

Abstract
We previously reported the construction of a family of reagentless fluorescent biosensor proteins by the structure-based design of conjugation sites for a single, environmentally sensitive small molecule dye, thus providing a mechanism for the transduction of ligand-induced conformational changes into a macroscopic fluorescence observable. Here we investigate the microscopic mechanisms that may be responsible for the macroscopic fluorescent changes in such Fluorescent Allosteric Signal Transduction (FAST) proteins. As case studies, we selected three individual cysteine mutations (F92C, D95C, and S233C) of Escherichia coli maltose binding protein (MBP) covalently labeled with a single small molecule fluorescent probe, N-((2-iodoacetoxy)ethyl)-N-methyl)amino-7-nitrobenz-2-oxa-1,3-diazole (NBD), each giving rise to a robust FAST protein with a distinct maltose-dependent fluorescence response. The fluorescence emission intensity, anisotropy, lifetime, and iodide-dependent fluorescence quenching were determined for each conjugate in the presence and absence of maltose. Structure-derived solvent accessible surface areas of the three FAST proteins are consistent with experimentally observed quenching data. The D95C protein exhibits the largest fluorescence change upon maltose binding. This mutant was selected for further characterization, and residues surrounding the fluorophore coupling site were mutagenized. Analysis of the resulting mutant FAST proteins suggests that specific hydrogen-bonding interactions between the fluorophore molecule and two tyrosine side-chains, Tyr171 and Tyr176, in the open state but not the closed, are responsible for the dramatic fluorescence response of this construct. Taken together these results provide insights that can be used in future design cycles to construct fluorescent biosensors that optimize signaling by engineering specific hydrogen bonds between a fluorophore and protein.
Keywords: biosensors, protein design, periplasmic binding proteins, maltose binding protein, fluorescence, allosteric mechanism
Facile detection of small molecules in situ has applications in such diverse areas as cell biology, medicine, environmental sciences, and defense (Turner 1994; Wolfbeis 2002). Central to this analytical technology is a molecular recognition element that binds to a specified analyte and transduces this binding event into a readily detectable physical observable such as a change in fluorescence or electrochemical behavior (Turner 2000). A variety of different molecular scaffolds have been explored for this purpose, including macrocycle chemosensors (deSilva et al. 2001), imprinted polymers (McQuade et al. 2000), coupled enzymes (Scheller et al. 2001; Subrahmanyam et al. 2002), immunoassays (Luppa et al. 2001), RNA aptamers (Burgstaller et al. 2002), transmembrane pores (Bayley and Martin 2000), metallo-enzymes (Thompson et al. 1998, 2002), bacterial receptor proteins (Gilardi et al. 1994; Brune et al. 1998; Tolosa et al. 1999; Dattelbaum and Lakowicz 2001; de Lorimier et al. 2002; Shrestha et al. 2002; Salins et al. 2004), and macromolecular assemblies (Miyawaki and Tsien 2000; Fehr et al. 2002).
Ideally, a molecular sensor is reagentless, such that the sensor composition remains invariant during the measurement, which is not the case for sensors based on enzymes or competitive binding assays. Systematic development of reagentless protein sensors requires that two fundamental mechanistic problems in structural biochemistry be solved. First, a thermodynamic linkage relationship between two sterically separated sites must be established to couple a reporter group to a ligand-binding site. Second, the ligand-binding affinity and specificity must be manipulated to create a sensor or family of sensors with appropriate dynamic range for the specified application and little or no interference from nontarget ligands (Hellinga and Marvin 1998). The optimization of biosensor ligand-binding affinity and specificity has been addressed in some detail (Gilardi et al. 1994; Dattelbaum and Lakowicz 2001; Marvin et al. 2001; Millet et al. 2003), including the radical computational redesign (Marvin and Hellinga 2001; Dwyer et al. 2003; Looger et al. 2003; Allert et al. 2004) or de novo design (Hellinga et al. 1991) of ligand-binding sites. The construction of a thermodynamic linkage between ligand binding and fluorescence (Gilardi et al. 1994; de Lorimier et al. 2002; Fehr et al. 2002) or electrochemical (Benson et al. 2001) properties has been facilitated by the determination of both apo- and ligand-bound protein structures, which allows the ranking of residues in terms of ligand-dependent distance changes (Marvin et al. 1997), or changes in solvent accessibility (Dattelbaum and Lakowicz 2001). However, most successful biosensor linkages in terms of both attachment site and fluorophore choice have been discovered by inspection or serendipity.
The Escherichia coli maltose binding protein (MBP) is a monomeric, soluble periplasmic protein involved in the transport of and chemotaxis toward maltose (Gerstein et al. 1994; Quiocho and Ledvina 1996). MBP is a member of the bacterial periplasmic binding (PBP) superfamily, which includes proteins with specificity for carbohydrates, amino acids, vitamins, oligopeptides, metals, and anions (Higgins 1992; Dwyer and Hellinga 2004). Previously, we reported the construction of rationally designed mutants of MBP, in which the behavior of a covalently attached fluorophore is allosterically coupled to ligand binding (Marvin et al. 1997). Proteins exhibiting such a Fluorescent Allosteric Signal Transduction (FAST) mechanism were constructed through the in vitro modification of single-cysteine mutants. Here, we further elucidate the microscopic mechanisms by which these fluorophores may transduce ligand-binding events into macroscopic observables. Site-directed mutagenesis and fluorescence data were used to investigate how local conformational changes at three distinct fluorophore coupling sites in MBP affect the environmentally sensitive fluorophore IANBD. Study of the underlying molecular mechanisms by which ligand-mediated conformational changes affect the behavior of reporter group conjugates will help to complete a protein design cycle, in which experimental data and molecular modeling together may be used to improve the future rational design of fluorescent protein biosensors that incorporate conjugated fluorophores.
