

Abstract
The synucleins are a family of intrinsically disordered proteins involved in various human diseases. α-Synuclein has been extensively characterized due to its role in Parkinson's disease where it forms intracellular aggregates, while γ-synuclein is overexpressed in a majority of late-stage breast cancers. Despite fairly strong sequence similarity between the amyloid-forming regions of α- and γ-synuclein, γ-synuclein has only a weak propensity to form amyloid fibrils. We hypothesize that the different fibrillation tendencies of α- and γ-synuclein may be related to differences in structural propensities. Here we have measured chemical shifts for γ-synuclein and compared them to previously published shifts for α-synuclein. In order to facilitate direct comparison, we have implemented a simple new technique for re-referencing chemical shifts that we have found to be highly effective for both disordered and folded proteins. In addition, we have developed a new method that combines different chemical shifts into a single residue-specific secondary structure propensity (SSP) score. We observe significant differences between α- and γ-synuclein secondary structure propensities. Most interestingly, γ-synuclein has an increased α-helical propensity in the amyloid-forming region that is critical for α-synuclein fibrillation, suggesting that increased structural stability in this region may protect against γ-synuclein aggregation. This comparison of residue-specific secondary structure propensities between intrinsically disordered homologs highlights the sensitivity of transient structure to sequence changes, which we suggest may have been exploited as an evolutionary mechanism for fast modulation of protein structure and, hence, function.
Keywords: SNCG, NMR, residual structure, secondary chemical shifts, chemical shift re-referencing
In recent years, an increasing number of proteins have been identified as being intrinsically disordered, meaning they lack stable folded tertiary structure under normal conditions (Dyson and Wright 2005). Recent predictions suggest that nearly 50% of eukaryotic proteins contain disordered regions longer than 30 residues, with even higher predictions in the subset of human cancer-associated and signaling proteins (80% and 67%, respectively) (Iakoucheva et al. 2002). Despite their lack of folded, globular structure, intrinsically disordered states of proteins possess significant amounts of nonrandom structure (Lacy et al. 2004; Dedmon et al. 2005). Although typically referred to as “residual structure,” we prefer to use the terms “transient structure” and “structural propensities,” since “residual” implies the existence of a state with fully formed structure, which is not necessarily the case for intrinsically disordered proteins.
One question that has not yet been sufficiently explored is the sensitivity of structural propensities in disordered states to changes in sequence. Whereas the structures of folded proteins are generally considered to be fairly robust to changes in sequence, with homologs as low as 40% sequence identity still expected to be reasonably well described by homology models (Sanchez and Sali 1997), very little research has addressed this issue for disordered proteins. The widely varying probabilities of different amino acids to form different types of secondary structure (Muñoz and Serrano 1994b) suggest that secondary structure propensities would be fairly sensitive to at least certain sequence changes. Disease-causing point mutations in α-synuclein have been shown to cause changes in 13Cα chemical shifts, suggesting local modulation of secondary structure propensities (Bussell and Eliezer 2001), and a single point mutation in a marginally stable SH3 domain is thought to stabilize the folded state by disrupting nonnative helical propensity in the unfolded state (Mok et al. 2001). In addition, Wirmer et al. (2006) looked at chemical shifts and 15N relaxation in the urea-denatured states of homologous proteins and found some significant differences. On the other hand, Ackerman and Shortle (2002) suggested that transient long-range structure is quite robust to changes in amino acid sequence based upon residual dipolar couplings, although recent work has questioned the interpretation of these results (Mohana-Borges et al. 2004).
The synucleins are a family of intrinsically disordered proteins associated with very different human diseases. α-Synuclein is well known for its role in Parkinson's disease, where it forms intracellular aggregates, and α-synuclein mutations are associated with familial Parkinson's disease (Bennett 2005). In addition, an α-synuclein fragment is found in amyloid plaques associated with Alzheimer's disease (Bennett 2005). There is evidence that β-synuclein mutations may predispose to dementia with Lewy bodies (Ohtake et al. 2004). γ-Synuclein (also known as SNCG or breast cancer specific gene 1 [BCSG1]) is known primarily for its overexpression in late-stage breast cancer, where it can cause increased metastasis (Jia et al. 1999; Liu et al. 2005) and resistance to certain chemotherapeutic drugs (Pan et al. 2002; Zhou et al. 2006). Although the normal biological functions of the synucleins are unclear, recent evidence points to chaperone activity, with α-synuclein assisting in the refolding of SNARE proteins (Chandra et al. 2005) and γ-synuclein participating in the heat shock protein–based multichaperone complex involved in estrogen receptor-α activation (Jiang et al. 2004).
There has been much recent interest in α-synuclein amyloid formation and its relation to neurodegenerative disorders. Interestingly, the strong fibrillation propensity of α-synuclein is not shared by β- or γ-synuclein. Uversky et al. (2002) found that β-synuclein did not aggregate at all, while γ-synuclein would only form amyloid fibrils at much higher concentrations and at a much slower rate than α-synuclein. In addition, both β- and γ-synuclein were shown to inhibit α-synuclein fibrillation in vitro (Uversky et al. 2002). α-Synuclein amyloid has been shown to involve residues 39–101 (Del Mar et al. 2005), and the N-terminal 87 residues are sufficient for fibrillation (Serpell et al. 2000). The failure of β-synuclein to aggregate can be explained by an 11-residue deletion in the amyloid-forming region (Fig. 1). γ-Synuclein, however, is very similar to α-synuclein over the entire N-terminal 100 residues (66% sequence identity, 74% similarity). We therefore hypothesize that the strong aggregation propensity of α-synuclein relative to γ-synuclein may be due to differences in secondary structure propensities. To address this, we have compared secondary structure propensities calculated from chemical shifts for α- and γ-synuclein. In addition to providing insight into synuclein aggregation, this has allowed us to compare residue-specific secondary structure propensities between homologous intrinsically disordered proteins.
