

Abstract
Amide hydrogen-deuterium exchange has proven to be a powerful tool for detecting and characterizing high-energy conformations in protein ensembles. Since interactions with ligands can modulate these high-energy conformations, hydrogen exchange appears to be an ideal experimental probe of the physical mechanisms underlying processes like allosteric regulation. The chemical mechanism of hydrogen exchange, however, can complicate such studies. Here, we examine hydrogen exchange rates in a simple model system, the c-Src SH3 domain interacting with a short peptide ligand. Addition of ligand slows the rates of hydrogen exchange at nearly every amide for which we can obtain data. Careful analysis, however, reveals that this slowing is due primarily to a reduction in the population of free protein in the system, and not to any specific property of the complex. We present a method to separate the contributions of free and bound protein to the exchange kinetics that has allowed us to identify the subset of amides where exchange arises directly from the complex. These results demonstrate that the slowing of hydrogen exchange induced by ligand interactions should be interpreted with caution, and more extensive experiments are required to correlate changes in hydrogen exchange with changes in structure or internal dynamics.
Keywords: SH3, hydrogen exchange, ligand binding, protein dynamics
Proteins are highly dynamic molecules. A variety of experimental techniques have revealed protein motions ranging from sidechain rotations to large domain rearrangements, on timescales from nanoseconds to years. Some of these motions can be thought of as transitions between the most populated, native conformation and less populated high-energy states. Under conditions that favor the native conformations, myriad high-energy forms are also sampled, ranging from small local fluctuations to complete unfolding. At equilibrium, the relative population of each conformation is determined by its energy according to a Boltzmann distribution.
Although the existence of high-energy forms in the native state ensemble is well established, the functional significance of these conformations in many proteins remains unclear. In allosteric regulation, they may be extremely important. Models of allostery postulate an equilibrium between inactive, tense states and high-energy, active, relaxed states (Monod et al. 1965; Koshland et al. 1966). Positive effectors preferentially bind the active ensemble, shifting the equilibrium and activating the enzyme. This model is well recognized for large, multimeric proteins like hemoglobin and allosteric enzymes. In addition, phosphorylation of the bacterial signaling molecule NtrC has been shown to alter a preexisting equilibrium between low- and high-energy states, favoring active conformations (Volkman et al. 2001). Computer modeling has implicated a similar mechanism for allosteric control in the monomeric enzyme dihydrofolate reductase (Pan et al. 2000), and has suggested that this mechanism may be broadly applicable to many important aspects of protein function, including ligand binding, enzymatic catalysis, and signal transduction (Luque and Freire 2000). Investigations of high-energy states, and the modulation of these states, are likely to be very important in identifying structure–function relationships in many proteins.
Because they are by nature sparsely populated, high-energy conformations are extremely difficult to study. In the case of NtrC, the high-energy state was sufficiently populated without phosphorylation to allow detection by NMR and chemical exchange (Volkman et al. 2001). However, many functionally important states may have populations so low that they are not detectable by most techniques. Amide hydrogen-deuterium exchange provides an ideal probe of these conformations, because this method is sensitive to rare fluctuations in protein structure. Many studies have used hydrogen exchange to characterize rare conformations associated with protein folding and unfolding (Clarke and Itzhaki 1998; Raschke and Marqusee 1998; Englander 2000), as well as those associated with allosteric transitions in molecules like hemoglobin (Englander and Englander 1994). More recently, hydrogen exchange has been used to study changes in complex molecules like signaling kinases in response to nucleotide binding and phosphorylation (Andersen et al. 2001; Hoofnagle et al. 2001; Hamuro et al. 2002). Hydrogen exchange has also been used to study protein–protein interactions, identifying binding interfaces (Werner and Wemmer 1992; Mandell et al. 1998) and possible structural changes (Mayne et al. 1992; Engen et al. 1999) induced by ligand binding.
Although the chemistry of hydrogen exchange is quite straightforward in a monomeric protein, the dynamic equilibrium that exists between bound and free species in a protein–ligand mixture complicates the analysis of exchange kinetics for multicomponent systems. Despite the potential complications in analysis, alterations in fast-exchanging amides upon ligand binding have been used effectively to map binding sites in a variety of systems, including a complex between cytochrome c and an antibody (Mayne et al. 1992) and two protease–inhibitor complexes (Werner and Wemmer 1992; Mandell et al. 1998). In many cases, however, binding also results in a decrease in exchange rates in regions distant from the binding site. While it is common to attribute these changes to changes in internal dynamics of the protein upon ligand binding, this connection is not necessarily always correct. Hydrogen exchange in ribonuclease S (RNase S) is a good example. Ribonuclease A (RNase A) can be cleaved by subtilisin into two fragments that, when mixed, form an active, heterodimeric enzyme called RNase S. Hydrogen exchange in RNase S is much faster than in RNase A, an observation that could be attributed to increased flexibility or dynamics in the complex. However, it has been shown that the faster exchange is due to small populations of free fragments (the S-protein and S-peptide) in solution with RNase S, and that the internal dynamics of RNase S do not differ significantly from RNase A (Chakshusmathi et al. 1999). In general, the equilibrium between free and bound protein can be a major influence on hydrogen exchange rates.
