

Abstract
Improving the affinity of a high-affinity protein–protein interaction is a challenging problem that has practical applications in the development of therapeutic biomolecules. We used a combination of structure-based computational methods to optimize the binding affinity of an antibody fragment to the I-domain of the integrin VLA1. Despite the already high affinity of the antibody (Kd ∼7 nM) and the moderate resolution (2.8 Å) of the starting crystal structure, the affinity was increased by an order of magnitude primarily through a decrease in the dissociation rate. We determined the crystal structure of a high-affinity quadruple mutant complex at 2.2 Å. The structure shows that the design makes the predicted contacts. Structural evidence and mutagenesis experiments that probe a hydrogen bond network illustrate the importance of satisfying hydrogen bonding requirements while seeking higher-affinity mutations. The large and diverse set of interface mutations allowed refinement of the mutant binding affinity prediction protocol and improvement of the single-mutant success rate. Our results indicate that structure-based computational design can be successfully applied to further improve the binding of high-affinity antibodies.
Keywords: antibody, affinity maturation, computational protein design, protein–protein interactions, binding energy prediction
Computational techniques for small molecule design have recently become an established part of the drug discovery process, and many studies have been published in which structure-based design has led to high-affinity molecules (Jorgensen 2004). In contrast, there has been considerably less usage of computational design techniques in the field of protein engineering. This is due in part to the effectiveness of directed evolution experimental techniques (Crameri et al. 1996; Hanes et al. 1998), the computational complexity of treating full proteins, and the relative scarcity of structural information on engineered proteins. Very recently there have been a number of successes in computational protein design, such as the redesign of an internal domain–domain interface of an endonuclease (Chevalier et al. 2002), the design of a novel protein fold (Kuhlman et al. 2003), the design of specific enzymatic activity into a periplasmic binding protein (Dwyer et al. 2004), and alteration of DNase-inhibitor pair binding specificity (Kortemme et al. 2004). It is now foreseeable that biomolecule therapeutic design could be addressed using computational techniques.
Antibodies are the most widely used format for protein therapeutic applications for a variety of reasons, including high affinity and the ability to trigger immune responses (Brekke and Sandlie 2003). Traditionally, monoclonal antibodies are produced by immunization of mice, construction of hybridomas, and selection of single clones expressing the desired antibody (Kohler and Milstein 1975). More recently, directed evolution techniques such as phage-display and related in vitro library display methods have become commonly used (Crameri et al. 1996; Hanes et al. 1998). Either in vivo or in vitro techniques can produce high-affinity antibodies for most targets (Kretzschmar and Von Ruden 2002; van den Beucken et al. 2003). Further affinity enhancement using directed evolution techniques has been shown to be quite effective (Daugherty et al. 2000; Midelfort et al. 2004). In this report, we investigate the applicability of structure-based computational design to improving the affinity of a mature antibody. The antibody optimized in this work is specific for the I domain of human integrin (VLA1) (Karpusas et al. 2003). This integrin is a cell-surface receptor for collagen and laminin and is present on some T-cells. Anti-VLA1 is a potential therapeutic intended to inhibit the entry of activated T-cells and monocytes to sites of inflammation and may have uses in the treatment of arthritis (Ben-horin and Bank 2004).
In essence, computational protein design rests on techniques to sample a large number of designs and the ability to accurately predict the properties of the designs. Sampling of amino acid types and side chain rotamers can be done efficiently using algorithms such as dead-end elimination (DEE) (Desmet et al. 1992) and its refinements (Goldstein 1994; Pierce et al. 2000; Looger and Hellinga 2001), Monte Carlo-based searches (Kuhlman and Baker 2000), or combinations (Shah et al. 2004). Using these methods, a very large number of residue types and conformations at many selected positions can be screened in silico using fast evaluations of energetic properties. Equally important is the quality of the energy evaluations, especially the treatment of the solvent and electrostatic interactions. For these energy terms, the highest-quality methods, such as region-dependent dielectric constants (Wisz and Hellinga 2003) or numerical solution of the Poisson-Boltzmann equation (Marshall et al. 2005), are only now becoming compatible with the exhaustive search algorithms. In principle, the ability of computational methods to find the best designs in a virtual library of around 1040 sequences within a few days is a major advantage over directed evolution methods, which explore on the order of 1010 sequences within a time frame of weeks to months. For example, computational methods can exhaustively test all sequence combinations for a system in which 20 residues are allowed to vary (2020 ∼ 1026), whereas directed evolution can only explore a tiny fraction.
Computational protein design has been applied to the redesign of protein cores for the purpose of making proteins more thermostable or for exploring new protein folds. Several successful core redesigns have been reported (Dahiyat and Mayo 1997; Dantas et al. 2003), but significant challenges still exist (Dantas et al. 2003; Mooers et al. 2003). Compared with core redesign, there is considerably less experience with protein–protein interfaces. Interface surfaces, particularly antibody–antigen interfaces, are different from protein cores because they may be more flexible and their residue composition is often substantially more polar and diverse (Lo Conte et al. 1999). They must also have good solubility and resist aggregation when unbound. Additionally, interfaces usually contain more bound water molecules, which suggests that continuum treatments of solvation are more likely to fail.
In this work we use structure-based computational design to enhance the binding affinity of the anti-VLA1 antibody AQC2 and determined experimental binding affinities for >80 designed variants. Mutations have been made to nearly every antigen-contacting residue position using suggestions from computational methods. A number of multiple-mutant variations have been constructed, and crystallographic data for one of the best, a quadruple mutant, have been collected. This represents the first example in which structural verification has been obtained for a protein–protein complex designed to have increased affinity. Previous examples of computationally designed higher-affinity binders used only charge optimizations and lacked structural verification (Selzer et al. 2000; Marvin and Lowman 2003). The extent and the variety of the mutations allow conclusions about the general applicability of this technology and provide a large enough test set to further advance the quality of mutant binding energy predictions.
Results
Our general approach to improve the antibody's affinity was first to computationally design and then to express single and double mutants. We intentionally did not restrict the expressed mutants to those we thought most likely to succeed. The possibility of learning from mutation effects also factored into the decisions. After identification of mutants with improved affinity, we expressed combination mutants to further increase the affinity. Here, we first give a general overview of the results, followed by sections that focus on mutants designed by side chain repacking, by electrostatic optimization, and by combination of single mutations.
General overview
Based on the results of the calculations, nearly all antibody residue positions in contact with the antigen were varied. Figure 1 provides a view of the mutated positions on the interface. Residue H:Asp101 (101 in heavy chain), which contacts the manganese ion in the antigen, was not mutated because it is experimentally known to be critical for binding (Karpusas et al. 2003), and calculations show that it is already optimal (Sherman 2004). It is clear from the figure that most of the beneficial mutations were found at the periphery of the antibody–antigen interface. The exception is the H:T50V mutation, which is located relatively deep in the antibody.
