

Abstract
Membrane proteins are regulated by the lipid bilayer composition. Specific lipid–protein interactions rarely are involved, which suggests that the regulation is due to changes in some general bilayer property (or properties). The hydrophobic coupling between a membrane-spanning protein and the surrounding bilayer means that protein conformational changes may be associated with a reversible, local bilayer deformation. Lipid bilayers are elastic bodies, and the energetic cost of the bilayer deformation contributes to the total energetic cost of the protein conformational change. The energetics and kinetics of the protein conformational changes therefore will be regulated by the bilayer elasticity, which is determined by the lipid composition. This hydrophobic coupling mechanism has been studied extensively in gramicidin channels, where the channel–bilayer hydrophobic interactions link a “conformational” change (the monomer↔dimer transition) to an elastic bilayer deformation. Gramicidin channels thus are regulated by the lipid bilayer elastic properties (thickness, monolayer equilibrium curvature, and compression and bending moduli). To investigate whether this hydrophobic coupling mechanism could be a general mechanism regulating membrane protein function, we examined whether voltage-dependent skeletal-muscle sodium channels, expressed in HEK293 cells, are regulated by bilayer elasticity, as monitored using gramicidin A (gA) channels. Nonphysiological amphiphiles (β-octyl-glucoside, Genapol X-100, Triton X-100, and reduced Triton X-100) that make lipid bilayers less “stiff”, as measured using gA channels, shift the voltage dependence of sodium channel inactivation toward more hyperpolarized potentials. At low amphiphile concentration, the magnitude of the shift is linearly correlated to the change in gA channel lifetime. Cholesterol-depletion, which also reduces bilayer stiffness, causes a similar shift in sodium channel inactivation. These results provide strong support for the notion that bilayer–protein hydrophobic coupling allows the bilayer elastic properties to regulate membrane protein function.
Keywords: gramicidin A, bilayer material properties, bilayer deformation energy, hydrophobic coupling, lipid–protein interactions
INTRODUCTION
Regulation of membrane protein function by the host bilayer lipid composition has long been enigmatic. There are numerous examples of such regulation (e.g., Bienvenüe and Marie, 1994; Dowhan, 1997; Killian, 1998; Lee, 2003), but the underlying mechanisms remain unclear, and specific lipid–protein interactions have been identified only in a few cases (e.g., Awasthi et al., 1971; Robinson, 1982; Hilgemann and Ball, 1996; Balla et al., 2000; Hilgemann et al., 2001; Hla et al., 2001). At ∼100 K, lipids can bind so tightly to membrane proteins that they are identified in crystal structures (Iwata et al., 1995; Luecke et al., 1999b; McAuley et al., 1999; Valiyaveetil et al., 2002). At ∼300 K, however, the residence time of most lipids in the annulus surrounding a protein is 10−7 to 10−6 s, and the specificity of lipid–protein interactions tends to be modest (Marsh and Horváth, 1998). Consistent with this modest specificity, accumulating evidence show that protein function may be regulated by the bilayer-collective properties such as bilayer thickness and monolayer equilibrium curvature (for reviews see Epand, 1997; Brown, 1997; Andersen et al., 1999; Bezrukov, 2000). This “nonspecific” regulation of protein function could result from protein–bilayer hydrophobic interactions that couple protein conformational changes to elastic deformations of the surrounding bilayer. This would provide a mechanistic basis for the regulation of protein function by the bilayer collective properties. In the present study we investigate whether voltage-dependent sodium channels expressed in HEK293 cells are regulated by such a mechanism.
The “nonspecific” bilayer regulation of protein function has been elucidated in studies on simple systems such as gramicidin and alamethicin channels. In these simple channels, it is well established that channel function is regulated by the elastic properties of the bilayer in which they are imbedded (Sawyer et al., 1989; Andersen et al., 1992; Keller et al., 1993; Lundbæk and Andersen, 1994, 1999; Lundbæk et al., 1996, 1997; Bezrukov et al., 1998). The situation is best understood in gramicidin channels. The structure of the gA channel is known (Arseniev et al., 1986; Ketchem et al., 1997; Townsley et al., 2001; Allen et al., 2003); gA channels are miniproteins formed by transmembrane dimerization of β6.3-helical subunits residing in each bilayer leaflet (O'Connell et al., 1990) (see Fig. 1 A), and the channel structure, specifically the helical pitch, does not vary when the bilayer thickness is varied (Katsaras et al., 1992).
Figure 1.

Hydrophobic coupling between channel conformational changes and lipid bilayer deformations/perturbations. (A) Formation of a gA channel by the trans-bilayer association of two monomers causes a bilayer deformation that involves the compression and bending of the bilayer leaflets adjacent to the channel. These deformations incur energetic costs with energy densities of 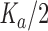 ·
·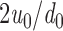 2 and
2 and 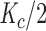 ·▽2
u
0−c
0
2, respectively. K
a and K
c denote the bilayer compression and bending moduli, 2u
0 the bilayer deformation − the difference between the actual bilayer thickness (l) and the (average) thickness of the unperturbed bilayer (d
0) − and c
0 the monolayer equilibrium curvature. The bilayer deformation energy (ΔG
def
0) is obtained by integrating the energy density from the channel/bilayer boundary to infinity (Huang, 1986; Helfrich and Jakobsson, 1990; Nielsen and Andersen, 2000). (B) The hydrophobic coupling between a membrane protein and the surrounding bilayer means that a protein conformational change will be associated with a deformation of the surrounding bilayer. The total free energy change associated with the protein conformational change is the sum of the free energy change associated with the intrinsic rearrangement of the protein and the free energy change associated with the bilayer deformation.
·▽2
u
0−c
0
2, respectively. K
a and K
c denote the bilayer compression and bending moduli, 2u
0 the bilayer deformation − the difference between the actual bilayer thickness (l) and the (average) thickness of the unperturbed bilayer (d
0) − and c
0 the monolayer equilibrium curvature. The bilayer deformation energy (ΔG
def
0) is obtained by integrating the energy density from the channel/bilayer boundary to infinity (Huang, 1986; Helfrich and Jakobsson, 1990; Nielsen and Andersen, 2000). (B) The hydrophobic coupling between a membrane protein and the surrounding bilayer means that a protein conformational change will be associated with a deformation of the surrounding bilayer. The total free energy change associated with the protein conformational change is the sum of the free energy change associated with the intrinsic rearrangement of the protein and the free energy change associated with the bilayer deformation.
gA channel formation and disappearance (the transmembrane monomer↔dimer transitions) represent well-defined “conformational” changes (e.g., Koeppe and Andersen, 1996). When the channel's hydrophobic length (l) is less than the hydrophobic thickness of an unperturbed bilayer (d 0), channel formation causes the bilayer hydrophobic core to adjust (or deform) in an attempt to match the channel's hydrophobic exterior (Elliott et al., 1983; Huang, 1986; Helfrich and Jakobsson, 1990; Lundbæk and Andersen, 1999) (see Fig. 1 A). Conversely, channel dissociation causes a release of the bilayer deformation.
Lipid bilayers are liquid crystals, with well-defined elastic properties, such as monolayer equilibrium curvature and compression and bending moduli (Helfrich, 1973; Evans and Hochmuth, 1978; Gruner, 1985; Evans and Needham, 1987; Rand and Parsegian, 1997). The energetic cost of a bilayer deformation therefore can be analyzed using the theory of elastic bilayer deformations (Huang, 1986; Helfrich and Jakobsson, 1990; Nielsen et al., 1998; Dan and Safran, 1998; Nielsen and Andersen, 2000; Partenskii and Jordan, 2002). In simple cases, such as the gA channel where the protein can be approximated as a cylinder, a mismatch between d 0 and l will induce a bilayer deformation that involves compression and bending of the two monolayers (Fig. 1 A). When the equilibrium monolayer curvature (c 0) is zero, the energetic cost of this deformation is equivalent to the deformation of a linear spring, and the deformation-free energy (ΔG def 0) is given by, e.g., Nielsen et al. (1998):
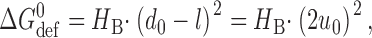 |
(1) |
where H B is a phenomenological spring constant whose magnitude is determined by the bilayer elastic moduli (the compression and bending moduli, K a and K c), d 0 and the protein radius, r 0. 2u 0 is the hydrophobic mismatch (2u 0 = d 0 − l). In case c 0 ≠ 0, the unperturbed bilayer (where 2u 0 = 0) will possess a curvature frustration free energy that contributes to ΔG def 0 (Gruner, 1985). In this case, the expression for ΔG def 0 becomes (Nielsen and Andersen, 2000):
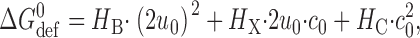 |
(2) |
where the coefficients H X and H C, like H B, are determined by d 0, K a, K c, and r 0 (Nielsen and Andersen, 2000).1 Both elastic moduli, as well as c 0, are functions of the profile of intermolecular interactions among the bilayer-forming lipids (Helfrich, 1981; Seddon, 1990; Petrov, 1999). Explicit expressions have proven difficult to obtain (e.g., Helfrich, 1981), but molecular dynamics simulations predict moduli that are in reasonable agreement with experimental values (Lindahl and Edholm, 2000).
Alterations in the intermolecular interactions among the bilayer lipids, and the lipids and imbedded proteins, will express themselves in terms of altered moduli (and curvature)—or altered coefficients in Eqs. 1 and 2, i.e., as changes in bilayer elasticity. The energetic consequences of altered interactions between integral, membrane-spanning proteins and their host bilayer can be described also using other terms, such as: changes in bilayer compression (Mouritsen and Bloom, 1984) or curvature frustration (Gruner, 1985; Epand, 1998) energy, which were combined using the theory of elastic bilayer deformations (Huang, 1986); changes in acyl chain packing (Fattal and Ben-Shaul, 1993), the lateral pressure profile across the bilayer (Cantor, 1997), or lipid-packing stress (Bezrukov, 2000); and changes in bilayer-free volume (Mitchell et al., 1990; Booth et al., 2001). Though couched in different terms, all the above descriptions represent different approaches to parameterize the lateral interactions among the bilayer-forming lipids and between the lipids and imbedded membrane proteins (see Lundbæk et al., 1997; Dan and Safran, 1998; Bezrukov, 2000; Nielsen and Andersen, 2000). The advantage of using the theory of elastic bilayer deformations, as formulated in Eqs. 1 and 2, is that the coefficients H B, H X, and H C can be evaluated knowing K a, K c, d 0, and r 0 (Nielsen and Andersen, 2000).
As ΔG def 0 contributes to the energetic cost of gA channel formation, changes in bilayer elasticity are reflected in channel appearance rate and lifetime, as well as in channel activity (the number of conducting channels in the bilayer, a measure of the channel dimerization constant [Sawyer et al., 1989]). When bilayer elasticity is evaluated from changes in gA channel lifetime as a function of d 0, the results conform to the predictions of Eq. 1 and the experimentally determined H B is in good agreement with predictions based on the theory of elastic bilayer deformations (Lundbæk and Andersen, 1999). The effects of amphiphiles on gA channel function agree with their effects on bilayer elasticity: micelle-forming amphiphiles, which decrease bilayer “stiffness”, increase gA channel appearance rate and lifetime; cholesterol, which increases bilayer stiffness, has the opposite effect (Sawyer et al., 1989; Lundbæk and Andersen, 1994; Lundbæk et al., 1996, 1997). The hydrophobic coupling between a gA channel and the surrounding bilayer thus enables bilayer elasticity to regulate channel function in a well-defined manner. Moreover, it is possible to obtain direct in situ measurements of bilayer elasticity using gA channels as molecular force transducers (Andersen et al., 1999; Lundbæk and Andersen, 1999)
Are integral membrane proteins regulated by bilayer elasticity? Quite apart from the extensive literature on bilayer regulation of protein function, membrane proteins would be expected to be subject to regulation by bilayer elasticity if their function depends on conformational changes that involve the protein/bilayer boundary. There is substantial evidence that integral membrane proteins undergo such changes. Early, low resolution structures of gap junction channels (Unwin and Ennis, 1984) and the nicotinic acetylcholine receptor (Unwin et al., 1988) showed that protein function is associated with changes in quaternary structure (changes in subunit tilt within the bilayer). Similar subunit rearrangements have been proposed based on high-resolution structures of the extra-membranous domains of glutamate-activated channels (Mayer et al., 2001). High-resolution structures of several transmembrane domains also provide evidence for reorganization of the transmembrane domains of H+- and Ca2+-gated potassium channels (Perozo et al., 1999; Jiang et al., 2002), bacteriorhodopsin (Luecke et al., 1999a; Vonck, 2000), the stretch-activated MscL channel (Chang et al., 1998; Perozo et al., 2002), the sarcoplasmic Ca2+-ATPase (Toyoshima and Nomura, 2002), as well as members of the major facilitator superfamily of transport proteins (Abramson et al., 2003; Hirai et al., 2003; Huang et al., 2003). Chemical cross-linking and spectroscopic studies similarly provide evidence for the movement of membrane-spanning α-helices relative to each other (Sakmar, 1998; Kaback et al., 2001; Sukharev et al., 2001). Moreover, conformational changes in membrane proteins modulate the bilayer structure. Rhodopsin activation, for example, changes the organization of the adjacent phospholipids (Isele et al., 2000).
As for gA channels, the hydrophobic coupling between a membrane protein and the surrounding bilayer will tend to cause the bilayer's hydrophobic core to adjust in an attempt to match the protein's hydrophobic exterior (Owicki et al., 1978; Mouritsen and Bloom, 1984)—and protein conformational changes that involve the protein/bilayer boundary will perturb the bilayer. In Fig. 1 B the protein conformational change involves a change in hydrophobic length as well as shape—and the bilayer perturbation is represented by a local bilayer thinning (and monolayer bending). As is the case for “simple” length changes, perturbations in local lipid packing due to changes in “shape” also will incur an energetic cost due to altered lipid–lipid and lipid–protein interactions (Dan and Safran, 1998). The free energy change associated with the bilayer deformation (ΔG def 0) contributes to the total free energy change associated with protein conformational change (ΔG tot 0). To the extent that ΔG def 0 is significant—on the order of a few kJ/mol, or larger—its contribution to ΔG tot 0 will be important for the distribution between protein conformational states.
