

Abstract
Probing structures and dynamics within biomolecules using ensemble and single-molecule fluorescence resonance energy transfer requires the conjugation of fluorophores to proteins in a site-specific and thermodynamically nonperturbative fashion. Using single-molecule fluorescence-aided molecular sorting and the chymotrypsin inhibitor 2–subtilisin BPN′ complex as an example, we demonstrate that protein–protein interactions can be exploited to afford site-specific labeling of a recombinant double-cysteine variant of CI2 without the need for extensive and time-consuming chromatography. The use of protein–protein interactions for site-specific labeling of proteins is compatible with and complementary to existing chemistries for selective labeling of N-terminal cysteines, and could be extended to label multiple positions within a given polypeptide chain.
Keywords: protein labeling, protein folding, protein–protein interaction, fluorescence resonance energy transfer (FRET), single molecule spectroscopy, alternating laser excitation, fluorescence-aided molecular sorting
Fluorescence resonance energy transfer (FRET) between a single donor (D) fluorophore and a complementary single acceptor (A) fluorophore (single-pair FRET, or spFRET) is a particular powerful and sensitive method for monitoring conformational dynamics in biomolecules, and protein folding in particular, at ensemble and single-molecule resolution (Selvin 2000;Weiss 2000; Deniz et al. 2001; Ha 2001, 2004). The FRET-efficiency E is a sensitive function of the D/A-distance R, as E=[1+(R/R0)6]−1. R0 is a constant that corresponds to a D/A-distance at which E=50% (Clegg 1992). Because of its dependence on the distance R, spFRET can be used as a distance ruler to track intrachain-conformational dynamics in polypeptide chains in the 2–8 nm range (Stryer and Haugland 1967).
A critical component in a spFRET protein folding experiment is the ability to label a polypeptide chain with a unique D/A-pair in a controlled and site-specific way. In the past, single-molecule spFRET folding studies have been performed with chemically synthesized polypeptides (Jia et al. 1999; Deniz et al. 2000; Talaga et al. 2000). Chemical synthesis of polypeptides has the advantage that side-chain protecting groups can be exploited to facilitate site-specific two-color labeling, but the labeling of proteins of >100 amino acids in length are difficult to achieve.
Recombinant expression of proteins offers more flexibility with respect to chain size. Cysteine (Cys) residues are statistically underrepresented in protein sequences, and many proteins are either devoid of Cys or intrinsic Cys can be removed by site-directed mutagenesis. A unique pair of Cys can then be (re)introduced into the protein at carefully selected surface accessible positions for conjugation with thiol-specific fluorophores. For stoichiometric labeling of double-Cys proteins with a D/A-pair, a two-step protocol is usually employed (Sinev et al. 2000; Ratner et al. 2002; Schuler et al. 2002; Rhoades et al. 2003). First, the protein is reacted with a single fluorophore, added at (sub)stoichiometric ratio to minimize double labeling. Singly modified protein molecules are then separated chromatographically from unreacted or doubly labeled molecules and reacted with the second, complementary, dye. Unfortunately, this two-step sequential labeling is not strictly site-specific. Unless the dye-accessibility of the two thiol groups differs drastically, the first added fluorophore can be attached to either of the two sulfhydryl groups, giving rise to mixtures of D/A-labeled molecules and the dye-permutated, A/D analogs (Ratner et al. 2002). Such mixtures can lead to unwanted sample heterogeneity, as the conjugated dyes can exert a positional-dependent perturbation of the folding free energy of the modified protein. Also, heterogeneities in photophysical properties of the fluorophores due to different local environments (local charge, pH, or hydrophobicity) could complicate the interpretation of spFRET measurements (Moerner and Orrit 1999; Brasselet and Moerner 2000). Last, strict site-specificity of labeling is absolutely mandatory for more sophisticated three- or multicolor FRET experiments (Liu and Lu 2002; Watrob et al. 2003; Hohng et al. 2004).
