

Abstract
A central feature of transmissible spongiform encephalopathies (TSE or prion diseases) involves the conversion of a normal, protease-sensitive glycoprotein termed prion protein (PrP-sen) into a pro-tease-resistant form, termed PrP-res. The N terminus of PrP-sen has five copies of a repeating eight amino acid sequence (octapeptide repeat). The presence of one to nine extra copies of this motif is associated with a heritable form of Creutzfeld-Jakob disease (CJD) in humans. An increasing number of octapeptide repeats correlates with earlier CJD onset, suggesting that the rate at which PrP-sen misfolds into PrP-res may be influenced by these mutations. In order to determine if octapeptide repeat insertions influence the rate at which PrP-res is formed, we used a hamster PrP amyloid-forming peptide (residues 23–144) into which two to 10 extra octapeptide repeats were inserted. The spontaneous formation of protease-resistant PrP amyloid from these peptides was more rapid in response to an increased number of octapeptide repeats. Furthermore, experiments using full-length glycosylated hamster PrP-sen demonstrated that PrP-res formation also occurred more rapidly from PrP-sen molecules expressing 10 extra copies of the octapeptide repeat. The rate increase for PrP-res formation did not appear to be due to any influence of the octapeptide repeat region on PrP structure, but rather to more rapid binding between PrP molecules. Our data from both models support the hypothesis that extra octapeptide repeats in PrP increase the rate at which protease resistant PrP is formed which in turn may affect the rate of disease onset in familial forms of CJD.
Keywords: prion, fibril, amyloid, kinetics, octapeptide repeat, insertional mutations
Transmissible spongiform encephalopathies (TSE or prion diseases) are a unique category of fatal neurodegenerative disorders that affect humans and other mammals (Prusiner 1998; Chesebro 2003; Aguzzi and Polymenidou 2004; Collins et al. 2004). A critical component of the disease process is the conversion of prion protein (PrP-sen) into an insoluble and protease resistant isoform, termed PrP-res or PrPSc (Bendheim et al. 1988). Human TSEs occur in sporadic, acquired and hereditary forms (Wadsworth et al. 2003). Sporadic Creutzfeld-Jakob disease (CJD) occurs in about one out of a million people annually without clear evidence of exposure to an infectious agent. In acquired TSE diseases, exposure to the TSE agent can occur iatrogenically, such as during neurosurgery, or via ingestion (Will et al. 1996).
There are at least 14 known PrP mutations that have been linked to hereditary TSE disease (Gambetti et al. 2003). One of these is an insertional mutation in the N terminus of PrP-sen whereby pathology is associated with extra copies of an eight amino acid repetitive motif comprised of the residues P(H/Q)GGG(−/ G)WGQ (octapeptide repeat). Normal PrP-sen contains five copies of this region. Most PrP insertional mutations consist of one to nine additional octapeptide repeats, resulting in a disease phenotype similar to sporadic CJD (Goldfarb et al. 1991; Owen et al. 1992; Krasemann et al. 1995; Campbell et al. 1996; Capellari et al. 1997; Vital et al. 1999; Yanagihara et al. 2002; Lewis et al. 2003; Pietrini et al. 2003). Patients with PrP insertional mutations usually display signs of illness early in life and die after a clinical course spanning several years while brain material from these patients can transmit TSE disease to non-human primates (Goldfarb et al. 1991). Interestingly, amyloid plaques have been observed in patients with more than six additional octapeptide repeats, suggesting that the formation of amyloid is also exacerbated by insertional mutations (Vital et al. 1998). A similar phenomenon is observed with Huntington’s disease, in which the largest expansions of a repeating polyglutamine sequence are associated with aggregation, amyloid formation, and an earlier onset of pathology (Penney et al. 1997). However, in the absence of a known exposure to TSE infectivity, it is unclear how PrP-res formation is triggered in patients with insertional PrP mutations.
It has been suggested that mutations in the PrP gene render an organism more susceptible to TSE disease by causing dysfunctional interactions between PrP-sen and molecules such as chaperones (DebBurman et al. 1997) or copper ions (Brown et al. 1997), or by increasing the population of partially unfolded species needed for PrP-res formation. Cell culture experiments have demonstrated that PrP-sen containing seven to 14 copies of the octapeptide repeat region displayed increased protease resistance and detergent insolubility compared with wild-type PrP-sen (Lehmann and Harris 1996a,b; Priola and Chesebro 1998; Narwa and Harris 1999; Ivanova et al. 2001). Studies in transgenic mice containing PrP genes with either four or nine additional octapeptide repeats developed progressive neurodegeneration, suggesting that the mutation is neurotoxic (Chiesa et al. 1998, 2000, 2001; Harris et al. 2003). While mutant PrP isolated from these transgenic mice exhibited certain characteristics reminiscent of PrP-res, it did not trigger a TSE infection when injected into wild-type mice (Priola and Chesebro 1998; Chiesa et al. 2003; Harris et al. 2003). Thus, none of the cell-culture, in vitro, or transgenic mouse experiments to date have demonstrated that, in the absence of TSE, insertional mutants of PrP-sen spontaneously refold into PrP-res and produce TSE infectivity.
One possibility, which has not been directly addressed experimentally, is that additional octapeptide repeat insertions in PrP-sen may act to enhance susceptibility to TSE disease by influencing the rate at which PrP-res is formed. Consistent with this hypothesis is the observation that earlier CJD onset correlates with an increasing number of octapeptide repeat insertions in the PrP gene (Capellari et al. 1997; Gambetti et al. 2003; Croes et al. 2004). Using an amyloidogenic PrP peptide that is associated with a heritable form of human TSE disease, we now show that the formation of protease resistant PrP amyloid is enhanced by an increased number of octapeptide repeats. Despite the observed differences in reaction rate, the overall three-dimensional fold of the amyloid core was not influenced by the mutations. Additionally, in a cell-free assay for PrP-res formation using full-length glycosylated PrP-sen, we demonstrate that PrP-res formation also occurs more rapidly in a PrP-sen mutant containing 10 extra copies of the octapeptide repeat and that this effect correlates with the amount of PrP-sen bound to PrP-res. Prion insertional mutations may therefore influence the rate of spontaneous TSE pathogenesis not by influencing the final conformation of protease resistant PrP, but rather by promoting PrP binding, thereby lowering the kinetic barrier for the formation of both PrP-res and PrP amyloid.