It is, in this context, important that the volumetric compressibility modulus of proteins (Gekko and Noguchi, 1979) is one to two orders of magnitude higher than that of lipids (Liu and Kay, 1977), and bilayer-spanning α-helices tend to be fairly rigid, compared with lipid bilayers, meaning that they will remain in an α-helical conformation irrespective of any hydrophobic mismatch (Zhang et al., 1992). The bilayer-protein hydrophobic coupling therefore will have little impact on any given protein conformation; but if two protein conformations differ in free energy (ΔG tot 0) by a few kJ/mole, changes in ΔG def 0 will be important for protein function (Sackmann, 1984; Gruner, 1991; Andersen et al., 1992; Lundbæk and Andersen, 1994). The protein–bilayer hydrophobic coupling thus provides a mechanistic link between bilayer lipid elasticity, which varies with bilayer composition (Evans and Needham, 1987; Evans and Rawicz, 1990; Rawicz et al., 2000; Brown et al., 2002) and membrane protein function. In general, however, there is not sufficient information to evaluate a priori how changes in bilayer properties should alter ΔG def 0 and protein function (Perozo et al., 2002). It thus becomes necessary to correlate changes in membrane protein function to changes in bilayer properties, using another (reference) system, and thereby determine whether there could be a causal link between changes in bilayer properties and membrane protein function.
We have previously provided qualitative support for such a link, as the effects of amphiphiles on N-type calcium channels correlate with the changes in bilayer elasticity, evaluated using gA channels (Lundbæk et al., 1996). Two amphiphiles that increase gA channel appearance rate and lifetime, β-octyl-glucoside (βOG) and Triton X-100 (TX100) (see Fig. 2) cause a hyperpolarizing shift in steady-state N-type calcium channel availability.
Figure 2.

The molecules used in this study. (Top) Micelle-forming amphiphiles promote positive monolayer equilibrium curvature by increasing the average volume of the lipid head groups in a monolayer. The shading of the head group regions GX100, TX100 and rTX100 denotes the approximate region filled by the polyoxyethylene chain (estimate of radius based on Kuga, 1981). (Bottom) Cholesterol promotes negative curvature by decreasing the volume of the head group region. The stippled lines illustrate the hydrophilic-hydrophobic interface.
Conversely, cholesterol, which decreases gA channel lifetime, causes a depolarizing shift in steady-state channel availability (Lundbæk et al., 1996).
The present study provides a quantitative evaluation of the effects of bilayer elasticity on membrane protein function. This evaluation was done by comparing the effects of amphiphiles on the function of gA channels and skeletal-muscle voltage-dependent sodium channels, which have a simpler subunit composition and regulation than N-type calcium channels. The experimental design does not assume similarity of the conformational changes in gA and sodium channels; but if the effects of structurally different micelle-forming amphiphiles, on sodium channel function, correlate quantitatively with changes in gA lifetime—and further these effects are the opposite of those caused by cholesterol—it would provide strong support for the importance of bilayer elasticity. Given the complexity of bilayer–membrane protein interactions, we varied both the hydrophobic and the hydrophilic ends of the amphiphiles, but limited the variations to the compounds shown in Fig. 2.
We show: first, that βOG and TX100, as in N-type calcium channels, cause a hyperpolarizing shift in steady-state channel availability, whereas the voltage-dependent activation appears to be unaltered; second, that these effects are reproduced by reduced Triton X-100 (rTX100) and Genapol X-100 (GX100) (Fig. 2), two other micelle-forming amphiphiles that increase gA channel duration; third, that cholesterol causes a depolarizing shift in channel availability; fourth, that there is a quantitative correlation between the effects on gA channels in planar bilayers and on voltage-dependent sodium channels in living cells; and fifth, that the effects of the amphiphiles on the properties of plasma membranes in living cells correlate with their effects on planar, lipid bilayers. We conclude that sodium channel function most likely is regulated by the bilayer elasticity, and that gramicidin channels provide a tool for quantitative studies of the role of bilayer elasticity (and hydrophobic coupling) for membrane protein function.
MATERIALS AND METHODS
Transfection of HEK293 Cells
Full-length cDNA encoding the muscle sodium channel μ1 α-subunit (Trimmer et al., 1989) was a generous gift from G. Mandel (State University of New York Stony Brook, NY). The cDNA was inserted into a pBK-CMV vector (Stratagene) and transfected into HEK293 cells using the Lipofectamine method (Life Technologies). HEK293 cells were grown on coverslips in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (GIBCO BRL), 100 U/ml penicillin (GIBCO BRL), and 100 μg/ml streptomycin (GIBCO BRL), at 37°C ambient atmosphere with 5% CO2. Stable clones expressing the sodium channel gene were selected using 0.5 mg/ml G418 (GIBCO BRL). In successfully transfected cells the average peak current was 1,560 ± 210 pA (mean ± SEM, n = 10). The depolarization induced membrane currents were inhibited >99% by 1 μM TTX. In nontransfected cells the average peak current was 20 ± 10 pA (mean ± SEM, n = 22), which is ∼1% of the peak currents in the transfected cells.
Whole-cell Voltage-clamp Experiments
Sodium channel currents were studied using whole-cell voltage clamp (Hamill et al., 1981) at room temperature (21–24°C). Patch pipettes had a tip resistance of 2–4 MΩ. Voltage-pulse generation and data acquisition were controlled using an Axopatch 200A amplifier and pClamp 6.0 (Axon Instruments, Inc.). Linear leakage corrections were done online using a P/4 pulse protocol (Armstrong and Bezanilla, 1974) or during subsequent analysis using the leakage currents induced by a step in membrane potential from −80 to −90 mV. Unless otherwise noted, the currents were filtered at 10 kHz and sampled at 40 kHz. Pipette and membrane capacitances were electronically compensated for. Only experiments with a series resistance below 4 MΩ (after 80% compensation) and a compensated voltage drop across the series resistance of less than 5 mV were used for analysis. The bathing solution was: 140 mM NaCl, 4 mM KCl, 2 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 10 mM glucose, adjusted to pH 7.4 with NaOH. The electrode solution was: 140 mM CsCl, 20 mM HEPES, 11 mM EGTA, 1 mM CaCl2, 1.8 mM MgATP, 0.46 mM Na3GTP, adjusted to pH 7.3 with CsOH. Test solutions of βOG (>97% purity), GX100 (protein grade), TX100 (protein grade), and rTX100 (protein grade), all from Calbiochem, were prepared by dissolving the compounds directly into the bathing solution. Control or test solutions were applied to the cells using a fast superfusion system (Konnerth et al., 1987). The cell-membrane leak conductance in control cells (0.6 ± 0.2 nS, n = 12) was not significantly different from that in the presence of 30 μM TX100 (1.0 ± 0.4 nS, n = 6), 2.5 mM βOG (0.3 ± 0.1 nS, n = 6), or in cholesterol-enriched (0.7 ± 0.3 nS, n = 8) or cholesterol-depleted cells (0.9 ± 0.2 nS, n = 7) (P > 0.05). “Timed control experiments”, done at a time establishing the whole-cell configuration that matched that in experiments using amphiphiles, were done for all experimental protocols.
Manipulation of Cellular Cholesterol Content
Cell membrane cholesterol content was increased by exposure to cholesterol complexed with methylated β-cyclodextrin (MβCD) (Christian et al., 1997). 5 mM MβCD–cholesterol complex (MβCD–cholesterol ratio 10:1) was prepared by adding cholesterol (Sigma-Aldrich) from a stock solution in chloroform–methanol 1:1 (vol:vol) to a glass test tube and evaporating the solvent under nitrogen. MβCD (average MW 1338; Cyclodextrin Technologies Development) dissolved in 5 ml growth medium was added to the glass tube. The solution was vortexed, sonicated for 3 min, and incubated in a rotating water bath at 37°C overnight. Prior to use, the solution was filtered through a 0.45-μm syringe filter to remove excess cholesterol crystals. The cells were exposed to this solution for 21 h. In a second group of cells, the membrane cholesterol content was decreased by exposure to 5 mM cholesterol-free MβCD for 21 h. (This incubation time was chosen for practical reasons, shorter exposures have been used previously [Christian et al., 1997].) That the effects of cholesterol-depletion, in the present study, were reversed by exposure to MβCD–cholesterol shows that they are not due to irreversible changes in cell function.) Experimental conditions in these experiments were as described above, except that MβCD without cholesterol was dissolved in growth medium. In a third group of cells, cholesterol-depletion induced by exposure to cholesterol-free MβCD for 21 h was reversed by a final 2-h exposure to 5 mM MβCD–cholesterol complex (10:1) prepared as described above, but dissolved in the bathing solution used in the patch clamp experiments.
To measure the cell-cholesterol content the growth medium was removed and cell monolayers on coverslips were washed three times with Hanks' balanced salt solution. Cell lipid was extracted with isopropanol for 30 min and cell protein in the lipid-extracted monolayer was recovered with NaOH (McCloskey et al., 1987). The cholesterol content was measured using the “cholesterol CHOD-PAP method” (Boehringer). Cell protein was measured photometrically (Pesce and Strande, 1973).
Data Analysis
All curve fitting was done using the nonlinear least-squares algorithm in Origin 7.0 (Microcal Software). The voltage dependence of channel activation and inactivation was obtained by fitting Hodgkin-Huxley (HH)-type relations (e.g., Hille, 2001) to the peak currents. The voltage dependence of activation was determined by fitting the relation:
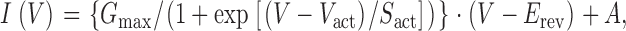 |
to the results. I(V) is the peak current at the test potential V, G max the maximal conductance, V act the voltage of half maximal activation, S act a slope factor, E rev the reversal potential, and A an offset determined by the peak-to-peak noise. (This offset appears because the baseline current is determined as the mean current, whereas the fitting was to the lower envelope of the current trace, which produced a systematic offset equal to half the peak-to-peak current noise.) The voltage dependence of the steady-state inactivation was obtained by fitting a two-state Boltzmann distribution to the peak currents:
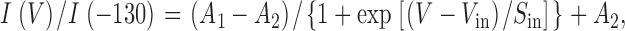 |
where I(V) is the peak current after a prepulse V, I(−130) the peak current at a prepulse of −130 mV, V in the voltage of half maximal inactivation, A 1 the maximum value in the fit (A 1 ∼ 1), A 2 a constant determined by noninactivating currents and noise, and S in a slope factor.
gA Channels in Planar Lipid Bilayers
gA channels were studied using the bilayer punch method (Andersen, 1983) at 25 ± 1°C. The bilayers were formed from a dioleoylphosphatidylcholine (DOPC)/n-decane solution (2% wt/vol). The electrolyte solution was unbuffered 1 M NaCl. gA was added to both aqueous solutions from an ethanolic stock; similarly, the micelle-forming amphiphiles were added from ethanolic stock solutions. The amount of ethanol in the chamber never exceeded 0.5% (vol:vol), which has no effect on channel properties (Sawyer et al., 1990). The current signal was recorded using a Dagan 3,900 patch-clamp amplifier (Dagan Instruments). The applied potential was ±200 mV. The current signal was filtered at 2,000 Hz, digitized and sampled at 20,000 Hz, digitally filtered at 500 Hz, and the current transitions were detected on-line (Andersen, 1983); single-channel current transition amplitudes and lifetimes were determined as described previously (Sawyer et al., 1989) with software written using AxoBasic (Axon Instruments, Inc.) or Visual Basic (Microsoft). The lifetime distributions were fitted by single exponential distributions, N(t) = N(0) exp(−t/τ), where N(t) is the number of channels with duration longer than t, N(0) the total number of channels, and τ the average channel lifetime.
gA Channels in HEK293 Cells
The effects of the amphiphiles on gA channels in the plasma membrane of native HEK293 cells were studied using whole-cell voltage clamp. The bathing and electrode solutions were the same as in the experiments with voltage-dependent sodium channels, except that 20 mM sucrose had been added to the bathing solution to prevent cell swelling. The membrane leak conductance in the native HEK293 cells (∼1.0 nS) was comparable to the value in transfected cells. gA from an ethanolic stock solution was added to the bathing solution in the experimental chamber to a nominal gA concentration of 10–100 μM, ∼106-fold higher than in the planar bilayer experiments. (The ethanol concentration in the experimental chamber never exceeded 0.2%, which has no effect on the membrane conductance.) When the membrane conductance in the presence of gA had increased ∼10-fold relative to the control value, βOG or TX100 was added by superfusing the cells with bathing solution, in which these compounds had been dissolved directly.
Statistical Analysis
Statistical analysis was done using SigmaStat 2.0 and either Student's t test or one way analysis of variance (ANOVA) with Dunnetts' method as post hoc test. Results are given as mean ± SEM, (n) where n is the number of cells.
Online Supplemental Material
A detailed kinetic analysis of channel gating at +20 mV is available at http://www.jgp.org/cgi/content/full/jgp.200308996/DC1.
RESULTS
βOG and TX100 Reversibly Promote Sodium Channel Inactivation
βOG and TX100 reversibly inhibit sodium currents, and the inhibition varies as a function of the prepulse potential. Fig. 3 A shows the changes in sodium currents in response to a 25-s application of 5 mM βOG or 30 μM TX100. After a 300-ms prepulse to either −130 or −60 mV, the cells were depolarized to +20 mV. Both βOG and TX100 decrease the current, but the effect is larger after the −60-mV prepulse than after the −130-mV prepulse (Fig. 3 A). There is little apparent effect on the time course of activation and inactivation at +20 mV (see online supplemental material). Fig. 3 B shows the effects on the normalized peak currents. Prior to the application of βOG or TX100, the peak currents after the −60- and −130-mV prepulse on average differed by a factor 2.
Figure 3.

Effects of βOG and TX100 on sodium channels. (A) Current traces showing the reversible inhibition of sodium currents by βOG and TX100. (B) The time course of peak current inhibition during and after a 25-s application of 30 μM TX100 or 5 mM βOG. Pulse protocol: every 5 s the cells were depolarized to +20 mV from a 300 ms prepulse to either −60 mV (•) or −130 mV (○). Holding potential −80 mV. The horizontal bar denotes the time period of application. Mean ± SEM (n = 3, 4, TX100, βOG). (C) Time course of peak current inhibition during 9-min application of 30 μM TX100 followed by a 5-min washout period (mean ± range, n = 2). Horizontal bars denote time periods of application and washout, respectively.
βOG reduced the normalized peak currents after a −60-mV prepulse by 60%, whereas the reduction was only 42% after a −130-mV prepulse. Using the same prepulse potentials, TX100 reduced the peak currents by 61% and 17%, respectively. That the inhibition after the −60-mV prepulse is larger than after the −130-mV prepulse suggests that inhibition is coupled to sodium channel inactivation, and that βOG and TX100 shifts the equilibrium between the closed and inactivated state(s) toward the inactivated state(s). The promotion of inactivation is reversible: one min after βOG or TX100 had been removed from the bathing solution, the ratio of the peak currents after the −60- and −130-mV prepulse had returned to the preapplication value (Fig. 3, A and B). In the more complex protocols, described below, the amphiphiles were applied for at least 5 min and the changes in peak current, and the ratio of the peak currents, were not always fully reversible.