To increase the site-specificity of sequential labeling, labeling chemistries have been developed that selectively modify N-terminal Cys residues. For example, N-terminal Cys specifically react with thioester-moieties into a stable amide bond (Dawson et al. 1994; Muir et al. 1998; Tolbert and Wong 2002). This chemistry has been exploited by Schuler and Pannell (2002) to label a short synthetic model peptide at the N terminus using a commercial fluorophore chemically modified with a thiobenzylester functionality. Other strategies involve the oxidation of an N-terminal serine (Ser) or threonine (Thr) to the corresponding aldehyde and subsequent coupling with fluorophore containing hydrazine, alkoxy-amine, or hydrazide functionalities (Geoghegan and Stroh 1992), or the specific reaction of an N-terminal Cys with aldehydes into thiazolidines, a reaction that has been utilized to label and immobilize peptides and proteins (Shao and Tam 1995; Zhang and Tam 1996; Guillaumie et al. 2002; Chelius and Shaler 2003).
Here we show that protein–protein interactions can be utilized to site-specifically label recombinantly expressed multi-Cys proteins. The new labeling method does not require a N-terminal Cys to afford site-specific labeling, and is thus complementary to the various chemistries developed for N-terminal Cys modification.
Results
Protein–protein interactions as a tool for site-specific labeling of proteins
Protein–protein interactions are among the most ubiquitous types of interactions in biological systems. Protein–protein interactions are generally characterized by large buried binding interfaces. An analysis of over 70 protein–protein complexes (Lo Conte et al. 1999; Chakrabarti and Janin 2002) showed that a typical interface buries on average about 1600 ± 400 Å2. Alanine scanning studies in 22 protein–protein complexes indicate that only a fraction of those residues constituting a binding interface are actually important to binding energetics (Bogan and Thorn 1998). Therefore, we rationalized that it should be possible to introduce a Cys residue into the binding regions of protein known to be engaged in high affinity protein interactions without severely compromising its binding affinity. Upon protein–protein interaction, the engineered Cys in the interface region will become surface-inaccessible and protected from conjugation with thiol-specific fluorophores, while a second, solvent-accessible Cys that is not part of the binding interface should be susceptible to labeling.
We tested the concept of selective protection of cysteines upon protein–protein interaction using chymotrypsin inhibitor 2 (CI2) and subtilisin BPN′ (Sbt) as model proteins. CI2, a small 64-residue single-domain protease inhibitor binds with high affinity (KD=3.0 × 10−12 M−1) to Sbt BPN′, a serine protease (Fig. 1 ▶). The recognition site of the CI2 inhibitor, known as the inhibitory loop, forms a single-patch interface of standard buried surface area (1630 Å2) with the active site region of Sbt (Radisky and Koshland 2002, 2003). As a bound inhibitor is cleaved by active Sbt and a long-lived acyl-enzyme intermediate is formed (Radisky and Koshland 2002), a catalytically impaired Sbt variant was used for labeling (Bryan et al. 1995). As wild type-CI2 (wt-CI2) is devoid of Cys residues, two Cys were engineered into the inhibitor sequence. The first Cys (dubbed Cys1′ to retain the numbering scheme of wt-CI2) was inserted between the initiation methionine (Met1) and lysine 2 (Lys2) at theNterminus. The second Cys substitutes a methionine (Met) residue at position 40 (Cys40) in the center of the inhibitory loop (Fig. 1 ▶). The side chain of Met40 is solvent-accessible in the unliganded CI2, but is completely buried in the CI2•Sbt complex. Site-directed mutagenesis data implies that Met40 is neither important for protein stability nor required for high-affinity complex formation (Jackson and Fersht 1991, 1994; Radisky et al. 2004).
Figure 1.