Figure 1.

Sequence alignment of α-, β-, and γ-synuclein showing the N-terminal, amyloid-forming, and C-terminal regions. Black shading highlights identical sequences among all three synucleins, while gray shading highlights identical shading among two of the synucleins.
Results and Discussion
Measurement and re-referencing of chemical shifts
A series of NMR experiments was performed to obtain complete 1HN, 15N, 13C′, 13Cα, and 13Cβ resonance assignments for γ-synuclein. α-Synuclein chemical shifts were available from a previous study (Bermel et al. 2006). The deviations of certain chemical shifts from their expected random coil values, known as secondary chemical shifts (Δδ = δobserved − δcoil), are a useful measure of secondary structure (Wishart and Sykes 1994a). In Figure 2, we show ΔδCα − ΔδCβ values for α- and γ-synuclein. This is a common method of reporting secondary chemical shifts and has the advantage of canceling out chemical shift referencing errors. Positive values indicate α-helical structure, and negative values indicate β-strand or extended structure, with consecutive values of about ±2 ppm typically interpreted as having fully formed secondary structure, and lower values indicating partially formed structure (i.e., secondary structure propensities). Although there appear to be some significant differences in secondary structure propensities between α- and γ-synuclein, the interpretation and comparison of these values are difficult because there is considerable variation in the sensitivity of specific chemical shifts from different amino acids for measuring α and β secondary structure (Wang and Jardetzky 2002). Thus, direct comparison of the α- and γ-synuclein secondary chemical shifts is complicated by the sequence differences between the two proteins and the choice of chemical shifts used. In addition, it is difficult to quantitatively interpret secondary chemical shifts alone because the expected values for fully formed secondary structure vary for different amino acids (Wang and Jardetzky 2002).
Figure 2.

ΔδCα − ΔδCβ secondary chemical shifts for γ-synuclein (A) and α-synuclein (B). Positive values indicate α-structure propensity and negative values indicate β-structure propensity.
Another problem with using chemical shifts to assess secondary structure is uncertainties regarding chemical shift referencing. This is especially important for disordered proteins that often have chemical shifts very close to their expected random coil values. Upon downloading α-synuclein chemical shifts from the BioMagResBank (BMRB) (Seavey et al. 1991), it was immediately clear from comparison of ΔδCα − ΔδCβ and ΔδCα plots that there was a significant referencing error. Indeed, many entries in the BMRB are incorrectly referenced, including some that claim to follow the proper DSS referencing protocol (Zhang et al. 2003).
We have implemented a very simple method for 13C chemical shift re-referencing, based only upon relative 13Cα and 13Cβ chemical shifts. We assume that, for a given amino acid, the relative difference between the ΔδCα value observed and expected for fully formed α- or β-structure should be similar to the relative difference between the ΔδCβ value observed and expected for fully formed α- or β-structure. Since the dependencies of ΔδCα and ΔδCβ values on α- and β-structure are inversely correlated, we adjust the chemical shift referencing offset until those differences are minimized. This is done according to Equation 1, where the term Z is minimized through iterative re-referencing; the summation is over all residues i; ΔδCi α/β obs are the observed secondary chemical shifts; and ΔδCi α/β α/β are the average secondary chemical shifts for fully formed α- or β-structure.
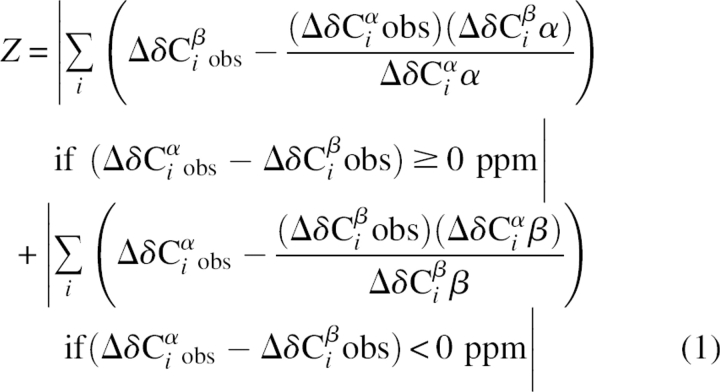 |
We tested this re-referencing procedure using RefDB, a database of uniformly referenced protein chemical shifts (Zhang et al. 2003) in which most of the chemical shifts are expected to be properly referenced. Figure 3 shows the distribution of referencing offsets calculated for 13Cα and 13Cβ chemical shifts with our procedure for a collection of proteins from RefDB. The average referencing offset was 0.06 ppm, and the standard deviation was 0.14 ppm. This is consistent with the stated referencing offset errors of about ±0.2 ppm for proteins in the RefDB (Zhang et al. 2003). We further tested the procedure on multiple disordered proteins studied in our lab with chemical shifts known to be properly DSS-referenced. In all cases, the calculated referencing offsets were <0.06 ppm (data not shown). γ-Synuclein had a calculated referencing offset of 0.03 ppm from its DSS-referenced 13C shifts. α-Synuclein, which we believed to be improperly referenced, had a calculated referencing offset of 0.39 ppm. The re-referenced α-synuclein 13Cα values agree very well with other published values (Eliezer et al. 2001), which differ by 0.36 ppm on average from the α-synuclein shifts used in this study (Bermel et al. 2006) before re-referencing. These results suggest our procedure is effective in properly re-referencing 13C chemical shifts for both folded and disordered proteins. This re-referencing procedure has been incorporated into the SSP program described below and as a standalone script, available at http://pound.med.utoronto.ca/software.html.