The decrease in the peak currents was qualitatively similar during longer applications. Fig. 3 C shows results from experiments where 30 μM TX100 was applied for 9 min. Every 10 s the cells were depolarized to +20 mV, from a −80-mV holding potential. The peak currents decreased monotonically, with a fast decay to 50% of the initial value over the first 10 s, and a slower decay to an approximately steady-state value over the next 5 min. After a 5 min washout period, the decrease in peak current amplitude could be reversed only partially (Fig. 3 C).
The effects of βOG and TX100 on sodium channel inactivation were investigated further (Fig. 4) . In both the absence and presence of these amphiphiles, the voltage dependence of the steady-state inactivation after a 300-ms prepulse to varying potentials was well fitted by a two-state Boltzmann distribution (Fig. 4, top and bottom, shows measured and normalized peak currents, respectively.)
Figure 4.

The voltage dependence of steady-state inactivation can be fitted by a two-state model in both the absence and the presence of the micelle-forming compounds. Peak currents (top) and normalized peak currents (bottom). Left: (•) control; (▾) 10 μM TX100; (▿) washout. Center: (•) control; (▾) 2.5 mM βOG; (▿) washout. Right: timed control experiments. (•) control 1; (▾) control 2; (▿) control 3. Pulse protocol: every 5 s the cells were depolarized to +20 mV from 300-ms prepulses to potentials varying from −130 to +50 mV. Holding potential −80 mV. Results from single experiments.
The effects of βOG and TX100 on the voltage of half-maximal inactivation (V in) and the slope factor of the inactivation curve (S in) are shown in Fig. 5 .
Figure 5.

Changes in channel steady-state availability induced by βOG and TX100. 10 μM TX100 (▾) and 2.5 mM βOG (▴) cause a negative shift in V in (top) and a positive shift in S in (bottom). (P < 0.05) For comparison (•) shows results obtained in timed control experiments. Pulse protocol as in Fig. 4. Mean ± SEM based on the changes in individual cells (n = 7, 6, 7, TX100, βOG, control).
In the control cells V in and S in were −61.2 ± 1.4 mV and 7.0 ± 0.4 mV, respectively. 2.5 mM βOG or 10 μM TX100 changed V in by −8.3 ± 1.6 mV or −9.8 ± 1.0 mV, and S in by +2.2 ± 0.6 mV or +1.7 ± 0.3 mV, respectively. (The changes are given as the average of the changes in individual cells.) These shifts in V in and S in are much larger than the shifts in timed control experiments. We conclude that βOG and TX100 promote steady-state inactivation.
In addition to the current inhibition that is coupled to inactivation, both amphiphiles inhibit the sodium currents by a mechanism that does not appear to be coupled to inactivation. Steady-state inactivation, in both the absence and presence of TX100 or βOG, is abolished after a 300-ms prepulse to −130 mV (Fig. 4); but both amphiphiles cause some current inhibition after such a prepulse (Figs. 3 and 4). We did not pursue the basis for this contribution to the current inhibition.
The effects of βOG and TX100 are reproduced by rTX100 and GX100, two other micelle-forming amphiphiles (Fig. 2) of different structure. Both compounds promote sodium channel inactivation in a reversible manner. Fig. 6 shows the concentration dependence of the effects of βOG, GX100, TX100, and rTX100 on V in; results from timed control experiments are shown for comparison.
Figure 6.

Concentration dependence of the effects of the amphiphiles on V in. Top left: Effects of 10, 30, and 100 μM TX100. Top right: Effects of 0.3, 1, and 2.5 mM βOG. Middle left: Effects of 10 and 30 μM rTX100. Middle right: Effects of 4.5 and 15 μM GX100. Bottom: Timed control experiments. Pulse protocol: every 5 s the cells were depolarized to +20 mV from a 20 ms prepulse to potentials varying from −130 to +50 mV. Holding potential −80 mV. Filter 10 kHz, sampling rate 20 kHz. Mean ± SEM. n = 4, 6, 4 (3, 10, 30 μM TX100); 3, 6, 7 (0.3, 1, 2.5 mM βOG); 4, 1 (10, 30 μM rTX100); 3, 5 (4.5, 15 μM GX100); 6 (timed control).
To keep the exposure to the compounds as short as possible, the prepulse duration was 20 ms, and the shift in V in was totally reversible. The adsorption coefficient of amphiphiles is expected to vary with their CMC (Bullock and Cohen, 1986; Sawyer et al., 1989; Heerklotz and Seelig, 2000). To achieve similar mole-fractions in the bilayer, the amphiphiles were applied at concentrations that are comparable relative to their CMC (βOG, 25 mM; GX100, 150 μM, TX100, 300 μM; rTX100, 250 μM; Neugebauer, 1987).2 These different micelle-forming amphiphiles all promote steady-state inactivation in a reversible, concentration-dependent manner, and their effects are comparable when considering the different CMCs.
Lack of Effects on Sodium Channel Voltage Activation
To test whether the changes in sodium channel inactivation reflect a general change in all aspects of channel function, we investigated the effects of βOG or TX100 on the channels' voltage activation (Fig. 7) .
Figure 7.
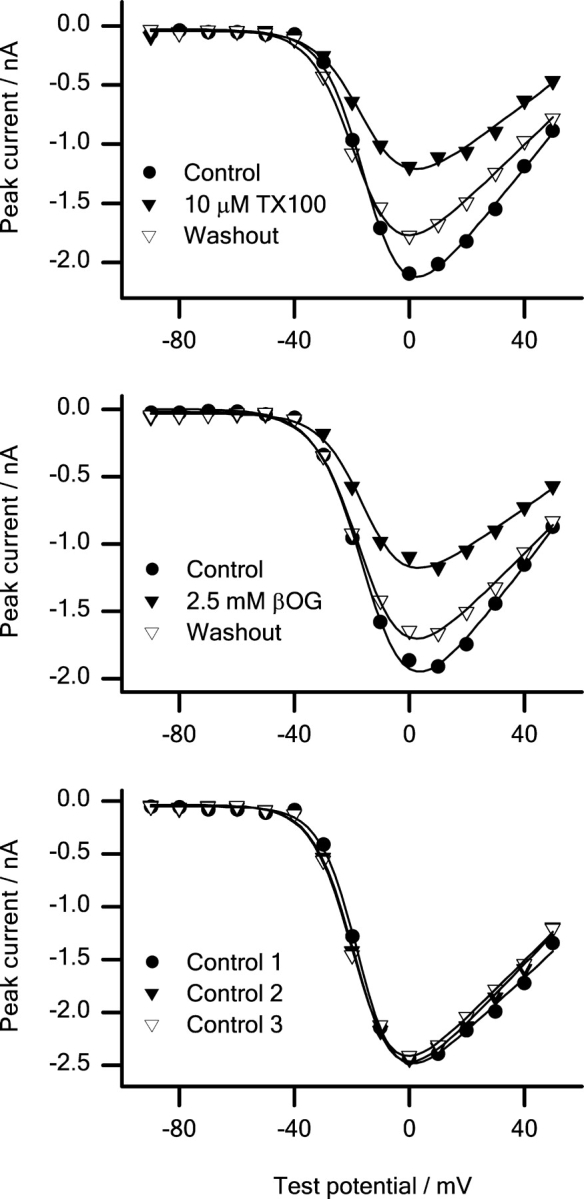
Peak sodium currents versus test potential in the absence and the presence of the micelle-forming amphiphiles. In all cases the peak current voltage relations were fitted with a two-state model (see materials and methods). Top: (•) control; (▾) 10 μM TX100; (▿) washout. Middle: (•) control; (▾) 2.5 mM βOG; (▿) Washout. Bottom: Timed control experiments. (•) Control 1; (▾) control 2; (▿) control 3. Pulse protocol: every 5 s the cells were depolarized from a holding potential of −80 mV to test potentials ranging from −90 to +50 mV. Results from single experiments.
The experiments were done only at 2.5 mM βOG and 10 μM TX10, as the current inhibition at higher amphiphile concentrations was so large that the analysis would be compromised by the noise in the current traces. In both the absence and presence of βOG or TX100, the current-voltage relation could be described by a conventional HH type relation (see materials and methods). In control cells, the voltage of half-maximal activation (V act), the slope factor of the voltage-activation relation (S act), and the reversal potential (E rev) were −13.5 ± 1.3 mV, −7.4 ± 0.3 mV, and 91.3 ± 5.2 mV, respectively. Neither 2.5 mM βOG nor 10 μM TX100 altered any of these values (Table I) .
TABLE I.
Sodium Channel Activation in Cells Modified by TX100 or βOG
| ΔV act/mV | ΔS act/mV | ΔE rev/mV | |
|---|---|---|---|
| 10 μM TX100 | −0.1 ± 0.6 | −0.5 ± 0.2 | −1.8 ± 1.4 |
| 2.5 mM βOG | 1.2 ± 0.5 | 0.4 ± 0.3 | 2.8 ± 3.0 |
| Timed control | −0.8 ± 0.9 | −0.1 ± 0.3 | −3.7 ± 4.2 |
Results are given as mean ± SEM of changes in individual cells (n = 3, 5, 4, TX100, βOG, and control). The changes induced by TX100 or βOG are not significantly different from the changes in timed control cells (P > 0.05).
The effects on channel activation are minimal despite the significant alterations in channel inactivation, this was confirmed in a kinetic analysis of channel activation at +20 mV (available as online supplemental material at http://www.jgp.org/cgi/content/full/jgp.200308996/DC1).
Effects on Inactivation Are Not Due to Unspecific Membrane Damage
βOG, GX100, TX100, and rTX100 are detergents that, at concentrations close to (and above) their CMC, may cause irreversible cell-membrane damage and membrane-protein denaturation (Weltzien, 1979). The promotion of sodium channel inactivation is not due to such extraneous, irreversible effects. First, the nominal concentrations of the amphiphiles always were well below their CMC. Second, the modulation of inactivation is reversible, at least for short applications (Figs. 3 and 6). (When the amphiphiles were applied for 5–15 min the changes in peak currents and midpoint potentials were not always completely reversed by a 5-min washout period. We did not investigate whether the apparently irreversible effects of longer applications could be reversed by even longer washouts.) Third, the membrane leak conductance is not increased by 30 μM TX100 or 2.5 mM βOG (see materials and methods). Fourth, the effects of the amphiphiles are specific in the sense that they depend on the physiological state of the channel (Fig. 3).
We also can exclude that promotion of inactivation is due to other irreversible, time-dependent processes caused by the whole-cell configuration (changes in bilayer–cytoskeleton or channel–cytoskeleton interactions or the loss of channel subunits) because: first, the changes in channel inactivation are reversible; and second, all experiments were compared with timed control experiments (which in their own right show only modest changes in V in; Figs. 5 and 6).
Membrane Cholesterol Inhibits Sodium Channel Inactivation
TX100 and other micelle-forming amphiphiles increase bilayer elasticity, as evaluated from changes in gA channel lifetimes (Sawyer et al., 1989); cholesterol decreases bilayer elasticity as evaluated from changes in bilayer elastic moduli (Evans and Rawicz, 1990; Needham and Nunn, 1990) and gA channel lifetimes (Lundbæk et al., 1996). Thus, if the amphiphile-induced changes in sodium channel gating arise simply from changes in the bilayer elasticity, cholesterol-depletion should to a first approximation have effects similar to those observed after adding the micelle-forming amphiphiles—and cholesterol enrichment should have the opposite effects.
The cholesterol content of HEK293 cells was modified using MβCD (see materials and methods). In control cells, the total cholesterol content was 25.0 ± 6.0 μg cholesterol/mg protein (n = 3), which is comparable to the value found in other cells types (Christian et al., 1997; Levitan et al., 2000). After incubation with 5 mM MβCD–cholesterol (10:1) for 21 h, the cholesterol content was increased to 53.0 ± 6.0 μg cholesterol/mg protein (n = 3). After incubation with cholesterol-free MβCD for 21 h, the cholesterol content was decreased to 7.2 ± 1.3 μg (n = 3) cholesterol/mg protein. When the cholesterol-depleted cells were incubated for 2 h with MβCD–cholesterol, the cholesterol content was increased to 41.6 ± 4.1 μg cholesterol/mg protein. (Cholesterol-depletion causes dissolution of sphingolipid- and cholesterol-enriched membrane rafts [e.g., Brown and London, 2000].) It is not known whether sodium channels reside in rafts; but, whether the channels reside in rafts or not, cholesterol-depletion will decrease the cholesterol content of the bilayer surrounding the channel.)
Fig. 8 shows the effects of changes in cell-cholesterol content on the voltage dependence of steady-state inactivation. In all cases, the voltage dependence of inactivation is well described by a two-state Boltzmann distribution (Fig. 8 A). Fig. 8 B shows the effects of modulation of cell cholesterol content on V in and S in. These experiments were done as a separate series, after the experiments with βOG and TX100, and the effects of cholesterol were compared with a separate control group investigated during the same time period. Further, as each cell cannot be used as its own control, the results for the cholesterol experiments are reported as absolute values rather than relative changes.
Figure 8.

Effect of changes in membrane cholesterol content on sodium channel steady-state availability. (A) Steady-state availability curves obtained in cells that are cholesterol-depleted or -enriched. At all cholesterol contents the voltage dependence of steady-state inactivation is fitted with a two-state model. (•) Control; (▾) cholesterol depleted; (▿) cholesterol enriched. Results from single experiments. (B) Effects of altering cell membrane cholesterol content on V in (Top) and S in (Bottom). Experimental conditions as in Fig. 4. Cholesterol-depletion changed both V in and S in compared with the control group (P < 0.05). Reversal of cholesterol-depletion again changed both V in and S in compared with the cholesterol-depleted group (P < 0.05). Mean ± SEM (n = 9, 8, 14, 6, cholesterol enriched, cholesterol-depleted, cholesterol-depletion reversed, timed control experiments).
In control cells, V in and S in were −61.1 ± 2.0 mV and 8.3 ± 0.4 mV, respectively. Increasing the cell cholesterol content altered neither V in nor S in. Decreasing cell cholesterol, however, caused a −8.9-mV shift in V in and a +2.1-mV shift in S in. The effects of cholesterol-depletion were partially reversed by subsequent cholesterol enrichment: a 2-h exposure of cholesterol-depleted cells to MβCD–cholesterol complex caused +5.9-mV and −2.3-mV shifts in V in and S in relative to the values in the cholesterol-depleted cells (Fig. 8 B). As MβCD is present both during depletion and enrichment of cell cholesterol, the effects of cholesterol-free MβCD are not due to a direct effect of MβCD itself.