A careful, quantitative treatment of hydrogen exchange kinetics in a protein–ligand mixture would help to clarify these issues. Here, we present such a treatment in a simple model system, the src homology 3 (SH3) domain of chicken c-Src in complex with a peptide ligand. This system has many advantages. It is highly amenable to NMR analysis, allowing residue-level resolution of hydrogen exchange. In addition, the folding kinetics and thermodynamics of the free protein and the thermodynamics of binding have been thoroughly characterized previously (Grantcharova and Baker 1997), making the interpretation of hydrogen exchange results relatively simple. We show that addition of peptide slows exchange at nearly every amide in SH3, and that this slowing can be attributed primarily to a reduction in the population of free protein in solution, not to any ligand-induced changes in internal dynamics. We also detect a ligand concentration-independent process that affects exchange at some amides in SH3. This process can be attributed to fluctuations or local unfolding in the SH3–ligand complex, without dissociation of ligand.
Results
Hydrogen exchange in apo SH3
To understand how peptide binding affects the mechanism of hydrogen exchange in the SH3 domain, it was first necessary to characterize the mechanism of exchange in the apo protein. Hydrogen exchange rates were determined in apo SH3 and analyzed as described in Materials and Methods. Exchange rate constants (kobs) were measured at several values of pH and guanidinium chloride (GdmCl) concentration to confirm the EX2 condition and determine the mechanism of exchange (Fig. 1 ▶). The log of the observed rate constant increased with pH, as expected for an EX2 exchange mechanism. Three lines of evidence suggest that virtually every amide in SH3 exchanges through global unfolding of the protein. First, the slope of log (kobs) versus pH is slightly less than 1.0 for most amides (Fig. 1A ▶), reflecting the slight increase in global stability over this pH range (data not shown). The reduced slope cannot be interpreted as a result of the exchange kinetics being intermediate between the EX1 and EX2 limits, because it holds true for nearly every amide, despite intrinsic exchange rates (krc) that span two orders of magnitude. Second, exchange at most sites displays a marked and uniform dependence on GdmCl concentration, indicating that exchange is accompanied by the exposure of a substantial amount of protein to solvent, consistent with global unfolding (Fig. 1B ▶). Finally, most amides have a ΔG°HX (see Materials and Methods) that is in very good agreement with a ΔG°unfolding of 4.8 kcal/mol measured by GdmCl unfolding in D2O monitored by fluorescence (Fig. 3 ▶, below).
Figure 1.
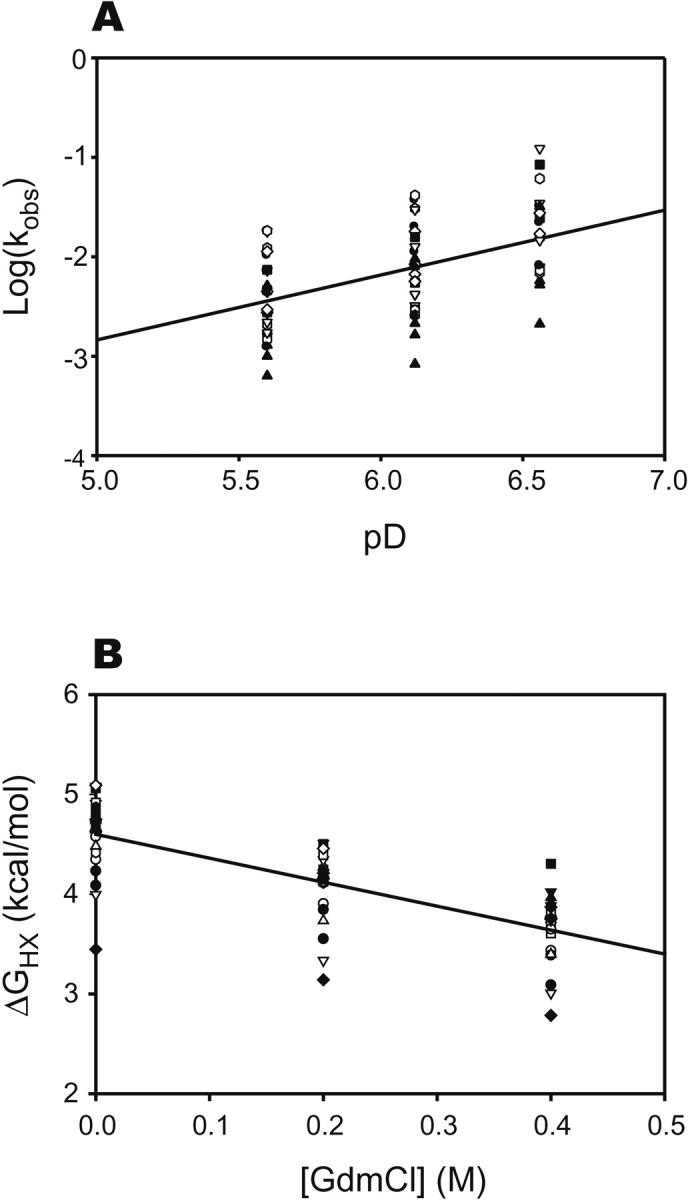
Summary of hydrogen exchange results for apo c-Src SH3. Symbols correspond to individual amides with measurable exchange rates. (A) Dependence of observed exchange rates on pD for each amide in SH3. A linear fit to all data gives a slope of 0.7. The reduced slope is due to a slight (0.2 kcal/mol) increase in the stability of SH3 over this pD range. The deviation from slope of 1 is uniform for all amides, regardless of krc, arguing against an exchange mechanism intermediate between EX1 and EX2. (B) Dependence of ΔG°opapo on GdmCl concentration. Exchange rates at most amides have a significant and a uniform denaturant dependence, suggesting that exchange proceeds through complete unfolding of the protein.
Figure 3.

