferi levels in each tissue were measured by qPCR. The passive transfer of either of the Bmp antibodies dramatically reduced B. burgdorferi levels in the joints, and to a lesser extent in skin, but not in other tissues, such as heart and bladder, at all time points. The data from days 15 and 18 are presented in Fig. 3. Because Lyme arthritis has been directly attributed to the presence of B. burgdorferi in joint tissue (1), we determined whether BmpA or BmpB antibodies influenced disease. Development of joint swelling in B. burgdorferi–infected mice immunized with glutathione S-transferase (GST) antisera was significantly higher (1.36 ± 0.25 mm; P < 0.02) than in groups of mice administered either BmpA (0.47 ± 0.13) or BmpB (0.65 ± 0.1) antibodies, suggesting a role for these proteins in the genesis of arthritis.
Figure 3.

Affinity-purified Bmp antibodies most dramatically affect B. burgdorferi persistence in mouse joints. Affinity-purified BmpA or B antibodies interfere with the persistence of B. burgdorferi in joints. Mice (15 animals/group) were infected with B. burgdorferi and administered affinity-purified BmpA (Affi-BmpA), BmpB (Affi-BmpB), or control GST antibodies. The B. burgdorferi level in mouse tissues was assessed by measuring flaB copies at days 15 and 18 after B. burgdorferi infections, and it is expressed as flaB/mouse β-actin. The B. burgdorferi load in joints and skin of mice receiving BmpA or BmpB antibodies was significantly lower than in mice receiving control GST sera. n = 3. P < 0.01–0.001. Bars represent the mean ± the SEM from two independent experiments and the spirochete burden of the control mice was considered as the 100% value.
bmpA/B-deficient B. burgdorferi persist poorly in joints and fail to induce arthritis
To directly study the importance of BmpA and BmpB in the spirochete persistence within joints, we created bmpA/B-deficient (bmpA/B−) B. burgdorferi. The mutant was constructed by exchanging a DNA fragment within the bmpAB gene with a kanamycin-resistance cassette via homologous recombination (Fig. 4 A). A suicide plasmid for recombination in the intended chromosomal locus was constructed and transformed into B. burgdorferi, and transformants were screened as detailed in the Materials and methods. PCR analysis indicated that the antibiotic cassette was appropriately inserted into the intended chromosomal locus and that the plasmid profile of the mutant spirochetes was similar to that of the wild-type organisms (unpublished data). RT-PCR showed that bmpA or bmpB mRNA was absent in the mutant, and, as expected, the mutant expressed both bmpC and bmpD mRNA (Fig. 4 B). In addition, qRT-PCR showed that both wild-type spirochetes and bmpA/B− B. burgdorferi produced similar levels of transcripts expressed from neighboring gene loci, such as bb0381, bmpC, and bmpD. The protein profiles of the bmpA/B− spirochetes and the wild-type organisms were identical (Fig. 4 C, left), except that the bmpA/B− B. burgdorferi did not produce BmpA (Fig. 4 C, top right) or BmpB protein (Fig. 4 C, bottom right).
Figure 4.

Construction and analysis of the bmpA/B− B. burgdorferi. (A) Schematic drawings of the wild-type isolate (WT) and the bmpA/B− mutant at the bmpAB locus. Genes bb0381, bmpB, bmpA, bmpC, and bmpD (white box-arrows) and the kanamycin-resistance cassette driven by the B. burgdorferi flaB promoter (flaB-Kan, black box-arrow) are indicated. Nucleotide positions of primers P1-P4 in the B. burgdorferi genomic database (www.tigr.org) are indicated within parentheses. The 5′ and the 3′ arms for homologous recombination, flanking up- and downstream of the bmpAB locus were amplified using primers P1-P2 and P3-P4 and ligated to the flaB-Kan cassette, as detailed in the Materials and methods. (B) RT-PCR analysis of the bmpA and B transcripts. Total RNA were isolated from either the wild-type (WT) or mutant (bmpA/B−) B. burgdorferi, converted to cDNA, and subjected to PCR analysis with flaB, bmpA, bmpB, bmpC, or bmpD primers and analyzed on a 2% agarose gel. (C) Protein analysis of wild-type (WT) or bmpA/B− spirochetes. Equal amounts of proteins from wild-type or bmpA/B− B. burgdorferi were separated on a SDS-PAGE gel, and either stained with Coomassie blue (left) or transferred onto a nitrocellulose membrane and probed with the affinity-purified BmpA (top right) or affinity-purified BmpB (bottom right) antibody. Migration of protein standards is shown to the left in kilodaltons. (D) Comparable levels of wild-type and bmpA/B− B. burgdorferi in the dermis of infected mice. Total DNA was isolated from the skin of mice 1 wk after B. burgdorferi challenge. The B. burgdorferi burden was analyzed by qPCR measurement of flaB copies and expressed as flaB/mouse β-actin. Wild-type (WT) and bmpA/B− B. burgdorferi were evident in similar numbers. n = 3. P > 0.5. (E) The bmpA/B− B. burgdorferi were capable of dissemination from the dermis of infected mice. Blood was collected from mice 1 wk after B. burgdorferi challenge, and spirochete burden was analyzed by qPCR measurement of flaB copies and expressed as flaB/mouse β-actin. Similar levels of wild-type (WT) and bmpA/B− B. burgdorferi were detected. n = 3. P > 0.5.