Decreasing the cell-cholesterol content has similar effects as adding micelle-forming amphiphiles: a hyperpolarizing shift in V in and a positive shift in S in. Increasing the cholesterol content of cholesterol-depleted cells has the opposite effects; but the effects of cholesterol on inactivation appear to saturate when the cholesterol content is increased above the level in control cells. This latter result is not due to sequestration of the added cholesterol into a pool that does not affect the sodium channels, as cholesterol enrichment causes a shift in the voltage dependence of channel activation (see below).
Effects of Cholesterol on Sodium Channel Voltage Activation
In all experiments where the cell cholesterol content was manipulated, the peak current-voltage relations were well described by an HH description. Table II summarizes the effects of the cholesterol manipulations on V act, S act, and E rev.
TABLE II.
Effects of Cholesterol on Sodium Channel Activation
| V act/mV | S act/mV | E rev/mV | |
|---|---|---|---|
| Control | −16.0 ± 1.6 | −6.6 ± 0.4 | 84.1 ± 2.6 |
| Cholesterol enriched | −10.5 ± 1.2a | −8.0 ± 0.2a | 79.7 ± 3.0 |
| Cholesterol depleted | −17.1 ± 1.9 | −7.0 ± 0.3 | 73.5 ± 2.8 |
| Cholesterol-depletion reversed | −9.6 ± 1.8a | −8.7 ± 0.3a | 85.9 ± 0.9a |
Mean ± SEM of changes in individual cells (n = 6, 9, 8, 6, control, cholesterol enriched, cholesterol-depleted, cholesterol-depletion reversed). Effects of cholesterol-depletion and enrichment were compared to control cells. Effects of reversal of cholesterol-depletion were compared to cholesterol-depleted cells.
Statistically significant changes (P < 0.05).
Decreasing cell cholesterol did not significantly alter any of the gating parameters in Table II. By contrast, increasing the cholesterol content in control or cholesterol-depleted cells caused a positive shift in V act and a negative shift in S act. Whereas V act and S act were similar in control and cholesterol-depleted cells, E rev became more positive when cell cholesterol was increased in cholesterol-depleted cells, but not in control cells. Cholesterol enrichment also decreased the peak current amplitudes (from −2,510 ± 550 pA to −1,050 ± 140 pA after a 300-ms prepulse at −130 mV, P < 0.05), whereas cholesterol-depletion had no effect on the peak current amplitudes (=2,330 ± 450 pA). The effects of cholesterol-depletion on inactivation are fairly straightforward, and consistent with the changes seen with TX100 and βOG. We do not understand why cholesterol enrichment alters channel activation with no effect on inactivation.
Kinetics of Inactivation
The effects of βOG, TX100, and cholesterol on the time course of inactivation were investigated at −60 mV, which is close to V in for the control situation (Figs. 4 and 8). The cells were depolarized from a holding potential of −110 to −60 mV, for 0.4 to 810 ms, and channel availability was determined using test pulses to +20 mV (Fig. 9) .
Figure 9.

Effects of amphiphiles on the time course of inactivation at −60 mV. Every 10 s the cells were depolarized from a holding potential of −110 mV to a prepulse of −60 mV for a time interval that varied from 0.4 to 811.2 ms before the channel availability was determined at a potential of +20 mV. (A) (•) Control; (▾) 30 μM TX100; (▿) washout. (B) (•) Control; (▾) 2.5 mM βOG; (▿) washout. (C) Timed control experiments. (•) Control 1; (▾) control 2; (▿) control 3. (D) (•) Control; (▾) cholesterol depleted; (▿) cholesterol enriched. Results are given as mean ± SEM of peak current values in individual cells. (The time to 75% channel availability determined from these mean values may differ from the mean of the time to 75% channel availability in the individual cells, which is given in the text.) (n = 6, 6, 5, 6, 7, and 7: TX100, βOG, timed controls, cholesterol enriched, cholesterol-depleted, and timed controls for cholesterol experiments).
The channel steady-state availability curves (Fig. 4) are well described by a simple two-state scheme. Nevertheless, the time course of inactivation was best fitted as a double exponential decay; and, in control cells, the steady-state channel availability was 0.45 ± 0.03. We therefore characterized the changes in inactivation time course by the time it takes to reach to a channel availability of 0.75 (t 0.75). Because the steady-state availability is ∼0.5, t 0.75 denotes the “half-time” for channel inactivation under control conditions. In control cells, t 0.75 = 12.0 ± 1.3 ms; in the presence of 2.5 mM βOG or 10 μM TX100, t 0.75 was 6.3 ± 0.9 ms or 7.6 ± 1.2 ms, respectively. In timed control experiments t 0.75 was unchanged. Both amphiphiles thus decreased t 0.75 relative to the value before application (P < 0.05). Cholesterol-depletion decreased t 0.75 to 3.5 ± 0.6 ms, and from 11.7 ± 1.1 ms in the corresponding control cells (P < 0.05). In cholesterol enriched cells t 0.75 was 19.7 ± 8.0 ms, which is not significantly different from the control value.
The effects of βOG, TX100, and cholesterol-depletion on the return from inactivation were investigated at −80 mV. After a 300-ms depolarization to +10 mV, the cells were repolarized to −80 mV for 1–128 ms, and channel availability was determined at +10 mV (Fig. 10) .
Figure 10.

Time course of recovery from inactivation. (A) (•) Control; (▾) 10 μM TX100; (▿) washout. (B) (•) Control; (▾) 2.5 mM βOG; (▿) washout. (C) Timed control experiments. (•) Control 1; (▾) control 2; (▿) control 3. (D) (•) Control; (▾) cholesterol depleted. Pulse protocol: every 5 s the cells were depolarized from −80 to +10 mV for 300 ms, followed by a repolarization to −80 mV for time intervals varying from 1 to 128 ms; finally the channel availability was determined at +10 mV. Mean ± SEM (n = 3, 3, 6, 6, and 5: TX100, βOG, timed controls, cholesterol-depleted, timed controls for cholesterol experiments).
10 μM TX100 and 2.5 mM βOG induced a reversible retardation of return from inactivation: after a 128-ms repolarization, the steady-state availability was 0.56 and 0.52 of the availability in control cells. The effects of cholesterol-depletion were more complex: the initial rate of return was accelerated relative to control cells; the later time course was similar to that in control cells. In summary, promotion of inactivation induced by βOG or TX100 results from a combination of an accelerated time course of inactivation and as a decelerated return from inactivation. Promotion of inactivation induced by cholesterol-depletion, in contrast, is primarily due to a faster time course of inactivation.
Effects on Gramicidin Channels
To determine whether the changes in sodium channel inactivation could be related to changes in bilayer elastic properties, we examined the effects of the micelle-forming amphiphiles and cholesterol on the function of gA channels in planar lipid bilayers (Fig. 11) and HEK293 cell membranes (Fig. 12) . We first studied the changes in gA channel function in dioleoylphosphatidylcholine (DOPC)/n-decane bilayers.
Figure 11.

Effects of βOG, GX100, TX100, rTX100, and cholesterol on gA channel activity and lifetimes. (A) Single-channel current traces recorded from the same bilayer before (top) and after (bottom) the addition of 0.3 mM βOG. (B) Lifetime distributions (plotted as normalized survivor plots) for gA channels in DOPC/n-decane bilayers at GX100 concentrations of 0, 4.5, and 15 μM. (C) Lifetime distributions in the absence and presence of cholesterol (1:2) in the bilayer-forming solution. (D) Concentration dependence of the variation in τ observed with βOG, GX100, TX100, and rTX100. The results are plotted as τ/τcntrl, where the subscript “cntrl” denotes the control situation versus the amphiphile concentration divided by the relevant CMC: (▾) βOG; (▪) GX100; (•) TX100; (▴) rTX100. Each data point denotes results (mean ± SEM) obtained in three or more separate experiments.
Figure 12.

Effects of 2.5 mM βOG on the conductance of a HEK293 cell plasma membrane in the presence of gA. After establishing the whole-cell configuration, 10 μM gA was added to the extracellular solution (first arrow). When the membrane conductance in the presence of gA had reached a steady level, βOG was added using a superfusion system as in the experiments with sodium channels (second arrow). Results from a single experiment.
Fig. 11 A shows current traces recorded before and after the addition of 0.3 mM βOG to both solutions bathing the bilayer. Inspection of the traces shows that the amphiphiles increase both the channel appearance rate and lifetime (τ). The changes in channel lifetime were evaluated from the lifetime distributions (e.g., Fig. 11 B for GX100 and Fig. 11 C for cholesterol). The micelle-forming amphiphiles cause a concentration-dependent increase in τ (=1/k d, the rate constant for channel dissociation). Fig. 11 D shows the results obtained with βOG, GX100, TX100, and rTX100 plotted as τ/τcntrl (=k d,cntrl/k d), where the subscript “cntrl” denotes the control situation, as a function of the relative amphiphile concentration (normalized to the respective CMC). At ∼10% of their CMC, the amphiphiles decrease k d 5- to 25-fold. In contrast, cholesterol (DOPC–cholesterol 1:2) increases k d fivefold (decreases τ fivefold, Fig. 11 C).
All four amphiphiles increase the overall channel activity (the average number of conducting channels in the bilayer, which is a measure of the channel dimerization constant) (e.g., Sawyer et al., 1989) (Fig. 11 A). Cholesterol decreases the channel-forming ability of gA (meaning that one needs to add more gA to observe similar channel appearance rates; unpublished data). The micelle-forming amphiphiles thus alter the monomer↔dimer equilibrium in favor of the membrane-spanning dimers (channels); cholesterol has the opposite effect.
βOG and TX100 also modulate the gramicidin channel activity in the plasma membrane of native HEK293 cells. Fig. 12 shows results from an experiment with 2.5 mM βOG. The membrane conductance was determined using whole-cell voltage clamp and 5-s test pulses to +40 mV from a −70-mV holding potential.
The “background” membrane conductance, G m, in native HEK293 cells (∼1.0 nS) was comparable to the value in transfected cells (see materials and methods). After exposure to 10–100 μM gA, the membrane conductance increased ∼10-fold to a new a steady level, G m + gA. Addition of the amphiphile caused a further three- to fourfold conductance increase to G m + gA,test ∼30 nS. In the absence of gA, βOG or TX100 did not alter the membrane conductance (see materials and methods).
The gA single-channel conductance in HEK293 cells (3.0 ± 0.2 pS, n = 3) was not affected by 2.5 mM βOG or 10 μM TX100. The increase in the gA-induced conductance therefore can be equated with an increase in the number of gA channels in the HEK cell membrane. The relative increase in the number of conducting channels was determined as: (G m+gA,test − G m,test)/(G m+gA − G m), where G m,test (∼G m) is the membrane conductance in the presence of the test compounds (but absence of gA). 2.5 mM βOG or 10 μM TX100 increased gA channel activity by a factor 4.3 ± 1.7 (mean ± range, n = 2) or 2.7 ± 0.2 (mean ± range, n = 2), respectively. Though quantitatively smaller, the changes in gA channel activity in the HEK293 cell membrane are consistent with the changes observed in planar bilayers (Fig. 11 A). The quantitative differences may arise because gA crosses lipid bilayers slowly (O'Connell et al., 1990), such that the conductance increases in the HEK293 cells become limited by the amount of gA in the intracellular leaflet.
DISCUSSION
We have previously shown that TX100 and βOG promote inactivation of N-type calcium channels, whereas cholesterol addition has the opposite effect (Lundbæk et al., 1996). Based on the qualitative correlation with gA channel lifetime, we proposed that membrane proteins are generally regulated by the bilayer deformation energy associated with protein conformational change. The present study provides quantitative support for this hypothesis. βOG, GX100, TX100, and rTX100, which increase gA channel duration, promote skeletal muscle sodium channel inactivation in a concentration-dependent, reversible manner. We further show that βOG and TX100 increase gA channel activity in the plasma membrane of living cells. Cholesterol-depletion, also promotes sodium channel inactivation. As in N-type calcium channels, the effects are specific in that there is little change in channel activation. Nor is the channels' permeability properties altered, as there is little change in the reversal potential (Tables I and II). The different chemical structures of the bilayer-modifying amphiphiles (Fig. 2), and the fact that the same general changes are induced by adding modifiers or by removing a normal membrane constituent, suggest that the changes in sodium channel function are due to changes in a general bilayer property, the bilayer elasticity.
Voltage-dependent sodium channels are just one of many different types of membrane proteins whose function could be (are) altered by changes in bilayer properties, and we cannot exclude that some changes in sodium channel function are secondary to changes in other membrane proteins that activate intracellular biochemical events, which in turn modify the sodium channels and thereby alter their function (e.g., by phosphorylation/dephosphorylation [Catterall, 2000]). (Maneuvers that activate protein kinase C, for example, slow the inactivation rate of skeletal muscle channels [Numann et al., 1994; Bendahhou et al., 1995], but the regulation is complex, as it persists after eliminating the consensus phosphorylation site [Bendahhou et al., 1995].) But even if the effects were secondary to actions on (and of) other protein(s), the basic arguments would not be altered. We therefore will discuss our results as if the changes in sodium channel function were direct consequences of the imposed experimental maneuvers. It is in this context important that TX100 and βOG cause a hyperpolarizing shift in the steady-state availability of not only skeletal muscle but also cardiac muscle sodium channels (Leaf et al., 2003), as these channels differ in their regulation. Whereas protein kinase A activation reduces sodium channel currents in cardiac channels, skeletal muscle sodium channels are not affected (Smith and Goldin, 2000). Similarly, calmodulin causes a hyperpolarizing shift in the steady-state availability of skeletal but not of cardiac sodium channels (Deschênes et al., 2002).
We first discuss the changes in sodium channel gating induced by micelle-forming amphiphiles and cholesterol, including the possibility of specific binding interactions. We then discuss how altered gA channel function sheds light on the energetics of the protein–bilayer hydrophobic coupling—including a discussion of the relation between changes in equilibrium monolayer curvature and bilayer elastic moduli. Finally, we discuss briefly results on other membrane proteins, which similarly indicate that hydrophobic coupling between a membrane-spanning protein and the host bilayer provides a mechanistic basis for regulation of protein function by bilayer elasticity.
Effects of βOG, GX100, TX100, rTX100, and Cholesterol on Sodium Channel Function
In this study, voltage-dependent sodium channels represent a generic membrane-spanning protein with properties that allow for a fairly convenient determination of the distribution among different channel states. The molecular rearrangement(s) that occur in sodium channel activation and inactivation will differ from the rearrangement that occurs in the gA channel monomer↔dimer equilibrium; but, as noted in the Introduction, rearrangement of transmembrane domains is emerging as being a norm for integral membrane proteins involved in transmembrane solute movement.