Comparison of hydrogen exchange free energy measured directly from apo SH3 (ΔG°opapo, black bars) and calculated from the peptide concentration dependence of exchange rates in the SH3–peptide complex (ΔG°opF, gray bars). The horizontal line indicates the ΔG°unfolding for apo SH3 measured by GdmCl denaturation followed by fluorescence.
Previous results (Grantcharova and Baker 1997) have shown that the protection factors in SH3 are remarkably uniform, also suggesting that most exchange occurs through a single opening mechanism. These studies also indicated that a number of amides in SH3 have ΔG°HX significantly greater than ΔG°unfolding, a phenomenon termed superprotection and interpreted as evidence for residual protection in the thermodynamically unfolded state. In our studies, however, we saw no evidence for such superprotection.
Hydrogen exchange is slowed by peptide in a concentration-dependent manner
Addition of peptide slows all measurable hydrogen exchange rates in SH3 (Fig. 2A ▶). This slowing is a result of a more complex mechanism of exchange in the presence of a reversibly bound ligand (Mayne et al. 1992; Chakshusmathi et al. 1999). Instead of a simple equilibrium between closed and open forms, several pathways govern exchange rates as shown in Scheme 1 ▶. KopB and KopF are the equilibrium constants for opening of the bound and the free states, respectively, Kd and Kd′ are the dissociation constants for the open and closed states, and krc is the intrinsic exchange rate from a random coil.
Figure 2.
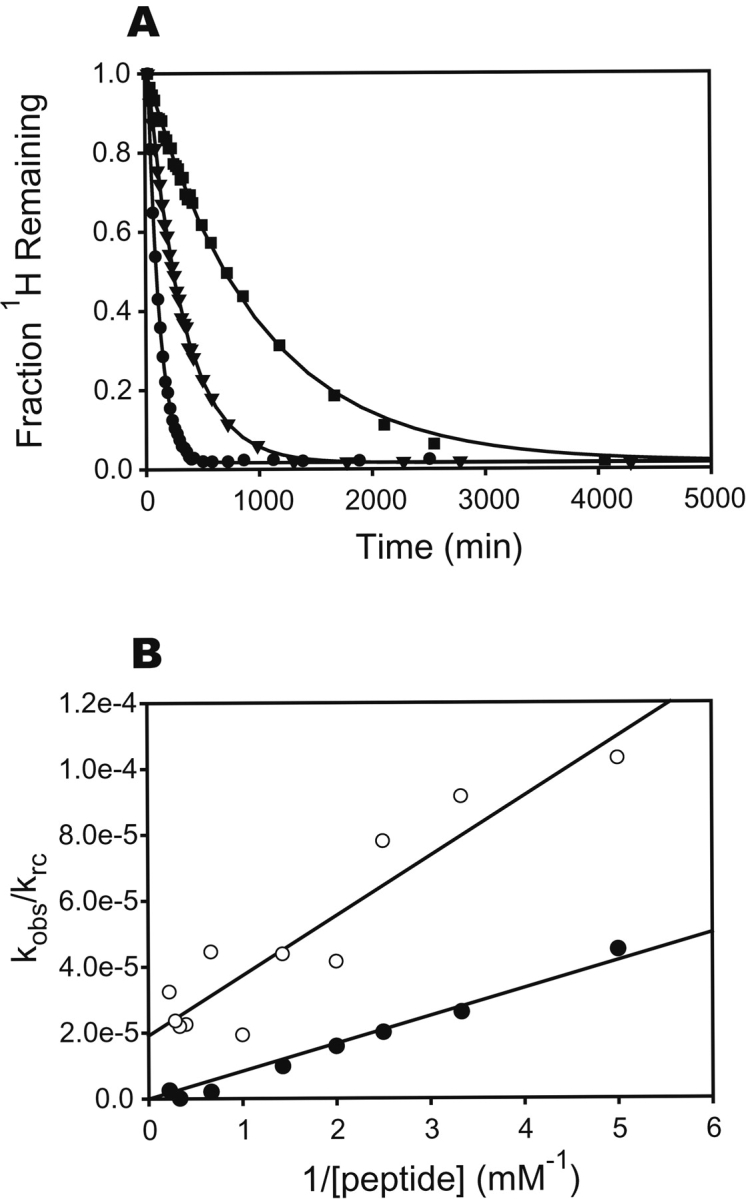
The effect of peptide binding on hydrogen exchange rates in the c-Src SH3 domain at differing concentrations of peptide ligand. (A) Exchange curves for Ser 44 with 0.3 mM (•), 0.7 mM (▴), and 3 mM (▪) free peptide. Solid lines are fits to a single-exponential decay. (B) Concentration dependence of the hydrogen exchange rates. Data from Val 11 (○) and Tyr 14 (•) are shown, with fits to equation 2. The t statistic was used to determine if the y-intercept differed significantly from zero, and KopB is reported only for those fits where P < 0.01.
Scheme 1.

For a given amide, opening may occur from either the bound or the free state. If the equilibration between the bound and free states, as well as the closing reaction, are fast compared to krc (analogous to the EX2 condition), the general expression for the observed exchange rate constant as a function of the concentration of free ligand is given in equation 1.
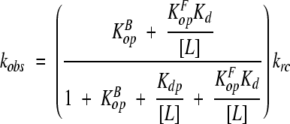 |
(1) |
If KopB ≪ 1, KopF ≪1, and [L] ≫ Kd, this equation can be simplified to a linear relationship between the ratio of the observed and intrinsic rates and the reciprocal of the ligand concentration, given in equation 2.