Figure 1.

Visualization of the mutated positions (yellow and orange) on the antibody–antigen interface. The view looks down through the antigen onto the complementary determining region (CDR) loops of the antibody. Only residues with an atom within 5 Å of the opposite side of the interface and mutation positions are shown. The antigen residues are colored purple, the light chain is green, and the heavy chain is blue. Positions of beneficial mutations are shown in orange. Expressed mutants at each position are noted to the right of the position number. All structural figures have been made using PyMOL (DeLano Scientific).
We found 10 single-point mutants with measured affinities better than wild type from the 83 constructed mutants, a success hit rate of 12% uninfluenced by binding energy predictions. Of the 10, nine were found in the first two passes of design calculations, which included only 40 mutants (23% hit rate). Further design rounds contributed only one additional successful mutant and were done to test new methods. All mutations and the measured binding affinities are shown in Table 1. EC50 values were measured for all mutants using competition ELISA, which is a solid-phase assay. Mutants with apparent affinities near or better than wild type were also tested using a more reliable solution-phase assay (KinExA) (see Materials and Methods). An example binding curve is given in Figure 2b. A decomposition of the calculated binding energies is given in the Supplemental Material.
Table 1.
Experimental and calculated binding affinity information
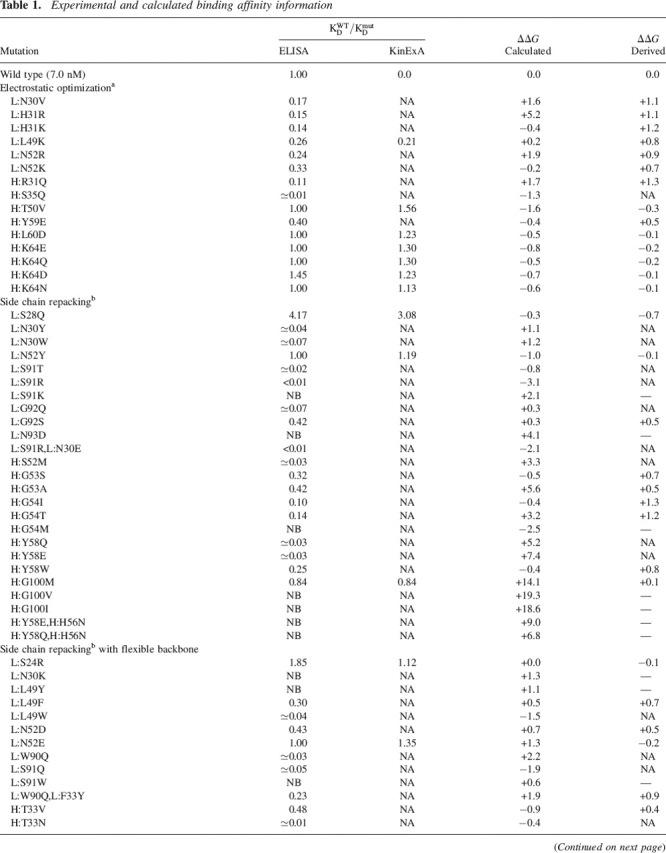
Table 1.
Continued
Binding affinities are reported with respect to the wild type as ratios (see Materials and Methods). Derived ΔΔG (kcal/mol) numbers are from the ratio of the wild-type affinity to the mutant affinity (ΔΔG = −RTln(KWTD/KmutD)) and are from the KinExA results if available. No numbers are derived for mutants for which the ratio had to be estimated. Single-structure calculations (see Materials and Methods) with minimal perturbation relative to the crystal structure yielded the calculated ΔΔG numbers. Many mutants were not tested (NA) or show no binding (NB) in the competition ELISA assay.
a Kangas and Tidor 1998; ICE software (D.F. Green, E. Kangas, and B. Tidor [MIT]).
b Benson et al. 2000; Looger and Hellinga 2001.
c Hanf 2002.
Figure 2.
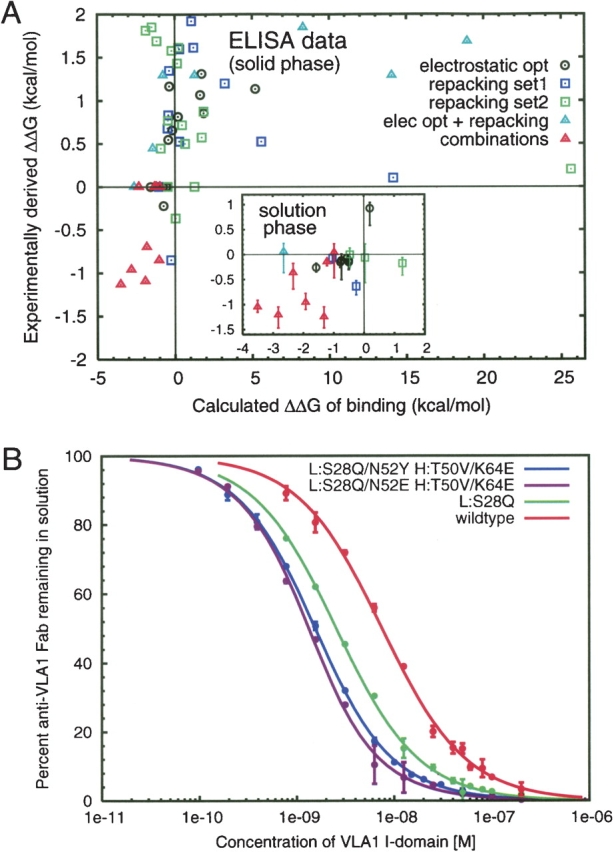
Changes in binding for the mutants. (A) Comparison of calculated with experimental ΔΔG of binding. All mutants with competition ELISA EC50s near wild type were reevaluated using the solution-based KinExA assay (inset). (B) Examples of the solution-phase (KinExA) binding curves for wild type and mutants. Experimental numbers were derived from the ratio of the wild-type affinity to the mutant affinity (ΔΔG) = −RTln(KWTD/KmutD). All nonbinders and mutants for which only an estimate is available are not shown.
The large number of mutations and their complex character has allowed us to refine our protocol for predicting ΔΔG’s of binding (see Materials and Methods). This protocol is distinct from the search algorithms used to find the mutations and was applied after the mutations were expressed and tested. Only a single false negative (L:N52E) resulted from the predictions. Rigorous testing of binding energy prediction methods is outside the focus of this work. However, it is worth mentioning that inclusion of more than local flexibility resulted in a higher number of false positives. A comparison of calculated and experimental binding energies is shown in Figure 2. If we retroactively apply the results from this protocol, we would only have made 35 of the 83 mutations, resulting in nine mutations with affinity higher than wild type and a corresponding hit rate of 26% (48% when restricted to the first two passes).