Protein–protein interactions as a tool for site-specific labeling of recombinant multi-Cys proteins. Chymotrypsin inhibitor 2 (CI2) (PDB entry 1LW6) is represented as Cα-backbone with side chains not shown explicitly and colored in magenta. Sbt (PDB entry 1LW6) is shown in light gray and depicted with the solvent-accessible surface area shown explicitly. The position of the engineered Cys (Cys1′, Cys40) in CI2 is indicated by filled blue spheres overlaid onto the Cα-backbone of the inhibitor. The donor (D) and acceptor (A) fluorophores are depicted as small circles and are color-coded green and orange, respectively.
Selective labeling of the two engineered Cys in CI2 is achieved in three steps: First, the CI2•Sbt complex is assembled at micromolar concentrations to protect Cys40 in the binding interface. Second, A647 is added to label the exposed thiol-moiety of the solvent-accessible Cys1′ residue. Excess unreacted dye is removed by gel filtration. Third, the CI2•Sbt complex is denatured in the presence of high concentrations of denaturant to expose and label the buried Cys40 by addition of a 10-fold stoichometric access of A488. A/D-labeled CI2 was separated from the protease by capturing the His6-tagged Sbt on an immobilized metal affinity chromatography (IMAC) resin under denaturing conditions. A variant of CI2, in which the position of the dyes is permutated (D/Alabeled CI2–Cys1′/Cys40), has been prepared analogously by changing the order of the added dye.
Labeling specificity in the CI2•Sbt complex
To demonstrate that Cys40 in the inhibitory loop is protected in the Sbt•CI2 complex, we performed a dye-accessibility experiment. First, we incubated CI2– Cys40 with or without a twofold stoichometric excess of Sbt (Cys-free). A 10-fold excess of A647 was added to the solution to quantitatively label any accessible thiol moieties. Excess dye was removed by filtration and the extent of A647-labeling was estimated from absorption spectra taken with the protein solution. Figure 2a ▶ shows that while the sample containing the CI2–Cys40 variant (but no Sbt) was labeled by A647 (as demonstrated by the A647-typical absorption band with a maximum around 647 nm and a shoulder at 600 nm), no measurable dye absorbance was detectable when A647 was added to the preassembled Sbt•CI2 complex. The minor peak at 280 nm observed in the spectrum of the Sbt•CI2 complex can be attributed to the high content of tyrosine (Tyr) and tryptophan (Trp) residues in Sbt (Sbt: 3 Trp, 10 Tyr; CI2: 1 Trp, 1 Tyr). We conclude that complex formation renders the buried Cys40 inaccessible to the added fluorophore and no unspecific labeling of either CI2 or Sbt has occurred (the sample containing the Sbt•CI2–Cys40 complex shows no absorbance at 647 nm). Experiments performed with A488 gave comparable results (data not shown).
Figure 2.

Thiol-accessibility assay of the engineered Cys1′ and Cys40 residues in CI2. Absorbance spectra obtained with (a) CI2–Cys40 and (b) CI2–Cys1′/Cys40 in the absence (black spectrum) and presence (gray spectrum) of a twofold stoichometric excess of Sbt after addition of a 10-fold stoichometric excess of A647 and incubation for 4 h at 25°C (excess free dye removed by gel filtration). (c) Absorbance spectrum of A/D-labeled CI2–Cys1′/Cys40, prepared according to Figure 1 ▶. (d) Fluorescence emission spectrum of the sample shown in c under native conditions (0 M GdnCl, gray spectrum) and strongly denaturing conditions (6 M GdnCl, black spectrum).
Next, we repeated the experiment with the CI2–Cys1′/Cys40 variant. In the presence of a twofold stoichometric excess of Sbt, the absorbance spectrum was essentially superimposable to the spectrum obtained with CI2–Cys40 in the absence of Sbt (Fig. 2b ▶). We conclude that Cys1′ at the N terminus is labeled with A647, while the buried Cys40 remains protected. In the absence of Sbt, the integrated area under the absorbance spectrum (500–760 nm) is approximately twice as high, an indication that both Cys1′ and Cys40 are efficiently labeled.