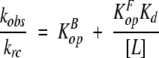 |
(2) |
To investigate the relative contributions of free and bound protein to hydrogen exchange in SH3 (KopB and KopF), we measured exchange rates over a range of free peptide concentrations from 0.2 mM to 4.5 mM and fit the data to equation 2. Representative fits are shown in Figure 2B ▶. Two classes of amides emerge from the exchange data in the presence of peptide. Most amides show a linear dependence of kobs/krc on 1/[peptide] that intercepts the Y-axis very close to zero, indicating that all detectable exchange occurs from the free protein after peptide dissociation. The exchange protection for these amides can be attributed entirely to a single opening equilibrium, KopF. For other amides, the line has a nonzero y-intercept, showing that exchange occurs by two competing pathways—one dependent on ligand dissociation, and the other not. For these, protection is due to a sum of the two equilibria, KopF and KopB.
The bottom pathway in Scheme 1 ▶ is the same as that described in equation 3 in Materials and Methods. Thus, the KopF term calculated from the concentration dependence of exchange rates using equation 2 should be identical to Kopapo, the equilibrium constant for opening when peptide is not present. We calculated KopF using a value of 19 μM for the Kd, based on previously reported titration calorimetry under similar conditions (Wang et al. 2001). Figure 3 ▶ shows a comparison of the free energy associated with these two equilibria, calculated using the relationship ΔG = −RTlnK. The generally close agreement between the opening free energies measured directly on apo SH3 and from the dependence of exchange rates on peptide concentration indicates that Scheme 1 ▶ closely models the actual exchange mechanism in the SH3–peptide complex. Only the value of ΔG°opF for Ser 22 is anomalous, because exchange at this position is dominated by ΔG°opB at all peptide concentrations, placing a high degree of uncertainty on the estimate of ΔG°opF. In the subsequent discussion, we will use the hydrogen exchange free energies, ΔG°opF and ΔG°opB, to denote exchange following dissociation and exchange from the bound complex. These values, measured at each slow-exchanging amide in SH3, are listed in Table 1.
Table 1.
Thermodynamic parameters obtained from hydrogen exchange data
| Residuea | ΔG°opapo (kcal/mol) | ΔG°opF (kcal/mol) | ΔG°opB (kcal/mol) | 2° Structure |
| Phe 10b | 4.1 ± 0.014d | — | β1 | |
| Val 11 | 4.3 ± 0.008 | 4.1 ± 0.08 | 6.4 ± 0.18 | β1 |
| Ala 12 | 5.1 ± 0.006 | 4.8 ± 0.05 | β1 | |
| Leu 13b | 4.0 ± 0.013 | — | β1 | |
| Tyr 14 | 4.8 ± 0.008 | 4.5 ± 0.03 | β1 | |
| Asp 15c | — | 3.3 ± 0.26 | 4.7 ± 0.09 | β1 |
| Tyr 16 | 4.7 ± 0.010 | 4.8 ± 0.15 | β1 | |
| Asp 23c | — | 2.1 ± 0.20 | 4.4 ± 0.18 | β2 |
| Leu 24 | 3.4 ± 0.013 | 3.1 ± 0.04 | β2 | |
| Ser 25 | 4.6 ± 0.008 | 5.7 ± 0.75 | 5.5 ± 0.07 | β2 |
| Phe 26 | 4.7 ± 0.011 | 4.5 ± 0.05 | β2 | |
| Lys 27 | 4.7 ± 0.011 | 4.6 ± 0.09 | 6.9 ± 0.17 | β2 |
| Lys 28 | 4.7 ± 0.012 | 4.4 ± 0.05 | β2 | |
| Gly 29 | 4.4 ± 0.016 | 4.6 ± 0.51 | 5.6 ± 0.08 | β2 |
| Glu 30 | 4.2 ± 0.007 | 4.0 ± 0.05 | β3 | |
| Leu 32 | 4.6 ± 0.009 | 4.4 ± 0.05 | β3 | |
| Gln 33 | 4.7 ± 0.010 | 4.5 ± 0.06 | 7.6 ± 0.65 | β3 |
| Trp 43 | 4.9 ± 0.019 | 4.2 ± 0.05 | β4 | |
| Ala 45 | 5.1 ± 0.010 | 4.8 ± 0.05 | β4 | |
| His 46 | 4.9 ± 0.047 | 4.8 ± 0.06 | β4 | |
| Ser 47 | 4.6 ± 0.104 | 5.2 ± 0.05 | 8.7 ± 0.23 | β4 |
| Gln 52c | — | 4.3 ± 0.48 | 4.4 ± 0.04 | β5 |
| Gly 54 | 4.9 ± 0.013 | 4.7 ± 0.05 | β5 | |
| Tyr 55 | 4.7 ± 0.012 | 4.5 ± 0.05 | β5 | |
| Ile 56 | 5.1 ± 0.021 | 4.8 ± 0.30 | 8.1 ± 1.16 | β5 |
| Ser 58 | 4.6 ± 0.054 | 5.0 ± 0.12 | 310 | |
| Asn 59c | — | 3.8 ± 0.85 | 6.7 ± 0.43 | 310 |
| Tyr 60 | 4.3 ± 0.014 | 3.8 ± 0.06 | 310 | |
| Val 61 | 4.5 ± 0.009 | 4.1 ± 0.03 | β6 | |
| Ala 62 | 4.9 ± 0.008 | 4.5 ± 0.06 | 7.5 ± 0.51 | β6 |
a Residues are numbered as in Feng et al. 1994.
b Exchange not measured in ligand complex due to spectral overlap.
c Exchange in apo SH3 is too rapid to measure accurately.
d Errors given are calculated from the standard error of the fit, and are intended only to indicate the quality of the data. Uncertainty in krc introduces an error of approximately 0.4 kcal/mol in all values of ΔG°.