Figure 3.

Distribution of referencing offsets for 13Cα and 13Cβ chemical shifts calculated for 247 proteins from RefDB.
It should be noted that two alternative procedures also exist for re-referencing chemical shifts. The first, by Wang and Wishart (2005), is not appropriate for disordered proteins since it assumes they exist as random coils; thus the presence of structural propensities would cause improper calculation of referencing offsets. Another procedure, by Wang et al. (2005), called linear analysis of chemical shifts (LACS)—which we discovered only after implementation of our method—is similar to ours in that it relies on relative 13Cα and 13Cβ chemical shifts. Calculation of referencing offsets with LACS using the RefDB data set from above gave similar results to our method (average offset, 0.02 ppm; standard deviation, 0.17 ppm). While our method is likely of a similar accuracy to LACS, ours is simpler since it does not require linear regression and has the advantage of being available as a standalone script.
Secondary structure propensities of α- and γ-synuclein
In order to better quantify and compare secondary structure propensities, we have developed a method that combines chemical shifts from different nuclei into a single secondary structure propensity (SSP) score representing the expected fraction of α- or β-structure. The contributions of different chemical shifts are weighted by their sensitivity to α- and β-structure. Equation 2 shows the SSP score for a given residue i, where Δδ Xjobs is the observed secondary chemical shift, Δδ Xjα/β are the average secondary chemical shifts of fully formed secondary structure (α or β), and σjα/β are the standard deviations of Δδ Xjα/β. The summation over X is for all chemical shifts used and can include 13Cα, 13Cβ, 13C′, 1Hα, 1HN, and 15N. However, for the most part, only 13Cα, 13Cβ, and 1Hα seem to be very useful for disordered proteins. In addition, since our re-referencing method only works for 13C chemical shifts, other chemical shifts should probably not be used unless they are known to be properly DSS-referenced. Equation 2 includes a weighted averaging over five residues, although this can be adjusted within the program. This weighted averaging serves to minimize the contribution from chemical shifts that are poor measures of secondary structure. For example, since glycine has no Cβ, and 13Cα chemical shifts are a poor measure of β-structure, glycines can often give extreme values in the β-structure region. However, with weighted averaging, the contribution from these residues becomes negligible.
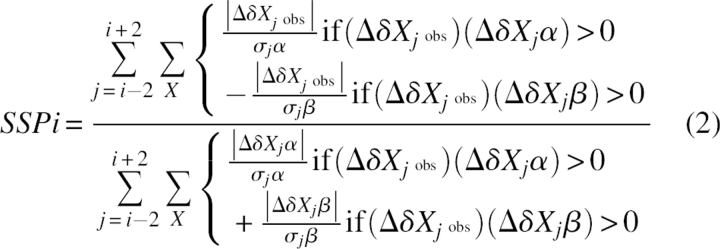 |
A SSP score at a given residue of 1 or −1 reflects fully formed α- or β-structure, respectively, while a score of 0.5 indicates that 50% of the conformers in the disordered state ensemble are helical at that position. This is based upon the assumption that a given secondary chemical shift as a fraction of the average secondary chemical shift for fully formed secondary structure corresponds to the fraction of secondary structure present at that position (Wishart and Sykes 1994a). Although SSP scores are not rigorously quantitative, due to the known variation of average random coil and secondary structure chemical shifts, they can be expected to roughly correspond to the secondary structure propensity at a given position. The SSP program is freely available at http://pound.med.utoronto.ca/software.html.
While the idea of combining multiple secondary chemical shifts into a single score is similar to the Chemical Shift Index (Wishart and Sykes 1994b) and the probability-based secondary structure identification of Wang and Jardetzky (2002), our objective is to calculate secondary structure propensities rather than the probability of fully formed secondary structure. Since our method weights individual chemical shifts by their reliability for detecting secondary structure, and because it attempts to provide a quantitative estimate of secondary structure propensities, we suggest that our method of displaying SSP scores be used as an alternative to simply showing secondary chemical shifts. This is especially so for the common practice of reporting only ΔδCα values, which provide a good measure of α-structure but are very poor at distinguishing β-structure from random coil (Wang and Jardetzky 2002).
SSP scores for α- and γ-synuclein are shown in Figure 4. The differences in secondary structure propensities between the two are much clearer than in the ΔδCα − ΔδCβ plots shown in Figure 2. Most strikingly, from residues 49–99 in the amyloid-forming region (53% sequence identity, 65% similarity), γ-synuclein has propensity for α-structure of up to ∼10%, while α-synuclein appears closer to random coil with some β-structure propensity. The N-terminal region (79% sequence identity, 82% similarity) is highly disordered for both proteins, although γ-synuclein does have a region of ∼15% β-propensity near the N terminus. Interestingly, this region maps to sites of interaction for multiple γ-synuclein binding partners (J.A. Marsh and J.D. Forman-Kay, in prep.). Both α- and γ-synuclein have up to 20% or 30% β-structure propensities in their C-terminal regions, although there are large differences between the two, as expected since this region is not conserved.
Figure 4.

Secondary structure propensity (SSP) scores for α-synuclein (dotted lines) and γ-synuclein (dashed lines) calculated using 13Cα and 13Cβ chemical shifts. Positive values represent α-structure propensity and negative values represent β-structure propensity. Upper and lower lines represent chemical shifts adjusted ±0.2 ppm from the calculated referencing offset for each protein.