Combination of four of the higher affinity mutations, S28Q and N52E in the light chain and T50V and K64E in the heavy chain, resulted in a quadruple mutant with an affinity of 850 pM, which is an ∼10-fold improvement over wild type. For one of the combination mutants (H:T50V/K64E, L:S28Q/N52Y), we solved the X-ray structure in complex with the VLA1 I-domain. There is no change in binding epitope between the original complex structure (Protein Data Bank [PDB] ID 1MHP) and the quadruple mutant, but some interesting structural changes occurred, which are described in the following sections.
Side chain repacking mutants
A set of designs was proposed using a side chain repacking algorithm developed by Hellinga and coworkers (Looger and Hellinga 2001) that uses DEE to find optimal sequences for a structure with a fixed backbone conformation. Solvation effects are approximated by using region-dependent dielectric constants in the evaluation of electrostatic energy terms (Wisz and Hellinga 2003). One advantage of this DEE implementation is its ability to quickly and exhaustively explore variations of all of the antigen-contacting residues simultaneously.
We generated a large number (500) of alternative structures, each with a different sequence. The structural solutions were filtered by comparing each new sequence to wild type and looking for promising variations at each position. To account for cooperativity, we looked for residue changes that were always accompanied by mutations at neighboring positions. These predicted single- or double-point mutants were inspected visually, and if we observed an improvement in packing, additional intermolecular hydrogen bonds, or an increase of intermolecular hydrophobic contacts, we decided to make and express the mutants.
Our first set of mutations generated our best single-point mutant (L:S28Q), which by itself improves affinity by almost a factor of 3. Another mutant (L:N52Y) was found with a small but not statistically significant improvement over wild type by itself. Both of these mutants are present in the quadruple mutant crystal structure and allow comparison between prediction and experiment.
Figure 3A shows the crystal structure of the quadruple mutant (dark gray) in the region around the best single-point mutation (L:S28Q). The longer glutamine residue at this position stacks against the aromatic ring of Tyr264 on the antigen. The tyrosine residue is rotated inward toward the bulk of the antigen. Comparison with the wild-type crystal structure (data not shown) indicates that the tyrosine residue changed conformation, flipping inward to make the interaction with the glutamine. The protocol used to evaluate the binding energies for Figure 2 predicts a small (−0.3 kcal/mol) improvement in the binding energy relative to experiment (−0.7 kcal/mol). It fails to rotate the tyrosine inward as observed in the crystal structure. Consequently, it does not pick up the correct glutamine rotamer. Allowing a small degree of backbone flexibility in the vicinity of the mutation substantially improves the predicted binding energy and allows the correct rotamer to be found and the correct stacking interaction to be formed (shown in light gray). However, only if the backbone of the quadruple mutant is used in the calculations is the tyrosine conformation correctly predicted.
Figure 3.

Visualization of the quadruple mutant crystal structure (dark gray) near side chain repacking mutants. (A) Comparison to the predicted structure (light gray) in the vicinity of the S28Q mutation in the light chain. The predicted structure forms a similar stacking-like interaction between Tyr264 on antigen and the glutamine. The electron density (2Fo − Fc, σ = 1.1) indicates that the tyrosine has swung inward toward the bulk of the antigen. (B) Comparison of the wild-type (light gray) and quadruple mutant crystal (dark gray) structures in the vicinity of the N52Y mutation in the light chain.
Figure 3B focuses on the L:N52Y mutation, which is part of the quadruple mutant for which the crystal structure was solved. The tyrosine side chain extends toward Ser295 on the antigen. A long (4.3 Å) distance between the serine and the tyrosine oxygens means that direct hydrogen bonding cannot account for the apparent increase in affinity. As described below, we also designed and tested an N52E mutant, which also resulted in a small binding-affinity improvement. Aspartate, arginine, or lysine substitutions at this position did not produce an improvement in affinity.
The possibility of filling a cavity in the neighboring antigen makes the glycine at position 100 in the heavy chain an interesting position for mutations. However, all attempts to increase affinity by filling the cavity were unsuccessful. As shown in Figure 4, the cavity borders the metal ion position in the antigen, is lined with polar residues, and contains water molecules that were not visible in the original structure. Although the the glycine has glycine-only φ–ψ backbone angles, the repacking software suggested a number of drastic substitutions at this position. Experimentally, mutation of the glycine to a methionine gives essentially no change from wild-type affinity (+0.1 kcal/mol) and is the best substitution at this position. Substitution to phenylalanine or serine reduces affinity by 60%, leucine drops the affinity by 1 order of magnitude, and isoleucine or valine fail to show binding in the competition ELISA assay.
Figure 4.

The quadruple mutant crystal structure in the vicinity of the metal-contacting H:Asp101 residue. The H:Gly100 position on the antibody is colored yellow. A pocket in the antigen (purple) lined with polar residues and water molecules (red spheres) is visible below the metal ion (orange sphere). A new residue at the G100 position would extend its Cβ atom toward the water molecules on the right.
An alternative design set using the repacking methodology was generated using a simple modification to include backbone flexibility (see Materials and Methods). Multiple starting backbone conformations were generated using molecular dynamics and run through the same procedure used for the single-crystal structure backbone. The wild-type residue appeared much more frequently than in the single-structure calculations, which was encouraging. However, a larger fraction of the resulting designs showed no binding in the competition ELISA assay. One new mutation (L:N52E) is apparently better than wild type, and another mutation (H:F99W) is a wild-type equivalent.
Another set of designs was proposed using a combination of high-quality electrostatic evaluations and a repacking algorithm (see multilevel DEE/A* in Materials and Methods). Some advantages of this method are its detailed Poisson-Boltzmann treatment of solvation and electrostatics and its use of both binding and folding energies in selecting promising designs. Compared with the side chain repacking algorithm used in the previous section, this method is best used to redesign small clusters of residues. We used this methodology to design an additional 10 mutants, of which three were double mutants and two were quadruple mutants. Two of the single-residue mutations were found to have wild-type-equivalent affinity, but no additional higher-affinity mutants were found.
Electrostatic optimization mutants
Another set of mutations took advantage of a charge optimization algorithm based on the electrostatic potential generated from solving the Poisson-Boltzmann equation (PBE) (Kangas and Tidor 1998). Single residue replacements that better meet the optimized charge distribution are manually selected, modeled, and reevaluated computationally to find mutations that improve electrostatic interactions and desolvation energies at the interface while not adversely affecting packing interactions. The specific rationale for making the individual electrostatic mutations is presented elsewhere (Sherman 2004).