We then compared the infectivity between bmpA/B− B. burgdorferi and the wild-type spirochete in vivo using the mouse model of Lyme borreliosis. In nature, ticks transmit B. burgdorferi infection to mammals; however, to ensure an identical inoculation dose of wild-type and mutant spirochetes in the mouse host, we infected mice with a needle inoculum containing a specific amount of B. burgdorferi. Groups of 10 C3H/HeN mice were inoculated intradermally on the hind legs with 104 wild-type or bmpA/B− B. burgdorferi. Both wild-type and mutant spirochetes were readily cultured from skin, and qPCR of mouse skin biopsy (ear) specimens or blood (Fig. 4, D and E) collected 1 wk after infection showed similar pathogen levels. When ticks were allowed to engorge on mice infected with bmpA/B− B. burgdorferi, the mutant spirochetes were able to migrate into fed ticks, albeit at a lower level than the wild-type spirochetes (unpublished data). Because bmpA/B− B. burgdorferi were as infectious as the isogenic wild-type spirochetes in the mice, we assessed whether BmpA and BmpB were required for B. burgdorferi to effectively colonize mouse joint tissue. The spirochete loads were analyzed in joints or in control tissue locations (skin and bladder) 14 and 18 d after B. burgdorferi infection. qPCR indicated that bmpA/B− B. burgdorferi were not detected in joints, but were evident in the skin and bladder tissue at all time points, such as day 14 or 18 (unpublished data) after infection. Severe joint swelling developed in mice infected with wild-type B. burgdorferi, but not in animals infected with bmpA/B− B. burgdorferi. Collectively, these data suggest that BmpA and BmpB may be essential for B. burgdorferi to colonize joints and induce Lyme arthritis.
Complementation restores the ability of bmpA/B− B. burgdorferi to colonize joints and cause arthritis
To exclude the possibility that the failure of the bmpA/B− B. burgdorferi to persist within joints was caused by an anomalous effect of transformation, we complemented, in trans, the bmpA/B−spirochetes with a wild-type copy of the bmpA and bmpB gene under the control of native promoter. To construct complemented isolates of B. burgdorferi, we transformed the mutant with the recombinant shuttle plasmid pKFSS1 carrying a wild-type copy of the bmpA and bmpB gene (Fig. 5 A) placed upstream of a streptomycin-resistance gene. As controls for the complementation experiments, additional mock-complemented isolates were also constructed by transforming the wild-type spirochetes and bmpA/B− B. burgdorferi with the empty pKFSS1vector. PCR analysis confirmed that, similar to the wild-type B. burgdorferi, the mock-complemented and bmpAB-complemented spirochetes retained all of the B. burgdorferi plasmids (unpublished data). Immunoblotting showed that the bmpAB-complemented mutant, but not the mock-complemented mutant, produced both BmpA and BmpB protein (Fig. 5 B).
Figure 5.

Complementation of bmpA/B− B. burgdorferi with bmpA and bmpB restores the ability of spirochetes to survive in joints and induces greater levels of arthritis. (A) The bmpA and B gene, including its native promoter region was amplified and cloned into the shuttle vector pKFSS1 and transformed into the bmpA/B− spirochetes. (B) Western blot demonstrates production of the BmpA and BmpB proteins by the complemented B. burgdorferi. Equal amounts of proteins from the mock-complemented bmpA/B− spirochetes (bmpA/B−), mock-complemented wild-type (WT), or complemented spirochetes (bmpAB Com) were separated on a SDS-PAGE gel, which was either stained with Coomassie blue (left) or transferred to nitrocellulose membrane. Individual Bmp proteins were probed with the affinity-purified BmpA (top right) or BmpB (bottom right) antibodies. (C) The B. burgdorferi burden in mice infected with mock-complemented wild-type (gray bar), mock complemented bmpA/B− mutant (black bar), and bmpA/B− mutant complemented with bmpA and bmpB (white bar). Groups of mice (10 animals/group) were infected with wild-type or genetically manipulated isolates of B. burgdorferi, and the spirochete burden was analyzed at day 14, 21, and 28 by measuring copies of the B. burgdorferi flaB gene. Amounts of mouse β-actin were determined in each sample and used to normalize the quantities of spirochete DNA. Mutant or complemented spirochete levels in the bladder and skin (ear) tissue were similar. n = 3. P > 0.5. Levels of bmpAB complemented isolates were significantly higher in the joints than bmpA/B− mutants. Error bars represent the mean ± the SEM of relative tissue levels of B. burgdorferi. n = 3. *, P < 0.05. (D) Severity of joint swelling in B. burgdorferi–infected mice. Groups of mice (10 animals/group) were separately infected with mock complemented wild-type B. burgdorferi (gray bar), mock complemented bmpA/B− mutant (black bar), and bmpA/B− spirochetes complemented with bmpA and bmpB gene (white bar). Arthritis was evaluated by the assessment of development of joint swelling after 14, 21, and 28 d of spirochete challenge using a digital caliper. Bars represent the mean measurements (± the SEM) from three independent experiments. Significant differences in the inflammation level were noticed between groups of mice infected with wild-type isolates with the bmpA/B− mutant at all time points. **, P < 0.001 - 0.04. Highly significant differences between joint inflammation was also noted in groups of mice infected with bmpA/B− spirochetes and bmpA/B− spirochetes complemented with bmpAB at day 21. **, P < 0.001.
We then characterized the ability of the complemented spirochetes to establish infection and induce joint inflammation in mice. Groups of 10 C3H/HeN mice were inoculated intradermally in the hind legs with 104 wild-type spirochetes, mock-complemented bmpA/B− B. burgdorferi, or bmpAB-complemented bmpA/B− B. burgdorferi. The spirochete burden in the joint, bladder, or skin (ear) tissues and the joint swelling were evaluated 14, 21, and 28 d after B. burgdorferi infection. The bmpA/B− mutant and the complemented isolates all readily colonized the bladder and skin tissues. Although the bmpA/B− mutant was not detected in the joints, the bmpAB-complemented B. burgdorferi were able to persist in the joints at all times during peak infection, between days 14–21, albeit at lower levels than wild-type isolates (Fig. 5 C). At day 28, as the host immune response controlled the spirochete load, the B. burgdorferi levels in all tissues declined significantly. Joint swelling was minimal in mice infected with the bmpA/B− B. burgdorferi at 7, 14, 21, or 28 d, demonstrating that the kinetics of joint disease development was abrogated, and not merely delayed, in mice infected with the mutant. In agreement with earlier observations (14, 24), wild-type B. burgdorferi induced severe ankle swelling that is most dramatic at day 21 and is consistent with histopathological signs of arthritis (Fig. 5 D [P < 0.001] and Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20070962/DC1). Joint inflammation in bmpAB-complemented bmpA/B− B. burgdorferi-infected mice were lower than that induced by the wild-type spirochetes, however, at the peak period of joint inflammation, day 21, the bmpAB-complemented isolates induced significantly higher swelling of the joints compared with bmpA/B− B. burgdorferi (Fig. 5 D [P < 0.001] and Fig. S1).