By averaging the α- and β-regions of the calculated SSP scores, we calculate an overall total of 0.7% α-structure and 5.5% β-structure for α-synuclein, and 2.3% α-structure and 4.2% β-structure for γ-synuclein. This is consistent with the finding of Uversky et al. (2002) that α- and γ-synuclein had nearly identical far-UV circular dichroism spectra. However, using Fourier transform infrared (FTIR) spectroscopy, they also observed significant β-structure in γ-synuclein that was not present in α-synuclein. They speculated that this was due to a concentration-dependent oligomerization of the protein. We observe no chemical shift changes or resonance broadening in the 1H–15N HSQC spectra of γ-synuclein from 30 to 800 μM, which indicates that there is no concentration-dependent oligomerization over this range. We suggest that their results may be due to oligomerization or aggregation of the protein under FTIR conditions that does not occur under solution NMR conditions.
Although the α- and γ-synuclein chemical shifts were recorded under slightly different conditions (5°C vs. 15°C, pH 7.4 vs. 6.8), we have observed γ-synuclein 1HN and 15N chemical shifts to be fairly insensitive to buffer conditions from pH 5.4 to 7.4, NaCl from 0 to 250 mM, and from 5° to 37°C, suggesting that there are no significant changes in structural propensities over these conditions. A figure showing the 1H–15N HSQC spectra of γ-synuclein at 5° and 15°C and chemical shift changes is provided in the Electronic Supplemental Material. In addition, the α-synuclein 13Cα chemical shifts used here are very similar to those of Eliezer et al. (2001), which were recorded at 10°C (pH 7.4). SSP scores calculated using only the 13Cα chemical shifts from these two studies have a correlation coefficient (r) of 0.91, suggesting that structural differences are negligible. McNulty et al. (2006) have suggested that α-synuclein structure is sensitive to temperature based upon broadening of 1H–15N HSQC resonances at higher temperatures. We also observe broadening in γ-synuclein at 25°C, but close analysis indicates that all threonines, serines, and glycines are broadened while most other resonances are not. In addition, this broadening disappears when the pH is lowered to 7.0. This strongly suggests that the observed broadening is amide-proton exchange, which is accelerated at high pH and is highly sequence-dependent (Molday et al. 1972). Thus the possibility that raising the pH from 7.0 to 7.4 or increasing the temperature causes large conformational changes seems unlikely. The fact that γ-synuclein chemical shifts do not show any evidence of conformational change over a range of temperature and buffer conditions and the high similarity between α-synuclein chemical shifts from two studies at 10° and 15°C in different buffers strongly supports the validity of our comparison of α- and γ-synuclein chemical shifts.
The increased α-structure in the amyloid-forming region of γ-synuclein is either due to the intrinsic helix-forming propensity of the sequence or is induced through tertiary contacts. GOR4 (Garnier et al. 1996) secondary structure predictions suggest a moderately stronger helical propensity for γ-synuclein than α-synuclein in this region (Fig. 5). AGADIR (Muñoz and Serrano 1994a) gives similar predictions (not shown). The correlation coefficients (r) between the GOR4 predicted helicity and calculated SSP scores are 0.23 and 0.48 for the N-terminal 100 residues of α- and γ-synuclein, respectively. The correlation is much worse with the C-terminal region, not surprisingly since it contains β-structure propensity. While the increased α-structure of γ-synuclein could be accounted for by intrinsic secondary structure propensities, there are still very large differences between predicted and calculated secondary structure propensities pointing to nonlocal effects from tertiary contacts. The lower correlation of α-synuclein SSP scores with predicted helicity suggests that α-synuclein may possess more tertiary structure than γ-synuclein, consistent with the identification of tertiary contacts involving the C terminus of α-synuclein (Bernadó et al. 2005; Dedmon et al. 2005), which is not conserved with γ-synuclein and is 13 residues longer.
Figure 5.

GOR4 helix predictions for α-synuclein (dotted line) and γ-synuclein (dashed line).
Implications for synuclein fibrillation
α-Synuclein fibrillation is believed to progress through a soluble oligomeric intermediate rich in β-structure (Kaylor et al. 2005). The difference in secondary structure propensities between α- and γ-synuclein therefore provides a possible explanation for their differential fibrillation: The increased α-helical propensity in the amyloid-forming region of γ-synuclein may stabilize the intrinsically disordered state relative to the β-structured intermediate, thus inhibiting oligomerization. Although it is possible that the different aggregation propensities are due to the nonconserved C-terminal region, the fact that residues C-terminal to 101 are not required for or involved in α-synuclein amyloid-formation argues against this.
The hypothesis that increased α-helical propensity in the amyloid-forming region correlates with a reduced fibrillation propensity is supported by the work of Bussell and Eliezer (2001), who reported ΔδCα values for wild-type α-synuclein and the early-onset Parkinson's disease mutants A53T and A30P. Although we would have liked to directly compare SSP values, the ΔδCα values are supplied only in low-resolution graph format, making conversion to SSP values difficult. The A53T mutation, which has a significantly increased fibrillation rate relative to wild-type α-synuclein (Conway et al. 1998), had lower ΔδCα values in the amyloid-forming region from approximately residues 50–80, suggesting lower α-propensity or increased β-propensity in this region. In contrast, the A30P mutant has similar ΔδCα values to wild-type α-synuclein in the amyloid-forming region (although there were significant differences near the site of the mutation) and a reduced fibrillation propensity relative to the wild-type protein (Conway et al. 2000). However, the oligomerization rate of A30P was very similar to wild-type (Conway et al. 2000), suggesting that there are differences between synuclein oligomerization and fibril formation. In this paper, we talk about the relationship between secondary structure propensities and fibrillation because most papers have only looked at overall fibrillation rates. However, it seems likely that the structural propensities of intrinsically disordered synucleins may be more important to the oligomerization step, which, while being related to the overall fibrillation rate, is distinct from the fibril formation step.