One obvious benefit to the electrostatic optimizations is the ability to design for long-range interactions. The H:K64E change is a charge reversal mutation that occurs 8.4 Å from the nearest positively charged atom on the antigen. By itself, it does not produce a clear increase in affinity, but in combination it seems to consistently improve affinities (see last portion of Table 1). A Coulombic potential model truncated at 7 Å would have missed this mutation.
Though less significant than other mutations of the quadruple mutant in improving the binding affinity, H:T50V is interesting because it induces local hydrogen bonding rearrangements. It is also the only mutation buried in the interface rather than on the periphery. Figure 5 compares the quadruple mutant structure to the wild-type crystal structure in the vicinity of the mutation. A side chain rotation to a different rotamer relative to the threonine is seen in the valine mutant. The predicted structure captures this rotation but is not shown for clarity. Most interesting is the movement of the nearby tryptophan residue. The wild-type threonine was making a hydrogen bond with Ser35. This hydrogen bond is critical for maintaining affinity; mutation of Ser35 to alanine decreases affinity by ∼1 order of magnitude. Removal of hydrogen bonding possibilities with the threonine at position 50 leaves an unsatisfied hydroxyl group and causes the tryptophan nitrogen to move 0.9 Å toward the serine oxygen to allow hydrogen bonding. There is a clear and surprisingly large backbone movement of ∼0.7 Å at the tryptophan Cα. In this case, the environment at the mutation site clearly rearranges to eliminate an unsatisfied hydrogen bond.
Figure 5.

Comparison of the wild-type (light gray) and quadruple mutant (dark gray) crystal structures in the vicinity of the H:T50V mutation. The wild-type threonine hydrogen bonds with the tryptophan. When substituted with a valine, the local environment rearranges to eliminate an unsatisfied hydrogen bond.
Combination mutants
Construction of combination mutants serves two purposes. In some cases, it allows the production of a combined mutant with much higher affinity than any of its constitutive single point mutations (see Fig. 2B). The highest-affinity mutant was a combination of S28Q and N52E in the light chain and T50V and K64E in the heavy chain with ∼10 times the affinity of the wild type. It is also possible to examine the cooperativity of mutations by measuring affinities for the combination mutants. Combining the K64E and L60D mutations in the heavy chain does not lead to an apparent affinity increase, presumably because they are nearby (see Fig. 1) and both introduce a similar redundant negative charge. The small absolute value of many of the binding energy changes makes extensive consideration of the cooperativity speculative, but it should be noted that significant cooperativity effects have been observed for mutations on antibody–antigen interfaces (Sundberg and Mariuzza 2003).
The mutations primarily improve affinity by decreasing the dissociation rates. This is a significant contrast to previous work, which explicitly targeted association rate improvements through electrostatic optimization (Selzer et al. 2000; Marvin and Lowman 2003). Table 2 compares the wild-type kinetic rates to both the isolated S28Q mutation and a combination of the S28Q, N52Y, and T50V mutations. Affinities (Kd) from both the surface plasmon resonance measurements (Biacore) and the solution-based assay (KinExA) agree well.
Table 2.
Association and dissociation kinetics measured using surface plasmon resonance (Biacore) for the wild type and two variants

The calculated equilibrium dissociation constants are similar to those observed from the solution-based equilibrium assay (KinExA).
The effect of the antigen-binding loop conformation was indirectly probed by comparison with a variant with a different supporting framework. During the humanization of the anti-VLA1 antibody, several variants were made that contained fewer antibody framework back mutations but had identical CDRs. These variants had significantly lower affinity (∼1 μM) than the humanized antibody described in this work (M. Jarpe, unpubl.). We attempted to “salvage” the affinity of one of the low-affinity humanization variants by applying the affinity-enhancing mutants that were found in this work. When the N52Y, S28Q, and T50V mutations were applied, they improved the affinity to 140 nM. Adding the additional K64E mutation brought the affinity to 110 nM. With the K64E mutation and substituting N52E for N52Y, the affinity became 100 nM, or an order of magnitude improvement, just as observed for the wild-type reference used in this work. This shows that beneficial mutants can be transferred to another base wild-type anti-VLA1 with a similar net affinity improvement.
Discussion
The results presented in this paper indicate that it is possible using rational structure-based design to significantly enhance the affinity of an antibody produced by traditional in vivo immunization methods. Side chain repacking calculations found the most effective single mutation and electrostatic optimization methods provided additional affinity-enhancing mutations. Combination of separately beneficial mutations yields the best binders and validates the search-and-combine strategy.
Although we were able to increase the affinity by an order of magnitude, many of the designs were not successful. Computational design relies strongly on the accuracy of the structure, and the relatively low resolution (2.8 Å) likely contributed to inaccurate predictions. We have seen here that most beneficial mutations, at least for this in vivo optimized interface, tend to have <1 kcal/mol effect on the binding energy. Predicting such small binding energy changes is difficult and our results highlight the requirement for further improvement, a need that is shared with the field of small molecule computational design. Areas for improvement include better treatments of solvation effects, protein flexibility, entropic contributions, and the unbound states of the molecules in the complex. The crystal structure of the quadruple mutant allows us to comment on two additional important factors: explicit water molecules and hydrogen bonding networks.
Explicit consideration of molecular water positions during the design process is an important contemporary goal in structure-based drug design. There are two clear examples in this work where water mediates the antibody–antigen interaction and is likely to contribute strongly to the affinity. The L:N52Y and the L:N52E mutations marginally increase the affinity of the antibody, although the L:N52Y mutation does not make direct contact with the antigen (see Fig. 3B). A contact through a single bridging water molecule is structurally possible and is likely to be present despite not being visible in the crystal structure.
The Gly100 position in the heavy chain (Fig. 4) shows an unusually broad range of mutation effects. It borders the polar cavity on the antigen side that contains the metal ion and buried water molecules. Although the glycine occupies a glycine-only region in the Ramachandran plot, substitution for methionine or phenylalanine produces near-wild-type affinities. Strikingly worse effects are seen for less extended residues such as serine, isoleucine, or valine. This behavior suggests that longer hydrophobic residues (methionine or phenylalanine) can gain enough interactions in the cavity to offset the costs of desolvating the cavity. The residues with branches at the Cβ position (isoleucine or valine) presumably introduce steric restrictions and may cause backbone reorientations. Expulsion of water from the cavity by the longer residues and the resulting entropy gain would explain why the measured binding is much more favorable than that predicted (wild type vs. +14.1 kcal/mol for G100M).