Voltage-dependent sodium channel gating can be summarized as:
Each of the three functional “states” in the scheme may represent a number of distinct molecular states. The effects of micelle-forming amphiphiles and cholesterol-depletion are primarily on the transition(s) to and from the inactivated state(s), as the steady-state inactivation curve is shifted toward hyperpolarized potentials (Figs. 4, 5, and 8). There are minimal changes in channel activation (Tables I and II; see also online supplemental material available at http://www.jgp.org/cgi/content/full/jgp.200308996/DC1). To a first approximation we therefore can summarize the changes in channel function as being due to a shift in the closed/open↔inactivated distribution toward the inactivated state(s). This is not the well-known time-dependent shift in sodium channel inactivation (Fernandez et al., 1984; Kimitsuki et al., 1990). First, the shift is reversible, and second it is much larger than in timed control experiments (Figs. 5 and 6).
Though models of voltage-dependent gating are in a state of flux, e.g., Miller (2003), the conformational changes may involve a rearrangement of elements at the channel–bilayer interface (Elinder et al., 2001; Jiang et al., 2003a,b)—and even the core of Shaker potassium channels (corresponding to transmembrane segments S5 and S6 plus the intervening selectivity filter) may be in direct contact with the bilayer lipids (Li-Smerin and Swartz, 2000). Moreover, though fast inactivation involves residues in the cytoplasmic linker between domains III and IV (Patton et al., 1992; West et al., 1992), this process is coupled to charge movement in domain IV (Chen et al., 1996), which could perturb the channel–bilayer interface. In any case, though the relative magnitudes of the changes in channel structure associated with activation and inactivation remain unknown, the present results suggest that the major change in bilayer deformation energy (lipid reorganization) occurs in the transition to the inactivated state.
The Changes in Sodium Channel Gating Are Unlikely to Result from Specific Interactions
According to the conventional view of pharmacologically induced changes in protein function, the changes in sodium channel function described in this article would be ascribed to specific binding. The shifts in V in induced by amphiphiles or by cholesterol-depletion indeed are similar to those caused by local anesthetics (e.g., Hille, 2001), which bind to a site in the S6 segment of the rat brain type IIA channel (Yarov-Yarovoy et al., 2001). This site usually is accessible (to local anesthetics) only from the intracellular solution—when the channel is open (see Hille, 2001). The present results are unlikely to result from direct interactions with this local anesthetic binding site. First, local anesthetics usually are aromatic amines, or have other complex ring structures (see Hille, 2001). None of the compounds used in this study are amines, and whereas TX100 has an aromatic ring, rTX100 does not—and βOG and GX100 have unbranched aliphatic chains. Three of the compounds, GX100, TX100, and rTX100, possess a polyoxyethylene chain; βOG does not (Fig. 2). The compounds thus differ in both their nonpolar and polar moieties, which suggests that they would not bind to the same site. Second, the effects of cholesterol-depletion (Fig. 8) are difficult to explain by a mechanism that involves binding to the local anesthetic binding site. Third, the effect of the prepulse potential on the βOG- or TX100-induced inhibition of the peak currents (Fig. 3) is difficult to explain by these compounds' binding to (and block of) the open channel. Fourth, the similar effects of the structurally dissimilar micelle-forming amphiphiles versus the opposite effects of cholesterol, as well as the correlation between the effects of these micelle-forming compounds on gA channels and on sodium channels (Fig. 13 , below), suggest that the changes in V in are not due to specific interactions. Though none of the individual arguments is conclusive, together they provide strong evidence that the changes in sodium channel gating are not due to specific amphiphile–channel interactions.
Figure 13.

Effects of βOG, GX100, TX100, and rTX100 on sodium channel inactivation (ΔV in) as a function of their effects on gA channel lifetime. (▾) βOG; (▪) GX100; (•) TX100; (▴) rTX100. The results are plotted as ΔV in = ΔV in (in the presence of the amphiphile) − ΔV in (in timed control cells) as a function of ln{k d/k d,cntrl} determined at the different amphiphile concentrations. Sodium channel inactivation was studied using the pulse protocol described in Fig. 4. Results obtained with all the compounds, in concentrations below 0.1 CMC, were fitted to a straight line. βOG, GX100, TX100, and rTX100 where used in concentrations of 0.3, 1, 2.5 mM; 4.5, 15 μM; 10, 30, 100 μM; and 10, 30, 300 μM, respectively.
The Changes in Sodium Channel Gating Are Not Due to a Bilayer-couple Mechanism
In whole-cell experiments, the amphiphiles were added only to the extracellular side of the plasma membrane. If the rate at which the amphiphiles cross the membrane were slow, compared with the time of application, the amphiphiles might establish a bilayer-couple (Sheetz and Singer, 1974), in which the extracellular leaflet expands so as to stretch the intracellular leaflet, which would establish a lateral pressure profile across the bilayer and thereby alter sodium channel gating. This scenario is unlikely to apply. First, nonionic detergents equilibrate across (cholesterol-free) lipid bilayers within a second or so (le Maire et al., 1987; Heerklotz and Seelig, 2000), meaning that the amphiphile asymmetry, which would be required to establish a bilayer couple, would be minimal. Second, the effects on sodium channel inactivation are qualitatively similar for applications lasting only 10 s and >5 min (compare Fig. 3, A and B, with Figs. 3 C, 5, and 6). Moreover, the peak-current amplitude decreases monotonically during applications lasting >5 min (Fig. 3 C). These experimental results provide no evidence for the “release” of a bilayer couple—due to amphiphile equilibration across the plasma membrane—during prolonged amphiphile application. We conclude that changes in lateral pressure, due to the creation of a bilayer couple, cannot be a major cause of the changes in sodium channel function.
Energetic Consequences of Protein–Bilayer Coupling
Sodium channels.
Though the structures of an increasing number of membrane proteins are known at atomic resolution, it remains difficult to assess how membrane-protein function is regulated by protein–bilayer interactions (see Perozo et al., 2002). The difficulties arise for several reasons: first, usually only a single conformation of a protein is known; second, there usually is not sufficient structural information to deduce the changes in lipid packing that are associated with changes in protein conformation; and third, even if this information is available, it is difficult to deduce the energetic consequences of the changes in lipid packing. That said, the energetic consequences of the hydrophobic bilayer–protein coupling may be considerable (Nielsen et al., 1998; Nielsen and Andersen, 2000). Approximating a protein's bilayer-spanning domain by a smooth cylinder, and using the theory of elastic bilayer deformations (Huang, 1986), a 1% change in protein hydrophobic length (the length change associated with the open↔closed transition in gap junction channels; Unwin and Ennis, 1984) in a 1-stearoyl-2-oleoyl-phosphatidylcholine (SOPC):cholesterol (1:1) bilayer is associated with a ΔG
def
0 ∼5 kJ/mol (Nielsen and Andersen, 2000). In a two-state closed/open↔inactivated scheme, a hydrophobic coupling energy of this magnitude will shift V
in by 14 mV relative to the situation where there is no channel–bilayer coupling (V
in=S
in·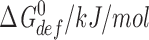 ).
).
Cholesterol also is implicated in the formation and maintenance of plasma membrane lipid domains (Brown and London, 1998; Simons and Toomre, 2000; Maxfield, 2002). If sodium channels reside in such domains, which are disrupted by cholesterol removal, the depletion would still alter the elastic properties of the bilayer surrounding the channel (Lundbæk et al., 2003). Also, whereas cholesterol-depletion may disrupt or reorganize the plasma membrane domain structure (Ilangumaran and Hoessli, 1998; Hao et al., 2001), TX100 promotes raft-like domain formation (Heerklotz, 2002). Thus, the changes in sodium channel function are unlikely to be direct consequences of plasma membrane domain disruptions.
In any case, the magnitude of the observed changes in sodium channel function (Figs. 4–6 and 8) is consistent with changes in bilayer elasticity. To further investigate the possibility of cause and effect, it becomes necessary to relate changes in protein function to changes in bilayer properties (determined independently); the latter can be assessed using gA channels (Lundbæk and Andersen, 1994; Lundbæk et al., 1997).
gA channels and micelle-forming amphiphiles
When amphipathic substances adsorb at a bilayer–solution interface they generally will alter the monolayer equilibrium curvature (Cullis and de Kruijff, 1979; Seddon, 1990) and bilayer elastic moduli (Safinya et al., 1989; Duwe et al., 1990; Evans et al., 1995; McIntosh et al., 1995; Otten et al., 2000; Brown et al., 2002). Micelle-forming amphiphiles will tend to promote a positive monolayer curvature (Cullis and de Kruijff, 1979) and decrease the area compression and monolayer bending moduli (McIntosh et al., 1995; Brown et al., 2002). The decrease in the moduli occurs, at least in part, because the (reversible) adsorption of water-soluble amphiphiles varies as function of bilayer tension (Evans et al., 1995) and curvature (Epand and Epand, 1994). This means that the energetic cost of a bilayer compression/expansion will be reduced (relative to the situation where no amphiphile is present) by the varying mole-fraction of the amphiphile in the bilayer. Both the changes in curvature and moduli will tend to decrease the cost of the bilayer deformation associated with gA channel formation. gA channel activity and lifetime therefore are increased (Fig. 11).
gA channels and cholesterol.
Compared with the micelle-forming amphiphiles, cholesterol changes all the bilayer properties defining ΔG def 0 in the opposite direction. Cholesterol tends to promote a negative monolayer curvature (Seddon, 1990) and increase the area compression and monolayer bending moduli (Evans and Rawicz, 1990; Needham and Nunn, 1990; McIntosh et al., 1995; Meleard et al., 1997). These changes all increase the cost of the bilayer deformation associated with gA channel formation. Channel activity and lifetime therefore are decreased (Fig. 11).
Cholesterol also changes the acyl chain order parameter (Oldfield et al., 1978; Sankaram and Thompson, 1990), which has led to the notion that changes in cell-cholesterol content could regulate membrane protein function by changing cell membrane “fluidity” (e.g., Yeagle, 1985). Changes in bilayer fluidity, however, cannot account for the effects of cholesterol on gA or sodium channel function. Changes in the equilibrium distribution between different conformations of a bilayer-embedded protein (i.e., the gA monomer↔dimer equilibrium or the steady-state inactivation of sodium channels) reflect changes in the standard free energy difference between the different protein conformations. These changes in free energy cannot be direct consequences of changes in fluidity (Lee, 1991). The bilayer fluidity may well be altered by the same changes in intermolecular interactions that alter the bilayer elastic properties (e.g., Brown et al., 2002), but the mechanistic significance of changes in bilayer fluidity, in a liquid-crystalline bilayer, remains obscure.
Relation between sodium and gA channel function.
The changes in gA channel lifetime (Fig. 11) can be understood in terms of altered bilayer elasticity. If the changes in sodium channel function also were due to altered bilayer elasticity, the changes in sodium channel function should be correlated to the changes in gA channel lifetime. Indeed, the shifts in V in, induced by the amphiphiles, can be related to changes in bilayer elasticity, as monitored by changes in the gA-channel dissociation rate constant (1/lifetime). Fig. 13 shows the changes in V in plotted as a function of ln{k d/k d,cntrl}, which provides a measure of the change in the disjoining force pulling the dimer apart (Lundbæk and Andersen, 1999).
Though the amphiphiles differ in structure, the correlations between the effects in the two systems are striking. First, they all modulate sodium channel function at the same concentrations where gA channels are affected. Second, at concentrations <0.1 CMC, the changes in V in are linear functions of ln{k d/k d,cntrl}. Third, despite the ∼1,000-fold ratio of amphiphile concentrations, the correlations are very similar. At concentrations <0.1 CMC, the results for all compounds can be fitted by a straight line with a slope of 5.2 ± 0.4 mV (r = 0.96) (Fig. 13). The correlation is remarkable, in the sense that it is possible to use changes in gA channel lifetimes to predict how structurally different molecules (Fig. 2) alter sodium channel inactivation. This suggests that bilayer elasticity is the relevant variable. (For all the amphiphiles the results at ∼0.1 CMC deviate from the linear relation. We have no explanation for this deviation, but the adsorption coefficient, and the effects on bilayer elasticity, is likely to be complex at high amphiphile concentrations. Moreover, it is not clear, however, to what extent the correlation can be extended to molecules with very different structures, which may be localized differently relative to the bilayer–solution interface.)
Monolayer Equilibrium Curvature Versus Elastic Moduli—Effects of Polyunsaturated Fatty Acids
Fig. 13 provides support for the notion that sodium channel function is subject to regulation by bilayer elasticity. But an apparent inconsistency arises if we compare the effects of the micelle-forming amphiphiles with those of polyunsaturated fatty acids (PUFAs), which also shift sodium channel inactivation in the hyperpolarizing direction and slow the rate of recovery from inactivation (Xiao et al., 1998). The similar direction of the shifts is surprising because, after suggestions by Cullis and de Kruijff (1979) and Gruner (1985), bilayer-mediated effects of amphiphiles on membrane protein function tend to be interpreted in terms of changes in bilayer curvature stress (e.g., Epand, 1997)—with the implicit assumption that PUFAs and micelle-forming compounds, which promote negative (Seddon, 1990; Tate et al., 1991) and positive (Israelachvili et al., 1976; Vinson et al., 1989; Heerklotz et al., 1997; Fuller and Rand, 2001) equilibrium curvatures, respectively, should have opposite effects on protein function. Given that micelle-forming amphiphiles promote sodium channel inactivation by changing the bilayer properties, PUFAs would thus be expected to cause a depolarizing shift in sodium channel inactivation—contrary to what is observed. The effects of PUFAs could result from specific interactions; but, like the micelle-forming amphiphiles, PUFAs alter the function of numerous ion channels and other integral membrane proteins (Hwang et al., 1990; Ordway et al., 1991; Wallert et al., 1991; Meves, 1994; Leaf et al., 1999, 2003; Patel et al., 2001), which suggests that they also may act through a “nonspecific” bilayer-dependent mechanism.
To address this question, we have examined the effects of the PUFA and docosahexaenoic acid (DHA) on gA channel function in lipid bilayers (Bruno et al., 2003): in DOPC/n-decane bilayers 10 μM DHA increases the gA channel lifetime ∼50%, whereas the channel appearance rate increases more than 10-fold. (In dieicosaenoylphosphatidylcholine/n-decane bilayers, the lifetime increase is almost fourfold.) Similarly, in HEK293 membranes, 10 μM DHA causes a three- to fourfold increase in gA channel activity (unpublished data). That is, even though DHA promotes a negative monolayer curvature, it stabilizes gramicidin channels; the general correlation between changes in gA channel lifetime and shifts in V in (compare Fig. 13) thus is preserved. Taken together with the similar effects of βOG, TX100, and PUFAs on cardiac sodium channels (Leaf et al., 2003), these results suggest that the DHA-induced changes in elastic moduli are quantitatively more important than the change in curvature. (As was noted above for the micelle-forming amphiphiles, DHA is unlikely to act by a bilayer-couple mechanism [Sheetz and Singer, 1974] because the transbilayer movements of fatty acids is fast [Hamilton, 2003].)