We provide further support for our hypothesis by comparing average GOR4 helix predictions in the amyloid-forming regions of α-, β-, and γ-synucleins; mouse α-synuclein; and several α-synuclein point mutations with their relative fibrillation propensities (Table 1). Although we cannot quantify fibrillation, we can assign relative fibrillation propensities based upon the results of various studies. Interestingly, there is a very strong correlation between GOR4 predicted helicity and relative fibrillation propensity. For example, mouse α-synuclein, which was reported to fibrillate faster than any human α-synuclein mutants, has the lowest predicted helicity. On the other hand, γ- and β-synucleins have the highest predicted helicities and the lowest fibrillation propensities. This strongly supports the idea that synuclein fibrillation is correlated with amyloid-forming region helical propensities.
Table 1.
Relative fibrillation propensities and average GOR4 helix predictions for the amyloid-forming regions of various synucleins and mutants

The lower level of γ-synuclein fibrillation leads to the question of why α-synuclein retains its aggregation propensity when evolution could have reduced this deleterious tendency via subtle amino acid substitutions. One possible explanation is that there is a positive effect from α-synuclein oligomerization. For example, there is recent evidence suggesting that neuronal protein aggregation may actually be a protective response against neurotoxic stress (Lee et al. 2006). Alternatively, the α-synuclein sequence that leads to fibrillation could be important for its chaperone activity or other functions by determining lipid- or protein-binding specificities, thus providing an evolutionary restraint maintaining the fibrillation propensity.
Evolution of secondary structure propensities in disordered regions
In this work, we compared residue-specific secondary structure propensities of intrinsically disordered homologs and have shown that there are significant differences between α- and γ-synuclein. One possible explanation for this is that disordered regions simply evolve faster than folded regions due to their reduced tertiary contacts. A recent analysis by Chen et al. (2006) has found that sequence conservation, as measured by normalized Shannon's entropy values (Shannon 1948), is similar in conserved predicted disordered regions (0.35 ± 0.15) and conserved predicted ordered regions (0.34 ± 0.14), where the given errors are standard deviations. Smaller Shannon's entropy values indicate increasing sequence conservation, with a value of 0 being completely conserved and 1 being completely nonconserved. This implies that, on average, the sequences of conserved disordered regions evolve at a similar, although possibly slightly faster, rate than conserved ordered regions. In Figure 6, we show the sequence conservation of the synucleins analyzed by calculation of normalized Shannon's entropy values for 27 sequences (see Materials and Methods). The N-terminal, amyloid-forming, and C-terminal regions have average normalized Shannon's entropy values of 0.09, 0.27, and 0.42, respectively. Although direct comparison of Shannon's entropy values can be difficult because they depend on the choice of sequence alignments, even more conservative synuclein sequence alignments that removed closely related sequences gave lower than average values in the N-terminal and amyloid-forming regions (see Electronic Supplemental Material). This demonstrates that the N-terminal and amyloid-forming regions are more conserved than the average conserved disordered and ordered regions, and thus have evolved at a slower rate.
Figure 6.

Sequence conservation in the synucleins as represented by normalized Shannon's entropy calculated from a multiple sequence alignment of 27 synucleins.
The sequence conservation of the N-terminal and amyloid-forming regions of the synucleins and the significant differences in secondary structure propensities strongly suggest that structural propensities in disordered states are quite sensitive to changes in sequence. As discussed earlier, this is not surprising given the known tendencies of different amino acids to form α and β secondary structure (Muñoz and Serrano 1994b). Based upon these results, it is tempting to speculate that intrinsic disorder provides a simple evolutionary mechanism for fast modulation of protein structure and, hence, function. Specifically, since transient structure in disordered regions is sensitive to sequence, the affinity and specificity of protein interactions can be more easily modulated (i.e., requiring less changes in sequence) than for folded proteins.
The strong correlation between disordered regions and complex eukaryotic regulatory pathways strongly supports a functional importance for disorder. In addition, protein disorder has significant biological costs, providing another strong argument for its importance. For example, several diseases are associated with protein disorder, either through aggregation (Uversky and Fink 2004) or chromosomal translocations (Dyson and Wright 2005). Surely disorder would have been selected against if it did not provide some benefit. Several functional advantages of disorder, relating mostly to protein interactions, have been proposed, including binding diversity (Kriwacki et al. 1996), enhanced binding kinetics (Shoemaker et al. 2000), precise thermodynamic control of interactions (i.e., high specificity with low affinity) (Zhou et al. 2001), and larger binding surfaces (Gunasekaran et al. 2003). The ability of intrinsically disordered regions to modulate their structural propensities faster than folded regions on an evolutionary timescale provides another functional benefit of disorder, regardless of whether this is due to the sensitivity of structural propensities to sequence changes or faster evolution due to reduced constraints from tertiary structure.
Recent evidence strongly suggests that the synucleins function as molecular chaperones (Souza et al. 2000; Jiang et al. 2004; Chandra et al. 2005). Interestingly, chaperones have been shown to have a very high frequency of intrinsic disorder (Tompa and Csermely 2005). Consistent with having chaperone function, the synucleins are known to participate in a large number of interactions (Zhou et al. 2004). Although some interactions are shared by different synucleins (Payton et al. 2004), others are specific for individual synucleins (J.A. Marsh and J.D. Forman-Kay, in prep.). It is likely that the functional differences between the synucleins arise from their different interactions. Therefore, the intrinsically disordered nature of the synucleins may have helped in the differentiation of synuclein interactions, leading to their current unique (but overlapping) cellular functions and their association with widely different diseases.