The H:T50V mutation shown in Figure 5 is an excellent example of the importance of maintaining hydrogen bond networks. As described in the results, the T50V mutation disrupts a hydrogen bond that existed between Thr50 and Ser35. Our calculations assumed that the backbone conformation would not change significantly, but in the mutant crystal structure the local environment adjusted backbone and side chains to enable a new hydrogen bonding network. If a mutation leads to the introduction of unsatisfied hydrogen bonds, it is likely that the environment rearranges to fix the hydrogen bonding network. Similar effects have been seen elsewhere (Takano et al. 1999) and it is an important factor to take into account when designing protein mutants.
A couple of questions arise regarding the affinity-enhancing mutations that were found. Why didn't nature find these mutations when the antibody was matured in the mouse, and have all the beneficial mutations been found? Nature is capable of discriminating high affinity from low affinity during the affinity maturation process up to ∼0.1 nM (Foote and Eisen 1995; Batista and Neuberger 1998). The original affinity of the anti-VLA1 antibody was 7–10 nM, putting it well inside the discrimination range of the naive B-cell receptor. For the best single mutation that we found (L:S28Q), more than one base mutation is needed, making it more difficult to find via random mutagenesis. A definitive answer to the first question would require more immunizations and careful selection. Determining whether better solutions exist would require the use of a set of library display experiments, similar to those used to mature other antibodies to very high affinity (Midelfort et al. 2004).
The type and locations of solutions are worth considering. Figure 1 shows that all affinity-improving mutations except T50V are on the periphery of the antibody–antigen interface. This could be a side effect of the computational methodology that we use. Incomplete optimization of the periphery could also be a general feature of antibody–antigen interfaces (Li et al. 2003). At the periphery, one needs to worry less about repacking a set of cooperatively interacting side chains. It is sufficient to add new interactions across the interface by changing a single residue. The new residue is therefore less likely to disturb existing interactions and finding these solutions is more amenable to our present search strategy. It is also possible that optimizing the core of the interface could require relative reorientation of the antibody and antigen or sizable backbone movements, neither of which is handled in most of the methodologies we used. As our ability to predict binding energy changes evolves, it will be possible to trust more of the multiple mutation designs and find solutions anywhere at the antibody–antigen interface.
In conclusion, we have shown that structure-based design can be used to further affinity mature antibodies of high affinity. One of the advantages of computational design is that, in principle, the smallest set of mutations required for optimization can be determined. Consequently, experimental follow-up by site-directed mutagenesis or a directed library approach can be done faster than through general directed evolution methods. It is likely that through improvements in computational methodology, structure-based protein design will become an important industrial approach for the optimization of protein properties such as affinity, stability, and solubility.
Materials and methods
Computational design methods
The starting complex structure at 2.8 Å was obtained from the PDB (1MHP chains B, X, and Y) and chains X and Y were relabeled as H and L. Asparagine, glutamine, and histidine side chain flips and histidine protonation states were corrected using suggestions generated by the WHATIF software (Vriend 1990). We patched the N terminus of the light chain with an acetyl group because the first residue was not visible in the 1MHP structure. The constant domains from the antibody's heavy and light chains were removed and the C termini patched with N-methylamide groups. The structure was minimized using harmonic constraints (10 kcal/mol/Å2 on all heavy atoms) to remove major steric clashes.
The calculated binding energies given in Table 1 and shown graphically in Figure 2 were calculated using a combination of electrostatics from the Poisson-Boltzmann equation and van der Waals and multi-body terms from the CHARMM22 force field (MacKerell et al. 1998). These calculations are distinct from the search algorithms that help to find the mutations. All binding energy calculations were done using CHARMM (Brooks et al. 1983; MacKerell et al. 1998), with the exception of the electrostatic interactions and desolvation energies which were calculated with the ICE software (Kangas and Tidor 1998; D.F. Green, E. Kangas, and B. Tidor, MIT). For each binding energy evaluation the mutant structure was generated from the same starting structure using a custom rotamer search method. An identical protocol was applied to regenerate the wild-type structure at the mutant position. CHARMM was used to scan the χ angles of each rotamer exhaustively in 60° increments using a script adapted from one written by D.F. Green (D.F. Green, E. Kangas, and B. Tidor, MIT). From each starting conformation, the rotamer and all side chains with atoms within 5 Å of the Cα position were minimized and the lowest energy conformation retained. The protocol uses the rigid binding approximation, no relaxation of the proteins in an unbound state or the backbone was allowed.
The electrostatic optimization is implemented in the ICE software from the Tidor group (Kangas and Tidor 1998; D.F. Green, E. Kangas, and B. Tidor, MIT). It uses a numerical solution of the Poisson-Boltzmann equation to calculate the optimal charge distribution on a set of atomic centers. Empirical group-based PARSE charges with polar hydrogens derived from experimental desolvation measurements are used (Sitkoff et al. 1994). The DEZYMER software (Benson et al. 2000; Looger and Hellinga 2001) developed by the Hellinga laboratory was used for the side chain repacking calculations. It uses a custom force field based on region-dependent dielectric constants, Lennard-Jones van der Waals interactions, explicit hydrogen bonding, a pair-wise decomposable surface area term, and approximations for rotamer entropy (Wisz and Hellinga 2003). Van der Waals interactions were cut at 7.0 Å and the well width expanded from the conventional Lennard-Jones form to shrink the hard-sphere radii by a factor of 1.35 without changing the well depth.
The flexible backbone calculations were performed by starting the side chain repacking runs from 20 unique backbone configurations. The configurations were snapshots taken every 10 psec from a constrained molecular dynamics simulation using the CHARMM19 force field and a 4r dielectric. Harmonic constraints of 1.0 kcal/mol/Å2 were applied to each Cα atom to prevent large departures from the crystal structure.
The multilevel DEE/A* (Hanf 2002) protein design method uses the dead-end elimination (Desmet et al. 1992; Lasters and Desmet 1993; Goldstein 1994; Pierce et al. 2000) and A* (Winston 1992; Leach and Lemon 1998) discrete search algorithms to find conformations of a set of mobile residues that minimize the sum of the binding and folding energies. The method is novel in its use of three levels of increasingly detailed discrete searching (sequence, fleximers [Mendes et al. 1999; Caravella 2002], and rotamers [Dunbrack and Karplus 1993]), and its use of multiple hierarchical energy functions. After the multilevel DEE/A*, a more accurate energy function, including finite-difference Poisson-Boltzmann (FDPB) electrostatics (Gilson et al. 1988; Sharp and Honig 1990) was used to further screen the best candidate structures found by DEE/A*. Mutant sequences considered for synthesis were those that had a structure with better predicted binding energy, and predicted folding energy no more than 1 kcal/mol worse, than the wild-type structure.