Discussion
Reduced rates of hydrogen exchange in the presence of ligands at sites distant from the binding interface have been interpreted as evidence of changes in protein dynamics (Engen et al. 1999), backbone hydrogen bonding (Wang et al. 2001), and core packing (Williams et al. 2003) induced by ligand binding. However, as we demonstrate here, the dynamic equilibrium that exists between free and bound states in a protein–ligand mixture can lead to reduced exchange rates that reveal little about these features of protein structure.
In the SH3 domain, the majority of slow-exchanging amides require complete unfolding of the protein in order to exchange. The peptide-binding surface in SH3 is made up of a set of residues distant in primary structure but localized in the folded protein. It is reasonable, therefore, to expect that SH3 in its unfolded state has little affinity for peptide. Thus, unfolding and ligand dissociation are linked; unfolding cannot occur in the ligand-bound state. Because of the selective binding to the folded state, addition of ligand decreases the unfolded state concentration in a ligand concentration-dependent manner. This effect will result in a ligand-dependent slowing of hydrogen exchange, even though the addition of ligand has not perturbed the mechanism of exchange from that of the apo protein. Processes like this are reflected in hydrogen exchange rates that are ligand concentration-dependent. This type of protection, denoted here by ΔG°opF, reports only on the overall affinity of protein for ligand (Kd), and provides no new information about the dynamics or physics of the ligand–protein interaction.
Perhaps counterintuitively, it is the ligand concentration-independent term (ΔG°opB) of the equation for the observed exchange rate (equation 2) that reports on fluctuations in the protein–ligand complex. This term is more informative than ΔG°opF, because it potentially contains residue-specific information about dynamics in the SH3–ligand complex. The free energy of these fluctuations reports on high-energy states that may be unique to the SH3–peptide complex, and therefore induced by binding.
Structural mapping of exchange mechanisms
Since ΔG°opB reports on exchange from the ligand-bound conformation, the distribution of amides with measurable ΔG°opB may provide information about regions in the SH3 structure that are modulated by ligand binding. Figure 4 ▶ shows the amide probes in our studies mapped onto the NMR solution structure of the SH3–peptide complex (Feng et al. 1994) and colored according to exchange mechanism. There is no apparent structural clustering of amides that exchange from the bound conformation or amides that exchange only after ligand dissociation. In fact, both types of amides appear evenly distributed throughout the structure. Further investigation reveals no apparent correlation between amides that exchange from the bound conformation with other known changes in SH3 that occur with ligand binding, including alterations in backbone motions probed by 15N relaxation (Wang et al. 2001) and ligand-induced hydrogen bond strain revealed by h3JNC′ scalar couplings (Cordier et al. 2000). Furthermore, there is no general sequence conservation of residues that show only ΔG°opF or those that show both ΔG°opF and ΔG°opB (Larson and Davidson 2000). These results suggest that changes in hydrogen exchange in this system do not correlate with previously characterized changes in the protein induced by ligand binding.
Figure 4.
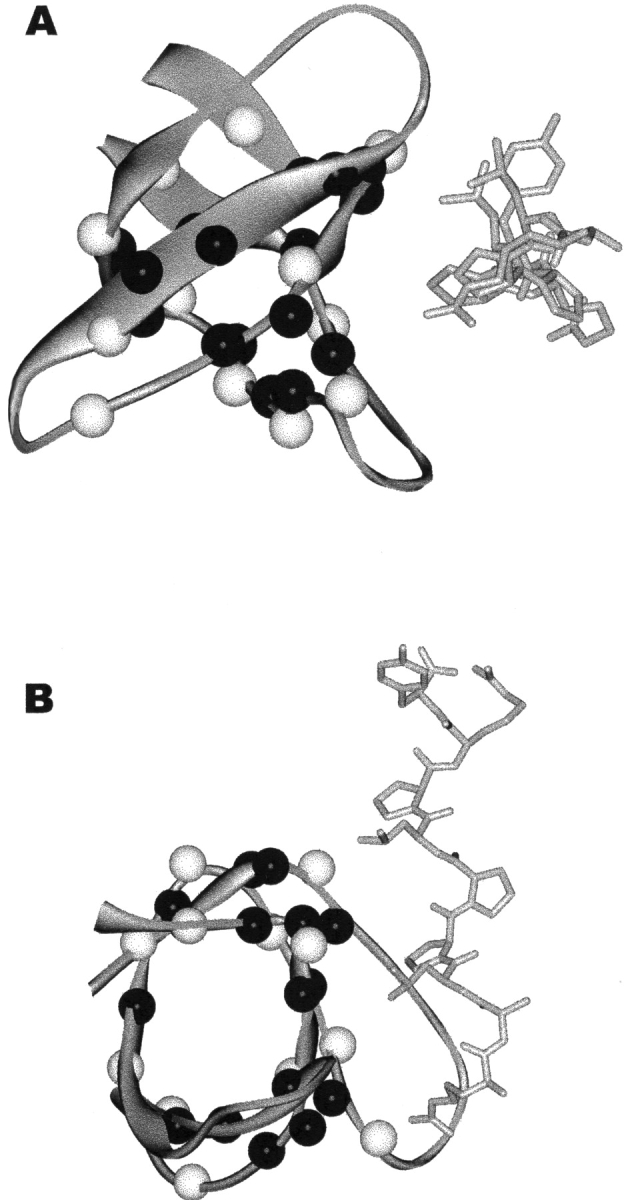
Locations of hydrogen exchange probe amides in the NMR solution structure of the src SH3 domain in complex with peptide. White spheres indicate probes where some exchange occurs without ligand dissociation (ΔG°opB is measurable), while black spheres indicate probes where all measurable exchange occurs from free protein. (A) View down the helical axis of the peptide. (B) Molecule in A rotated forward 90°.