Materials and methods
Protein preparation
Isotopically labeled GST-tagged human γ-synuclein was expressed in Escherichia coli BL21 cells in 2 l M9 minimal media containing 1 g/L 15N-ammonium chloride and 2 g/L 13C-glucose and induced with 0.25 g/L IPTG for 3 h at 37°C. The cell pellet was stored at −20°C until purification and lysed by sonication in 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM K2HPO4) at pH 7.4. Cell lysate was mixed with glutathione sepharose 4B (GE Healthcare), washed extensively, and cleaved with thrombin on the resin. Protein was further purified via Mono Q anion-exchange chromatography (Pharmacia). The final NMR sample for assignment contained 0.35 mM 13C/15N–γ-synuclein in 1× PBS (pH 7.4), 10% D2O.
NMR spectroscopy
NMR experiments were performed on 500 and 800 MHz Varian Inova spectrometers at 5°C. Resonances were assigned using HNCACB, CBCA(CO)NH, HNCO, HN(CA)CO, and CCC-TOCSY experiments (Cavanagh et al. 1995). Spectra were processed with NMRPipe (Delaglio et al. 1995) and assigned with NMRView (Johnson and Blevins 1994).
Chemical shift re-referencing and secondary structure calculations
To test our re-referencing procedure, we selected 247 proteins from RefDB (Zhang et al. 2003) that had at least 50 13Cβ chemical shifts, were not from TROSY experiments, and also had LACS referencing offsets available at http://bija.nmrfam.wisc.edu/MANI-LACS (Wang et al. 2005). In addition, we did not use proteins that had obvious chemical shift misassignments (chemical shifts >15 ppm from random coil value). Cysteines and residues immediately preceding proline were ignored during re-referencing. Random coil, α-helical and β-strand chemical shifts, and standard deviations came from Zhang et al. (2003). Residues immediately preceding proline were ignored in secondary chemical shift calculations. SSP scores were calculated using 13Cα and 13Cβ chemical shifts, five-residue weighted averaging, and all other default parameters (see the SSP program and accompanying documentation for more details).
Sequence alignments and calculation of normalized Shannon's entropy
Synuclein sequences were selected with BLAST (Altschul et al. 1997) searches for human α-, β-, and γ-synucleins against the UniProt database (Apweiler et al. 2004), retaining unique full-length sequences with E-values <0.0001. Also excluded were sequences containing <90 residues. Finally, sequences with <10 amino acid differences from another sequence were excluded, leaving 27 total synuclein sequences. Sequence alignments were performed with ClustalW (Thompson et al. 1994). Normalized Shannon's entropy values were calculated as in Chen et al. (2006). Insertions relative to human α-synuclein in the N-terminal and amyloid-forming regions were ignored.
Deposition of data
The 1H, 15N, and 13C chemical shift assignments for γ-synuclein have been deposited in the BioMagResBank (accession no. 7244).
Electronic supplemental material
Provided are 1H–15N HSQC spectra of γ-synuclein at 5° and 15°C and chemical shift changes over this range (Fig. S1); proteins from RefDB used to test our re-referencing procedure (Table S1); sequences (Table S2) and the sequence alignment (Fig. S2) used for calculation of normalized Shannon's entropy; and a discussion of how changing sequence alignments affects normalized Shannon's entropy (Fig. S3).
Acknowledgments
This research was supported by grants from the Canadian Institutes of Health Research (to J.D.F.-K.). J.A.M. is the recipient of a University of Toronto Fellowship.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Julie D. Forman-Kay, Structural Biology and Biochemistry, Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada; e-mail: forman@sickkids.ca; fax: (416) 813-5022.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.062465306.
References
- Ackerman, M.S. and Shortle, D. 2002. Robustness of the long-range structure in denatured staphylococcal nuclease to changes in amino acid sequence. Biochemistry 41: 13791–13797. [DOI] [PubMed] [Google Scholar]
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apweiler, R., Bairoch, A., Wu, C.H., Barker, W.C., Boeckmann, B., Ferro, S., Gasteiger, E., Huang, H., Lopez, R., and Magrane, M., et al. 2004. UniProt: The Universal Protein knowledgebase. Nucleic Acids Res. 32: D115–D119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M.C. 2005. The role of α-synuclein in neurodegenerative diseases. Pharmacol. Ther. 105: 311–331. [DOI] [PubMed] [Google Scholar]
- Bermel, W., Bertini, I., Felli, I.C., Lee, Y.M., Luchinat, C., and Pierattelli, R. 2006. Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J. Am. Chem. Soc. 128: 3918–3919. [DOI] [PubMed] [Google Scholar]
- Bernadó, P., Bertoncini, C.W., Griesinger, C., Zweckstetter, M., and Blackledge, M. 2005. Defining long-range order and local disorder in native α-synuclein using residual dipolar couplings. J. Am. Chem. Soc. 127: 17968–17969. [DOI] [PubMed] [Google Scholar]
- Bussell, R. and Eliezer, E. 2001. Residual structure and dynamics in Parkinson's disease-associated mutants of α-synuclein. J. Biol. Chem. 276: 45996–46003. [DOI] [PubMed] [Google Scholar]
- Cavanagh, J., Fairbrother, W.J., Palmer, A.G., and Skelton, N.J. 1995. Protein NMR spectroscopy: Principles and practice. Academic Press, San Diego CA.