Protein production
Escherichia coli expressed his-tagged Fab fragments of anti-VLA1 and the described variants were used for this study. The plasmid consisting of the Fab fragment (Abraham et al. 2004), a 6-His tag, and OmpA and PhoA periplasmic localization signals in a bicistronic arabinose-inducible vector was constructed. Amino acid substitutions were introduced into the anti-VLA1 Fab by QuikChange site-directed mutagenesis (Stratagene). Expression of the antibody variants was carried out in super broth using induction with 0.002% arabinose. Cells were pelleted, resuspended in 30 mM Tris, 20% sucrose with gentle mixing, repelleted, and resuspended in 5 mM MgSO4 with vigorous mixing. The cells were again pelleted, and the supernatant periplasmic lysate fraction was filtered. An alternative cell line was used to produce Fabs for crystallography (see Supplemental Material).
The various His-tagged mutants of anti-VLA1 were purified by passing the periplasmic supernatant over a 1-mL HisTrap HP nickel chelate column (Pharmacia), which had been equilibrated with 50 mM sodium phosphate, 300 mM NaCl, 20 mM imidazole, 0.05% Tween-20 (pH 8.0). After 10 column washings, the Tween-20 was removed from the buffer and then the imidazole concentration was increased to 250 mM for elution. The proteins were analyzed by PAGE, UV scan, mass spectrometry, and SEC/light scattering to look for aggregates and multimers.
Recombinant α1β1 integrin I-domain (humanized rat) was expressed in E. coli as a GST-fusion protein. The I domain was cleaved from the purified GST-fusion protein with thrombin and further purified as described previously (Gotwals et al. 1999). A 1.2 molar excess purified I-domain was mixed with purified anti-VLA1 Fab and the complex purified by size exclusion chromatography on a Superdex 200 (Amersham) in 50 mM HEPES, 150 mM NaCl, 2 mM DTT (pH 7.4). The complex was buffer-exchanged into 20 mM Tris, 0.04% NaN3, 2 mM DTT, and further concentrated to 11 mg/mL using a centricon-10 ultrafiltration device (Millipore).
Production of Fabs for crystallography
For crystallography, Fabs are expressed using the strain W3110ara, a Δara derivative of W3110. A 100-mL culture containing the expression plasmid, at a density of >109 cells/mL, was used to inoculate (1:100) a 5-L fermenter containing fermentation medium containing 2% fructose and 100 μg of ampicillin. The fermentation culture was grown overnight at a pH of 7.0 with the DO2 maintained at 30% by fructose titration. The culture was induced at 15 OD with a final concentration of 0.02% arabinose. At the time of induction 250 mL of induction medium (Amisoy, 80 g/L; yeast extract, 20 g/L; L-proline, 12 g/L; L-leucine, 12 g/L; tryptophan, 6 g/L) was added. The culture was harvested 3 h after induction at an OD600 of ∼25–30.
Binding affinity measurements
To estimate the fold change in affinity of mutant anti-VLA1 proteins, we used a competition ELISA. In this assay, GST I-domain fusion protein was coated onto an ELISA plate. A dilution series of anti-VLA1 Fab samples was incubated with 10 nM biotin anti-VLA1 Fab on the plate for 2 h at room temperature in HEPES buffer with 150 mM NaCl, 1 mM MgCl2, 0.05% Tween-20, and 1% BSA. The plate was washed and the amount of biotinylated anti-VLA1 Fab bound was determined using streptavidin HRP as a secondary. The fold change in affinity versus wild type was determined by comparing the EC50 of binding to wild-type Fab measured on the same plate.
To measure the solution phase affinity we employed the KinExA 3000 (Sapidyne Instruments) and surface plasmon resonance (Biacore) (see Supplemental Material for details). Polystyrene beads were coated with GST I-domain fusion protein by passive adsorption. Purified anti-VLA1 Fab fragment is flowed through the column to bind to the I-domain on the bead. The Fab is detected with a secondary anti-mouse IgG F(ab′)2 fragment specific antibody conjugated with the fluorescent dye Cy5. A dilution series of soluble I-domain protein with 3-h equilibration is used. The amount of free anti-VLA1 Fab that remains in solution is determined by the intensity of the fluorescence signal. A nonlinear regression curve fit gives a Kd value.
Surface plasmon resonance measurements
Binding kinetic constants were obtained using the surface plasmon resonance technique on a Biacore apparatus (Biacore USA). Research-grade CM5 sensor chips, N-hyroxysuccinimide (NHS), N-ethyl-N′-(3-diethylaminopropyl) carbodiimide (EDC), and ethanolamine-HCl were purchased from the manufacturer. All buffers were filtered and degassed daily. For amine coupling, carboxymethylated dextran surfaces on CM5 chips were activated using the standard NHS/EDC procedure with 7-min contact times. Anti-GST proteins were diluted to no less than 20 nM in 10 mM sodium acetate (pH 5). The diluted proteins were injected over the activated surface until the desired surface densities were achieved. Activated, coupled surfaces were then quenched of reactive sites with 1 M ethanolamine (pH 8) for 7 min. Anti-GST surfaces were prepared according to the manufacturer's recommendations. Low and ultra-low surfaces were prepared; reference surfaces consisted of activated CM dextran, subsequently blocked with ethanolamine. Data were analyzed from both the low and ultra-low surface capacities.
All data were collected at 10 Hz using at least three to four replicate injections for each determination. Regeneration of active surfaces (when necessary) was carried out by 3-sec injection of 100 mM HCl. All final data sets were double-referenced. Sensorgram data sets were analyzed using the Scrubber software (Myszka and Morton 1998; Myszka 1999). The data obtained fit globally to the binding models. The raw experimental data were corrected for instrumental and bulk solvent artifacts by double-referencing using both activated and blocked surfaces as controls. The data were globally fit to the replicate data for each ligand but allowed all the ligands to have independent Rmax, ka, and kd. Each was also allowed an independent bulk refractive index change. All responses were fit with one global km since they should share the same molecular weight.
Crystallography
One microliter of the protein was mixed with an equal amount of reservoir buffer containing 12%–14%Â Peg 20,000, 100 mM Mes (pH 5.5), 10 mM DTT using vapor diffusion techniques. Crystals typically appeared after 1–2 d with microseeding and grew to full size (0.5 × 0.1 × 0.025 mm) in 1–2 wk. For data collection, crystals were typically broken from a larger mass, incubated in mother liquor with 25% ethylene glycol, and flash-frozen in liquid nitrogen prior to data collection. Data were collected at −160°C using 1.1 Å X-rays at the National Synchrotron Light Source (X25; Brookhaven National Laboratories) using a Quantum315 CCD detector (ADSC). Data processing and reduction was performed using HKL2000 (Otwinowski and Minor 1997). Molecular replacement was performed using MOLREP (Vagin and Teplyakov 1997) using one monomer of the published wild-type structure (PDB ID 1MHP). Iterative rounds of refinement and model building utilized CNX (Brunger et al. 1998) and O (Jones et al. 1991). The Ramachandran plot shows that 87.4% of all residues are in the most favored regions. Data collection and refinement statistics are given in Table 2 of the Supplemental Material. The coordinates have been deposited at the PDB (PDB ID 2B2X).