The PUFA results demonstrate the limitations of unimodal attempts to relate changes in bilayer properties to changes in membrane protein function. There is no information on the effects of DHA on the bilayer elastic moduli, but the PUFAs adsorb reversibly to the bilayer, which mean that they will decrease the monolayer compression and bending moduli (see above). Whereas micelle-forming amphiphiles will reduce both the compression and bending, as well as the curvature-contribution to ΔG def 0, associated with gA channel formation, the effects of PUFAs on these contributions may have opposite signs. The final outcome, whether the gA channels are destabilized or stabilized, will depend on the relative magnitudes of these changes. This fundamental uncertainty will arise whenever changes in the different contributions to ΔG def 0 are of opposite sign. To complicate the situation further, the relative changes in the different contributions to ΔG def 0 are likely to vary as a function of the membrane lipid composition.
The last point may be important also for the effects of n-alkanols on membrane protein function. Long-chain n-alkanols promote a negative equilibrium curvature (Seddon, 1990; Tate et al., 1991)—and reduce the lifetime of gA channels (Elliott et al., 1985), which would suggest that the curvature-dependent changes in ΔG def 0 dominate the moduli-dependent changes. However, the effects of n-alkanols on sodium channel inactivation vary among different preparations—with no effect in the squid axon (Armstrong and Binstock, 1964; Haydon and Urban, 1983) and a hyperpolarizing shift in dorsal root ganglia (Elliott and Elliott, 1989).
Bilayer Deformation Energy and Membrane Protein Function
The focus of this study is voltage-dependent sodium channels, as a mere example of a membrane-spanning protein whose function is comparatively simple to monitor (a channel or receptor, which allows for the determination of the distribution between different functional states [Andersen et al., 1998]). We would expect that the experimental maneuvers used in this study will alter the function of many integral membrane proteins—to varying degrees. Table III shows examples of reversible effects of TX100 on a wide variety of membrane proteins.
TABLE III.
Membrane Proteins Reversibly Modulated by TX100
| Voltage-dependent potassium channels |
Hollerer-Beitz and Heinemann, 1998 |
|---|---|
| N-type calcium channel | Lundbæk et al., 1996 |
| KATP channel | Smith and Proks, 1998 |
| Nicotinic acetylcholine receptor | Kasai et al., 1970 |
| Bacteriorhodopsin | Mukhopadhyay et al., 1996 |
| Na+,K+-ATPase | Huang et al., 1985 |
| Ca2+-ATPase | McIntosh and Davidson, 1984 |
Moreover, the reciprocal effects of cholesterol and micelle-forming compounds on sodium channel function have been observed also in other, structurally unrelated membrane proteins, such as the nicotinic acetylcholine receptor (nAChR) and the sarcoplasmic Ca2+-ATPase (SERCA). TX100 promotes desensitization of nAChR (Kasai et al., 1970), whereas cholesterol promotes channel activation (Fong and McNamee, 1986; Sunshine and McNamee, 1992). Furthermore, solubilization of nAChR in βOG or TX100 promotes a change in structure (as monitored by changes in photoaffinity labeling) that is similar to the one induced by long-term exposure to acetylcholine, i.e., the desensitized state (McCarthy and Moore, 1992). Similarly, though TX100-solubilized SERCA is catalytically active (Lund et al., 1989), TX100 inhibits the activity of bilayer-incorporated SERCA and reduces the cooperativity of Ca2+ binding (McIntosh and Davidson, 1984). Cholesterol, in contrast, increases the stoichiometry of Ca2+ binding (Lee et al., 1994). The bilayer (deformation energy) thus appears to be a general modulator of membrane protein function (Gruner, 1991; Andersen et al., 1992).
Hydrophobic Coupling, Bilayer Elasticity, and Deformation Energy
The present results demonstrate both the simplicity and the complexity of the interactions between membrane proteins and lipid bilayers. A priori arguments—based on notions of bilayer thickness, elastic moduli, and monolayer curvature—provide general understanding of how water-soluble amphiphiles can alter protein function by altering bilayer elasticity. These parameters cannot be varied separately, however, meaning that any experimental maneuver will alter many, if not all, of the component energies (in the same or in opposite directions). The energetic consequences of changing any of the above parameters therefore are not separable, as demonstrated previously (Nielsen et al., 1998; Nielsen and Andersen, 2000). Further, the use of continuum elastic moduli, obtained in protein-free systems, to evaluate protein–bilayer interactions may be problematic because the relevant geometries and length scales differ (Helfrich, 1981; Partenskii and Jordan, 2002).
It thus becomes important to have systems, such as the gA channels, that provide for an in situ measure of changes in the bilayer deformation energy associated with a change in bilayer molecular composition. Given the structural difference between gA and sodium channels, the expected complexity of membrane protein structure and conformational changes, and the heterogeneous molecular composition of cell membranes, the correlation between the changes in gA and sodium channel function (Fig. 13) is surprisingly simple.
How can this correlation be so simple? ΔG def 0 associated with a conformational change in a bilayer-spanning domain will vary with both the global bilayer elastic properties and the local lipid packing around the protein. Integral membrane proteins are not smooth cylinders, but irregularly shaped bodies with crevices between the bilayer-spanning α-helices (e.g., Doyle et al., 1998), or the subunits or domains (e.g., Li-Smerin and Swartz, 2000). Generally therefore, amphiphiles could alter membrane protein function by four mechanisms, which are not mutually exclusive: first, by binding to sites formed by the protein itself—at the protein/aqueous solution interface; second, by binding in crevices at the protein/bilayer boundary; third, by accumulating at this boundary and thereby altering the local constraints on lipid packing; and fourth by altering the global bilayer elasticity. The first two mechanisms will exhibit selectivity—in the classic sense that binding will depend on the detailed structure of the amphiphile. The third also will exhibit selectivity because the local accumulation of a given amphiphile (and its consequences for the deformation energy) will vary among proteins. Only the fourth mechanism is likely to be truly nonspecific. But the latter three mechanisms will all express themselves by altering the energetic cost of the bilayer reorganization associated with a protein conformational change—and a given compound may alter a protein's function by any combination of the four mechanisms.
The striking correlation between the changes in V in for the sodium channels and the lifetime of gA channels in planar bilayers (Fig. 13) suggests that changes in bilayer elasticity are a major common factor in terms of modulating sodium channel function, but it does not necessarily mean that changes in global, continuum contribution to the bilayer deformation energy are the mechanistically most important, as the latter two, and maybe the latter three, of the four contributions listed above are likely to scale similarly (e.g., Nielsen et al., 1998; Partenskii and Jordan, 2002). The overall picture becomes that sodium channel inactivation involves a change in the channel's hydrophobic domain. Our results thus lend support to the proposal that mechanosensitivity is a general property of membrane proteins (Gu et al., 2001), and more generally that bilayer–protein hydrophobic coupling represents a general organizing principle for the regulation of membrane protein function by lipid bilayer elasticity.
Figure .

Supplemental Material
Acknowledgments
We thank Dr. G. Mandel for the generous gift of rat skeletal muscle cDNA, and Drs. L.G. Palmer and T. Werge for helpful discussions.
This work was supported by NIH Grants GM21342 (O.S. Andersen) and GM34968 and RR15569 (R.E. Koeppe and D.V. Greathouse), as well as grants from the Danish Medical Research Council (J.A. Lundbæk), the Danish Natural Science Research Council, and the Carlsberg Foundation (C. Nielsen).
Henry A. Lester served as guest editor.
The online version of this article contains supplemental material.
Abbreviations used in this paper: βOG, β-octyl-glucoside; CMC, critical micellar concentration; DHA, docosahexaenoic acid; DOPC, dioleoylphosphatidylcholine; gA, gramicidin A; GX100, Genapol X-100; HH, Hodgkin-Huxley; MβCD, methylated β-cyclodextrin; TX100, Triton X-100; rTX100, reduced TX100.
Footnotes
Though ΔG
def
0 is determined by K
a, K
c, c
0, d
0, and r
0, the precise value will be a function also of the constraints on lipid packing around the protein (Helfrich and Jakobsson, 1990; Nielsen and Andersen, 2000), as well as any spatial variation in the elastic moduli adjacent to the protein (Partenskii and Jordan, 2002). Further, although monolayer compression and bending are independent modes of deformation; the elastic moduli associated with these two modes of deformation are related by K
c=K
a· (Evans and Skalak, 1979), where b ∼24 (see Rawicz et al., 2000).
(Evans and Skalak, 1979), where b ∼24 (see Rawicz et al., 2000).
The CMC varies as a function of salt concentration (Walter et al., 2000). The quoted values are for 0.1–0.2 M NaCl solutions.
References
- Abramson, J., I. Smirnova, V. Kasho, G. Verner, H.R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- Allen, T.W., O.S. Andersen, and B. Roux. 2003. The structure of gramicidin A in a lipid bilayer environment determined using molecular dynamics simulations and solid-state NMR data. J. Am. Chem. Soc. 125:9868–9878. [DOI] [PubMed] [Google Scholar]
- Andersen, O.S. 1983. Ion movement through gramicidin A channels. Single-channel measurements at very high potentials. Biophys. J. 41:119–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen, O.S., C. Nielsen, A.M. Maer, J.A. Lundbæk, M. Goulian, and R.E. Koeppe II. 1998. Gramicidin channels: molecular force transducers in lipid bilayers. Biol. Skr. Dan. Vid. Selsk. 49:75–82. [Google Scholar]
- Andersen, O.S., C. Nielsen, A.M. Maer, J.A. Lundbæk, M. Goulian, and R.E. Koeppe, II. 1999. Ion channels as tools to monitor lipid bilayer-membrane protein interactions: gramicidin channels as molecular force transducers. Methods Enzymol. 294:208–224. [DOI] [PubMed] [Google Scholar]
- Andersen, O.S., D.B. Sawyer, and R.E. Koeppe, II. 1992. Modulation of channel function by the host bilayer. Biomembrane Structure and Function. K.R.K. Easwaran and B. Gaber, editors. Adenine Press, Schenectady, NY. 227–244.
- Armstrong, C.M., and F. Bezanilla. 1974. Charge movement associated with the opening and closing of the activation gates of the Na+ channels. J. Gen. Physiol. 63:533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and L. Binstock. 1964. The effects of several alcohols on the properties of the squid giant axon. J. Gen. Physiol. 48:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arseniev, A.S., A.L. Lomize, I.L. Barsukov, and V.F. Bystrov. 1986. Gramicidin A transmembrane ion-channel. Three-dimensional structure reconstruction based on NMR spectroscopy and energy refinement. Biol. Membr. 3:1077–1104. [Google Scholar]
- Awasthi, Y.C., T.F. Chuang, T.W. Keenan, and F.L. Crane. 1971. Tightly bound cardiolipin in cytochrome oxidase. Biochim. Biophys. Acta. 226:42–52. [DOI] [PubMed] [Google Scholar]
- Balla, T., T. Bondeva, and P. Varnai. 2000. How accurately can we image inositol lipids in living cells? Trends Pharmacol. Sci. 21:238–241. [DOI] [PubMed] [Google Scholar]
- Bendahhou, S., T.R. Cummins, J.F. Potts, J. Tong, and W.S. Agnew. 1995. Serine-1321-independent regulation of the μ1 adult skeletal muscle Na+ channel by protein kinase C. Proc. Natl. Acad. Sci. USA. 92:12003–12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezrukov, S., R.P. Rand, I. Vodyanoy, and V.A. Parsegian. 1998. Lipid packing stress and polypeptide aggregation: alamethicin channel probed by proton titration of lipid charge. Faraday Discuss. 111:173–183. [DOI] [PubMed] [Google Scholar]
- Bezrukov, S.M. 2000. Functional consequences of lipid packing stress. Curr. Opin. Coll. Interface Sci. 5:237–243. [Google Scholar]
- Bienvenüe, A., and J.S. Marie. 1994. Modulation of protein function by lipids. Curr. Top. Membr. 40:319–354. [Google Scholar]
- Booth, P.J., R.H. Templer, W. Meijberg, S.J. Allen, A.R. Curran, and M. Lorch. 2001. In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol. Biol. 36:501–603. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- Brown, D.A., and E. London. 2000. Structure and function of sphingolipid-and cholesterol-rich membrane rafts. J. Biol. Chem. 275:17221–17224. [DOI] [PubMed] [Google Scholar]
- Brown, M.F. 1997. Influence of nonlamellar-forming lipids on rhodopsin. Curr. Topics Membr. Transp. 44:285–356. [Google Scholar]
- Brown, M.F., R.L. Thurmond, S.W. Dood, D. Otten, and K. Bayer. 2002. Elastic deformation of membrane bilayers probed by deuterium NMR relaxation. J. Am. Chem. Soc. 124:8471–8484. [DOI] [PubMed] [Google Scholar]
- Bruno, M.J., H. Sun, D.V. Greathouse, R.E. Koeppe, II, and O.S. Andersen. 2003. Modification of gramicidin channel function by poly-unsaturated fatty acids. Biophys. J. 84:512a. [Google Scholar]
- Bullock, J.O., and F.S. Cohen. 1986. Octyl glucoside promotes incorporation of channels into neutral phospholipid bilayers. Studies with colicin. Biochim. Biophys. Acta. 856:101–108. [DOI] [PubMed] [Google Scholar]
- Cantor, R. 1997. Lateral pressures in cell membranes: a mechanism for modulation of protein function. J. Physiol. Chem. B. 101:1723–1725. [Google Scholar]
- Catterall, W.A. 2000. From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron. 26:13–25. [DOI] [PubMed] [Google Scholar]
- Chang, G., R.H. Spencer, A.T. Lee, M.T. Barclay, and D.C. Rees. 1998. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 282:2220–2226. [DOI] [PubMed] [Google Scholar]
- Chen, L.Q., V. Santarelli, R. Horn, and R.G. Kallen. 1996. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J. Gen. Physiol. 108:549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian, A.E., M.P. Haynes, M.C. Phillips, and G.H. Rothblat. 1997. Use of cyclodextrins for manipulating cellular cholesterol content. J. Lipid Res. 38:2264–2272. [PubMed] [Google Scholar]
- Cullis, P.R., and B. de Kruijff. 1979. Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta. 559:399–420. [DOI] [PubMed] [Google Scholar]
- Dan, N., and S.A. Safran. 1998. Effect of lipid characteristics on the structure of transmembrane proteins. Biophys. J. 75:1410–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschênes, I., N. Neyroud, D. DiSilvestre, E. Marban, D.T. Yue, and G.F. Tomaselli. 2002. Isoform-specific modulation of voltage-gated Na+ channels by calmodulin. Circ. Res. 90:E49–E57. [DOI] [PubMed] [Google Scholar]
- Dowhan, W. 1997. Molecular basis for membrane phospholipid diversity: why are there so many lipids? Annu. Rev. Biochem. 66:199–232. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J. Morais Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Duwe, H.P., J. Kaes, and E. Sackmann. 1990. Bending elastic moduli of lipid bilayers: modulation by solutes. J. Physiol. France. 51:945–962. [Google Scholar]
- Elinder, F., P. Århem, and H.P. Larsson. 2001. Localization of the extracellular end of the voltage sensor S4 in a potassium channel. Biophys. J. 80:1802–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, A.A., and J.R. Elliott. 1989. The role of inactivation in the effects of n-alkanols on the sodium current of cultured rat sensory neurones. J. Physiol. 415:19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, J.R., D. Needham, J.P. Dilger, O. Brandt, and D.A. Haydon. 1985. A quantitative explanation of the effects of some alcohols on gramicidin single-channel lifetime. Biochim. Biophys. Acta. 814:401–404. [DOI] [PubMed] [Google Scholar]
- Elliott, J.R., D. Needham, J.P. Dilger, and D.A. Haydon. 1983. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Biophys. Acta. 735:95–103. [DOI] [PubMed] [Google Scholar]
- Epand, R. 1997. Lipid Polymorphism and Membrane Properties. Acad. Press, San Diego.