What structural features can lead to a change in the exchange process in the ligand-bound state? Because unfolding and ligand dissociation are coupled, exchange from the bound conformation must occur through a local fluctuation or partial unfolding mechanism. Local fluctuations are more likely to lead to exchange for amides that are close to solvent; deeply buried amides are thought to require large-scale rearrangements to become exchange-competent (Milne et al. 1998). Are the amides with measurable ΔG°opB less deeply buried than those without? To measure burial, we made use of the Born radius, a value that quantitatively represents the distance of an atom from solvent in a folded protein, with smaller Born radii corresponding to less deeply buried atoms (Pokala and Handel 2004). In general, amides with measurable ΔG°opB have smaller Born radii (Fig. 5 ▶), consistent with this value representing local fluctuations in the protein–ligand complex. Given this, it must be noted that the random coil exchange rate constants, krc, may not be valid for a small fluctuation, where the open state does not closely resemble a random coil (Maity et al. 2003). This may introduce significant uncertainty in the value of ΔG°opB. As with the local fluctuations observed in native state hydrogen exchange experiments, ΔG°opB gives a qualitative picture of local motions in the native state, but the exact values of these free energies should be interpreted with caution.
Figure 5.

Histograms of the Born radii of probe amides. Hatched bars indicate amides for which only ΔG°opF was observed (mean radius = 4.44). Filled bars are amides with measured ΔG°opB (mean radius = 3.53). The difference in Born radius between the two groups is highly significant (P < 0.001). Lines indicate fit to a Gaussian curve.
What is the functional significance of the newly observed local fluctuations in the bound state? It is important to note that these newly uncovered high-energy fluctuations may not actually be specific to the bound state. Hydrogen exchange kinetics are dominated by the most accessible open state of a protein. For example, in many proteins in the absence of denaturants, some amides exchange with solvent by a local fluctuation. The free energies, and thus the populations, of local fluctuations are relatively insensitive to denaturants because they expose little protein surface to solvent. The free energy of unfolding, in contrast, is very sensitive to denaturants. As denaturants are added, the free energy of unfolding decreases, while the free energy of fluctuations remains constant. At some concentration of denaturant unfolding requires less energy than fluctuation, and exchange proceeds through unfolding. This does not mean that the high-energy fluctuation no longer exists; only that unfolding is sufficiently facile that all exchange proceeds through that pathway, and fluctuations are masked.
In the apo state of SH3, any fluctuations higher in energy than global unfolding will be masked. Indeed, by looking in the EX1 regime, such fluctuations were found recently for several amides in domain 1 of rat CD2 (Cliff et al. 2004). The exchange we observe from the ligand-bound form of SH3 could be an example of the same phenomenon. Apo SH3 is relatively unstable toward global unfolding, and the most accessible open state for most slow-exchanging amides is the completely unfolded protein. Addition of ligand effectively decreases the population of unfolded SH3 in a concentration-dependent way; as more ligand is added, unfolded protein becomes more rare. If binding does not similarly affect the population of a high energy locally open state, then when KopB exceeds KopF Kd/[L], the high-energy local fluctuation becomes more accessible to exchange than the unfolded state. Thus, ΔG°opB may reflect exchange from high-energy states that are present in both free and bound protein, but are masked in the free protein due to its low global stability. Because the observed ΔG°opB are greater than the stability of the apo-protein, it is impossible to tell at this point whether the exchange observed in the ligand-bound conformation arises from ligand-specific fluctuations.
Initially, it seems surprising that this detailed hydrogen exchange study revealed few, if any, changes in local dynamics induced by binding. It is worth noting, however, that there is little evidence for conformational changes or allosteric behavior in the c-Src SH3 domain upon ligand binding (Feng et al. 1994). The results from this study of such a simple system are an excellent example of why caution must be exercised when interpreting changes in hydrogen exchange rates induced by ligand binding. Binding perturbs the equilibria that determine amide hydrogen exchange rates in proteins, and this perturbation can lead to a reduction in hydrogen exchange rates that reports on nothing more than the fact that the protein binds ligand. When investigating a reduction in hydrogen exchange rates induced by ligand, it is necessary to separate the contribution of the minute population of free protein that exists at any ligand concentration from the contribution of bound protein. This can be done quite easily by examining the dependence of hydrogen exchange rates on ligand concentration using the method we present here. Using this technique, it should be possible to investigate the energetics of structural fluctuations in protein–ligand complexes with atomic resolution.
Materials and methods
Protein expression and purification
The SH3 domain of chicken c-Src was overexpressed with an N-terminal 6-his tag and TEV protease cleavage site using a T7 promoter system in Escherichia coli BL21(DE3) pLysS cells. Isotopic labeling with 15N was carried out by transferring cells at OD600 of 0.6 from LB media to labeled M9 minimal media as described (Marley et al. 2001). Cells were lysed by sonication in 50 mM sodium phosphate (pH 8.0), 300 mM NaCl, and the lysate was clarified by centrifugation. Protein was purified on a Ni-NTA agarose column (Qiagen Inc.) and the tag was cleaved by overnight digestion at room temperature with TEV protease containing the stabilizing mutation S219V (expressed and purified as described) (Kapust et al. 2001). Residual tagged protein and protease were removed with a second pass over Ni-NTA. Purified protein, comprising residues 79–146 of full-length c-Src with Gly-Ala on the N terminus left by tag cleavage, was dialyzed into 50 mM potassium phosphate (pH 6.0), concentrated to 1.5 mM in a Centriprep centrifugal concentrator (Millipore Corp.), and stored frozen at −80°C. Purity was determined to be greater than 95% by SDS-PAGE and mass spectrometry. Protein concentration was determined by UV absorbance using a calculated extinction coefficient of 16,500 M−1cm−1 at 280 nm (Gill and von Hippel 1989).