Results
Steady-state fluorescence of allosterically linked sites
Analysis of the high-resolution crystal structures of the maltose-free (apo) and maltose-bound (holo) forms of MBP has identified three sites on the surface of the protein that undergo local structural changes in concert with ligand-mediated global domain movement (Fig. 1 ▶; Marvin et al. 1997). Phe92, Asp95, and Ser233 are sterically separated from the maltose-binding site and reside in surface crevices located at the interface of the N- and C- terminal domains. By using site-directed mutagenesis, three single cysteine mutants were constructed at each position. Upon covalent coupling of the environmentally sensitive fluorophore NBD, changes in the fluorescence properties were observed upon titration with increasing amounts of maltose (0–200μM) (Fig. 2 ▶; Marvin et al. 1997). While the F92C-NBD protein displays a 20% decrease in fluorescence emission intensity upon the addition of maltose, mutations at D95C or S233C produce an increase of 440% and 200%, respectively. No effect on NBD fluorescence was measured following incubation of a free fluorophore NBD-DTT adduct with 200 μM maltose, indicating that the observed changes in fluorescence are protein mediated.
Figure 1.

Three-dimensional X-ray crystal structures of the E. coli MBP. The structures of apo (PDB code 1OMP) and maltose-bound (1ANF) MBP are shown on the left and right, respectively. N-terminal domain indicated by green; C-terminal domain, blue; hinge residues, purple; and Phe92, Asp95, and Ser233, red sticks. Figure generated with PyMOL (DeLano 2002).
Figure 2.
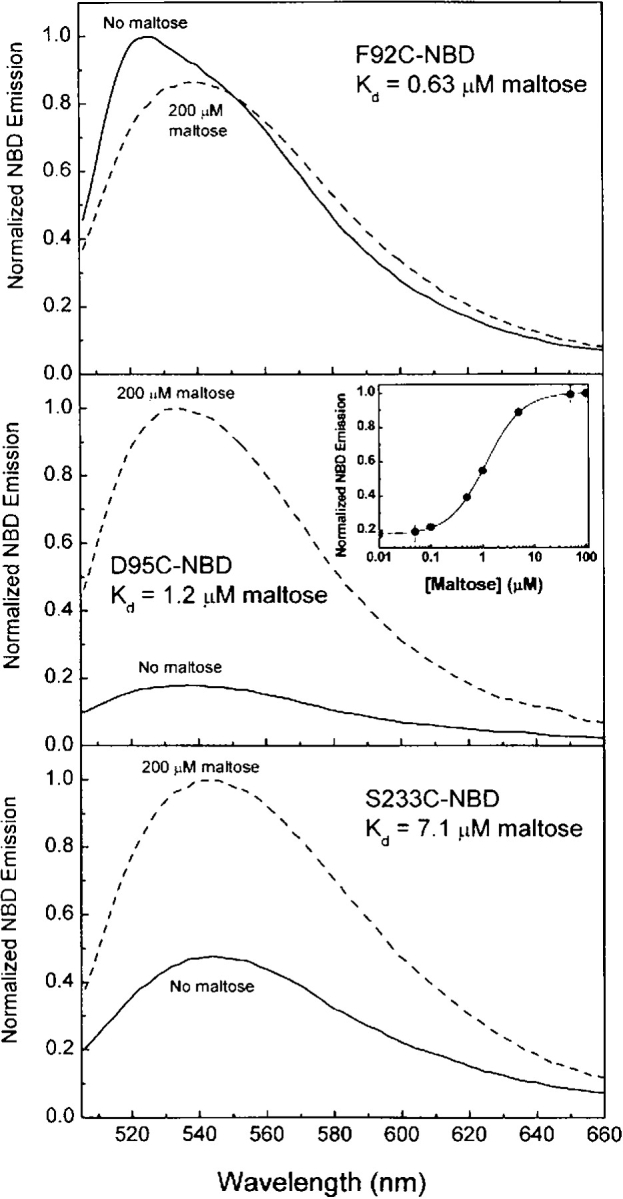
Normalized fluorescence spectra of F92C-NBD, D95C-NBD, and S233C-NBD FAST proteins: no maltose, solid line; 200 μM maltose, dashed line. Measurements were performed in 20 mM MOPS (pH 6.9) and 150 mM NaCl at 25°C.
Table 1 summarizes the fluorescence and ligand-binding properties for each of the three fluorescently labeled proteins in the presence and absence of maltose. The maltose-binding affinities of the three FAST proteins fall in the range of 0.6–7 μM, consistent with previously determined values (Marvin et al. 1997). While F92C-NBD displays only a modest decrease in emission intensity upon the addition of 200 μM maltose, it produces the largest red shift (12 nm) in emission maximum.
Table 1.