DISCUSSION
B. burgdorferi alters its gene expression profile as it cycles between arthropods and mammals (29, 30), and specific genes help maintain the spirochete in nature (20, 24, 31–35). I. scapularis ticks, while feeding on a mammalian host, deposit a heterogeneous population of B. burgdorferi in the dermis (36), which eventually spread to multiple organs (2). Selected B. burgdorferi genes may be preferentially expressed in different locations (22); however, it is not known if the gene products that are induced in tissues can directly contribute to disease. We show that both genes of the B. burgdorferi bmpA/B operon undergo selective up-regulation in mouse joints (37), and that the targeted deletion of bmpA/B specifically decreases B. burgdorferi persistence in this tissue.
Immune sera from a B. burgdorferi–infected host (38, 39), or specific antisera (40–42), is protective when administered before spirochete challenge. The same antisera does not eradicate B. burgdorferi when transferred after challenge, but can modulate arthritis (39), showing that the borreliacidal and disease-modifying components in immune sera are different. Antisera against ubiquitously expressed spirochetal antigens, such as Arp (15, 16, 43) and DbpA (43), can modify arthritis with no effect on spirochete levels in tissues. Functions of BmpA and BmpB proteins in the B. burgdorferi natural cycle, and their correlation with the induction of arthritis, are currently unknown. In our study, both BmpA and BmpB antisera most dramatically inhibited the colonization of spirochetes in the joints, and to a lesser extent, in the skin, but not in other tissues. Although BmpA, DbpA, OspC, and FlaB antibodies have been identified in immune sera that has arthritis-resolving properties (12, 39), the passive transfer of BmpA antisera did not previously influence the genesis of arthritis (39). Differences in study design, spirochete isolates, such as N40 versus B31, used in the current experiments, or the quality of the antisera can potentially influence such results. Although the N40 isolate has been well known for its capability to induce arthritis in mice (12, 39), the limited genomic information on this isolate compared with B31 (25), and more importantly, the extreme difficulties in the genetic transformation of N40 (44), makes B31 preferable for these genetic studies. The current experiments conclusively demonstrate that loss of both bmpA and bmpB influence the selective survival of spirochetes in joints and severely impeded the development of inflammation. Complementation of the bmpA/B− B. burgdorferi with both bmpA and bmpB partially reconstituted the wild-type arthritis phenotype, further supporting a role for both BmpA and BmpB in Lyme arthritis. Differences between the complemented and wild-type spirochetes could be caused by the expression of bmpA/B on a shuttle vector in the complemented isolate, rather than from their native position on the B. burgdorferi chromosome, among other possibilities.
We have also examined several human samples to extend our observations beyond mice. bmpA and bmpB levels were measured in skin biopsies from five patients with early Lyme disease and erythema migrans, and synovial fluid from four patients with acute Lyme arthritis. qRT-PCR showed significantly higher levels (P < 0.03–0.009) of bmpA and bmpB expression per spirochete in the synovial fluid compared with the skin. In the joints, 21.4 ± 10.8 ng bmpA/ng flaB was noted compared with 0.36 ± 0.2 ng bmpA/ng flaB in the skin. Similarly, 11.0 ± 3.0 ng bmpB/ng flaB was present in the joints, compared with 0.34 ± 0.17 ng bmpB/ng flaB in the skin. We recognize that the skin and joint samples come from different patients; however, simultaneous skin and joint specimens from the same patient are rarely, if ever, obtained. Moreover, the spirochete in each sample may not be identical, even though the vast majority of organisms in the New York/Connecticut region are B. burgdorferi sensu stricto. Nevertheless, these data provide a first suggestion that our mouse data may be applicable to human disease as well.
In nature, B. burgdorferi survives in an enzootic cycle where immature ticks acquire the pathogen from wild rodents, and then maintain B. burgdorferi and subsequently transmit the spirochetes back to mice (3). As the tick bite site is primarily located in the mouse dermis, the population of B. burgdorferi that persistently resides in skin is most likely to complete the tick–mouse–tick life cycle, and should have a selective advantage in nature. The adaptive significance of dissemination of spirochetes to deeper host organs (37), such as the joints, where they are unlikely to be acquired by ticks, is not clear, although certain sites may afford survival advantages because of protection from host responses, binding ligands, or nutrients present in the microenvironment.
Lyme arthritis is the direct result of a localized host inflammatory response (1), presumably against antigens that are abundant on the surface of the invading spirochete. Joint-induced, surface-exposed, and highly antigenic B. burgdorferi proteins, such as BmpA and B could potentially initiate the complex cascade of host inflammatory responses that lead to arthritis. However, as the spirochete is coated with many proinflammatory lipoproteins, it is more likely that BmpA and B have a role in spirochete survival in the joints, and that the lack of arthritis in the bmpA/B− B. burgdorferi is caused by the inability of these spirochetes to colonize and persist in joint tissue. In summary, we present direct evidence for a dynamic transcriptome of B. burgdorferi in vivo and show that differentially up-regulated genes, such as bmpA and bmpB in the joints, play a role in spirochete persistence in a specific host environment. This information contributes to our understanding of the adaptive strategies of a pathogen that is able to survive in numerous tissues, and may lead to new ways to modulate Lyme arthritis.
MATERIALS AND METHODS
B. burgdorferi and mice.
A low passage (passage 3) virulent isolate of B. burgdorferi B31, clone 5A11, was used throughout this study (45). Mice were infected with a single inoculum of 104–105 spirochetes/mouse by intradermal injection to the hind leg. 4–6-wk-old C3H/HeN mice were purchased from the National Institutes of Health. All animal procedures were performed in compliance with the guidelines and with the approval of the Institutional Animal Care and Use Committee.
Selective amplification of B. burgdorferi RNA and microarray analysis.