- Epand, R.M. 1998. Lipid polymorphism and protein-lipid interactions. Biochim. Biophys. Acta. 1376:353–368. [DOI] [PubMed] [Google Scholar]
- Epand, R.M., and R.F. Epand. 1994. Calorimetric detection of curvature strain in phospholipid bilayers. Biophys. J. 66:1450–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., and D. Needham. 1987. Physical properties of surfactant bilayer membranes: thermal transitions, elasticity, rigidity, cohesion, and colloidal interactions. J. Physiol. Chem. 91:4219–4228. [Google Scholar]
- Evans, E., and W. Rawicz. 1990. Entropy-driven tension and bending elasticity in condensed fluid membranes. Physiol. Rev. Lett. 64:2094–2097. [DOI] [PubMed] [Google Scholar]
- Evans, E.A., and R. Skalak. 1979. Mechanics and thermodynamcs of biomembranes: part 1. CRC Crit. Rev. Bioeng. 3:181–330. [PubMed] [Google Scholar]
- Evans, E., W. Rawicz, and A.F. Hofmann. 1995. Lipid bilayer expansion and mechanical disruption in solutions of water-soluble bile acid. Bile Acids in Gastroenterology Basic and Clinical Advances. A.F. Hofmann, G. Paumgartner, and A. Stiehl, editors. Kluwer Academic Publishers, Dordrecht, Netherlands. 59–68.
- Evans, E.A., and R.M. Hochmuth. 1978. Mechanochemical properties of membranes. Curr. Top. Membr. Transp. 10:1–64. [Google Scholar]
- Fattal, D.R., and A. Ben-Shaul. 1993. A molecular model for lipid-protein interaction in membranes: the role of hydrophobic mismatch. Biophys. J. 65:1795–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez, J.M., A.P. Fox, and S. Krasne. 1984. Membrane patches and whole-cell membranes: a comparison of electrical properties in rat clonal pituitary (GH3) cells. J. Physiol. 356:565–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, T.M., and M.G. McNamee. 1986. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry. 25:830–840. [DOI] [PubMed] [Google Scholar]
- Fuller, N., and R.P. Rand. 2001. The influence of lysolipids on the spontaneous curvature and bending elasticity of phospholipid membranes. Biophys. J. 81:243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gekko, K., and H. Noguchi. 1979. Compressibility of globular proteins in water at 25°C. J. Physiol. Chem. 83:2706–2714. [Google Scholar]
- Gruner, S.M. 1985. Intrinsic curvature hypothesis for biomembrane lipid composition: a role for nonbilayer lipids. Proc. Natl. Acad. Sci. USA. 82:3665–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner, S.M. 1991. Lipid membrane curvature elasticity and protein function. Biologically Inspired Physics. L. Peliti, editor. Plenum Press, NY. 127–135.
- Gu, C.X., P.F. Juranka, and C.E. Morris. 2001. Stretch-activation and stretch-inactivation of Shaker-IR, a voltage-gated K+ channel. Biophys. J. 80:2678–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill, O.P., A. Marty, E. Neher, B. Sakmann, and F.J. Sigworth. 1981. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391:85–100. [DOI] [PubMed] [Google Scholar]
- Hamilton, J.A. 2003. Fast flip-flop of cholesterol and fatty acids in membranes: implications for membrane transport proteins. Curr. Opin. Lipidol. 14:263–271. [DOI] [PubMed] [Google Scholar]
- Hao, M., S. Mukherjee, and F.R. Maxfield. 2001. Cholesterol depletion induces large scale domain segregation in living cell membranes. Proc. Natl. Acad. Sci. USA. 98:13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydon, D.A., and B.W. Urban. 1983. The action of alcohols and other non-ionic surface active substances on the sodium current of the squid giant axon. J. Physiol. 341:411–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerklotz, H. 2002. Triton promotes domain formation in lipid raft mixtures. Biophys. J. 83:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerklotz, H., H. Binder, G. Lantsch, and G. Klose. 1997. Lipid/detergent interaction thermodynamics as a function of molecular shape. J. Physiol. Chem. B. 101:639–645. [Google Scholar]
- Heerklotz, H., and J. Seelig. 2000. Correlation of membrane/water partition coefficients of detergents with the critical micelle concentration. Biophys. J. 78:2435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich, P., and E. Jakobsson. 1990. Calculation of deformation energies and conformations in lipid membranes containing gramicidin channels. Biophys. J. 57:1075–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfrich, W. 1973. Elastic properties of lipid bilayers: theory and possible experiments. Z. Naturforsch. 28C:693–703. [DOI] [PubMed] [Google Scholar]
- Helfrich, W. 1981. Amphiphilic mesophases made of defects. Physique des Défauts (Physics of Defects). R. Balian, M. Kléman, and J.-P. Poirier, editors. North-Holland Publishing Company, NY. 716–755.
- Hilgemann, D.W., and R. Ball. 1996. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 273:956–959. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., S. Feng, and C. Nasuhoglu. 2001. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE 2001:RE19. [DOI] [PubMed]
- Hille, B. 2001. Ion Channels of Excitable Membranes. Sinauer Associates, Inc., Sunderland, MA.
- Hirai, T., J.A.W. Heymann, P.C. Maloney, and S. Subramaniam. 2003. Structural model for 12-helix transporters belonging to the major facilitator superfamily. J. Bacteriol. 185:1712–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollerer-Beitz, G., and S.H. Heinemann. 1998. Influence of detergents on the function of cloned potassium channels. Receptors Channels. 5:61–78. [PubMed] [Google Scholar]
- Hla, T., M.-J. Lee, N. Ancellin, J.H. Paik, and M.J. Kluk. 2001. Lysophospholipids–receptor revelations. Science. 294:1875–1878. [DOI] [PubMed] [Google Scholar]
- Huang, H.W. 1986. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 50:1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, W.H., S.S. Kakar, and A. Askari. 1985. Mechanisms of detergent effects on membrane-bound (Na+ + K+)-ATPase. J. Biol. Chem. 260:7356–7361. [PubMed] [Google Scholar]
- Huang, Y., M.J. Lemieux, J. Song, M. Auer, and D.-N. Wang. 2003. Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science. 301:616–620. [DOI] [PubMed] [Google Scholar]
- Hwang, T.-C., S.E. Guggino, and W.B. Guggino. 1990. Direct modulation of secretory chloride channels by arachidonic and other cis unsaturated fatty acids. Proc. Natl. Acad. Sci. USA. 87:5706–5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilangumaran, S., and D.C. Hoessli. 1998. Effects of cholesterol depletion by cyclodextrin on the sphingolipid microdomains of the plasma membrane. Biochem. J. 335:433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isele, J., T.P. Sakmar, and F. Siebert. 2000. Rhodopsin activation affects the environment of specific neighboring phospholipids: an FTIR spectroscopic study. Biophys. J. 79:3063–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelachvili, J.N., D.J. Mitchell, and B.W. Ninham. 1976. Theory of self-assembly of hydrocarbon amphiphiles into micelles and bilayers. J. Chem. Soc. Faraday Trans. 272:1525–1568. [Google Scholar]
- Iwata, S., C. Ostermeier, B. Ludwig, and H. Michel. 1995. Structure at 2 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 376:660–669. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, M. Cadene, B.T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., A. Lee, J. Chen, V. Ruta, M. Cadene, B.T. Chait, and R. MacKinnon. 2003. a. X-ray structure of a voltage-dependent K+ channel. Nature. 423:33–41. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., V. Ruta, J. Chen, A. Lee, and R. MacKinnon. 2003. b. The principle of gating charge movement in a voltage-dependent K+ channel. Nature. 423:42–48. [DOI] [PubMed] [Google Scholar]
- Kaback, H.R., M. Sahin-Toth, and A.B. Weinglass. 2001. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2:610–620. [DOI] [PubMed] [Google Scholar]
- Kasai, M., T.R. Podleski, and J.-P. Changeux. 1970. Some structural properties of excitable membranes labeled by fluorescent probes. FEBS Lett. 7:13–19. [DOI] [PubMed] [Google Scholar]
- Katsaras, J., R.S. Prosser, R.H. Stinson, and J.H. Davis. 1992. Constant helical pitch of the gramicidin channel in phospholipid bilayers. Biophys. J. 61:827–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, S.L., S.M. Bezrukov, S.M. Gruner, M.W. Tate, I. Vodyanoy, and V.A. Parsegian. 1993. Probability of alamethicin conductance states varies with nonlamellar tendency of bilayer phospholipids. Biophys. J. 65:23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketchem, R.R., B. Roux, and T.A. Cross. 1997. High-resolution polypeptide structure in a lamellar phase lipid environment from solid state NMR derived orientational constraints. Structure. 5:1655–1669. [DOI] [PubMed] [Google Scholar]
- Killian, J.A. 1998. Hydrophobic mismatch between proteins and lipids in membranes. Biochim. Biophys. Acta. 1376:401–415. [DOI] [PubMed] [Google Scholar]
- Kimitsuki, T., T. Mitsuiye, and A. Noma. 1990. Negative shift of cardiac Na+ channel kinetics in cell-attached patch recordings. Am. J. Physiol. 258:H247–H254. [DOI] [PubMed] [Google Scholar]
- Koeppe, R.E., II, and O.S. Andersen. 1996. Engineering the gramicidin channel. Annu. Rev. Biophys. Biomol. Struct. 25:231–258. [DOI] [PubMed] [Google Scholar]
- Konnerth, A., H.D. Lux, and M. Morad. 1987. Proton-induced transformation of calcium channel in chick dorsal root ganglion cells. J. Physiol. 386:603–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga, S. 1981. Pore size distribution analysis of gel substances by size exclusion chromatography. J. Chromatogr. 206:449–461. [Google Scholar]
- le Maire, M., J.V. Møller, and P. Champeil. 1987. Binding of a nonionic detergent to membranes: flip-flop rate and location on the bilayer. Biochemistry. 26:4803–4810. [DOI] [PubMed] [Google Scholar]
- Leaf, A., J.X. Kang, Y.-F. Xiao, G.E. Billman, and R.A. Voskuyl. 1999. The antiarrhythmic and anticonvulsant effects of dietary N-3 fatty acids. J. Membr. Biol. 172:1–11. [DOI] [PubMed] [Google Scholar]
- Leaf, A., Y.-F. Xiao, J.X. Kang, and G.E. Billman. 2003. Prevention of sudden cardiac death by n-3 polyunsaturated fatty acids. Pharm. Therapeut. 98:355–377. [DOI] [PubMed] [Google Scholar]
- Lee, A.G. 1991. Lipids and their effects on membrane proteins: evidence against a role for fluidity. Prog. Lipid Res. 30:323–348. [DOI] [PubMed] [Google Scholar]
- Lee, A.G. 2003. Lipid-protein interactions in biological membranes: a structural perspective. Biochim. Biophys. Acta. 1612:1–40. [DOI] [PubMed] [Google Scholar]
- Lee, A.G., A.P. Starling, J. Ding, J.M. East, and M. Wictome. 1994. Lipid-protein interactions and Ca2+-ATPase function. Biochem. Soc. Trans. 22:821–826. [DOI] [PubMed] [Google Scholar]
- Levitan, I., A.E. Christian, T.N. Tulenko, and G.H. Rothblat. 2000. Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J. Gen. Physiol. 115:405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li-Smerin, Y.H.D.H., and K.J. Swartz. 2000. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 25:411–423. [DOI] [PubMed] [Google Scholar]
- Lindahl, E., and O. Edholm. 2000. Mesoscopic undulations and thickness fluctuations in lipid bilayers from molecular dynamics simulations. Biophys. J. 79:426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, N., and R.L. Kay. 1977. Redetermination of the pressure dependence of the lipid bilayer phase transition. Biochemistry. 16:3484–3486. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H.T. Richter, J.P. Cartailler, and J.K. Lanyi. 1999. a. Structural changes in bacteriorhodopsin during ion transport at 2 Å resolution. Science. 286:255–261. [DOI] [PubMed] [Google Scholar]
- Luecke, H., B. Schobert, H.T. Richter, J.P. Cartailler, and J.K. Lanyi. 1999. b. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291:899–911. [DOI] [PubMed] [Google Scholar]
- Lund, S., S. Orlowski, B. de Foresta, P. Champeil, M. le Maire, and J. Møller. 1989. Detergent structure and associated lipid as determinants in the stabilization of solubilized Ca2+-ATPase from sarcoplasmic reticulum. J. Biol. Chem. 264:4907–4915. [PubMed] [Google Scholar]
- Lundbæk, J.A., and O.S. Andersen. 1994. Lysophospholipids modulate channel function by altering the mechanical properties of lipid bilayers. J. Gen. Physiol. 104:645–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbæk, J.A., and O.S. Andersen. 1999. Spring constants for channel-induced lipid bilayer deformations - estimates using gramicidin channels. Biophys. J. 76:889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbæk, J.A., O.S. Andersen, T. Werge, and C. Nielsen. 2003. Cholesterol-Induced Protein Sorting: An Analysis of Energetic Feasibility. Biophys. J. 84:2080–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundbæk, J.A., P. Birn, J. Girshman, A.J. Hansen, and O.S. Andersen. 1996. Membrane stiffness and channel function. Biochemistry. 35:3825–3830. [DOI] [PubMed] [Google Scholar]
- Lundbæk, J.A., A.M. Maer, and O.S. Andersen. 1997. Lipid bilayer electrostatic energy, curvature stress, and assembly of gramicidin channels. Biochemistry. 36:5695–5701. [DOI] [PubMed] [Google Scholar]
- Marsh, D., and L.I. Horváth. 1998. Structure, dynamics and composition of the lipid-protein interface. Perspectives from spin-labeling. Biochim. Biophys. Acta. 1376:267–296. [DOI] [PubMed] [Google Scholar]
- Maxfield, F.R. 2002. Plasma membrane microdomains. Curr. Opin. Cell Biol. 14:483–487. [DOI] [PubMed] [Google Scholar]
- Mayer, M.L., R. Olson, and G. Gouaux. 2001. Mechanisms for ligand binding to GluRO Ion channels: Crystal structures of the glutamate and serine complexes and a closed Apo state. J. Mol. Biol. 311:815–836. [DOI] [PubMed] [Google Scholar]
- McAuley, K., P.K. Fyfe, J.P. Ridge, N.W. Isaacs, R.J. Cogdell, and M.R. Jones. 1999. Structural details of an interaction between cardiolipin and an integral membrane protein. Proc. Natl. Acad. Sci. USA. 96:14706–14711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, M.P., and M.A. Moore. 1992. Effects of lipids and detergents on the conformation of the nicotinic acetylcholine receptor from Torpedo californica. J. Biol. Chem. 267:7655–7663. [PubMed] [Google Scholar]
- McCloskey, H.M., G.H. Rothblat, and J.M. Glick. 1987. Incubation of acetylated low-density lipoprotein with cholesterol-rich dispersions enhances cholesterol uptake by macrophages. Biochim. Biophys. Acta. 921:320–332. [DOI] [PubMed] [Google Scholar]
- McIntosh, D.B., and G.A. Davidson. 1984. Effects of nonsolubilizing and solubilizing concentrations of Triton X-100 on Ca2+ binding and Ca2+-ATPase activity of sarcoplasmic reticulum. Biochemistry. 23:1959–1965. [DOI] [PubMed] [Google Scholar]
- McIntosh, T.J., S. Advani, R.E. Burton, D.V. Zhelev, D. Needham, and S.A. Simon. 1995. Experimental tests for protrusion and undulation pressures in phospholipid bilayers. Biochemistry. 34:8520–8532. [DOI] [PubMed] [Google Scholar]
- Meleard, P., C. Gerbeaud, T. Pott, L. Fernandez-Puente, I. Bivas, M. Mitov, J. Dufourcq, and P. Bothorel. 1997. Bending elasticities of model membranes: influences of temperature and sterol content. Biophys. J. 72:2616–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meves, H. 1994. Modulation of ion channels by arachidonic acid. Prog. Neurobiol. 43:175–186. [DOI] [PubMed] [Google Scholar]
- Miller, C. 2003. A charged view of voltage-gated ion channels. Nat. Struct. Biol. 10:422–424. [DOI] [PubMed] [Google Scholar]
- Mitchell, D.C., M. Straume, J.L. Miller, and B.J. Litman. 1990. Modulation of metarhodopsin formation by cholesterol-induced ordering of bilayer lipids. Biochemistry. 29:9143–9149. [DOI] [PubMed] [Google Scholar]
- Mouritsen, O.G., and M. Bloom. 1984. Mattress model of lipid-protein interactions in membranes. Biophys. J. 46:141–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay, A.K., S. Dracheva, S. Bose, and R.W. Hendler. 1996. Control of the integral membrane proton pump, bacteriorhodopsin, by purple membrane lipids of Halobacterium halobium. Biochemistry. 35:9245–9252. [DOI] [PubMed] [Google Scholar]
- Needham, D., and R.S. Nunn. 1990. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys. J. 58:997–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer, J. 1987. A Guide to the Properties and Uses of Detergents in Biology and Biochemistry. Calbiochem, San Diego, CA.