Spectroscopic properties of FAST proteins
| λmaxEm(nm)a | rssa,b | ||||
| Mutation | No mal | Sat. mald | No mal | Sat. mald | Kd (μM)c |
| F92C | 526 | 538 | 0.283 | 0.266 | 0.63 |
| D95C | 536 | 534 | 0.220 | 0.265 | 1.2 |
| S233C | 544 | 540 | 0.181 | 0.188 | 7.1 |
a Fluorescence emission (λ) and steadystate anisotropy (rss) were measured as described in Materials and Methods.
b Measurements are averages of three trials. The error for rss is ±0.004 units.
c Calculation of maltose-binding affinity was performed using steady-state intensity data (Fig. 3 ▶) fit to a single-site binding isotherm (de Lorimier et al. 2002).
d 200 μM maltose.
Conformational flexibility of covalently attached NBD in the labeled MBP mutants
For fluorophores covalently bound to proteins, fluorescence anisotropy may be used to compare the steady-state (equilibrium) distribution of different conformational states (Lakowicz 1999). Additionally, anisotropy is a useful observable for biosensing strategies, because it is inherently ratiometric (Thompson et al. 1998). The steady-state anisotropy (rss) was measured for the labeled MBP mutants with and without saturating maltose (Table 1). Sites F92C and S233C exhibit small changes in NBD anisotropy; the D95C-NBD protein displays the largest increase in anisotropy (0.045) upon the addition of maltose, indicating that the NBD molecule has a significantly lower rate or extent of rotational diffusion, consistent with decreased conformational flexibility in the apo state, relative to the holo in this mutant.
Solvent accessibility of MBP allosteric mutants
The choice of fluorophore attachment sites in this study was guided by molecular modeling studies, through analysis of the inter-Cα distance changes between the open and closed forms of MBP (Marvin et al. 1997). We and others have shown that residues that experience large conformational changes in response to ligand binding typically demonstrate significant changes in the fluorescent properties of probes covalently tethered at such locations (Dwyer and Hellinga 2004). In order to develop predictive methods for the rational selection of fluorophore attachment sites, we investigated the extent to which solvent accessibility may contribute to the allosteric linkage of ligand binding and to the fluorescence properties of environmentally sensitive dyes at each of these sites. By using the Lee-Richards algorithm (Lee and Richards 1971; Richards 1977) as implemented in the DEZYMER software suite, the solvent accessible surface area (SASA) for each residue was calculated in each conformational state (Table 2). While moderate increases in SASA were modeled for F92C and D95C in the closed, relative to the open, state, a significant decrease in solvent accessibility was calculated for S233C.
Table 2.
Solvent accessibility of the FAST proteins
| Mutation | Calc. SASA (Å2) | KSV (M−1) | <τ0> (ns)a | kq (M−1 s−1 × 109)b |
| F92C | 195 | 0.96 | 1.22 | 0.8 |
| F92C + mal | 219 | 2.78 | 1.36 | 2.0 |
| D95C | 171 | 2.6 | 1.9 | 1.4 |
| D95C + mal | 207 | 3.17 | 1.7 | 1.8 |
| S233C | 222 | 2.72 | 0.23 | 11.8 |
| S233C + mal | 154 | 3.54 | 0.64 | 5.5 |
a The error in the measurements was taken to be δφ = 0.06 and δm = 0.007 for the phase and modulation, respectively. Average lifetimes were calculated using <τ0> = flτl.
b The bimolecular quenching constant, kq, was calculated according to: kq = KSV/<τ0>.
Solvent effects dominate the signaling of F92C-NBD and S233C-NBD
Stern-Volmer (SV) iodide quenching experiments were used to correlate experimental and predicted changes in solvent accessibility for each FAST protein (Fig. 3 ▶; Table 2). F92C-NBD displays a 12-m red shift in emission maximum upon the addition of maltose (Table 1), the most significant spectral shift of the three proteins. The SV- and bimolecular-quenching constants also increase significantly upon maltose addition (Table 2). The observed spectral data (red shift, emission intensity decrease, increased bimolecular-quenching constant) are therefore consistent with a predicted change in solvent accessibility for this mutant (12% increase), suggesting a more solvent-exposed position for the fluorophore in the closed, relative to the open, state.
Figure 3.