Results
The octapeptide repeat region influences the kinetics of PrP amyloid formation
Amyloid fibrils and plaques are associated with at least 20 protein misfolding diseases (Sipe and Cohen 2000), including TSE diseases, where PrP-res can be found in both amyloid and non-amyloid forms (Chesebro et al. 2005). Since PrP-res amyloid has been found in CJD patients whose PrP molecules contained extra copies of the octapeptide repeat region (Vital et al. 1998), we reasoned that the number of repeats might influence the formation of PrP amyloid. However, as fibrillization of full length PrP (residues 23–231) requires denaturing conditions (Bocharova et al. 2005), we chose to analyze PrP amyloid formation using a HaPrP peptide with a stop codon inserted at residue 145. In the human PrP gene, the 145stop mutation codes for a truncated PrP (residues 23–144) that has been associated with a form of Gerstmann-Straussler-Scheinker (GSS) disease characterized by neurofibrillary lesions and severe amyloid deposition in the central nervous system (Kitamoto et al. 1993; Ghetti et al. 1996). Importantly, the 145stop peptide has also been shown to form amyloid fibrils in vitro under non-denaturing conditions (Kundu et al. 2003; Vanik et al. 2004; Jones and Surewicz 2005). The HaPrP 145stop peptide was thus used in this study as a model PrP molecule to examine the effect of octapeptide repeat insertions upon the rate of PrP amyloid formation.
Purified 145stop PrP peptides containing five to 15 copies of the octapeptide repeat region were >99% pure (Fig. 1). After fibrillization, each of the 145stop peptides fulfilled the generally recognized criteria for amyloid, including fibrils visualized by electron microscopy (Fig. 2), reaction with ThT (Fig. 3), increased resistance to digestion by proteinase K (Figs. 4, 5), and the acquisition of β-sheet secondary structure (Fig. 6).
Figure 1.

Purity of hamster 145stop peptides containing five to 15 octapeptide repeats. Samples were analyzed by 18% SDS-PAGE on Tris-glycine gels stained with Coomassie Blue (left panel) or Western blot using the hamster PrP-specific monoclonal antibody 3F4 at a 1:20,000 dilution (right panel). The preparations were ~99% pure by SDS-PAGE analysis. Molecular mass markers in kilodaltons are shown to the left.
Figure 2.
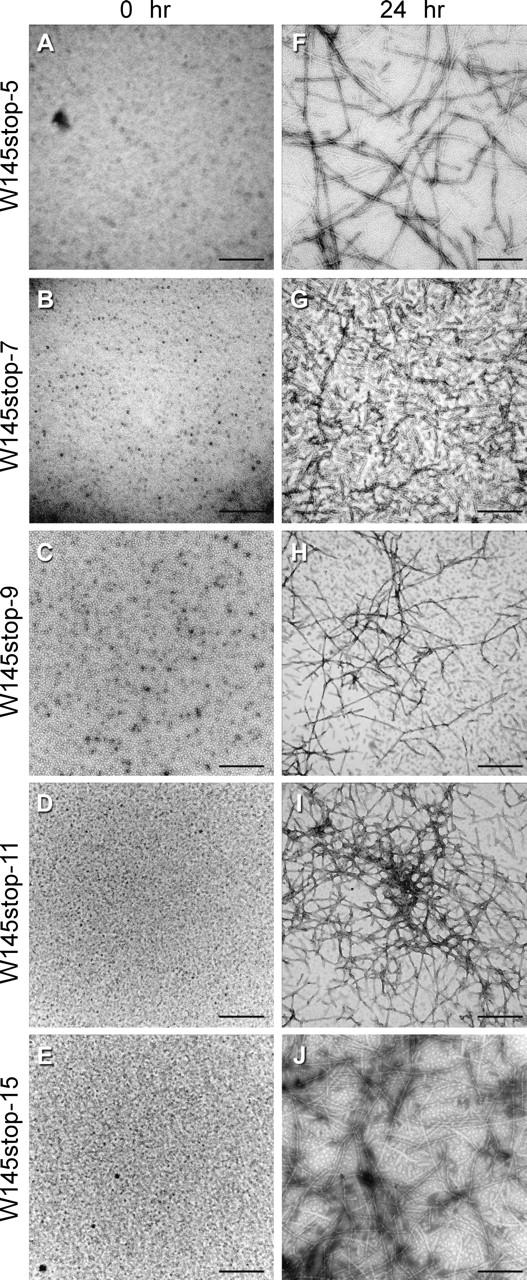
Hamster 145stop peptides containing five to 15 repeats form fibrils. Samples were analyzed at 0 h (A–E) and after completion of the fibrillization reactions by 24 h (F–J). The micrographs are at 100,000 × magnification. Representative micrographs are shown. No consistent morphological differences were observed between fibrils containing five to 15 repeat motifs. Bars, 500 nm.
Figure 3.

Amyloid formation occurs more rapidly as the number of octapeptide repeats increases. (A) Hamster PrP 145stop peptides were vigorously agitated in 25 mM BisTris Propane (pH 7.0), and the emission spectrum of ThT at 482 nm was recorded over 18 h. A representative experiment is shown. (B) The duration of the lag phase, defined as the length of time in which no significant change in the background level of ThT fluorescence was observed, was used to compare the kinetics of fibril formation between peptides. The bars represent an average of three samples ±SEM. (*) P < 0.01 when compared with 145stop-5 as the control using 1-way ANOVA with Dunnett’s post test.
Figure 4.

Amyloid isoforms of 145stop-5 and 145stop-11 are more protease-resistant than their non-fibrillar counterparts. 145stop-5 (A) and 145stop-11 (B) peptides at a concentration of 75 μM were subjected to PK digestion as described in the Materials and Methods. The non-digested portion of each peptide was calculated from the intensity of the Western blot bands with digitizing software. Each digestion was done in triplicate. The error bars indicate the mean ± SEM. (*) P = 0.02 and (**) P = 0.007 when the protease resistance of the amyloid form was compared with the non-amyloid form using the unpaired student’s t-test. There was no significant difference in protease resistance between the amyloid forms of 145stop-5 and 145stop-11 (P > 0.2 using the unpaired student’s t-test).
Figure 5.

Each 145stop octapeptide repeat mutant contains the same protease resistant core. Coomassie Blue stained SDS-PAGE gel showing the protease-resistant fragment obtained from the partial PK digestion of fibrillized 145stop peptides containing five to 15 copies of the octapeptide repeat region. Each peptide was digested at 100 μM concentration in either the absence (−) or presence (+) of 1 μg/ mL PK for 1 h at 37C. For each peptide, PK digestion yielded a protease-resistant fragment with the N-terminal sequence (QWNKPS…) corresponding to PK cleavage at residue 98 of hamster PrP.
Figure 6.

Second derivative ATR-FTIR spectra of the 145stop peptides show remarkably similar β-sheet architecture. Amyloid pellets in aqueous slurries were scanned from 1400–1800 cm−1, as described in Materials and Methods. The spectral peaks at 1627 and 1637 cm−1 represent the acquisition of significant β-sheet structure upon formation of amyloid from natively unstructured precursors.