Acknowledgments
We thank Graham Farrington, Ellen Garber, Juswinder Singh, and Fred Taylor for useful discussions and suggestions. This work was made possible partially through support from the Biogen Idec post-doctoral fellow program.
Footnotes
Supplemental material: see www.proteinscience.org
Reprint requests to: Louis A. Clark, Biogen Idec, Inc., 14 Cambridge Center, Cambridge, MA 02142, USA; e-mail: louie@alumni.northwestern.edu; fax: (617) 679-4998.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.052030506.
References
- Abraham W.M., Ahmed A., Serebriakov I., Carmillo A.N., Ferrant J., De Fougerolles A.R., Garber E.A., Gotwals P.J., Koteliansky V.E., Taylor F.et al. 2004. A monoclonal antibody to α1β1 blocks antigen-induced airway responses in sheep Am. J. Respir. Crit. Care Med. 169: 97–104. [DOI] [PubMed] [Google Scholar]
- Batista F.D. and Neuberger M.S. 1998. Affinity dependence of the B cell response to antigen: A threshold, a ceiling, and the importance of off-rate Immunity 8: 751–759. [DOI] [PubMed] [Google Scholar]
- Ben-horin S. and Bank I. 2004. The role of very late antigen-1 in immune-mediated inflammation Clin. Immunol. 113: 119–129. [DOI] [PubMed] [Google Scholar]
- Benson D.E., Wisz M.S., Hellinga H.W. 2000. Rational design of nascent metalloenzymes Proc. Natl. Acad. Sci. 97: 6292–6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brekke O.H. and Sandlie I. 2003. Therapeutic antibodies for human diseases at the dawn of the twenty-first century Nat. Rev. Drug Discov. 2: 52–62. [DOI] [PubMed] [Google Scholar]
- Brooks B.R., Bruccoleri R.E., Olafson B.D., States D.J., Swaminathan S., Karplus M. 1983. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations J. Comput. Chem. 4: 187–217. [Google Scholar]
- Brunger A.T., Adams P.D., Clore G.M., Delano W.L., Gros P., Grosse-Kunstleve R.W., Jiang J.S., Kuszewski J., Nilges M., Pannu N.S.et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination Acta Crystallogr. D Biol. Crystallogr. 54: 905–921. [DOI] [PubMed] [Google Scholar]
- Caravella J.A. “Electrostatics and packing in biomolecules: Accounting for conformational change in protein folding and binding.” Ph.D. thesis 2002. Massachusetts Institute of Technology, Cambridge, MA.
- Chevalier B.S., Kortemme T., Chadsey M.S., Baker D., Monnat R.J., Stoddard B.L. 2002. Design, activity, and structure of a highly specific artificial endonuclease Mol. Cell 10: 895–905. [DOI] [PubMed] [Google Scholar]
- Crameri A., Cwirla S., Stemmer W.P. 1996. Construction and evolution of antibody-phage libraries by DNA shuffling Nat. Med. 2: 100–102. [DOI] [PubMed] [Google Scholar]
- Dahiyat B.I. and Mayo S.L. 1997. De novo protein design: Fully automated sequence selection Science 278: 82–87. [DOI] [PubMed] [Google Scholar]
- Dantas G., Kuhlman B., Callender D., Wong M., Baker D. 2003. A large scale test of computational protein design: Folding and stability of nine completely redesigned globular proteins J. Mol. Biol. 332: 449–460. [DOI] [PubMed] [Google Scholar]
- Daugherty P.S., Chen G., Iverson B.L., Georgiou G. 2000. Quantitative analysis of the effect of the mutation frequency on the affinity maturation of single chain fv antibodies Proc. Natl. Acad. Sci. 97: 2029–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet J., De Maeyer M., Hazes B., Lasters I. 1992. The dead-end elimination theorem and its use in protein side-chain positioning Nature 356: 539–542. [DOI] [PubMed] [Google Scholar]
- Dunbrack R.L. Jr. and Karplus M. 1993. Backbone-dependent rotamer library for proteins. Application to side-chain prediction J. Mol. Biol. 230: 543–574. [DOI] [PubMed] [Google Scholar]
- Dwyer M.A., Looger L.L., Hellinga H.W. 2004. Computational design of a biologically active enzyme Science 304: 1967–1971. [DOI] [PubMed] [Google Scholar]
- Foote J. and Eisen H.N. 1995. Kinetic and affinity limits on antibodies produced during immune responses Proc. Natl. Acad. Sci. 92: 1254–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M.K., Sharp K.A., Honig B.H. 1988. Calculating the electrostatic potential of molecules in solution: Method and error assessment J. Comput. Chem. 9: 327–335. [Google Scholar]
- Goldstein R.F. 1994. Efficient rotamer elimination applied to protein side-chains and related spin glasses Biophys. J. 66: 1335–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotwals P.J., Chi-rosso G., Ryan S.T., Sizing I., Zafari M., Benjamin C., Singh J., Venyaminov S.Y., Pepinsky R.B., Koteliansky V. 1999. Divalent cations stabilize the α1 β1 integrin i domain Biochemistry 38: 8280–8288. [DOI] [PubMed] [Google Scholar]
- Hanes J., Jermutus L., Weber-bornhauser S., Bosshard H.R., Plückthun A. 1998. Ribosome display efficiently selects and evolves high-affinity antibodies in vitro from immune libraries Proc. Natl. Acad. Sci. 95: 14130–14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanf K.J.M. “Protein design with hierarchical treatment of solvation and electrostatics.” Ph.D. thesis 2002. Massachusetts Institute of Technology, Cambridge, MA.