Whole-genome nylon membrane arrays containing 1,697 putative ORFs encoded by B. burgdorferi B31 were used in this study (46). Selective amplification of B. burgdorferi transcripts in the infected mouse host was performed using the DECAL approach, as previously detailed (22, 47). Groups of C3H/HeN mice (10 mice/group) were injected with B. burgdorferi (104–105 spirochetes/mouse), and spirochetal infection was confirmed by PCR and culture (35). The mice were killed 15 d after spirochete inoculation, and total RNA was isolated from distal skin, joints (tibiotarsal), heart, and urinary bladder using the RNeasy RNA isolation kit (QIAGEN) as described by the manufacturer. 1 mg of total RNA was reverse transcribed into biotinylated cDNA using random hexamer primers, biotin-dNTP, and the SuperScript First Strand synthesis system (Invitrogen). Positive selection and amplification of spirochete transcripts was performed according to a published method (22), and the PCR products representing amplified B. burgdorferi mRNAs (skin-DECAL, joint-DECAL, heart-DECAL, and bladder-DECAL) were normalized using flaB-specific primers and labeled with a random priming method using FITC-labeled dNTPs (GE Healthcare). The labeled probes were further normalized based on their hybridization intensity to equal amounts of flaB PCR products blotted onto nylon membrane (unpublished data). The normalized probes were then used to hybridize duplicate microarrays using RapidHyb buffer (GE Healthcare) according to the manufacturer's instructions. After probing the arrays with DECAL-labeled cDNAs, the hybridization was scored visually, and spots were given a score ranging from 0 to 3 (in increments of 0.5) based on the intensity of hybridization, as previously described (22). A spot was considered positive when a minimum of twofold increase in hybridizations was noted (in joint), in comparison to similar spot hybridizations in other tissues (in skin, heart, and bladder).
PCR.
The nucleotide sequence of various primers used in specific PCR applications is indicated in Table S1 (available at http://www.jem.org/cgi/content/full/jem.20070962/DC1). RT-PCR or qRT-PCR analysis was performed as previously described (48) using iQ SYBR Green Supermix (Bio-Rad Laboratories) or using TaqMan probes synthesized by Applied Biosystems. All probes contained a 5′ reporter (FAM) and a 3′ quencher (TAMRA). The amounts of B. burgdorferi bmp transcripts in each sample were normalized to levels of flaB transcripts in qRT-PCR reactions. For quantitative detection of B. burgdorferi burdens within mouse tissue samples, qPCR was performed on an equal amount of isolated DNA using flagellin (flaB) as a surrogate marker (35). The mouse β-actin gene was amplified to normalize the amount of B. burgdorferi cDNA in the qPCR reaction. Skin biopsy specimens from five patients with erythema migrans, and synovial fluid from four patients with acute Lyme arthritis, were obtained at New York Medical College and Yale University School of Medicine. Skin biopsy specimens were immediately placed in RNALater (Ambion) and stored at −80°C; synovial fluid samples were rapidly frozen and stored at −20°C. Human samples were processed for cDNA synthesis and used for qRT-PCR, as described in this section.
Protein expression and preparation of affinity-purified polyclonal antibody.
Recombinant BmpA and BmpB proteins were produced in E. coli using the bacterial expression vector pGEX-6P1 (GE Healthcare). The list of primers used for directional cloning of Bmp proteins (without the N-terminal leader sequence) is indicated in Table S1. Expression, purification, and enzymatic cleavage of the GST fusion proteins were performed as previously described (48). To generate polyclonal antisera, BmpA or BmpB (without the GST tag) produced in E. coli was emulsified in complete Freund's adjuvant and injected into groups of 2–3 rabbits (100 μg/animal). The animals were boosted twice at 3 wk intervals with the same dose of antigen in incomplete Freund's adjuvant, and the sera were collected 2 wk after the second boost. Respective cross-reactivity of the BmpA or BmpB polyclonal antibodies against BmpB or BmpA were removed using the previously described affinity purification approach (28), with the following modifications. Recombinant BmpB (rBmpB) was expressed with a GST fusion protein, as described in this section, and bound to a Sephadex 4B column (GE Healthcare) according to the manufacturer's instructions. The column was washed with 10 bed volumes of PBS. To purify BmpA antibody, the antisera was diluted fivefold in PBS and cycled over the rBmpB-bound column by gravity flow for 5 cycles of 10 passages each, for a total of 50 passages. BmpB antibody was also purified in a similar fashion by passing the diluted antisera through rBmpA-bound column.
Characterization of antibody in vitro.
Affinity-purified BmpA and BmpB antibodies were tested for their bactericidal activity using a combination of two vital stains that specifically label live and dead spirochetes, as previously described (49). In brief, spirochetes (5 × 107/ml) were incubated in 15 μl BSK-H medium (Sigma-Aldrich) with the addition of equal volume of normal rabbit sera or purified Bmp antibodies for 48 h at 33°C. Spirochetes were labeled with the live/dead BacLight Viability kit (Invitrogen) according to manufacturer's instructions. Bactericidal activities of the antiserum were determined by counting the number of live and dead spirochetes from 10 random microscopic fields under high magnification, as previously described (49). Bactericidal rabbit antiserum against B. burgdorferi OspA was used as a control in the assay.
Antibody binding to unfixed B. burgdorferi was assessed by immunofluorescence microscopy, as previously described (32). Spirochetes (107/ml) were spotted on silylated glass slides (PGC Scientific), blocked with PST (0.05% Tween-20, 5% normal goat sera in PBS), and incubated with normal rabbit sera or affinity-purified BmpA or BmpB antibodies (1:500 dilution in PST). Samples were similarly incubated with secondary FITC-labeled goat anti–rabbit antibody (KPL) at a dilution of 1:100, and spirochetes were visualized using an epifluorescence microscope (Carl Zeiss, Inc.).
Western blotting was performed as previously described (48) using 1:5,000 dilutions of either of the affinity-purified BmpA or BmpB antibody and HRP-conjugated anti–rabbit secondary antibody (Sigma-Aldrich).
In vivo antibody-blocking studies.