A typical fibrillization reaction includes a lag phase (i.e., the last time point at which no significant change in the background level of ThT fluorescence is observed) followed by an abrupt rise in ThT fluorescence during which time fibrils are formed, followed by a decrease in ThT intensity. The decrease in ThT intensity is a common occurrence in fibrillization reactions and has been attributed to the formation of bundled aggregates of fibrils that lack a suitable surface for ThT binding (Nilsson 2004). A representative ThT curve for wild-type and mutant 145stop amyloid formation is shown in Figure 3A. The presence of fibrils at the end of each reaction was confirmed by electron microscopy (Fig. 2). When the length of the lag phase was compared to the number of octapeptide repeats in the 145stop peptide, it was found to be inversely related to the number of octapeptide repeat insertions. Thus, for any given experiment, the lag phase for 145stop-5 fibrillization was always longer than the lag phase for 145stop-11 (Fig. 3B). We were unable to assay the fibrillization kinetics for 145stop-15, as this peptide came out of solution so rapidly once agitation started that accurate lag phase measurements were not possible. However, analysis of the 145stop-15 reaction by electron microscopy showed that fibrils and aggregate were indeed formed in abundance (Fig. 2J). Overall, our results show that the tendency to aggregate and the rate of PrP amyloid formation increased when the number of octapeptide repeats in the molecule increased.
In order to assess whether or not the amyloid form of the 145stop peptide was more resistant to protease digestion than the non-amyloid form, the peptides were treated with successively higher concentrations of PK. After treatment with 0–10 μg/mL PK, the percentage of remaining full-length PrP was calculated for the amyloid and non-amyloid isoforms of 145stop-5 and 145stop-11. Non-amyloid 145stop-11 was significantly more resistant to PK-digestion than the non-amyloid 145stop-5 peptide (Fig. 4, P < 0.0001 by unpaired Student’s t-test). We attribute the higher PK resistance of 145stop-11 to the presence of the extra repeats. This is consistent with previous results using full length eukaryotic PrP from cell culture (Lehmann and Harris 1996a; Priola and Chesebro 1998). The amyloid forms of 145stop-5 (Fig. 4A) and 145stop-11 (Fig. 4B) were significantly more resistant to degradation than the non-amyloid forms. Thus, increasing the number of octapeptide repeats increased the overall protease resistance of the 145stop peptide.
The number of octapeptide repeats does not influence the overall structure of PrP amyloid
Partial proteolysis experiments have been used to examine the conformational states of proteins, including those that form amyloid (Fontana et al. 1997; Polverino de Laureto et al. 1999; Kheterpal et al. 2001; Spolaore et al. 2001). Additionally, unique three-dimensional conformations of PK-resistant PrP molecules purified from TSE-infected brain homogenates can often be differentiated by their molecular mass following PK treatment (Collinge et al. 1996; Piccardo et al. 2001; Lawson et al. 2004). In order to determine if the fundamental secondary structure of amyloid formed by 145stop peptides was influenced by the number of octapeptide repeats, fibrillized 145stop peptides were treated with PK. A 4.5 kDa PK-resistant fragment was isolated for each peptide (Fig. 5), suggesting that the fibrillized peptides were truncated at a similar point in the amino acid sequence. Consistent with this interpretation, N-terminal sequencing of the PK-resistant band revealed the sequence QWNKPS…, regardless of the number of octarepeat insertions in the molecule. This cleavage site corresponds to residue 98, demonstrating complete removal of the octapeptide repeat region. The consistency of the PK digestion patterns suggested that the overall fold of the protease resistant core did not change significantly because of the insertions.
In order to determine whether or not the 145stop peptides also had similar structures prior to PK digestion, each peptide was analyzed by attenuated total reflectance Fourier transform infrared (ATR-FTIR). ATR-FTIR is a method of choice for resolving differences in β-sheet character for aggregated proteins and amyloid since the structure of these compounds cannot be analyzed successfully by nuclear magnetic resonance or by X-ray crystallography (Calero and Gasset 2005). Prior to fibrillization, the 145stop peptides adopted a predominantly random coil formation (data not shown), consistent with published data for the PrP 23–144 sequence (Kundu et al. 2003; Jones and Surewicz 2005). In contrast, non-PK digested amyloid isoforms of the 145stop peptides containing five to 11 octapeptide repeat motifs were characterized by a strong β-sheet band at 1627–1628 cm−1 (Fig. 6). The FTIR spectra for the wild-type and mutant peptides were remarkably similar, despite the sequence variations in the N terminus. Thus, regardless of the number of repeats present, the structures of the final PK-resistant amyloid are likely very similar. It is, therefore, unlikely that insertional mutations lead to forms of protease-resistant PrP that are conformationally distinct from protease-resistant PrP formed from wild-type PrP-sen.
The octapeptide repeat region influences the kinetics of PrP-res formation
In order to determine if an increased number of octapeptide repeats also influenced the kinetics of PrP-res formation from PrP-sen molecules made in mammalian cells, the amount of full-length, glycosylated PrP-sen converted into PrP-res was assayed over time using a cell-free system of PrP-res formation (Kocisko et al. 1994). Radiolabeled HaPrP-sen molecules containing five (wild-type HaPrP-5) or 15 (HaPrP-15) copies of the octapeptide repeats were purified from mouse fibroblast cells and mixed with purified brain-derived 263K hamster PrP-res (HaPrP-res). The nascent 35S-HaPrP-res was then assayed over a 24-h period. For both constructs, new 35S-HaPrP-res was detectable by 2 h (Fig. 7A). However, PrP-res formation from HaPrP-15 reached maximal levels significantly more rapidly than wild-type HaPrP-5 (Fig. 7A). The relative rate at which hamster PrP-sen was converted into hamster PrP-res was therefore greater for HaPrP-15 than for wild-type hamster PrP.
Figure 7.