- Jones T.A., Zou J.Y., Cowan S.W., Kjeldgaard M. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models Acta Crystallogr. A 47: 110–119. [DOI] [PubMed] [Google Scholar]
- Jorgensen W.L. 2004. The many roles of computation in drug discovery Science 303: 1813–1818. [DOI] [PubMed] [Google Scholar]
- Kangas E. and Tidor B. 1998. Optimizing electrostatic affinity in ligand-receptor binding: Theory, computation, and ligand properties J. Chem. Phys. 109: 7522–7545. [Google Scholar]
- Karpusas M., Ferrant J., Weinreb P.H., Carmillo A., Taylor F.R., Garber E.A. 2003. Crystal structure of the α1β1 integrin i domain in complex with an antibody fab fragment J. Mol. Biol. 327: 1031–1041. [DOI] [PubMed] [Google Scholar]
- Kohler G. and Milstein C. 1975. Continuous cultures of fused cells secreting antibody of predefined specificity Nature 256: 495–497. [DOI] [PubMed] [Google Scholar]
- Kortemme T., Joachimiak L.A., Bullock A.N., Schuler A.D., Stoddard B.L., Baker D. 2004. Computational redesign of protein-protein interaction specificity Nat. Struct. Mol. Biol. 11: 371–379. [DOI] [PubMed] [Google Scholar]
- Kretzschmar T. and Von Ruden T. 2002. Antibody discovery: Phage display. Curr. Opin Biotechnol. 13: 598–602. [DOI] [PubMed] [Google Scholar]
- Kuhlman B. and Baker D. 2000. Native protein sequences are close to optimal for their structures Proc. Natl. Acad. Sci. 97: 10383–10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman B., Dantas G., Ireton G.C., Varani G., Stoddard B.L., Baker D. 2003. Design of a novel globular protein fold with atomic-level accuracy Science 302: 1364–1368. [DOI] [PubMed] [Google Scholar]
- Lasters I. and Desmet J. 1993. The fuzzy-end elimination theorem: Correctly implementing the side chain placement algorithm based on the dead-end elimination theorem Protein Eng. 6: 717–722. [DOI] [PubMed] [Google Scholar]
- Leach A.R. and Lemon A.P. 1998. Exploring the conformational space of protein side chains using dead-end elimination and the A* algorithm Proteins 33: 227–239. [DOI] [PubMed] [Google Scholar]
- Li Y., Li H., Yang F., Smith-gill S.J., Mariuzza R.A. 2003. X-ray snapshots of the maturation of an antibody response to a protein antigen Nat. Struct. Biol. 10: 482–488. [DOI] [PubMed] [Google Scholar]
- Lo Conte L., Chothia C., Janin J. 1999. The atomic structure of protein-protein recognition sites J. Mol. Biol. 285: 2177–2198. [DOI] [PubMed] [Google Scholar]
- Looger L.L. and Hellinga H.W. 2001. Generalized dead-end elimination algorithms make large-scale protein side-chain structure prediction tractable: Implications for protein design and structural genomics J. Mol. Biol. 307: 429–445. [DOI] [PubMed] [Google Scholar]
- MacKerell A.D., Brooks B., Brooks III C.L., Nilsson L., Roux B. 1998. CHARMM: The energy function and its parameterization In Encyclopedia of computational chemistry (ed. Schleyer P.v.R.) . pp. 271–277. Wiley, New York.
- Marshall S.A., Vizcarra C.L., Mayo S.L. 2005. One- and two-body decomposable Poisson-Boltzmann methods for protein design calculations Protein Sci. 14: 1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin J.S. and Lowman H.B. 2003. Redesigning an antibody fragment for faster association with its antigen Biochemistry 42: 7077–7083. [DOI] [PubMed] [Google Scholar]
- Mendes J., Baptista A.M., Carrondo M.A., Soares C.M. 1999. Improved modeling of side-chains in proteins with rotamer-based methods: A flexible rotamer model Proteins 37: 530–543. [DOI] [PubMed] [Google Scholar]
- Midelfort K.S., Hernandez H.H., Lippow S.M., Tidor B., Drennan C.L., Wittrup K.D. 2004. Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody J. Mol. Biol. 343: 685–701. [DOI] [PubMed] [Google Scholar]
- Mooers B.H., Datta D., Baase W.A., Zollars E.S., Mayo S.L., Matthews B.W. 2003. Repacking the core of T4 lysozyme by automated design J. Mol. Biol. 332: 741–756. [DOI] [PubMed] [Google Scholar]
- Myszka D.G. 1999. Improving biosensor analysis J. Mol. Recognit. 12: 279–284. [DOI] [PubMed] [Google Scholar]
- Myszka D.G. and Morton T.A. 1998. CLAMP: A biosensor kinetic data analysis program Trends Biochem. Sci. 23: 149–150. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z. and Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode Methods Enzymol. 276: 307–326. [DOI] [PubMed] [Google Scholar]
- Pierce N.A., Spriet J.A., Desmet J., Mayo S.L. 2000. Confomational splitting: A more powerful criterion for dead-end elimination J. Comp. Chem. 21: 999–1009. [Google Scholar]
- Selzer T., Albeck S., Schreiber G. 2000. Rational design of faster associating and tighter binding protein complexes Nat. Struct. Biol. 7: 537–541. [DOI] [PubMed] [Google Scholar]
- Shah P.S., Hom G.K., Mayo S.L. 2004. Preprocessing of rotamers for protein design calculations J. Comput. Chem. 25: 1797–1800. [DOI] [PubMed] [Google Scholar]
- Sharp K.A. and Honig B. 1990. Electrostatic interactions in macromolecules: Theory and applications Annu. Rev. Biophys. Biophys. Chem. 19: 301–332. [DOI] [PubMed] [Google Scholar]
- Sherman B.W. 2004. “Biomolecular ligand design: Enhancing binding affinity and specificity utilizing electrostatic charge optimization and packing techniques.” Ph.D. thesis Massachusetts Institute of Technology, Cambridge, MA.
- Sitkoff D., Sharp K.A., Honig B. 1994. Accurate calculation of hydration free energies using macroscopic solvent models J. Phys. Chem. 98: 1978–1988. [Google Scholar]
- Sundberg E.J. and Mariuzza R.A. 2003. Molecular recognition in antibody-antigen complexes Adv. Protein Chem. 61: 119–160. [DOI] [PubMed] [Google Scholar]
- Takano K., Yamagata Y., Funahashi J., Hioki Y., Kuramitsu S., Yutani K. 1999. Contribution of intra- and intermolecular hydrogen bonds to the conformational stability of human lysozyme Biochemistry 38: 12698–12708. [DOI] [PubMed] [Google Scholar]
- Vagin A. and Teplyakov A. 1997. MOLREP: An automated program for molecular replacement J. Appl. Crystallogr. 30: 1022–1025. [Google Scholar]
- van den Beucken T., Pieters H., Steukers M., van der Vaart M., Ladner R.C., Hoogenboom H.R., Hufton S.E. 2003. Affinity maturation of Fab antibody fragments by fluorescent-activated cell sorting of yeast-displayed libraries FEBS Lett. 546: 288–294. [DOI] [PubMed] [Google Scholar]
- Vriend G. 1990. What if: A molecular modeling and drug design program J. Mol. Graph. 8: 52–56. [DOI] [PubMed] [Google Scholar]
- Winston P.H. In Artificial intelligence . 1992. Addison-Wesley, Reading, Massachusetts.
- Wisz M.S. and Hellinga H.W. 2003. An empirical model for electrostatic interactions in proteins incorporating multiple geometry-dependent dielectric constants Proteins 51: 360–377. [DOI] [PubMed] [Google Scholar]