- Nielsen, C., M. Goulian, and O.S. Andersen. 1998. Energetics of inclusion-induced bilayer deformations. Biophys. J. 74:1966–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, C., and O.S. Andersen. 2000. Inclusion-induced bilayer deformations: effects of monolayer equilibrium curvature. Biophys. J. 79:2583–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numann, R., S.D. Hauschka, W.A. Catterall, and T. Scheuer. 1994. Modulation of skeletal muscle sodium channels in a satellite cell line by protein kinase C. J. Neurosci. 14:4226–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connell, A.M., R.E. Koeppe, II, and O.S. Andersen. 1990. Kinetics of gramicidin channel formation in lipid bilayers: transmembrane monomer association. Science. 250:1256–1259. [DOI] [PubMed] [Google Scholar]
- Oldfield, E., M. Meadows, D. Rice, and R. Jacobs. 1978. Spectroscopic studies of specifically deuterium labeled membrane systems nuclear magnetic resonance investigation of the effects of cholesterol in model systems. Biochemistry. 17:2727–2740. [DOI] [PubMed] [Google Scholar]
- Ordway, R.W., J.J. Singer, and J.V. Walsh, Jr. 1991. Direct regulation of ion channels by fatty acids. Trends Neurosci. 14:96–100. [DOI] [PubMed] [Google Scholar]
- Otten, D., M.F. Brown, and K. Beyer. 2000. Softening of membrane bilayers by detergents elucidated by deuterium NMR spectroscopy. J. Physiol. Chem. 104:12119–12129. [Google Scholar]
- Owicki, J.C., M.W. Springgate, and H.M. McConnell. 1978. Theoretical study of protein-lipid and protein-protein interactions in bilayer membranes. Proc. Natl. Acad. Sci. USA. 75:1616–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partenskii, M.B., and P.C. Jordan. 2002. Membrane deformation and the elastic energy of insertion: perturbation of membrane elastic constants due to peptide insertion. J. Chem. Physiol. 117:10768–10776. [Google Scholar]
- Patel, A.J., M. Lazdunski, and E. Honore. 2001. Lipid and mechano-gated 2P domain K+ channels. Curr. Opin. Cell Biol. 13:422–427. [DOI] [PubMed] [Google Scholar]
- Patton, D., J. West, W. Catterall, and A. Goldin. 1992. Amino acid residues required for fast Na+-channel inactivation: charge neutralizations and deletions in the III-IV linker. Proc. Natl. Acad. Sci. USA. 89:10905–10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, P. Sompornpisut, A. Kloda, and B. Martinac. 2002. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature. 418:942–948. [DOI] [PubMed] [Google Scholar]
- Pesce, M.A., and C.S. Strande. 1973. A new micromethod for determination of protein in cerebrospinal fluid and urine. Clin. Chem. 19:1265–1267. [PubMed] [Google Scholar]
- Petrov, A.G. 1999. The Lyotropic State of Matter—Molecular Physics and Living Matter Physics. Gordon & Breach, NY.
- Rand, R.P., and V.A. Parsegian. 1997. Hydration, curvature, and bending elasticity of phospholipid monolayers. Curr. Topics Mem. Transp. 44:167–189. [Google Scholar]
- Rawicz, W., K.C. Olbrich, T. McIntosh, D. Needham, and E. Evans. 2000. Effect of chain length and unsaturation on elasticity of lipid bilayers. Biophys. J. 79:328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, N.C. 1982. Specificity and binding affinity of phospholipids to the high-affinity cardiolipin sites of beef heart cytochrome c oxidase. Biochemistry. 21:184–188. [DOI] [PubMed] [Google Scholar]
- Sackmann, E. 1984. Physical basis of trigger processes and membrane structures. Biol. Membr. 5:105–141. [Google Scholar]
- Safinya, C.R., E.B. Sirota, D. Roux, and G.S. Smith. 1989. Universality in interacting membranes: The effect of cosurfactants on the interfacial rigidity. Phys. Rev. Lett. 62:1134–1137. [DOI] [PubMed] [Google Scholar]
- Sakmar, T.P. 1998. Rhodopsin: a prototypical G protein-coupled receptor. Prog. Nucleic Acid Res. Mol. Biol. 59:1–34. [DOI] [PubMed] [Google Scholar]
- Sankaram, M.B., and T.E. Thompson. 1990. Modulation of phospholipid acyl chain order by cholesterol. A solid-state 2H nuclear magnetic resonance study. Biochemistry. 29:10676–10684. [DOI] [PubMed] [Google Scholar]
- Sawyer, D.B., R.E. Koeppe, II, and O.S. Andersen. 1989. Induction of conductance heterogeneity in gramicidin channels. Biochemistry. 28:6571–6583. [DOI] [PubMed] [Google Scholar]
- Sawyer, D.B., R.E. Koeppe, II, and O.S. Andersen. 1990. Gramicidin single-channel properties show no solvent-history dependence. Biophys. J. 57:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seddon, J.M. 1990. Structure of the inverted hexagonal (HII) phase, and non-lamellar phase transitions of lipids. Biochim. Biophys. Acta. 1031:1–69. [DOI] [PubMed] [Google Scholar]
- Sheetz, M.P., and S.J. Singer. 1974. Biological membranes as bilayer couples. A molecular mechanism of drug-erythrocyte interactions. Proc. Natl. Acad. Sci. USA. 71:4457–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- Smith, P.A., and P. Proks. 1998. Inhibition of the ATP-sensitive potassium channel from mouse pancreatic b-cells by surfactants. Br. J. Pharmacol. 124:529–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, R.D., and A.L. Goldin. 2000. Potentiation of rat brain sodium channel currents by PKA in Xenopus oocytes involves the I-II linker. Am. J. Physiol. Cell Physiol. 278:C638–C645. [DOI] [PubMed] [Google Scholar]
- Sukharev, S., M. Betanzos, C. Chiang, and H.R. Guy. 2001. The gating mechanism of the large mechanosensitive channel MscL. Nature. 409:720–724. [DOI] [PubMed] [Google Scholar]
- Sunshine, C., and M.G. McNamee. 1992. Lipid modulation of nicotinic acetylcholine receptor function: the role of neutral and negatively charged lipids. Biochim. Biophys. Acta. 1108:240–246. [DOI] [PubMed] [Google Scholar]
- Tate, M.W., E.F. Eikenberry, D.C. Turner, E. Shyamsunder, and S.M. Gruner. 1991. Nonbilayer phases of membrane lipids. Chem. Phys. Lipids. 57:147–164. [DOI] [PubMed] [Google Scholar]
- Townsley, L.E., W.A. Tucker, S. Sham, and J.F. Hinton. 2001. Structures of gramicidins A, B, and C incorporated into sodium dodecyl sulfate micelles. Biochemistry. 40:11676–11686. [DOI] [PubMed] [Google Scholar]
- Toyoshima, C., and H. Nomura. 2002. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 418:605–611. [DOI] [PubMed] [Google Scholar]
- Trimmer, J.S., S.S. Cooperman, S.A. Tomiko, J. Zhou, S.M. Crean, M.B. Boyle, R.G. Kallen, Z.H. Sheng, R.L. Barchi, F.J. Sigworth, et al. 1989. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 3:33–49. [DOI] [PubMed] [Google Scholar]
- Unwin, N., C. Toyoshima, and E. Kubalek. 1988. Arrangement of the acetylcholine receptor subunits in the resting and desensitized states, determined by cryoelectron microscopy of crystallized Torpedo postsynaptic membranes. J. Cell Biol. 107:1123–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin, P.N.T., and P.D. Ennis. 1984. Two configurations of a channel-forming membrane protein. Nature. 307:609–613. [DOI] [PubMed] [Google Scholar]
- Valiyaveetil, F.I., Y. Zhou, and R. MacKinnon. 2002. Lipids in the structure, folding, and function of the KcsA K+ channel. Biochemistry. 41:10771–10777. [DOI] [PubMed] [Google Scholar]
- Vinson, P.K., Y. Talmon, and A. Walter. 1989. Vesicle-miceller transition of phosphatidylcholine and octyl glucoside elucidated by cryo-transmission electron microscopy. Biophys. J. 56:669–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonck, J. 2000. Structure of the bacteriorhodopsin mutant F219L N intermediate revealed by electron crystallography. EMBO J. 19:2152–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallert, M.A., M.J. Ackerman, D. Kim, and D.E. Clapham. 1991. Two novel cardiac atrial K+ channels, IK,AA and IK,PC. J. Gen. Physiol. 98:921–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, A., G. Kuehl, K. Barnes, and G. VanderWaerdt. 2000. The vesicle-to-micelle transition of phosphatidylcholine vesicles induced by nonionic detergents: effects of sodium chloride, sucrose and urea. Biochim. Biophys. Acta. 1508:20–33. [DOI] [PubMed] [Google Scholar]
- Weltzien, H.U. 1979. Cytolytic and membrane-perturbing properties of lysophosphatidylcholine. Biochim. Biophys. Acta. 559:259–287. [DOI] [PubMed] [Google Scholar]
- West, J.W., D.E. Patton, T. Scheuer, Y. Wang, A.L. Goldin, and W.A. Catterall. 1992. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. USA. 89:10910–10914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Y., S.N. Wright, G.K. Wang, J.P. Morgan, and A. Leaf. 1998. Fatty acids suppress voltage-gated Na+ currents in HEK293t cells transfected with the α-subunit of the human cardiac Na+ channel. Proc. Natl. Acad. Sci. USA. 95:2680–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarov-Yarovoy, V., J. Brown, E.M. Sharp, J.J. Clare, T. Scheuer, and W.A. Catterall. 2001. Molecular determinants of voltage-dependent gating and binding of pore blocking drugs in transmembrane segment IIIS6 of the Na+ channel α subunit. J. Biol. Chem. 276:20–27. [DOI] [PubMed] [Google Scholar]
- Yeagle, P.L. 1985. Cholesterol and the cell membrane. Biochim. Biophys. Acta. 822:267–287. [DOI] [PubMed] [Google Scholar]
- Zhang, Y.P., R.N. Lewis, R.S. Hodges, and R.N. McElhaney. 1992. Interaction of a peptide model of a hydrophobic transmembrane α-helical segment of a membrane protein with phosphatidylcholine bilayers: differential scanning calorimetric and FTIR spectroscopic studies. Biochemistry. 31:11579–11588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.