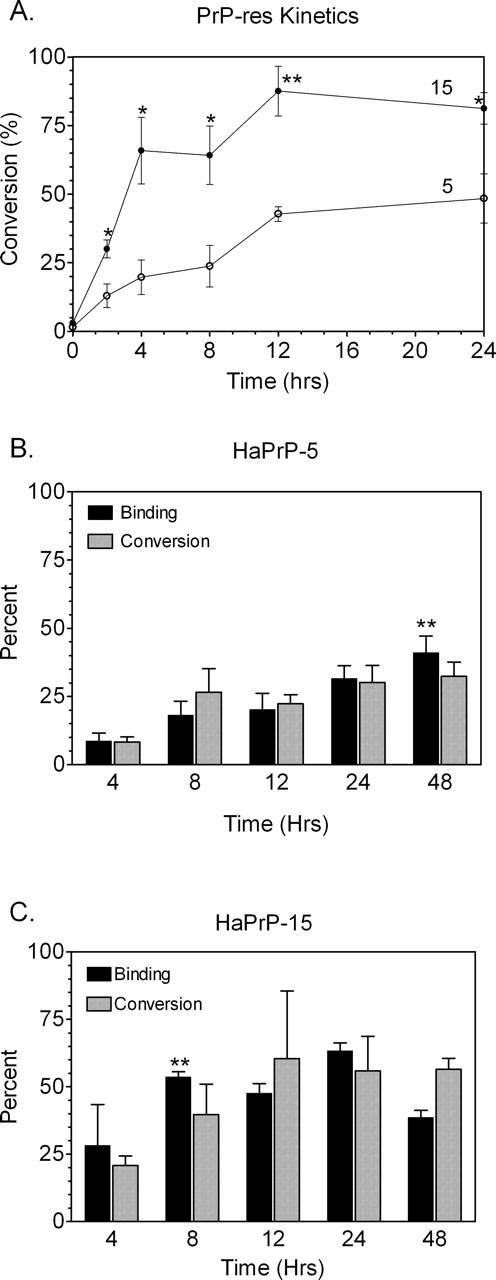
PrP-res formation and PrP-sen:PrP-res binding occur more rapidly in hamster PrP-sen with 15 copies of the octapeptide repeat. Immunoprecipitated 35S-HaPrP-sen containing five (HaPrP-5) or 15 (HaPrP-15) copies of the octapeptide repeat was incubated in the presence of HaPrP-res as described in Materials and Methods. The percentage of input 35S-labeled HaPrP-sen converted to 35S-HaPrP-res (% conversion) was determined over 24 h (A). Error bars represent the mean ± SEM for sample size N =4. (*) P =0.02 or (**) P =0.003 using the unpaired student’s t-test. HaPrP-5 (B) or HaPrP-15 (C) was incubated with hamster PrP-res in a cell-free conversion reaction. At the indicated time points, the amount of PrP-sen bound to PrP-res (black bars) and the amount of PrP-sen converted to PrP-res (gray bars) were quantified as a percentage of the input 35S-labeled HaPrP-sen. In order to correct for non-specific pelleting of radiolabeled HaPrP-5 and HaPrP-15 in the binding assay, the amount of radiolabeled PrP in the pellet in the absence of PrP-res was subtracted from the amount of radiolabeled PrP in the pellet in the presence of PrP-res. The columns and error bars show the mean ± SEM, respectively, for N =4–6 samples. The asterisksrepresentthelasttimepointatwhichtherewasnofurtherchangein the amount of PrP-sen bound to PrP-res using a 1-way ANOVA with Dunnett’s post test (**, P <0.01 when compared with the 4-h time point). For each data set at any given time point, there was no significant difference between the percentage of PrP-sen bound and the percentage of PrP-sen converted to PrP-res (P >0.1 using the paired student’s t-test).
Binding of 35S-HaPrP-sen to HaPrP-res is influenced by the octapeptide repeats
Formation of PrP-res is thought to occur in two broadly defined mechanistic steps: 1) binding of PrP-sen to PrP-res followed by 2) the conversion of PrP-sen to PrP-res (Horiuchi et al. 2000). Extra octapeptide repeats might influence one or both of these steps. In order to ascertain whether binding or conversion were enhanced by the number of octapeptide repeat motifs, at fixed time intervals we measured the extent to which 35S-HaPrP-sen bound to HaPrP-res in the cell-free reaction mixture. If binding was rate-limiting, then any change in binding behavior should affect the kinetics of PrP-res formation.
HaPrP-5 binding was gradual, with the amount bound to PrP-res at 48 h significantly higher than that at 4 h (Fig. 7B). In contrast, binding of HaPrP-15 to PrP-res was virtually complete by 8 h with only insignificant increases in the amounts of PrP-sen bound thereafter (Fig. 7C). Overall, the percentage of wild-type HaPrP-5 bound to PrP-res was significantly lower than that of HaPrP-15 up to 24 h (P = 0.02 to <0.0001 using the unpaired Student’s t-test). Non-linear regression analysis of the data in Figure 7, B and C, showed that 50% of PrP-sen was bound to PrP-res by either 4 or 11 h for HaPrP-15 and HaPrP-5, respectively. These data further support the conclusion that HaPrP-5 bound PrP-res at a slower rate than HaPrP-15. Thus, the binding kinetics of PrP-sen to PrP-res were affected by an increased number of octapeptide repeats.
In contrast, regardless of the number of octapeptide repeats, there was no significant difference between the amount of PrP-sen bound to PrP-res and the conversion of PrP-sen to PrP-res (Fig. 7B,C, cf. black bars with gray bars at each time point). This is consistent with published results using only wild-type hamster PrP-sen (Priola and Lawson 2001) and suggests that, under the conditions used, all of the bound PrP-sen was rapidly converted to PrP-res. These data suggest that the number of octapeptide repeats influenced the rate of PrP-res formation by influencing PrP-sen:PrP-res binding and not the actual conversion of PrP-sen to PrP-res. Thus, PrP binding was essentially the rate-limiting step in the reaction.
Discussion
Our data show that PrP insertional mutations affect PrP-res formation by promoting PrP binding relatively early in the mechanism, while the events associated with folding into a protease resistant isoform high in β-sheet structure remain relatively unaffected. Mechanistically, the number of repeats clearly affected PrP-sen:PrP-res binding within the first 24 h of PrP-res formation (Fig. 7). The percentage of PrP-sen bound to PrP-res was almost always equivalent to the percentage of PrP-sen converted to PrP-res, suggesting a cooperative process with binding increasing the probability of conversion. This is likely due to the fact that, in contrast to many enzyme or chemically mediated reactions, binding in the cell-free conversion does not appear to be a reversible equilibrium in which PrP-sen displays any tendency to disaggregate once it has bound to PrP-res (Callahan et al. 2001). This is in agreement with the observations that PrP-res is virtually always found in polymeric form and that therapeutic strategies for the reversal of in vivo plaque formation remain elusive. Our results are also consistent with studies in yeast, where the number of oligopeptide repeat expansions in N-terminal yeast prion domains influences the rate at which yeast prion protein forms (Liu and Lindquist 1999; Jiang et al. 2004). Thus, our data support the hypothesis that the conversion of PrP-sen to PrP-res is facilitated by the presence of an increased number of octapeptide repeats in a manner dependent upon their ability to lower the kinetic barrier for the rate-limiting binding step.
Our data also support the conclusion that extra copies of the octapeptide motif do not exert their effect by creating a conformationally distinct form of protease-resistant PrP. We could not use ATR-FTIR to assay the conformation of newly formed PrP-res in the cell-free conversion assay due to the high background of animal-derived PrP-res used to initiate the reaction. However, since the PrP amyloid was derived from purified protein, we were able to use the familial TSE 145stop mutants to analyze how the repeat motifs might influence the structure of a protease-resistant PrP molecule. ATR-FTIR analysis showed that the overall secondary structures of the amyloid formed were remarkably similar, regardless of the number of repeats (Fig. 6). Furthermore, treatment of the 145stop peptides with PK confirmed that wild-type and mutant peptides were cleaved at the same N-terminal residue (Fig. 5), suggesting that the structure of the protease resistant core was not significantly affected by the octapeptide motif. These PK cleavage data are consistent with earlier data demonstrating that PrP-res derived from PrP-sen mutants with five to 11 copies of the octapeptide repeat motif were similar in size (Priola and Chesebro 1998), data which imply that changing the number of repeats in PrP-sen does not significantly alter the conformation of PrP-res. They are also consistent with in vitro (Priola and Chesebro 1998) and in vivo (Chen et al. 1997; Chiesa et al. 2003) studies demonstrating that PrP-res can be derived from both wild-type PrP and PrP insertional mutants, data which provide compelling evidence that the mutant and wild-type PrP molecules are similar enough in structure to interact with each other to produce PrP-res. Overall, our studies strongly suggest that the influence of extra repeat regions in PrP-res formation, both non-amyloid and amyloid, is primarily kinetic and not conformational.