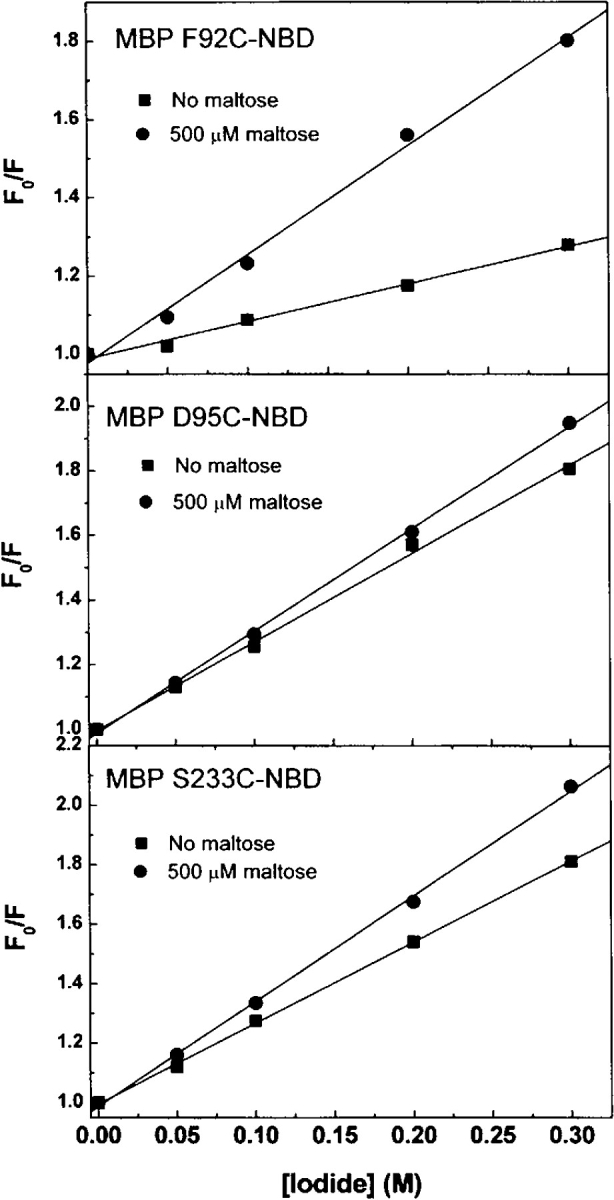
Stern-Volmer quenching plots for the apo- and maltose-bound MBP mutants. Iodide (0–300 mM) was used to quench fluorescence of NBD-labeled MBP mutants. Measurements were performed in 20 mM MOPS (pH 6.9) with constant ionic strength (500 mM), and 1 mM Na2S2O3 was used as reducing agent.
While the solvent accessibility of S233C-NBD was predicted to decrease by 31%, only minor changes were observed in emission maximum and in the SV-quenching constant. However, the average fluorescence lifetime of the NBD at this position increases threefold upon the addition of maltose, resulting in a halving of the bimolecular-quenching constant (Table 2). The experimental data (blue shift, emission intensity increase, decreased bimolecular-quenching constant) are therefore also consistent with a change (decrease) in solvent accessibility of the conjugated fluorophore upon ligand binding.
The D95C-NBD protein exhibits the largest increase in emission intensity upon maltose binding of the three mutants (Table 1) but shows no significant change in fluorescence lifetime, or SV- or bimolecular-quenching constants (Table 2). Taken together with the absence of a fluorescence wavelength shift (Table 1), these data suggest that the interactions between the fluorophore and its environment cannot be rationalized in terms of solvent exposure.
Hydrogen bonding interactions dominate the D95C-NBD signaling mechanism
To further investigate the mechanistic origin for the fluorescent response of MBP D95C-NBD, a simple model of the conformational flexibility of the NBD fluorophore, in the context of the open, and the closed, forms of MBP, was generated (Materials and Methods) using a van der Waals term (Street and Mayo 1999) to model steric interaction factors (Fig. 4 ▶). This model was generated using a simple representation of internal fluorophore flexibility, omitting protein main-chain flexibility, a rotameric model of side-chain flexibility at seven surrounding protein positions, and a rudimentary description of atomic interactions, and, as such, can be used only to identify putative interacting groups; a more realistic model of fluorophore-protein interactions will necessitate a more sophisticated treatment of conformational ensembles and interaction models. Within a 10 Å radius of its attachment site, side-chains that potentially interact with D95C-NBD include residues Tyr171 and Tyr176. The Cα-Cα distances of these residues to D95 increase by 8 Å and 4 Å, respectively, upon maltose binding (Fig. 4 ▶).
Figure 4.

Structural model of the D95C-NBD reporter site, in open and closed conformational states. The model of the NBD-cysteine conjugate is shown colored by atom type (carbon, black; oxygen, red; nitrogen, blue). Side-chains for Tyr171 and Tyr176 are shown in dark red; side-chains for Lys83, Gln86, Arg98, Lys256, and Glu328 are shown in gold. Minimum-energy conformers of the NBD-cysteine and the seven indicated side-chains were generated using a sterics model (see Materials and Methods). Models of the seven natural and one nonnatural side-chains are shown for the open (left) and closed (right) conformational states of maltose binding protein. Figure generated with PyMOL (DeLano 2002).
To study the effect of removing potential hydrogen-bonding groups, these two Tyr residues were mutated to Phe, individually and together. The fluorescence properties of the three mutants (Y171F, Y176F, Y171F/Y176F) were characterized as outlined below (Table 3). While the single Y171F and Y176F mutants still exhibit a maltose-dependent increase in NBD emission intensity, the magnitude of the increase is significantly reduced compared with the reference protein (D95C-NBD). As seen in Table 3, the largest effect is observed in the Y171F single mutant that shows a Δ ΔF of 4.3 and a large red shift in the emission maximum (Δλ = 14 nm) compared with D95C-NBD (Δλ = 2 nm). The construction of the Y171F/Y176F double mutant virtually abolishes the ligand-dependent change in NBD fluorescence intensity (Fig. 5 ▶; Table 3). These mutations dramatically increase the fluorescence of the maltose-free protein but have no effect on the maltose-bound form (Fig. 5 ▶; Table 3). The tryptophan fluorescence emission properties of these three mutants are identical to that of the wild-type protein (data not shown), indicating that these mutations do not disrupt the overall open-to-closed conformational change in any of the MBP mutants. Taken together, these results indicate that the hydroxyl moieties of these two side-chains, and the interactions that they make with the open form of MBP, account for the majority of the fluorescence change in the D95C-NBD protein. We propose that this response is mediated through quenching hydrogen bonds with the nitro group of the fluorophore in the open, but not the closed, state. It is also possible, however, that the effect of the tyrosine hydroxyls on the NBD is indirect, perhaps mediated through a hydrogen-bonding interaction with another side-chain, which orders the “flap” region (residues 165–187) of the protein in a conformation that quenches fluorescence. In either case, the interaction of the NBD fluorophore with specific atoms of the protein is consistent with the significant decrease in steady-state anisotropy upon protein closure.