In brief, C3H/HeN mice were infected with B. burgdorferi B31 (105 spirochetes/mouse) for 3 d and affinity purified Bmp or control (GST) antibodies were administered to groups of mice (15 animals/group, 100 μl antibody/mouse). To maintain effective concentrations of the antibody, the animals were treated with the respective antibody every 3 d for 2 wk. Parallel control studies confirmed that similar treatment of naive C3H mice with rabbit anti-BmpA/B sera do not result in joint pathology, as indicated by the assessment of arthritis at 2 and 4 wk after the initiation of antibody treatment (unpublished data). The mice were killed 12, 15, and 18 d after B. burgdorferi infections, and skin, heart, joint, and bladder samples were isolated and stored at –80°C. B. burgdorferi burdens in tissues samples were measured by qPCR analysis, as described in PCR.
Evaluation of joint inflammation.
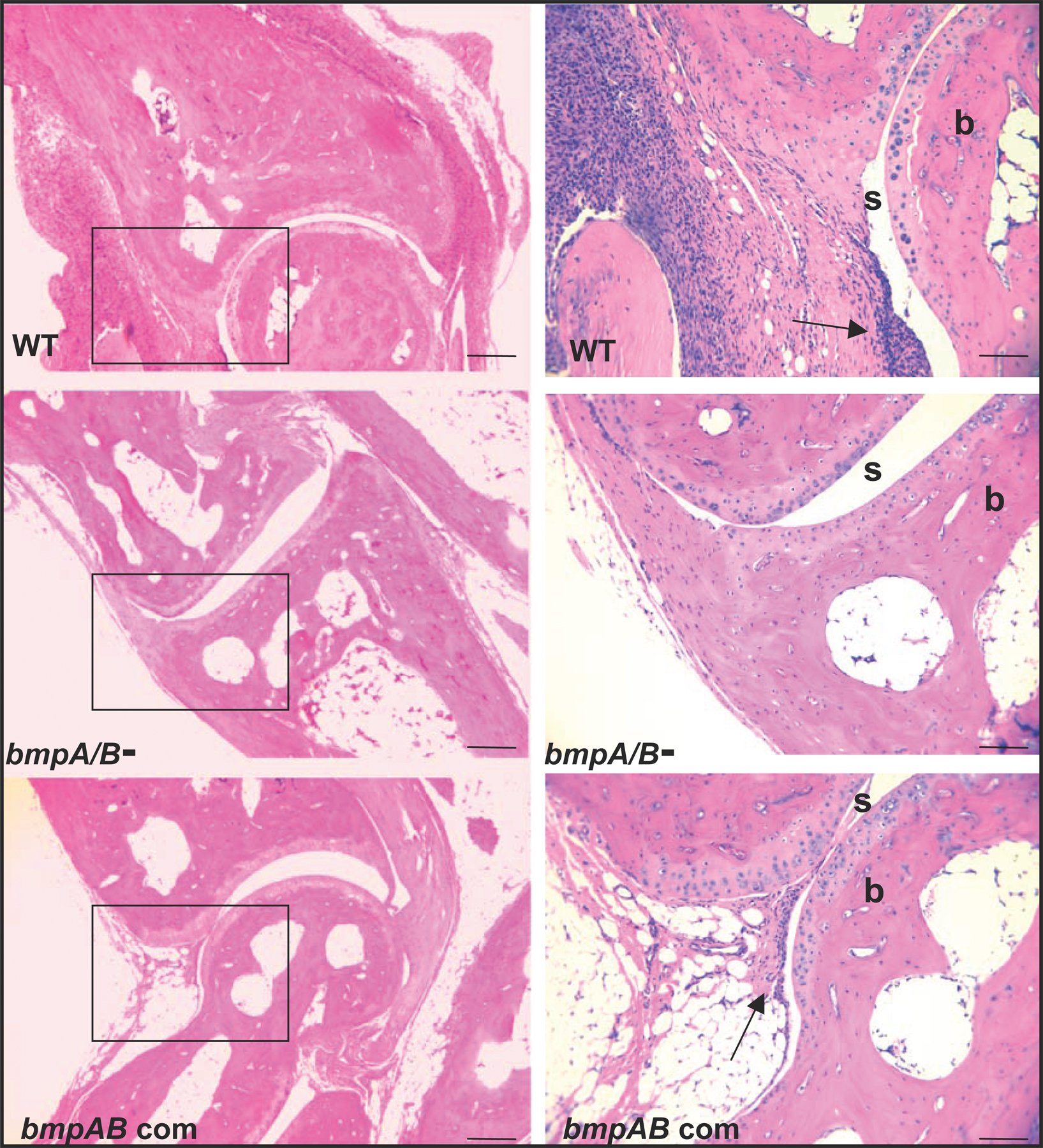
B. burgdorferi–infected mice were examined for swelling of the tibiotarsal joints, as previously detailed (12, 50, 51). Ankle joints from each of the rear legs of each mouse were measured using a precision metric caliper in blinded fashion. Thickest diameters of the tibiotarsal joints were measured in each mouse before B. burgdorferi infection, and the development of inflammation was monitored and tabulated on a weekly basis until the sacrifice of the mice. At least 5 ankle joints from each group of mice (10 animal/group) infected with the different strains were subjected to histological analyses. 20 randomly chosen sections from each of the mouse groups were assessed for histopathological comparisons. For histology, joints were fixed in 10% formalin, decalcified, and processed for hematoxylin and eosin staining. Overall signs of arthritis represented a combined assessment of histological parameters of B. burgdorferi–induced inflammation, such as exudation of fibrin and inflammatory cells into the joints, alteration in the thickness of tendons or ligament sheaths, and hypertrophy and hyperlexia of the synovium.
Generation and phenotypic analysis of bmpA/B mutant and genetic complementation of B. burgdorferi.