Peptide synthesis and purification
The SH3 ligand RALPPLPRY was synthesized using standard techniques (kindly provided by David King) and purified by reverse phase HPLC with a preparative-scale C18 column (Alltech). Peptide concentration was determined using a previously reported extinction coefficient of 911 M−1cm−1 at 280 nm (Wang et al. 2001).
Hydrogen exchange data collection
Exchange samples contained 1 mM SH3 protein, 0–5 mM peptide, 50 mM potassium phosphate, and 0.01% NaN3 in a total volume of 0.35 ml. The pH of each sample was adjusted to 6.0 with 50 mM H3PO4 or K3PO4. Samples were lyophilized and stored in a dessicator at 4°C. Amide hydrogen exchange was initiated by dissolving a sample in 0.35 ml D2O (Isotech). The sample was quickly transferred to an NMR tube and placed in the spectrometer. Total time between initiation of exchange and beginning of data collection averaged 25 min. Exchange at 25°C was monitored by 15N-1H heteronuclear single quantum coherence (HSQC) spectra using Bruker DMX 500 or DMX 600 NMR spectrometers. One hundred twenty-eight complex points were collected in the indirect dimension with four transients per point, for a total acquisition time of 21 min per spectrum. Spectra were collected at increasing time intervals for periods ranging from 24 h for the free protein to 3 mo for higher peptide concentrations. Sample pD was measured upon completion of exchange, and corrected for glass electrode isotope effects (Glasoe and Long 1960). Final, corrected pD ranged from 6.3 to 6.4 for all samples.
Spectra were processed using FELIX 97.0 (Accelrys). Peaks were identified using published assignments (Yu et al. 1993). Peak height as a function of time for each peak was fit to single exponential decay (see analysis below) using the program SigmaPlot (SPSS Inc.).
Analysis of hydrogen exchange rates
Amide hydrogen exchange is catalyzed by acid or base in aqueous solution. The first-order rate constant for an unstructured polypeptide (krc) depends on solution pH, temperature, and primary structure, and has been extensively calibrated with model compounds (Bai et al. 1993). Observed hydrogen exchange rates in proteins are often many orders of magnitude lower than the rate in a random coil, due to hydrogen bonding and/or burial in native protein structure. The hydrogen exchange reaction in a protein was modeled by a microscopic two-state equilibrium:
 |
(3) |
The observed rate constant (kobs) for a given amide can be expressed as a function of the opening and closing rate constants:
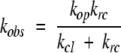 |
(4) |
In the EX2 limit (Hvidt and Nielsen 1966), where kcl ≫ krc, the rate is limited by the population of the open state:
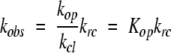 |
(5) |
The free energy difference between the closed state and the most accessible open state for each amide was calculated directly from the observed exchange rates using equation 3.
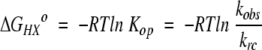 |
(6) |
Born radius calculations
Born radii were calculated with the program EGAD (Pokala and Handel 2004) using the minimized average NMR structure of the complex (PDB ID 1rlq).
Acknowledgments
This work was sponsored by the NIH, under contract GM50945. We thank David King for peptide synthesis, Jeff Pelton for assistance with NMR experiments, and Erik Miller, Chiwook Park, and Navin Pokala for helpful discussions and critical reading of the manuscript.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04990205.