The octapeptide repeat region does not appear to influence the final conformation of protease-resistant PrP (Figs. 5, 6) and is not absolutely required for PrP-res formation (Lawson et al. 2001), suggesting that this region is not required for TSE infectivity. Our data would thus predict that the octapeptide repeat region could also influence acquired forms of TSE disease, not by influencing susceptibility to TSE infection, but rather by influencing the tempo of TSE disease. Just as an increased number of octapeptide repeat motifs can increase the kinetics of PrP-res formation (Figs. 3, 7), a decreased number of repeats should negatively affect the rate of PrP-res formation. Indeed, a recent study showed that mice expressing a PrP gene in which the octapeptide repeat region was deleted were still susceptible to scrapie infection but demonstrated increased disease incubation times and reduced levels of PrP-res (Flechsig et al. 2000). Our data are compatible with this result and provide a mechanism for both the reduction of PrP-res and the observed increase in disease incubation times: The lack of the octapeptide repeat region leads to reduced PrP-sen:PrP-res binding, which results in a decreased rate of PrP-res formation and a longer disease incubation time.
Depending upon the TSE disease, protease resistant PrP can exist in amyloid as well as non-amyloid forms (Vital et al. 1998). An analysis of multiple cases of heritable TSE disease associated with octapeptide repeat insertions showed that 35% (7/20) of the cases were positive for amyloid plaques (Vital et al. 1998). All of these cases had 11 or more copies of the repeat motif, suggesting that a certain number of octapeptide repeats may predispose toward plaque formation (Vital et al. 1998). Our results with the 145stop peptide demonstrate that the octapeptide repeat region can indeed influence the rate of PrP amyloid formation. Furthermore, and similar to the in vivo observations, the influence of the octapeptide repeat on PrP amyloid formation was most significant only when nine or more copies of the motif were present (Fig. 3). Thus, our data are consistent with the idea that insertional mutations could promote amyloid formation in certain familial TSE diseases.
The idea that PrP insertional mutations influence the kinetics of PrP-res formation is compatible with the progressively earlier disease onset observed for those patients with increasing numbers of PrP octapeptide repeats (Croes et al. 2004). Patients with Huntington’s disease also succumb to illness influenced in part by the length of an abnormal polyglutamine repeat expansion in the Huntingtin gene (Penney et al. 1997). Thus, by influencing the rate at which protease-resistant and pathogenic PrP forms, expansions of the octapeptide repeat region may act in an analogous manner to the polyglutamine repeats in the Huntingtin gene and lead to an earlier onset of familial TSE disease.
Materials and methods
Clones and cell culture
Syrian hamster prion protein (HaPrP-sen) containing five octapeptide repeats (wild-type HaPrP-5) and a HaPrP-sen mutant containing 15 repeats (HaPrP-15) were constructed as previously reported (Priola and Chesebro 1998). Both HaPrP-sen constructs were expressed in fibroblast cells as previously described (Chesebro et al. 1993).
Kinetics of PrP-res formation
The cell-free conversion assay (Kocisko et al. 1994; Caughey et al. 1999) and cell-free conversion binding assays (Priola and Lawson 2001) were performed as described previously, with the following modifications. To assay PrP-res formation over time, 1.4 μg of hamster PrP-res purified from the brains of hamsters infected with the 263K strain of scrapie was incubated with 140,000 cpm of immunopurified trans35S (Perkin-Elmer) labeled HaPrP-sen (~1.4 ng) in cell-free conversion buffer (0.75 M guanidine hydrochloride, 1.25% sarkosyl, 5 mM cetyl pryidinium chloride, 50 mM sodium citrate buffer at pH 6.0; total volume of 140 μL). The reaction was incubated at 37°C, and at various time points a 20-μL aliquot was removed and assayed for the presence of 35S-HaPrP-res. Determination of the amount of 35S-HaPrP-sen bound to HaPrP-res over time (i.e., cell-free conversion binding assay) was done in the same way except that reaction volumes were doubled so that, at each time point assayed, two 20-μL samples could be removed. One sample was assayed for the presence of 35S-HaPrP-res while the second sample was used to assay the amount of 35S-HaPrP-sen bound to HaPrP-res, defined as the percentage of 35S-HaPrP-sen which pelleted with HaPrP-res, using centrifugation as described previously (Priola and Lawson 2001). Percent conversion was calculated based upon the percentage of input 35S-HaPrP-sen that became protease resistant (i.e., 35S-HaPrP-res). In all cases, 35S-HaPrP-sen or newly formed 35S-HaPrP-res was visualized by phosphorimaging with a Molecular Dynamics Storm scanner and quantified with ImageQuant software (Amersham/ General Electric).
Cloning of hamster PrP peptides
In humans, PrP-sen containing a 145stop mutation is associated with a form of Gerstmann-Straussler-Scheinker (GSS) syndrome resulting in severe deposition of amyloid (Kitamoto et al. 1993; Ghetti et al. 1996). The corresponding hamster PrP cDNA sequence was PCR amplified and ligated into the pET41 vector (EMD Biosciences) as NdeI/HindIII inserts, resulting in non-hexahistidine-tagged peptides containing 5, 7, 9, 11 or 15 octapeptide repeat regions flanked by wild-type residues 23–50 on the N-terminal side and 92–144 at the C terminus. The signal peptide residues 1–22 were replaced by methionine and tryptophan 145 was replaced by a stop codon. The constructs were sequenced using universal T7 forward and reverse primers to rule out the presence of spurious mutations (Seqwright). The peptides, termed 145stop-5, -7, -9, -11, or -15 were used to form amyloid, as described below.
Purification of hamster PrP 145stop peptides
Peptides containing five to 15 octapeptide repeats were expressed in Escherichia coli Rosetta cells and purified by a modification of the Zahn method (Zahn et al. 1997). In a typical procedure, 1 g of harvested cells was suspended in 10 mL of BugBuster with lysonase (EMD Biosciences) and EDTA-free protease inhibitors (Roche). The mixture was subjected to orbital rotation with sonication at 4°C and then centrifugation (20 min, 15,000g). The pellets were washed by suspension in BugBuster (5 mL) and centrifugation. The inclusion bodies were then dissolved in denaturing buffer (100 mM sodium phosphate at pH 8.0, 6 M guanidine hydrochloride, 10 mM Tris) and clarified by centrifugation. The peptide solutions were stirred for 1 h at room temperature with 2.5 mL of NiNTA resin (Qiagen) and loaded onto a 10-mL disposable column. Refolding was achieved with a linear gradient (20 mL, 1 h) into refolding buffer (100 mM sodium phosphate at pH 8.0, 10 mM Tris) with further incubation (3 h, 4°C) in the same buffer. Elution was effected with 100 mM sodium phosphate (pH 5.8), 10 mM Tris, 300 mM imidazole. Pooled fractions were dialyzed (4 L, overnight) against 15 mM sodium phosphate (pH 5.8), 0.5 mM EDTA and then against distilled Milli-Q water (2 L × 4, 1 h) at 4°C. The peptides were analyzed by SDS-PAGE and by Western blot (ECL) using the mouse-derived, hamster PrP specific monoclonal antibody 3F4 (Kascsak et al. 1987) at a 1:20,000 dilution.