Table 3.
Properties of D95C-NBD point mutants

Figure 5.

Normalized fluorescence spectra of D95C-NBD with mutations of either Y171F (dotted line), Y176F (dashed line), both (dashed/dotted line), or neither (solid line) in the absence of maltose. The emission spectra of all four proteins with 200 μM maltose overlay (solid line). Measurements were performed in 20 mM MOPS (pH 6.9) and 150 mM NaCl at 25°C.
Electrostatic contributions to D95C-NBD signaling
Potential contributions from electrostatic effects by other amino acids surrounding the attachment site was investigated by additional site-directed mutagenesis experiments. By using our model of D95C-NBD, six protein residues in close proximity to the modeled fluorophore (R83, Q86, R98, Y171, K256, E328) were mutagenized with each mutation either adding or removing a positively charged side-chain (Table 3). All mutants were expressed, purified, derivatized with the NBD fluorophore, and characterized as before. The calculated maltose-binding constants for the mutants were distributed over nearly an order of magnitude, ranging from 0.34–2.9 μM maltose (Table 3). Importantly, mutation of Tyr171 to Arg resulted in a reversal of the maltose-dependent change in emission intensity (ΔΔF = 8.4) (Table 3): While no change in emission maximum was observed with saturating maltose, the emission intensity decreased 46%. These data further demonstrate the importance of a specific interaction of amino acid 171 to the overall signaling of NBD at D95C. The effect of this mutation suggests that the phenylalanine side-chain still significantly quenches the fluorophore in the open state (aromatic solvent quenching of NBD has been observed) (Lin and Struve 1991), that the arginine side-chain quenches the fluorophore in the closed state, or that local conformational changes resulting from the mutation give rise to other effects, mediated either through protein or solvent.
Discussion
The rational design of attachment sites in proteins for environmentally sensitive small molecules dyes is a powerful approach for the development of a broad range of reagent-less biosensors (Hellinga and Marvin 1998). This may involve both the selection of protein residues for the introduction of thiol or other tethering groups, and the rational or computational design of other protein residues for optimization of FAST mechanistic properties. In general, to design such sensors, a fluorophore should be covalently attached to a site on the protein that undergoes a local conformational change upon ligand binding. While simple in principle, our work, as well as others, has demonstrated that in the design of protein biosensors, the response of different fluorophores (even those with similar structural and spectroscopic properties) coupled at a single site may vary widely, belying a simple explanation of the observed changes (Dwyer and Hellinga 2004). In order to improve the predictive power of the design approach, it would be advantageous to have a greater understanding of the microscopic mechanisms through which fluorophore, protein, and solvent together transduce a binding event. Here, we investigate the response of a single fluorophore, NBD, conjugated at three different locations in E. coli MBP.
The complex photo-physical properties of NBD have been studied with respect to interactions with the chemical moieties of amino acids. Specific quenching of NBD emission by the tryptophan analog, NATA, was observed by Lancet and Pecht (1977). Further study into the spectroscopic properties of NBD by Lin and Struve (1991) also suggested a direct interaction between this fluorophore and NATA in organic solutions. Using time-resolved fluorescence spectroscopy, Gilardi et al. (1997) have investigated the specific solvent mechanisms by which environmentally sensitive fluorophores may respond to ligand binding. Using SV fluorescence quenching with iodide, we have validated the use of solvent accessibility calculations as a potential predictor of fluorescence response, although the bio-sensor with the greatest change cannot be rationalized by this measure. We have shown that differential solvent accessibility need not be the only principle upon which a protein biosensor may be designed. Indeed, specific interactions between a fluorophore and protein are likely to provide the best signaling mechanism (D95C-NBD exhibits the largest maltose-dependent change in fluorescence). Our results identify a serendipitous quenching interaction between NBD and two nearby tyrosine residues, which results in a 440% increase in fluorescence intensity upon maltose binding, which is most likely to be mediated by two specific hydrogen bonds contributed by nearby tyrosine residues. To our knowledge this is the first report to identify a particular interaction between covalently tethered NBD and a specific tyrosine residue. Taken together, these results suggest that construction of a fluorophore “cage,” in which fluorescence is quenched in the open, but relieved in the closed state, through construction of differential hydrogen-bonding interactions in the two states is a very effective method for construction of optimal fluorescence signal transduction mechanisms in protein-based biosensors. It is likely that computational design techniques are well suited for this task.
Materials and methods
Preparation of MBP mutants
E. coli MBP was expressed with a C-terminal His6 tag and purified using immobilized metal affinity chromatography, as described previously (Marvin et al. 1997). Overlapping PCR site-directed mutagenesis (Ho et al. 1989) was used to construct point mutants of the malE gene, which were subsequently cloned into pET21a (Novagen), using the NdeI and EcoRI sites. Proteins were over-expressed in E. coli BL21(DE3) pLysS and were purified using Ni-NTA chelating resin (Amersham Biosciences) according to manufacturer protocols.