The oligonucleotide primers used for genetic manipulation of B. burgdorferi are indicated in Table S1. The bmpA/B mutant was constructed by exchanging the upstream of bmpA/B operon with a kanamycin-resistance cassette from the plasmid pXLF10601 (45) via homologous recombination. First, the 5′ and the 3′ arms flanking bmpA ORF (open reading frame) were PCR-amplified using primers P1-P4 and cloned into two multiple-cloning sites flanking the kanAn cassette in plasmid pXLF10601. Plasmid DNA of pXLF10601-bmpAB (30 μg) was electroporated into B. burgdorferi B31 clone 5A11 using the previously described protocol (45). Transformants were selected in BSK-H complete medium (Sigma-Aldrich) containing kanamycin (350 μg/ml). Spirochetes that survived antibiotic selection were further analyzed by PCR to confirm the desired integration of kanAn cassette, and all known endogenous plasmids that are present in parental B31 isolate were also assessed in the mutant clones, as previously described (32). One of the bmpA/B knockout clones that retained the same complete set of plasmids as the wild-type isolate was used in further experiments.
For genetic complementation of the bmpA/B mutant, we used the B. burgdorferi shuttle plasmid pKFSS1 (52) to deliver the wild-type copy of the bmpA and bmpB gene. The DNA fragment encompassing bmpA and bmpB gene and the native promoter (26) were PCR-amplified using primers (Table S1) and cloned into the BamHI and SalI sites of pKFSS1. 20 μg of the recombinant plasmid, designated pKFSS1-pbmpAB, were electroporated into the bmpA/B− B. burgdorferi, and complemented clones were selected using 50 μg/ml of streptomycin in the growth medium. As appropriate controls for complementation studies, additional mock-complemented isolates were also generated by transformation of the wild-type B. burgdorferi and bmpA/B− B. burgdorferi with the empty vector pKFSS1. PCR analyses were further performed to confirm that the mock or the bmpAB-complemented clones contained the respective plasmids, either the empty vector or pKFSS1-pbmpAB and also retained the same plasmid profile as the wild-type as described in the previous paragraph.
For phenotypic analysis of mock-complemented bmpA/B mutants and bmpAB-complemented isolates in vivo, B. burgdorferi isolates were injected into groups of mice (10 animals/group) via needle inoculation (104 spirochetes/mouse) intradermally on the hind legs. For efficient maintenance of the shuttle plasmid in vivo, the mice were injected with streptomycin (300 μg/animal) every 2 d after the spirochetal inoculum and until the sacrifice of the animals. Separate experiments indicated that the injected doses of streptomycin do not interfere with B. burgdorferi pathogenicity or infectivity in mice (unpublished data). For tick acquisition studies, groups of mice (5 animals/group, 20 ticks/mouse) were allowed to be fed by naive ticks after 1 wk of B. burgdorferi infection, as previously described (48). The ticks were allowed to feed to repletion and detach from the mice, which usually occurred between 72–96 h. Immediately after engorgement, fed ticks were analyzed for qRT-PCR measurement of bmp expression or B. burgdorferi burden, as detailed earlier. Guts from parallel group of nymphs were also dissected under a microscope in PBS (20 μl/gut) after engorgement, and analyzed for BmpA and BmpB localization in wild-type spirochetes using immunofluorescence studies with specific Bmp antibodies as described above. For mutagenesis studies, mice were killed at day 14 and 18, and for complementation experimentation the mice were killed 14, 21, and 28 d after the initial inoculum. Joint, skin (ear), and bladder samples were isolated from infected mice, and B. burgdorferi burdens in tissue samples were measured by qPCR analysis, as described in PCR. Development of joint swelling in the infected mice was also evaluated on day 7, 14, 21, and 28.
Statistical analysis.
Results are expressed as the mean ± the SEM. The significance of the difference between the mean values of the groups was evaluated by Student's t test with StatView software (SAS Institute).
Online supplemental material.
Histological images of the ankle joints of mice infected with the wild-type B. burgdorferi, bmpA/B mutant and bmpAB complemented isolates are shown in Fig. S1. The online version of this article is available at http://www.jem.org/cgi/content/full/jem.20070962/DC1.
Supplemental Material
Acknowledgments
We sincerely thank Deborah Beck for excellent technical help; Bossis Ioannis for histopathology; Xin Li, Nengyin Liu, and Sukanya Narasimhan for helpful discussion; and Radha Iyer and Dionysios Liveris for the handling and processing of clinical specimens.
This work was supported by grants from the National Institutes of Health and American Heart Association.
The authors declare that they have no competing financial interests.
Abbreviations used: DECAL, differential expression analysis using a custom-amplified library; GST, glutathione S-transferase; q, quantitative.
U. Pal, P. Wang, and F. Bao contributed equally to this paper.
U. Pal's present address is Department of Veterinary Medicine, University of Maryland, College Park, MD 20742.
F. Bao's present address is Department of Microbiology, Kunming Medical College, Kunming 650031, China.
References
- 1.Steere, A.C., and L. Glickstein. 2004. Elucidation of Lyme arthritis. Nat. Rev. Immunol. 4:143–152. [DOI] [PubMed] [Google Scholar]
- 2.Barthold, S.W., D.H. Persing, A.L. Armstrong, and R.A. Peeples. 1991. Kinetics of Borrelia burgdorferi dissemination and evolution of disease following intradermal inoculation of mice. Am. J. Pathol. 163:263–273. [PMC free article] [PubMed] [Google Scholar]
- 3.Nadelman, R.B., and G.P. Wormser. 1998. Lyme borreliosis. Lancet. 352:557–565. [DOI] [PubMed] [Google Scholar]
- 4.Steere, A.C., J. Coburn, and L. Glickstein. 2004. The emergence of Lyme disease. J. Clin. Invest. 113:1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Radolf, J. 2005. Posttreatment chronic Lyme disease–what it is not. J. Infect. Dis. 192:948–949. [DOI] [PubMed] [Google Scholar]
- 6.Barthold, S.W., D.S. Beck, G.M. Hansen, G.A. Terwilliger, and K.D. Moody. 1990. Lyme borreliosis in selected strains and ages of laboratory mice. J. Infect. Dis. 162:133–138. [DOI] [PubMed] [Google Scholar]
- 7.Barthold, S.W., M.S. deSouza, J.L. Janotka, A.L. Smith, and D.H. Persing. 1993. Chronic Lyme borreliosis in the laboratory mouse. Am. J. Pathol. 143:959–971. [PMC free article] [PubMed] [Google Scholar]
- 8.Moody, K.D., and S.W. Barthold. 1998. Lyme borreliosis in laboratory mice. Lab. Anim. Sci. 48:168–171. [PubMed] [Google Scholar]
- 9.Zeidner, N.S., B.S. Schneider, M.C. Dolan, and J. Piesman. 2001. An analysis of spirochete load, strain, and pathology in a model of tick-transmitted Lyme borreliosis. Vector Borne Zoonotic Dis. 1:35–44. [DOI] [PubMed] [Google Scholar]
- 10.Barthold, S.W., M. DeSouza, E. Fikrig, and D.H. Persing. 1992. Lyme borreliosis in the laboratory mouse. In Lyme disease. S.E. Schuster, editor. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 223-242.