Fibril growth
Directly after purification, the 145stop peptides were filtered (0.02 μm) and concentrated by centrifugal ultrafiltration (Amicon, MWCO 5000). The peptides were normalized to 125 with Milli-Q water and subjected to ultracentrifugation (256,000g) for 30 min immediately prior to use. Fibril growth was initiated by adjustment of the peptide solution to 25 mM Bis-Tris Propane (pH 7.0), followed by rapid agitation at 37°C for 18 h. Each reaction was done in triplicate and monitored hourly by thioflavin T (ThT) fluorescence (LeVine 1999). At fixed time intervals, an aliquot of each peptide was diluted to 5 μM in the presence of 10 μM ThT in 25 mM Bis-Tris Propane (pH 7.0) in a disposable Versafluor microcuvette (Bio-Rad). After 5 min of incubation at room temperature, the mixture was excited at 445 nm and the emission intensity was recorded at 482 nm using a Perkin-Elmer LS50B with 5 nm excitation slits and 10 nm emission slits.
Proteinase K digestion of HaPrP 145stop peptides
A 40-μL aliquot of each fibrillized peptide was treated for 1 h at 37°C with 0 or 1 μg/mL PK (Roche) and then boiled for 5 min with 40 μL 2× SDS-PAGE sample buffer. Ten-microliter portions of each mixture were analyzed by SDS-PAGE and transferred to PVDF membranes for N-terminal sequencing (Research Technology Branch, NIH). For comparison of relative protease resistance between the fibrillar and non-fibrillar isoforms of 145stop-5 and 145stop-11, 75-μM concentrations of each peptide in 25 mM Bis-Tris Propane (pH 7.0), were treated for 1 h at 37°C with 0, 0.1, 0.5, 1, and 10 μg/mL PK. The non-fibrillar peptides were subjected to ultracentrifugation (256,000g) prior to treatment with PK. Each reaction was done in triplicate and run on 18% TG gels, transferred to PVDF membrane and probed with 3F4 anti-HaPrP antibody. The intensity of the bands from the Western blot was digitized and quantified using UN-SCAN-IT software (Silk Scientific) and displayed graphically with Prism software (GraphPad Software).
FTIR spectroscopy
Amyloid fibrils were pelleted by centrifugation (15 min, 21,000g) and twice suspended and pelleted from 20 μL of sterile water. Slurried pellets were applied to a single reflection diamond attenuated total reflectance (ATR) unit and the sample was covered to prevent contamination with atmospheric water vapor and allowed to dry while being purged with dry air. A System 2000 FTIR instrument (Perkin Elmer) equipped with an nb1 MCT detector cooled by liquid nitrogen was used for data collection. Spectra were acquired from 500 cumulative scans at 1400–1800 cm−1 with 1 cm−1 resolution, 500 cm/sec OPD velocity, and a 0.5-cm−1 interval. Primary peptide spectra were obtained by sequential subtraction of buffer and water vapor spectra. Second-derivative spectra were calculated from primary spectra with Spectrum v5.01 (Perkin Elmer).
Electron microscopy
Droplets (3 μL) of suspended material were placed on 3% Parlodion (nitrocellulose)-coated, 300 mesh copper grids (Pelco) prepared by the method of Garon (1981). Samples were allowed to adsorb to the films for 60 min at room temperature. Excess fluids were removed from each film surface with a micropipette and then washed with pure water. No drying was allowed to occur in the course of these manipulations. Immediately upon removal of the last water rinse, the grids were stained with a preparation of 0.3% aqueous uranyl acetate (pH 3.9) for 30 sec. Excess stain was removed from the film surface by micropipette and the sample allowed to air dry. The samples were examined in a Hitachi 7500 electron microscope at 80 kV. Images were captured with a Hammamatsu CCD camera coupled with a digital imaging program (Advanced Microscopy Techniques Corp.).
Acknowledgments
We thank Drs. Byron Caughey, Sonja Best, and Lisa Kercher for critical reading of the manuscript, and Dr. Witold Surewicz for his generous advice on 145stop peptide purification and fibrillization. We also thank Anita Mora and Gary Hettrick for help in preparing the figures.
Abbreviations
PrP, prion protein
HaPrP, hamster prion protein
TSE, transmissible spongiform encephalopathies
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051822606.
References
- Aguzzi, A. and Polymenidou, M. 2004. Mammalian prion biology: One century of evolving concepts. Cell 116: 313–327. [DOI] [PubMed] [Google Scholar]
- Bendheim, P.E., Potempska, A., Kascsak, R.J., and Bolton, D.C. 1988. Purification and partial characterization of the normal cellular homologue of the scrapie agent protein. J. Infect. Dis. 158: 1198–1208. [DOI] [PubMed] [Google Scholar]
- Bocharova, O.V., Breydo, L., Parfenov, A.S., Salnikov, V.V., and Baskakov, I.V. 2005. In vitro conversion of full-length mammalian prion protein produces amyloid form with physical properties of PrP(Sc). J. Mol. Biol. 346: 645–659. [DOI] [PubMed] [Google Scholar]
- Brown, D.R., Qin, K., Herms, J.W., Madlung, A., Manson, J., Strome, R., Fraser, P.E., Kruck, T., von Bohlen, A., Schulz-Schaeffer, W., et al. 1997. The cellular prion protein binds copper in vivo. Nature 390: 684–687. [DOI] [PubMed] [Google Scholar]
- Calero, M. and Gasset, M. 2005. Fourier transform infrared and circular dichroism spectroscopies for amyloid studies. Methods Mol. Biol. 299: 129–151. [DOI] [PubMed] [Google Scholar]
- Callahan, M.A., Xiong, L., and Caughey, B. 2001. Reversibility of scrapie-associated prion protein aggregation. J. Biol. Chem. 276: 28022–28028. [DOI] [PubMed] [Google Scholar]
- Campbell, T.A., Palmer, M.S., Will, R.G., Gibb, W.R., Luthert, P.J., and Collinge, J. 1996. A prion disease with a novel 96-base pair insertional mutation in the prion protein gene. Neurology 46: 761–766. [DOI] [PubMed] [Google Scholar]
- Capellari, S., Vital, C., Parchi, P., Petersen, R.B., Ferrer, X., Jarnier, D., Pegoraro, E., Gambetti, P., and Julien, J. 1997. Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology 49: 133–141. [DOI] [PubMed] [Google Scholar]
- Caughey, B., Horiuchi, M., Demaimay, R., and Raymond, G.J. 1999. Assays of protease-resistant prion protein and its formation. Methods Enzymol. 309: 122–133. [DOI] [PubMed] [Google Scholar]