Preparation of reporter group conjugates
The unique cysteine residues introduced individually at positions F92, D95, and S233 were covalently labeled with the environmentally sensitive fluorophore, N-((2-iodoacetoxy)ethyl)-N-methyl) amino-7-nitrobenz-2-oxa-1,3-diazole (IANBD ester, Molecular Probes). Purified protein was incubated with a fivefold excess of reactive reporter group for 2 h at room temperature (20 mM MOPS at pH 6.9, 100 mM NaCl; 25°C). Unreacted fluorophore was removed by passing samples over an Econo-Pac 10 DG desalting column (Bio-Rad). Complete removal of maltose from MBP was ensured by rapid dialysis against reaction buffer (three buffer changes), using dialysis cartridges (Pierce Chemicals).
Steady-state fluorescence
All fluorescence measurements were recorded on a ThermoSpectronic AB2 spectrofluorometer. Fluorescence emission intensity from NBD-labeled protein conjugates (500 nM) in 20 mM MOPS (pH 6.9) and 100 mM NaCl was measured using 470-nm excitation at 25°C. To determine the maltose-binding affinity of each labeled mutant, changes in NBD fluorescence emission intensity were measured upon the addition of increasing concentrations of maltose (0–200 μM). Experimentally observed data were fit to a single-site binding isotherm (de Lorimier et al. 2002).
For iodide quenching of NBD-labeled proteins, the samples were incubated with increasing concentrations of NaI (0–300 mM), with NaCl added to achieve a final ionic strength of 500 mM. Samples also contained 1 mM Na2S2O3 to prevent iodine formation. For all measurements, excitation and emission polarizers were oriented 0° and 54.7° (“magic angle” conditions), respectively, to eliminate the intrinsic polarization of the system (Lakowicz 1999).
Time-resolved fluorescence
The fluorescence lifetimes of NBD-labeled proteins were measured using frequency domain instrumentation described previously (Thompson and Gratton 1988). The 442-nm output of a Kimmon HeCd laser was amplitude-modulated using an electro-optical pockel’s cell (ISS, Inc.). Rose Bengal in ethanol (τ = 0.9 nsec) was used as a lifetime standard. The data were analyzed using a multiexponential model and fit using nonlinear least squares, as previously described (Lakowicz and Gryczynski 1991).
Molecular modeling
The SASA of amino acid side-chain atoms was calculated using the Lee-Richards algorithm (Lee and Richards 1971; Richards 1977), as implemented in the DEZYMER software suite (Hellinga and Richards 1991; Looger et al. 2003). The solvent-accessible surface is defined as the locus of all points, which are a given probe radius (typically set to 1.4 Å) away from the set of van der Waals’ surfaces of all heavy atoms in the molecule. Per-residue SASA is computed as the area of that portion of the accessible surface that arises from a given side-chain. SASA was computed for the wild-type side-chain, in the crystallographically determined conformation, in either the open (Protein Data Bank [PDB] code 1OMP; Spurlino et al. 1991) or the closed (PDB code 1ANF; Quiocho et al. 1997) state.
A model of NBD-cysteine was created using Chem3D (http://www.cambridgesoft.com). An ensemble of low-energy conformers of the nonnatural side-chain, in vacuo, was computed using Monte Carlo sampling (Boltmann decision of e−ΔE/100) of the seven rotatable dihedral angles (each move a single change of dihedral angle taken randomly from the interval [−10°, 10°]), using a simple van der Waals term to model sterics (Street and Mayo 1999), as implemented in the DEZYMER suite. A total of 1,000,000 steps were performed, with the 5000 lowest-energy conformers constituting the ensemble of allowed NBD structures.
This ensemble of side-chain structures was placed at MBP position 95 (replacing all previous atoms contributed by the wild-type aspartate side-chain), in the open and the closed form, with the seven surrounding protein side-chains (residues 83, 86, 98, 171, 176, 256, and 328) truncated to alanine. NBD conformers with an interaction energy of >20 kcal/mol (using the same sterics potential) with this truncated protein scaffold were eliminated. Conformational states for the seven protein side-chains were taken from a high-quality rotamer library (Looger et al. 2003). The minimum-energy conformation of the seven natural and one nonnatural side-chains was determined by enumeration of all possibilities. The modeled minimum-energy conformation is shown for the open state (Fig. 4A ▶) and the closed state (Fig. 4B ▶) of MBP.
Structures were downloaded from the PDB (http://www.rcsb.org); molecular drawings were made with PyMOL (http:// www.pymol.org).
Acknowledgments
This work was supported by grants from the NIH (H.W.H.) and the Office of Naval Research (H.W.H.). D.E.B. was funded by a Fellowship Award (GM194559) from the NIH. The authors have no competing financial interests.
Abbreviations
DTT, 1,4-dithio-D,L-threitol
PBP, bacterial periplasmic binding protein
MBP, Escherichia coli maltose binding protein
IANBD, N-((2-(iodoacetoxy)-ethyl)-N-methyl)-amino-7-nitrobenz-2-oxa-1,3-diazole
ICT, intramolecular charge transfer
SASA, solvent accessible surface area
τ, average fluorescence lifetime
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.041146005.