- 11.Barthold, S.W., M. deSouza, and S. Feng. 1996. Serum-mediated resolution of Lyme arthritis in mice. Lab. Invest. 74:57–67. [PubMed] [Google Scholar]
- 12.Barthold, S.W., E. Hodzic, S. Tunev, and S. Feng. 2006. Antibody-mediated disease remission in the mouse model of Lyme borreliosis. Infect. Immun. 74:4817–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fikrig, E., S. Barthold, M. Chen, I. Grewal, and R. Flavell. 1996. Protective antibodies in Lyme disease arise independently of CD40 ligand. J. Immunol. 157:1–4. [PubMed] [Google Scholar]
- 14.Xu, Q., S.V. Seemanapalli, L. Lomax, K. McShan, X. Li, E. Fikrig, and F.T. Liang. 2005. Association of linear plasmid 28-1 with an arthritic phenotype of Borrelia burgdorferi. Infect. Immun. 73:7208–7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng, S., E. Hodzic, and S.W. Barthold. 2000. Lyme arthritis resolution with antiserum to a 37-kilodalton Borrelia burgdorferi protein. Infect. Immun. 68:4169–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feng, S., E. Hodzic, K. Freet, and S.W. Barthold. 2003. Immunogenicity of Borrelia burgdorferi arthritis-related protein. Infect. Immun. 71:7211–7214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Silva, A.M., and E. Fikrig. 1997. Arthropod- and host-specific gene expression by Borrelia burgdorferi. J. Clin. Invest. 99:377–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rosa, P.A., K. Tilly, and P.E. Stewart. 2005. The burgeoning molecular genetics of the Lyme disease spirochete. Nat. Rev. Microbiol. 3:129–143. [DOI] [PubMed] [Google Scholar]
- 19.Schwan, T.G., and J. Piesman. 2002. Vector interactions and molecular adaptations of Lyme disease and relapsing fever spirochetes associated with transmission by ticks. Emerg. Infect. Dis. 8:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fisher, M.A., D. Grimm, A.K. Henion, A.F. Elias, P.E. Stewart, P.A. Rosa, and F.C. Gherardini. 2005. Borrelia burgdorferi sigma54 is required for mammalian infection and vector transmission but not for tick colonization. Proc. Natl. Acad. Sci. USA. 102:5162–5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang, F.T., F.K. Nelson, and E. Fikrig. 2002. Molecular adaptation of Borrelia burgdorferi in the murine host. J. Exp. Med. 196:275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narasimhan, S., M.J. Caimano, F.T. Liang, F. Santiago, M. Laskowski, M.T. Philipp, A.R. Pachner, J.D. Radolf, E. Fikrig, and M.J. Camaino. 2003. Borrelia burgdorferi transcriptome in the central nervous system of non-human primates. Proc. Natl. Acad. Sci. USA. 100:15953–15958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alland, D., I. Kramnik, T.R. Weisbrod, L. Otsubo, R. Cerny, L.P. Miller, W.R. Jacobs Jr., and B.R. Bloom. 1998. Identification of differentially expressed mRNA in prokaryotic organisms by customized amplification libraries (DECAL): the effect of isoniazid on gene expression in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA. 95:13227–13232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Purser, J.E., M.B. Lawrenz, M.J. Caimano, J.K. Howell, J.D. Radolf, and S.J. Norris. 2003. A plasmid-encoded nicotinamidase (PncA) is essential for infectivity of Borrelia burgdorferi in a mammalian host. Mol. Microbiol. 48:753–764. [DOI] [PubMed] [Google Scholar]
- 25.Fraser, C.M., S. Casjens, W.M. Huang, G.G. Sutton, R. Clayton, R. Lathigra, O. White, K.A. Ketchum, R. Dodson, E.K. Hickey, et al. 1997. Genomic sequence of a Lyme disease spirochete, Borrelia burgdorferi. Nature. 390:580–586. [DOI] [PubMed] [Google Scholar]
- 26.Dobrikova, E.Y., J. Bugrysheva, and F.C. Cabello. 2001. Two independent transcriptional units control the complex and simultaneous expression of the bmp paralogous chromosomal gene family in Borrelia burgdorferi. Mol. Microbiol. 39:370–378. [DOI] [PubMed] [Google Scholar]
- 27.Ramamoorthy, R., N.A. McClain, A. Gautam, and D. Scholl-Meeker. 2005. Expression of the bmpB gene of Borrelia burgdorferi is modulated by two distinct transcription termination events. J. Bacteriol. 187:2592–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin, J.J., A.V. Bryksin, H.P. Godfrey, and F.C. Cabello. 2004. Localization of BmpA on the exposed outer membrane of Borrelia burgdorferi by monospecific anti-recombinant BmpA rabbit antibodies. Infect. Immun. 72:2280–2287. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Anguita, J., M.N. Hedrick, and E. Fikrig. 2003. Adaptation of Borrelia burgdorferi in the tick and the mammalian host. FEMS Microbiol. Rev. 27:493–504. [DOI] [PubMed] [Google Scholar]
- 30.Pal, U., and E. Fikrig. 2003. Adaptation of Borrelia burgdorferi in the vector and vertebrate host. Microbes Infect. 5:659–666. [DOI] [PubMed] [Google Scholar]