- Chen, S.G., Parchi, P., Brown, P., Capellari, S., Zou, W., Cochran, E.J., Vnencak-Jones, C.L., Julien, J., Vital, C., Mikol, J., et al. 1997. Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat. Med. 3: 1009–1015. [DOI] [PubMed] [Google Scholar]
- Chesebro, B. 2003. Introduction to the transmissible spongiform encephalopathies or prion diseases. Br. Med. Bull. 66: 1–20. [DOI] [PubMed] [Google Scholar]
- Chesebro, B., Wehrly, K., Caughey, B., Nishio, J., Ernst, D., and Race, R. 1993. Foreign PrP expression and scrapie infection in tissue culture cell lines. Dev. Biol. Stand. 80: 131–140. [PubMed] [Google Scholar]
- Chesebro, B., Trifilo, M., Race, R., Meade-White, K., Teng, C., LaCasse, R., Raymond, L., Favara, C., Baron, G., Priola, S., et al. 2005. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science 308: 1435–1439. [DOI] [PubMed] [Google Scholar]
- Chiesa, R., Piccardo, P., Ghetti, B., and Harris, D.A. 1998. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron 21: 1339–1351. [DOI] [PubMed] [Google Scholar]
- Chiesa, R., Drisaldi, B., Quaglio, E., Migheli, A., Piccardo, P., Ghetti, B., and Harris, D.A. 2000. Accumulation of protease-resistant prion protein (PrP) and apoptosis of cerebellar granule cells in transgenic mice expressing a PrP insertional mutation. Proc. Natl. Acad. Sci. 97: 5574–5579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiesa, R., Pestronk, A., Schmidt, R.E., Tourtellotte, W.G., Ghetti, B., Piccardo, P., and Harris, D.A. 2001. Primary myopathy and accumulation of PrPSc-like molecules in peripheral tissues of transgenic mice expressing a prion protein insertional mutation. Neurobiol. Dis. 8: 279–288. [DOI] [PubMed] [Google Scholar]
- Chiesa, R., Piccardo, P., Quaglio, E., Drisaldi, B., Si-Hoe, S.L., Takao, M., Ghetti, B., and Harris, D.A. 2003. Molecular distinction between pathogenic and infectious properties of the prion protein. J. Virol. 77: 7611–7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge, J., Sidle, K.C., Meads, J., Ironside, J., and Hill, A.F. 1996. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 383: 685–690. [DOI] [PubMed] [Google Scholar]
- Collins, S.J., Lawson, V.A., and Masters, C.L. 2004. Transmissible spongiform encephalopathies. Lancet 363: 51–61. [DOI] [PubMed] [Google Scholar]
- Croes, E.A., Theuns, J., Houwing-Duistermaat, J.J., Dermaut, B., Sleegers, K., Roks, G., Van den Broeck, M., van Harten, B., van Swieten, J.C., Cruts, M., et al. 2004. Octapeptide repeat insertions in the prion protein gene and early onset dementia. J. Neurol. Neurosurg. Psychiatry 75: 1166–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DebBurman, S.K., Raymond, G.J., Caughey, B., and Lindquist, S. 1997. Chaperone-supervised conversion of prion protein to its protease-resistant form. Proc. Natl. Acad. Sci. 94: 13938–13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flechsig, E., Shmerling, D., Hegyi, I., Raeber, A.J., Fischer, M., Cozzio, A., von Mering, C., Aguzzi, A., and Weissmann, C. 2000. Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 27: 399–408. [DOI] [PubMed] [Google Scholar]
- Fontana, A., Zambonin, M., Polverino de Laureto, L.P., De Fillipis, V., Clementi, A., and Scaramella, E. 1997. Probing the conformational state of apomyoglobin by limited proteolysis. J. Mol. Biol. 266: 223–230. [DOI] [PubMed] [Google Scholar]
- Gambetti, P., Parchi, P., and Chen, S.G. 2003. Hereditary Creutzfeldt-Jakob disease and fatal familial insomnia. Clin. Lab. Med. 23: 43–64. [DOI] [PubMed] [Google Scholar]
- Garon, C.F. 1981. Electron microscopy of nucleic acids. Gene Amplif. Anal. 2: 573–589. [PubMed] [Google Scholar]
- Ghetti, B., Piccardo, P., Spillantini, M.G., Ichimiya, Y., Porro, M., Perini, F., Kitamoto, T., Tateishi, J., Seiler, C., Frangione, B., et al. 1996. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: The phenotype of the stop codon 145 mutation in PRNP. Proc. Natl. Acad. Sci. 93: 744–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb, L.G., Brown, P., McCombie, W.R., Goldgaber, D., Swergold, G.D., Wills, P.R., Cervenakova, L., Baron, H., Gibbs Jr., C.J., and Gajdusek, D.C. 1991. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc. Natl. Acad. Sci. 88: 10926–10930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, D.A., Chiesa, R., Drisaldi, B., Quaglio, E., Migheli, A., Piccardo, P., and Ghetti, B. 2003. A murine model of a familial prion disease. Clin. Lab. Med. 23: 175–186. [DOI] [PubMed] [Google Scholar]
- Horiuchi, M., Priola, S.A., Chabry, J., and Caughey, B. 2000. Interactions between heterologous forms of prion protein: Binding, inhibition of conversion, and species barriers. Proc. Natl. Acad. Sci. 97: 5836–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova, L., Barmada, S., Kummer, T., and Harris, D.A. 2001. Mutant prion proteins are partially retained in the endoplasmic reticulum. J. Biol. Chem. 276: 42409–42421. [DOI] [PubMed] [Google Scholar]
- Jiang, Y., Li, H., Zhu, L., Zhou, J.M., and Perrett, S. 2004. Amyloid nucleation and hierarchical assembly of Ure2p fibrils. Role of asparagine/glutamine repeat and nonrepeat regions of the prion domains. J. Biol. Chem. 279: 3361–3369. [DOI] [PubMed] [Google Scholar]
- Jones, E.M. and Surewicz, W.K. 2005. Fibril conformation as the basis of species- and strain-dependent seeding specificity of mammalian prion amyloids. Cell 121: 63–72. [DOI] [PubMed] [Google Scholar]
- Kascsak, R.J., Rubenstein, R., Merz, P.A., Tonna-DeMasi, M., Fersko, R., Carp, R.I., Wisniewski, H.M., and Diringer, H. 1987. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J. Virol. 61: 3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheterpal, I., Williams, A., Murphy, C., Bledsoe, B., and Wetzel, R. 2001. Structural features of the Aβ amyloid fibril elucidated by limited proteolysis. Biochemistry 40: 11757–11767. [DOI] [PubMed] [Google Scholar]
- Kitamoto, T., Iizuka, R., and Tateishi, J. 1993. An amber mutation of prion protein in Gerstmann-Straussler syndrome with mutant PrP plaques. Biochem. Biophys. Res. Commun. 192: 525–531. [DOI] [PubMed] [Google Scholar]
- Kocisko, D.A., Come, J.H., Priola, S.A., Chesebro, B., Raymond, G.J., Lansbury, P.T., and Caughey, B. 1994. Cell-free formation of protease-resistant prion protein. Nature 370: 471–474. [DOI] [PubMed] [Google Scholar]