References
- Allert, M., Rizk, S.S., Looger, L.L., and Hellinga, H.W. 2004. Computational design of receptors for an organophosphate surrogate of the nerve agent soman. Proc. Natl. Acad. Sci. 101 7907–7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley, H. and Martin, C.R. 2000. Resistive-pulse sensing: From microbes to molecules. Chem. Rev. 100 2575–2594. [DOI] [PubMed] [Google Scholar]
- Benson, D.E., Conrad, D.W., de Lorimier, R.M., Trammell, S.A., and Hellinga, H.W. 2001. Design of bioelectronic interfaces by exploiting hinge-bending motions in proteins. Science 293 1641–1644. [DOI] [PubMed] [Google Scholar]
- Brune, M., Hunter, J.L., Howell, S.A., Martin, S.R., Hazlett, T.L., Corrie, J.E., and Webb, M.R. 1998. Mechanism of inorganic phosphate interaction with phosphate binding protein from Escherichia coli. Biochemistry 37 10370–10380. [DOI] [PubMed] [Google Scholar]
- Burgstaller, P., Jenne, A., and Blind, M. 2002. Aptamers and aptazymes: Accelerating small molecule drug discovery. Curr. Opin. Drug Discov. Dev. 5 690–700. [PubMed] [Google Scholar]
- Dattelbaum, J.D. and Lakowicz, J.R. 2001. Optical determination of glutamine using a genetically engineered protein. Anal. Biochem. 291 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano, W.L. 2002. The PyMOL Molecular Graphics System. Delano Scientific, San Carlos, CA.
- de Lorimier, R.M., Smith, J.J., Dwyer, M.A., Looger, L.L., Sali, K.M., Paavola, C.D., Rizk, S.S., Sadigov, S., Conrad, D.W., Loew, L., et al. 2002. Construction of a fluorescent biosensor family. Protein Sci. 11 2655–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deSilva, A.P., Fox, D.B., Moody, T.S., and Weir, S.M. 2001. The development of molecular fluorescent switches. Trends Biotechnol. 19 29–34. [DOI] [PubMed] [Google Scholar]
- Dwyer, M.A. and Hellinga, H.W. 2004. Bacterial periplasmic binding proteins: A versatile superfamily for protein engineering. Curr. Opin. Struct. Biol. 14 495–504. [DOI] [PubMed] [Google Scholar]
- Dwyer, M.A., Looger, L.L., and Hellinga, H.W. 2003. Computational design of a Zn2+ receptor that controls bacterial gene expression. Proc. Natl. Acad. Sci. 100 11255–11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehr, M., Frommer, W.B., and Lalonde, S. 2002. Visualization of maltose uptake in living yeast cells by fluorescent nanosensors. Proc. Natl. Acad. Sci. 99 9846–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstein, M., Lesk, A.M., and Chothia, C. 1994. Structural mechanisms for domain movements in proteins. Biochemistry 33 6739–6749. [DOI] [PubMed] [Google Scholar]
- Gilardi, G., Zhou, L.Q., Hibbert, L., and Cass, A.E. 1994. Engineering the maltose binding protein for reagentless fluorescence sensing. Anal. Chem. 66 3840–3847. [DOI] [PubMed] [Google Scholar]
- Gilardi, G., Mei, G., Rosato, N., Agro, A.F., and Cass, A.E. 1997. Spectroscopic properties of an engineered maltose binding protein. Protein Eng. 10 479–486. [DOI] [PubMed] [Google Scholar]
- Hellinga, H.W. and Marvin, J.S. 1998. Protein engineering and the development of generic biosensors. Trends Biotechnol. 16 183–189. [DOI] [PubMed] [Google Scholar]
- Hellinga, H.W. and Richards, F.M. 1991. Construction of new ligand binding sites in proteins of known structure, I: Computer-aided modeling of sites with pre-defined geometry. J. Mol. Biol. 222 763–785. [DOI] [PubMed] [Google Scholar]
- Hellinga, H.W., Caradonna, J.P., and Richards, F.M. 1991. Construction of new ligand binding sites in proteins of known structure, II: Grafting of a buried transition metal binding site into Escherichia coli thioredoxin. J. Mol. Biol. 222 787–803. [DOI] [PubMed] [Google Scholar]
- Higgins, C.F. 1992. ABC transporters: From microorganisms to man. Annu. Rev. Cell Biol. 8 67–113. [DOI] [PubMed] [Google Scholar]
- Ho, S.N., Hunt, H.D., Horton, R.M., Pullen, J.K., and Pease, L.R. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77 51–59. [DOI] [PubMed] [Google Scholar]
- Lakowicz, J.R. 1999. Principles of fluorescence spectroscopy, 2nd ed., p. 698. Plenum Publishers, New York.
- Lakowicz, J.R. and Gryczynski, I. 1991. Frequency domain fluorescence spectroscopy. In Topics in fluorescence spectroscopy (ed. J.R. Lakowicz), pp. 293–355. Plenum Press, New York.