- 31.Grimm, D., K. Tilly, R. Byram, P.E. Stewart, J.G. Krum, D.M. Bueschel, T.G. Schwan, P.F. Policastro, A.F. Elias, and P.A. Rosa. 2004. Outer-surface protein C of the Lyme disease spirochete: a protein induced in ticks for infection of mammals. Proc. Natl. Acad. Sci. USA. 101:3142–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pal, U., X. Yang, M. Chen, L.K. Bockenstedt, J.F. Anderson, R.A. Flavell, M.V. Norgard, and E. Fikrig. 2004. OspC facilitates Borrelia burgdorferi invasion of Ixodes scapularis salivary glands. J. Clin. Invest. 113:220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parveen, N., and J.M. Leong. 2000. Identification of a candidate glycosaminoglycan-binding adhesin of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:1220–1234. [DOI] [PubMed] [Google Scholar]
- 34.Schwan, T.G., and J. Piesman. 2000. Temporal changes in outer surface proteins A and C of the Lyme disease- associated spirochete, Borrelia burgdorferi, during the chain of infection in ticks and mice. J. Clin. Microbiol. 38:382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang, X.F., U. Pal, S.M. Alani, E. Fikrig, and M.V. Norgard. 2004. Essential role for OspA/B in the life cycle of the Lyme disease spirochete. J. Exp. Med. 199:641–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohnishi, J., J. Piesman, and A.M. de Silva. 2001. Antigenic and genetic heterogeneity of Borrelia burgdorferi populations transmitted by ticks. Proc. Natl. Acad. Sci. USA. 98:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hodzic, E., S. Feng, K.J. Freet, and S.W. Barthold. 2003. Borrelia burgdorferi population dynamics and prototype gene expression during infection of immunocompetent and immunodeficient mice. Infect. Immun. 71:5042–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barthold, S.W., and L.K. Bockenstedt. 1993. Passive immunizing activity of sera from mice infected with Borrelia burgdorferi. Infect. Immun. 61:4696–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Barthold, S.W., S. Feng, L.K. Bockenstedt, E. Fikrig, and K. Feen. 1997. Protective and arthritis-resolving activity in serum of mice actively infected with Borrelia burgdorferi. Clin. Infect. Dis. 25:S9–S17. [DOI] [PubMed] [Google Scholar]
- 40.Fikrig, E., S.W. Barthold, F.S. Kantor, and R.A. Flavell. 1990. Protection of mice against the Lyme disease agent by immunizing with recombinant OspA. Science. 250:553–556. [DOI] [PubMed] [Google Scholar]
- 41.Hanson, M.S., D.R. Cassatt, B.P. Guo, N.K. Patel, M.P. McCarthy, D.W. Dorward, and M. Hook. 1998. Active and passive immunity against Borrelia burgdorferi decorin binding protein A (DbpA) protects against infection. Infect. Immun. 66:2143–2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mbow, M.L., R.D. Gilmore Jr., and R.G. Titus. 1999. An OspC-specific monoclonal antibody passively protects mice from tick-transmitted infection by Borrelia burgdorferi B31. Infect. Immun. 67:5470–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKisic, M.D., and S.W. Barthold. 2000. T-cell-independent responses to Borrelia burgdorferi are critical for protective immunity and resolution of Lyme disease. Infect. Immun. 68:5190–5197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrenz, M.B., H. Kawabata, J.E. Purser, and S.J. Norris. 2002. Decreased electroporation efficiency in Borrelia burgdorferi containing linear plasmids lp25 and lp56: impact on transformation of infectious B. burgdorferi. Infect. Immun. 70:4798–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li, X., X. Liu, D.S. Beck, F.S. Kantor, and E. Fikrig. 2006. Borrelia burgdorferi lacking BBK32, a fibronectin-binding protein, retains full pathogenicity. Infect. Immun. 74:3305–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ojaimi, C., C. Brooks, D. Akins, S. Casjens, P. Rosa, A. Elias, A. Barbour, A. Jasinskas, J. Benach, L. Katonah, et al. 2002. Borrelia burgdorferi gene expression profiling with membrane-based arrays. Methods Enzymol. 358:165–177. [DOI] [PubMed] [Google Scholar]
- 47.Narasimhan, S., F. Santiago, R.A. Koski, B. Brei, J.F. Anderson, D. Fish, and E. Fikrig. 2002. Examination of the Borrelia burgdorferi transcriptome in Ixodes scapularis during feeding. J. Bacteriol. 184:3122–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pal, U., X. Li, T. Wang, R.R. Montgomery, N. Ramamoorthi, A.M. Desilva, F. Bao, X. Yang, M. Pypaert, D. Pradhan, et al. 2004. TROSPA, an Ixodes scapularis receptor for Borrelia burgdorferi. Cell. 119:457–468. [DOI] [PubMed] [Google Scholar]
- 49.Ledin, K.E., N.S. Zeidner, J.M. Ribeiro, B.J. Biggerstaff, M.C. Dolan, G. Dietrich, L. Vredevoe, and J. Piesman. 2005. Borreliacidal activity of saliva of the tick Amblyomma americanum. Med. Vet. Entomol. 19:90–95. [DOI] [PubMed] [Google Scholar]
- 50.Bolz, D.D., R.S. Sundsbak, Y. Ma, S. Akira, C.J. Kirschning, J.F. Zachary, J.H. Weis, and J.J. Weis. 2004. MyD88 plays a unique role in host defense but not arthritis development in Lyme disease. J. Immunol. 173:2003–2010. [DOI] [PubMed] [Google Scholar]
- 51.Wang, X., Y. Ma, J.H. Weis, J.F. Zachary, C.J. Kirschning, and J.J. Weis. 2005. Relative contributions of innate and acquired host responses to bacterial control and arthritis development in Lyme disease. Infect. Immun. 73:657–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frank, K.L., S.F. Bundle, M.E. Kresge, C.E. Eggers, and D.S. Samuels. 2003. aadA confers streptomycin rResistance in Borrelia burgdorferi. J. Bacteriol. 185:6723–6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}