- Krasemann, S., Zerr, I., Weber, T., Poser, S., Kretzschmar, H., Hunsmann, G., and Bodemer, W. 1995. Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Brain Res. Mol. Brain Res. 34: 173–176. [DOI] [PubMed] [Google Scholar]
- Kundu, B., Maiti, N.R., Jones, E.M., Surewicz, K.A., Vanik, D.L., and Surewicz, W.K. 2003. Nucleation-dependent conformational conversion of the Y145Stop variant of human prion protein: Structural clues for prion propagation. Proc. Natl. Acad. Sci. 100: 12069–12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson, V.A., Priola, S.A., Wehrly, K., and Chesebro, B. 2001. N-terminal truncation of prion protein affects both formation and conformation of abnormal protease-resistant prion protein generated in vitro. J. Biol. Chem. 276: 35265–35271. [DOI] [PubMed] [Google Scholar]
- Lawson, V.A., Priola, S.A., Meade-White, K., Lawson, M., and Chesebro, B. 2004. Flexible N-terminal region of prion protein influences conformation of protease-resistant prion protein isoforms associated with cross-species scrapie infection in vivo and in vitro. J. Biol. Chem. 279: 13689–13695. [DOI] [PubMed] [Google Scholar]
- Lehmann, S. and Harris, D.A. 1996a. Mutant and infectious prion proteins display common biochemical properties in cultured cells. J. Biol. Chem. 271: 1633–1637. [DOI] [PubMed] [Google Scholar]
- ———. 1996b. Two mutant prion proteins expressed in cultured cells acquire biochemical properties reminiscent of the scrapie isoform. Proc. Natl. Acad. Sci. 93: 5610–5614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeVine III, H. 1999. Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 309: 274–284. [DOI] [PubMed] [Google Scholar]
- Lewis, V., Collins, S., Hill, A.F., Boyd, A., McLean, C.A., Smith, M., and Masters, C.L. 2003. Novel prion protein insert mutation associated with prolonged neurodegenerative illness. Neurology 60: 1620–1624. [DOI] [PubMed] [Google Scholar]
- Liu, J.J. and Lindquist, S. 1999. Oligopeptide-repeat expansions modulate ‘protein-only’ inheritance in yeast. Nature 400: 573–576. [DOI] [PubMed] [Google Scholar]
- Narwa, R. and Harris, D.A. 1999. Prion proteins carrying pathogenic mutations are resistant to phospholipase cleavage of their glycolipid anchors. Biochemistry 38: 8770–8777. [DOI] [PubMed] [Google Scholar]
- Nilsson, M.R. 2004. Techniques to study amyloid fibril formation in vitro. Methods 34: 151–160. [DOI] [PubMed] [Google Scholar]
- Owen, F., Poulter, M., Collinge, J., Leach, M., Lofthouse, R., Crow, T.J., and Harding, A.E. 1992. A dementing illness associated with a novel insertion in the prion protein gene. Brain Res. Mol. Brain Res. 13: 155–157. [DOI] [PubMed] [Google Scholar]
- Penney Jr., J.B., Vonsattel, J.P., MacDonald, M.E., Gusella, J.F., and Myers, R.H. 1997. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann. Neurol. 41: 689–692. [DOI] [PubMed] [Google Scholar]
- Piccardo, P., Liepnieks, J.J., William, A., Dlouhy, S.R., Farlow, M.R., Young, K., Nochlin, D., Bird, T.D., Nixon, R.R., Ball, M.J., et al. 2001. Prion proteins with different conformations accumulate in Gerstmann-Straussler-Scheinker disease caused by A117V and F198S mutations. Am. J. Pathol. 158: 2201–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrini, V., Puoti, G., Limido, L., Rossi, G., Di Fede, G., Giaccone, G., Mangieri, M., Tedeschi, F., Bondavalli, A., Mancia, D., et al. 2003. Creutzfeldt-Jakob disease with a novel extra-repeat insertional mutation in the PRNP gene. Neurology 61: 1288–1291. [DOI] [PubMed] [Google Scholar]
- Polverino de Laureto, L.P., Scaramella, E., Frigo, M., Wondrich, F.G., De Fillipis, V., Zambonin, M., and Fontana, A. 1999. Limited proteolysis of bovine α-lactalbumin: Isolation and characterization of protein domains. Protein Sci. 8: 2290–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola, S.A. and Chesebro, B. 1998. Abnormal properties of prion protein with insertional mutations in different cell types. J. Biol. Chem. 273: 11980–11985. [DOI] [PubMed] [Google Scholar]
- Priola, S.A. and Lawson, V.A. 2001. Glycosylation influences cross-species formation of protease-resistant prion protein. EMBO J. 20: 6692–6699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner, S.B. 1998. Prions. Proc. Natl. Acad. Sci. 95: 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipe, J.D. and Cohen, A.S. 2000. Review: History of the amyloid fibril. J. Struct. Biol. 130: 88–98. [DOI] [PubMed] [Google Scholar]
- Spolaore, B., Bermejo, R., Zambonin, M., and Fontana, A. 2001. Protein interactions leading to conformational changes monitored by limited proteolysis: Apo form and fragments of horse cytochrome c. Biochemistry 40: 9460–9468. [DOI] [PubMed] [Google Scholar]
- Vanik, D.L., Surewicz, K.A., and Surewicz, W.K. 2004. Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol. Cell 14: 139–145. [DOI] [PubMed] [Google Scholar]
- Vital, C., Gray, F., Vital, A., Parchi, P., Capellari, S., Petersen, R.B., Ferrer, X., Jarnier, D., Julien, J., and Gambetti, P. 1998. Prion encephalopathy with insertion of octapeptide repeats: The number of repeats determines the type of cerebellar deposits. Neuropathol. Appl. Neurobiol. 24: 125–130. [DOI] [PubMed] [Google Scholar]
- Vital, C., Gray, F., Vital, A., Ferrer, X., and Julien, J. 1999. Prion disease with octapeptide repeat insertion. Clin. Exp. Pathol. 47: 153–159. [PubMed] [Google Scholar]
- Wadsworth, J.D., Hill, A.F., Beck, J.A., and Collinge, J. 2003. Molecular and clinical classification of human prion disease. Br. Med. Bull. 66: 241–254. [DOI] [PubMed] [Google Scholar]
- Will, R.G., Ironside, J.W., Zeidler, M., Cousens, S.N., Estibeiro, K., Alperovitch, A., Poser, S., Pocchiari, M., Hofman, A., and Smith, P.G. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347: 921–925. [DOI] [PubMed] [Google Scholar]
- Yanagihara, C., Yasuda, M., Maeda, K., Miyoshi, K., and Nishimura, Y. 2002. Rapidly progressive dementia syndrome associated with a novel four extra repeat mutation in the prion protein gene. J. Neurol. Neurosurg. Psychiatry 72: 788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn, R., von Schroetter, C., and Wuthrich, K. 1997. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 417: 400–404. [DOI] [PubMed] [Google Scholar]