- Lancet, D. and Pecht, I. 1977. Spectroscopic and immunochemical studies with nitrobenzoxadiazolealanine, a fluorescent dinitrophenyl analogue. Biochemistry 16 5150–5157. [DOI] [PubMed] [Google Scholar]
- Lee, B., and Richards, F.M. 1971. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 55 379–400. [DOI] [PubMed] [Google Scholar]
- Lin, S., and Struve, W.S. 1991. Time-resolved fluorescence of nitrobenzoxadiazole-aminohexanoic acid: Effect of intermolecular hydrogen-bonding on non-radiative decay. Photochem. Photobiol. 54 361–365. [DOI] [PubMed] [Google Scholar]
- Looger, L.L., Dwyer, M.A., Smith, J.J., and Hellinga, H.W. 2003. Computational design of receptor and sensor proteins with novel functions. Nature 423 185–190. [DOI] [PubMed] [Google Scholar]
- Luppa, P.B., Sokoll, L.J., and Chan, D.W. 2001. Immunosensors: Principles and applications to clinical chemistry. Clin. Chim. Acta 314 1–26. [DOI] [PubMed] [Google Scholar]
- Marvin, J.S. and Hellinga, H.W. 2001. Conversion of a maltose receptor into a zinc biosensor by computational design. Proc. Natl. Acad. Sci. 98 4955–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvin, J.S., Corcoran, E.E., Hattangadi, N.A., Zhang, J.V., Gere, S.A., and Hellinga, H.W. 1997. The rational design of allosteric interactions in a monomeric protein and its applications to the construction of biosensors. Proc. Natl. Acad. Sci. 94 4366–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade, D.T., Pullen, A.E., and Swager, T.M. 2000. Conjugated polymer-based chemical sensors. Chem. Rev. 100 2537–2574. [DOI] [PubMed] [Google Scholar]
- Miyawaki, A. and Tsien, R.Y. 2000. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327 472–500. [DOI] [PubMed] [Google Scholar]
- Quiocho, F.A. and Ledvina, P.S. 1996. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: Variation of common themes. Mol. Microbiol. 20 17–25. [DOI] [PubMed] [Google Scholar]
- Quiocho, F.A., Spurlino, J.C., and Rodseth, L.E. 1997. Extensive features of tight oligosaccharide binding revealed in high-resolution structures of the maltodextrin transport/chemosensory receptor. Structure 5 997–1015. [DOI] [PubMed] [Google Scholar]
- Richards, F.M. 1977. Areas, volumes, packing and protein structure. Annu. Rev. Biophys. Bioeng. 6 151–176. [DOI] [PubMed] [Google Scholar]
- Salins, L.L., Deo, S.K., and Daunert, S. 2004. Phosphate binding protein as the biorecognition element in a biosensor for phosphate. Sens. Actuators B Chem. 97 81–89. [DOI] [PubMed] [Google Scholar]
- Scheller, F.W., Wollenberger, U., Warsinke, A., and Lisdat, F. 2001. Research and development in biosensors. Curr. Opin. Biotechnol. 12 35–40. [DOI] [PubMed] [Google Scholar]
- Shrestha, S., Salins, L.L., Mark Ensor, C., and Daunert, S. 2002. Rationally designed fluorescently labeled sulfate-binding protein mutants: Evaluation in the development of a sensing system for sulfate. Biotechnol. Bioeng. 78 517–526. [DOI] [PubMed] [Google Scholar]
- Spurlino, J.C., Lu, G.Y., and Quiocho, F.A. 1991. The 2.3-Å resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J. Biol. Chem. 266 5202–5219. [DOI] [PubMed] [Google Scholar]
- Street, A.G. and Mayo, S.L. 1999. Computational protein design. Structure 7 R105–R109. [DOI] [PubMed] [Google Scholar]
- Subrahmanyam, S., Piletsky, S.A., and Turner, A.P. 2002. Application of natural receptors in sensors and assays. Anal. Chem. 74 3942–3951. [DOI] [PubMed] [Google Scholar]
- Thompson, R.B. and Gratton, E. 1988. Phase fluorometric method for determination of standard lifetimes. Anal. Chem. 60 670–674. [DOI] [PubMed] [Google Scholar]
- Thompson, R.B., Maliwal, B.P., Feliccia, V.L., Fierke, C.A., and McCall, K. 1998. Determination of picomolar concentrations of metal ions using fluorescence anisotropy: Biosensing with a “reagentless” enzyme transducer. Anal. Chem. 70 4717–4723. [DOI] [PubMed] [Google Scholar]
- Thompson, R.B., Peterson, D., Mahoney, W., Cramer, M., Maliwal, B.P., Suh, S.W., Frederickson, C., Fierke, C., and Herman, P. 2002. Fluorescent zinc indicators for neurobiology. J. Neurosci. Methods 118 63–75. [DOI] [PubMed] [Google Scholar]
- Tolosa, L., Gryczynski, I., Eichhorn, L.R., Dattelbaum, J.D., Castellano, F.N., Rao, G., and Lakowicz, J.R. 1999. Glucose sensor for low-cost lifetime-based sensing using a genetically engineered protein. Anal. Biochem. 267 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, A.P. 1994. Biosensors. Curr. Opin. Biotechnol. 5 49–53. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Tech.Sight: Biochemistry: Biosensors: Sense and sensitivity. Science 290 1315–1317. [DOI] [PubMed] [Google Scholar]
- Wolfbeis, O.S. 2002. Fiber-optic chemical sensors and biosensors. Anal. Chem. 74 2663–2677. [DOI] [PubMed] [Google Scholar]