- Chandra, S., Gallardo, G., Fernandez-Chacon, R., Schluter, O.M., and Sudhof, T.C. 2005. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 123: 383–396. [DOI] [PubMed] [Google Scholar]
- Chen, J.W., Romero, P., Uversky, V.N., and Dunker, A.K. 2006. Conservation of intrinsic disorder in protein domains and families: I. A database of conserved predicted disordered regions. J. Proteome Res. 5: 879–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, K.A., Harper, J.D., and Lansbury, P.T. 1998. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat. Med. 4: 1318–1320. [DOI] [PubMed] [Google Scholar]
- Conway, K.A., Lee, S.-J., Rochet, J.-C., Ding, T.T., Williamson, R.E., and Lansbury Jr., P.T. 2000. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. 97: 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedmon, M.M., Lindorff-Larsen, K., Christodoulou, J., Vendruscolo, M., and Dobson, C.M. 2005. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J. Am. Chem. Soc. 127: 476–477. [DOI] [PubMed] [Google Scholar]
- Delaglio, F., Grzesiek, S., Vuister, G.W., Zhu, G., Pfeifer, J., and Bax, A. 1995. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6: 277–293. [DOI] [PubMed] [Google Scholar]
- Del Mar, C., Greenbaum, E.A., Mayne, L., Englander, S.W., and Woods, V.L. 2005. Structure and properties of α-synuclein and other amyloids determined at the amino acid level. Proc. Natl. Acad. Sci. 102: 15477–15482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2005. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6: 197–208. [DOI] [PubMed] [Google Scholar]
- Eliezer, D., Kutluay, E., Bussell, R., and Browne, G. 2001. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 307: 1061–1073. [DOI] [PubMed] [Google Scholar]
- Garnier, J., Gibrat, J.F., and Robson, B. 1996. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 266: 540–543. [DOI] [PubMed] [Google Scholar]
- Greenbaum, E.A., Graves, C.L., Mizhizen-Eberz, A.J., Lupoli, M.A., Lynch, D.R., Englander, S.W., Axelsen, P.H., and Giasson, B.I. 2005. The E46K mutation in α-synuclein increases amyloid fibril formation. J. Biol. Chem. 280: 7800–7807. [DOI] [PubMed] [Google Scholar]
- Gunasekaran, K., Tsai, C.J., Kumar, S., Zanuy, D., and Nussinov, R. 2003. Extended disordered proteins: Targeting function with less scaffold. Trends Biochem. Sci. 28: 81–85. [DOI] [PubMed] [Google Scholar]
- Iakoucheva, L., Brown, C., Lawson, J., Obradovic, Z., and Dunker, A. 2002. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323: 573–584. [DOI] [PubMed] [Google Scholar]
- Jia, T., Liu, Y.E., Liu, J., and Shi, Y.E. 1999. Stimulation of breast cancer invasion and metastasis by synuclein γ. Cancer Res. 59: 742–747. [PubMed] [Google Scholar]
- Jiang, Y., Liu, Y.E., Goldberg, I.D., and Shi, Y.E. 2004. γ Synuclein, a novel heat-shock protein-associated chaperone, stimulates ligand-dependent estrogen receptor alpha signaling and mammary tumorigenesis. Cancer Res. 64: 4539–4546. [DOI] [PubMed] [Google Scholar]
- Johnson, B.A. and Blevins, R.A. 1994. NMRView: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 4: 603–614. [DOI] [PubMed] [Google Scholar]
- Kaylor, J., Bodner, N., Edridge, S., Yamin, G., Hong, D.-P., and Fink, A.L. 2005. Characterization of oligomeric intermediates in α-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F α-synuclein. J. Mol. Biol. 353: 357–372. [DOI] [PubMed] [Google Scholar]
- Kriwacki, R.W., Hengst, L., Tennant, L., Reed, S.I., and Wright, P.E. 1996. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: Conformational disorder mediates binding diversity. Proc. Natl. Acad. Sci. 93: 11504–11509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy, E.R., Filippov, I., Lewis, W.S., Otieno, S., Xiao, L., Weiss, S., Hengst, L., and Kriwacki, R.W. 2004. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 11: 358–364. [DOI] [PubMed] [Google Scholar]
- Lee, H., Zhu, X., Takeda, A., Perry, G., and Smith, M.A. 2006. Emerging evidence for the neuroprotective role of α-synuclein. Exp. Neurol. 200: 1–7. [DOI] [PubMed] [Google Scholar]
- Liu, H., Liu, W., Wu, Y., Zhou, Y., Xue, R., Luo, C., Wang, L., Zhao, W., Jiang, J.D., and Liu, J. 2005. Loss of epigenetic control of synuclein-γ gene as a molecular indicator of metastasis in a wide range of human cancers. Cancer Res. 65: 7635–7643. [DOI] [PubMed] [Google Scholar]
- McNulty, B.C., Tripathy, A., Young, G.B., Charlton, L.M., Orans, J., and Pielak, G.J. 2006. Temperature-induced reversible conformational change in the first 100 residues of α-synuclein. Protein Sci. 15: 602–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohana-Borges, R., Goto, N.K., Kroon, G.J., Dyson, H.J., and Wright, P.E. 2004. Structural characterization of unfolded states of apomyoglobin using residual dipolar couplings. J. Mol. Biol. 340: 1131–1142. [DOI] [PubMed] [Google Scholar]
- Mok, Y.K., Elisseeva, E.L., Davidson, A.R., and Forman-Kay, J.D. 2001. Dramatic stabilization of an SH3 domain by a single substitution: Roles of the folded and unfolded states. J. Mol. Biol. 307: 913–928. [DOI] [PubMed] [Google Scholar]
- Molday, R.S., Englander, S.W., and Kallen, R.G. 1972. Primary structure effects on peptide group hydrogen exchange. Biochemistry 11: 150–158. [DOI] [PubMed] [Google Scholar]