References
- Andersen, M.D., Shaffer, J., Jennings, P.A., and Adams, J.A. 2001. Structural characterization of protein kinase A as a function of nucleotide binding. Hydrogen-deuterium exchange studies using matrix-assisted laser desorption ionization-time of flight mass spectrometry detection. J. Biol. Chem. 276 14204–14211. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakshusmathi, G., Ratnaparkhi, G.S., Madhu, P.K., and Varadarajan, R. 1999. Native-state hydrogen-exchange studies of a fragment complex can provide structural information about the isolated fragments. Proc. Natl. Acad. Sci. 96 7899–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, J. and Itzhaki, L.S. 1998. Hydrogen exchange and protein folding. Curr. Opin. Struct. Biol. 8 112–118. [DOI] [PubMed] [Google Scholar]
- Cliff, M.J., Higgins, L.D., Sessions, R.B., Waltho, J.P., and Clarke, A.R. 2004. Beyond the EX1 limit: Probing the structure of high-energy states in protein unfolding. J. Mol. Biol. 336 497–508. [DOI] [PubMed] [Google Scholar]
- Cordier, F., Wang, C., Grzesiek, S., and Nicholson, L.K. 2000. Ligand-induced strain in hydrogen bonds of the c-Src SH3 domain detected by NMR. J. Mol. Biol. 304 497–505. [DOI] [PubMed] [Google Scholar]
- Engen, J.R., Gmeiner, W.H., Smithgall, T.E., and Smith, D.L. 1999. Hydrogen exchange shows peptide binding stabilizes motions in Hck SH2. Biochemistry 38 8926–8935. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. 2000. Protein folding intermediates and pathways studied by hydrogen exchange. Annu. Rev. Biophys. Biomol. Struct. 29 213–238. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. and Englander, J.J. 1994. Structure and energy change in hemoglobin by hydrogen exchange labeling. Methods Enzymol. 232 26–42. [DOI] [PubMed] [Google Scholar]
- Feng, S., Chen, J.K., Yu, H., Simon, J.A., and Schreiber, S.L. 1994. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3–ligand interactions. Science 266 1241–1247. [DOI] [PubMed] [Google Scholar]
- Gill, S.C. and von Hippel, P.H. 1989. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182 319–326. [DOI] [PubMed] [Google Scholar]
- Glasoe, P.K. and Long, F.A. 1960. Use of glass electrodes to measure acidities in deuterium oxide. J. Phys. Chem. 64 188–190. [Google Scholar]
- Grantcharova, V.P. and Baker, D. 1997. Folding dynamics of the src SH3 domain. Biochemistry 36 15685–15692. [DOI] [PubMed] [Google Scholar]
- Hamuro, Y., Wong, L., Shaffer, J., Kim, J.S., Stranz, D.D., Jennings, P.A., Woods Jr., V.L., and Adams, J.A. 2002. Phosphorylation driven motions in the COOH-terminal Src kinase, CSK, revealed through enhanced hydrogen-deuterium exchange and mass spectrometry (DXMS). J. Mol. Biol. 323 871–881. [DOI] [PubMed] [Google Scholar]
- Hoofnagle, A.N., Resing, K.A., Goldsmith, E.J., and Ahn, N.G. 2001. Changes in protein conformational mobility upon activation of extracellular regulated protein kinase-2 as detected by hydrogen exchange. Proc. Natl. Acad. Sci. 98 956–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hvidt, A. and Nielsen, S.O. 1966. Hydrogen exchange in proteins. Adv. Protein. Chem. 21 287–386. [DOI] [PubMed] [Google Scholar]
- Kapust, R.B., Tozser, J., Fox, J.D., Anderson, D.E., Cherry, S., Copeland, T.D., and Waugh, D.S. 2001. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14 993–1000. [DOI] [PubMed] [Google Scholar]
- Koshland Jr., D.E., Nemethy, G., and Filmer, D. 1966. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry 5 365–385. [DOI] [PubMed] [Google Scholar]
- Larson, S.M. and Davidson, A.R. 2000. The identification of conserved interactions within the SH3 domain by alignment of sequences and structures. Protein Sci. 9 2170–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luque, I. and Freire, E. 2000. Structural stability of binding sites: Consequences for binding affinity and allosteric effects. Proteins Suppl. 4: 63–71. [DOI] [PubMed]
- Maity, H., Lim, W.K., Rumbley, J.N., and Englander, S.W. 2003. Protein hydrogen exchange mechanism: Local fluctuations. Protein Sci. 12 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell, J.G., Falick, A.M., and Komives, E.A. 1998. Identification of protein–protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. 95 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marley, J., Lu, M., and Bracken, C. 2001. A method for efficient isotopic labeling of recombinant proteins. J. Biomol. NMR 20 71–75. [DOI] [PubMed] [Google Scholar]
- Mayne, L., Paterson, Y., Cerasoli, D., and Englander, S.W. 1992. Effect of antibody binding on protein motions studied by hydrogen-exchange labeling and two-dimensional NMR. Biochemistry 31 10678–10685. [DOI] [PubMed] [Google Scholar]
- Milne, J.S., Mayne, L., Roder, H., Wand, A.J., and Englander, S.W. 1998. Determinants of protein hydrogen exchange studied in equine cytochrome c. Protein Sci. 7 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod, J., Wyman, J., and Changeux, J.P. 1965. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 12 88–118. [DOI] [PubMed] [Google Scholar]
- Pan, H., Lee, J.C., and Hilser, V.J. 2000. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc. Natl. Acad. Sci. 97 12020–12025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokala, N. and Handel, T.M. 2004. Energy functions for protein design I: Efficient and accurate continuum electrostatics and solvation. Protein Sci. 13 925–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke, T.M. and Marqusee, S. 1998. Hydrogen exchange studies of protein structure. Curr. Opin. Biotechnol. 9 80–86. [DOI] [PubMed] [Google Scholar]
- Volkman, B.F., Lipson, D., Wemmer, D.E., and Kern, D. 2001. Two-state allosteric behavior in a single-domain signaling protein. Science 291 2429–2433. [DOI] [PubMed] [Google Scholar]
- Wang, C., Pawley, N.H., and Nicholson, L.K. 2001. The role of backbone motions in ligand binding to the c-Src SH3 domain. J. Mol. Biol. 313 873–887. [DOI] [PubMed] [Google Scholar]
- Werner, M.H. and Wemmer, D.E. 1992. Identification of a protein-binding surface by differential amide hydrogen-exchange measurements: Application to Bowman-Birk serine-protease inhibitor. J. Mol. Biol. 225 873–889. [DOI] [PubMed] [Google Scholar]
- Williams, D.H., Stephens, E., and Zhou, M. 2003. Ligand binding energy and catalytic efficiency from improved packing within receptors and enzymes. J. Mol. Biol. 329 389–399. [DOI] [PubMed] [Google Scholar]
- Yu, H., Rosen, M.K., and Schreiber, S.L. 1993. 1H and 15N assignments and secondary structure of the Src SH3 domain. FEBS Lett. 324 87–92. [DOI] [PubMed] [Google Scholar]