- Muñoz, V. and Serrano, L. 1994a. Elucidating the folding problem of helical peptides using empirical parameters. Nat. Struct. Biol. 1: 399–409. [DOI] [PubMed] [Google Scholar]
- Muñoz, V. and Serrano, L. 1994b. Intrinsic secondary structure propensities of the amino acids, using statistical ϕ/ψ matrices: Comparison with experimental scales. Proteins 20: 301–311. [DOI] [PubMed] [Google Scholar]
- Ohtake, H., Limprasert, P., Fan, Y., Onodera, O., Kakita, A., Takahashi, H., Bonner, L.T., Tsuang, D.W., Murray, I.V., and Lee, V.M., et al. 2004. β-synuclein gene alterations in dementia with Lewy bodies. Neurology 63: 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Z.Z., Bruening, W., Giasson, B.I., Lee, V.M., and Godwin, A.K. 2002. γ-Synuclein promotes cancer cell survival and inhibits stress- and chemotherapy drug-induced apoptosis by modulating MAPK pathways. J. Biol. Chem. 277: 35050–35060. [DOI] [PubMed] [Google Scholar]
- Payton, J.E., Perrin, R.J., Woods, W.S., and George, J.M. 2004. Structural determinants of PLD2 inhibition by α-synuclein. J. Mol. Biol. 337: 1001–1009. [DOI] [PubMed] [Google Scholar]
- Rochet, J.-C., Conway, K.A., and Lansbury Jr., P.T. 2000. Inhibition of fibrillization and accumulation of prefibrillar oligomers in mixtures of human and mouse α-synuclein. Biochemistry 39: 10619–10626. [DOI] [PubMed] [Google Scholar]
- Sanchez, R. and Sali, A. 1997. Advances in comparative protein-structure modelling. Curr. Opin. Struct. Biol. 7: 206–214. [DOI] [PubMed] [Google Scholar]
- Seavey, B.R., Farr, E.A., Westler, W.M., and Markley, J.L. 1991. A relational database for sequence-specific protein NMR data. J. Biomol. NMR 3: 217–236. [DOI] [PubMed] [Google Scholar]
- Serpell, L.C., Berriman, J., Jakes, R., Goedert, M., and Crowther, R.A. 2000. Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc. Natl. Acad. Sci. 97: 4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, C.E. 1948. A mathematical theory of communication. Bell Syst. Tech. J. 27: 623–656.
- Shoemaker, B., Portman, J., and Wolynes, P. 2000. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. 97: 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza, J.M., Giasson, B.I., Lee, V.M., and Ischiropoulos, H. 2000. Chaperone-like activity of synucleins. FEBS Lett. 474: 116–119. [DOI] [PubMed] [Google Scholar]
- Thompson, J.D., Higgins, D.G., and Gibson, T.J. 1994. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa, P. and Csermely, P. 2005. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 18: 1169–1175. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N. and Fink, A.L. 2004. Conformational constraints for amyloid fibrillation: The importance of being unfolded. Biochim. Biophys. Acta 1698: 131–153. [DOI] [PubMed] [Google Scholar]
- Uversky, V.N., Li, J., Souillac, P., Millett, I.S., Doniach, S., Jakes, R., Goedert, M., and Fink, A.L. 2002. Biophysical properties of the synucleins and their propensities to fibrillate. J. Biol. Chem. 277: 11970–11978. [DOI] [PubMed] [Google Scholar]
- Wang, Y. and Jardetzky, O. 2002. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 11: 852–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. and Wishart, D.S. 2005. A simple method to adjust inconsistently referenced 13C and 15N chemical shift assignments of proteins. J. Biomol. NMR 31: 143–148. [DOI] [PubMed] [Google Scholar]
- Wang, L., Eghbalnia, H.R., Bahrami, A., and Markley, J.L. 2005. Linear analysis of carbon-13 chemical shift differences and its application to the detection and correction of errors in referencing and spin system identifications. J. Biomol. NMR 32: 13–22. [DOI] [PubMed] [Google Scholar]
- Wirmer, J., Berk, H., Ugolini, R., Redfield, C., and Schwalbe, H. 2006. Characterization of the unfolded state of bovine α-lactalbumin and comparison of unfolded states of homologous proteins. Protein Sci. 15: 1397–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994a. Chemical shifts as a tool for structure determination. Methods Enzymol. 239: 363–392. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S. and Sykes, B.D. 1994b. The 13C chemical-shift index: A simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR 2: 171–180. [DOI] [PubMed] [Google Scholar]
- Zhang, H., Neal, S., and Wishart, D.S. 2003. RefDB: A database of uniformly referenced protein chemical shifts. J. Biomol. NMR 25: 173–195. [DOI] [PubMed] [Google Scholar]
- Zhou, P., Lugovskoy, A.A., McCarty, J.S., Li, P., and Wagner, G. 2001. Solution structure of DFF40 and DFF45 N-terminal domain complex and mutual chaperone activity of DFF40 and DFF45. Proc. Natl. Acad. Sci. 98: 6051–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Gu, G., Goodlet, D.R., Zhang, T., Pan, C., Montine, T.J., Montine, K.S., Aebersold, R.H., and Zhang, J. 2004. Analysis of α-synuclein-associated proteins by quantitative proteomics. J. Biol. Chem. 279: 39155–39164. [DOI] [PubMed] [Google Scholar]
- Zhou, Y., Inaba, S., and Liu, J. 2006. Inhibition of synuclein-γ expression increases the sensitivity of breast cancer cells to paclitaxel treatment. Int. J. Oncol. 29: 289–295. [PubMed] [Google Scholar]