Figure 2c ▶ shows a representative absorbance spectrum of A/D-labeled CI2–Cys1′/Cys40, prepared according to the scheme in Figure 1 ▶. The presence of two absorbance bands with maxima at 647 nm (typical for A647, molar extinction coefficient 262,000 M−1) and 488 nm (typical for A488; molar extinction coefficient 72,000 M−1) is evidence that singly labeled CI2–Cys1′/Cys40 can be released from the complex with Sbt and that the deprotected Cys40 can be modified by the second fluorophore added.
A fluorescence emission spectrum (excitation 470 nm) of the same sample is shown in Figure 2d ▶. The emission band with a maximum around 510 nm is due to A488 (D) emission. As direct excitation of A647 (A) is negligible at 470 nm, the additional red-shifted emission peak must result from A-emission, due to FRET between the D and A fluorophores. Unfolding of CI2 in the presence of 6 M GdnCl leads to a pronounced increase in D-fluorescence, and a decrease in A-fluorescence, consistent with an expected increase in the D/A-distance in the polypeptide chain.
Characterization of labeled products by mass spectrometry
To determine the labeling stoichometry of the various CI2 samples, aliquots of labeled products were subjected to MALDI-MS analysis (matrix-assisted laser desorption/ionization mass spectrometry). The following molecular weights were determined:
A488 (free dye in H2O): observed mass, 739.10 Da; expected mass, 720.48 Da (the difference is most likely due to hydrolysis of the maleimide moiety).
A647 (free dye in H2O): observed mass, 981 Da; expected mass, unknown (patent pending). As for A488, the mass obtained most likely represents hydrolyzed A647.
CI2-Cys1′/Cys40: expected mass, 7348.70 Da; observed mass, 7329.4 Da.
A/D (or D/A)–CI2–Cys1′/Cys40: observed mass, 9063.00 Da; expected mass, unknown (exact molecular weight of A647 unknown). Expected mass calculated from experimentally determined molecular weights of free A488 and A647 is 9068.70 Da. The mass agrees well with the mass expected from labeling of CI2 with a single D- and single A-fluorophore.
A–CI2–Cys1′/Cys40 (labeling in the presence of Sbt) (Figs. 1 ▶, 2a ▶): observed mass, 8318.23 Da; expected mass (calculated as in [4]), 8329.10 Da. The mass agrees well with labeling of only the N-terminal Cys1′ by A647 (Cys40 is thus protected in the Sbt– CI2 interface, consistent with Fig. 2a ▶). (6) D–CI2– Cys1′/Cys40 (labeling in the presence of Sbt [Fig. 1 ▶]): observed mass, 8053.17 Da; expected mass (calculated as in [4]), 8087.00 Da. The mass agrees well with labeling of only the N-terminal Cys1′ by A488 (Cys40 is thus protected in the Sbt–CI2 interface, consistent with Fig. 2a ▶).
Characterization of the labeled products using fluorescence-aided molecular sorting
The extent of labeling and sample heterogeneity was also addressed at the single molecule level. Alternatinglaser- excitation (ALEX) is a recently developed single molecule spectroscopy method that can be used for fluorescence-aided molecular sorting (FAMS) of freely diffusing molecules (Kapanidis et al. 2004). In ALEX-FAMS, single molecules are detected as bursts of fluorescence photons produced as they diffuse through a focused laser excitation volume. Both D and A in a D/A-labeled sample can be excited directly with an alternation period shorter than the diffusion time of the molecule through the excitation volume, allowing the determination of FRET-efficiencies (E) outside the dynamic range of the particular FRET-pair used (E<0.2 or E>0.9), due to coincidence detection of the D and A. ALEX-FAMS also provides information about labeling stoichiometry (i.e., D-only, A-only, or D/A-labeled subpopulations) through the ratio S (see Materials and Methods for details).
Figure 3 ▶ depicts representative 2D S–E histograms of D-only-labeled CI2–Cys40 (Fig. 3a ▶), A-only-labeled CI2– Cys1′ (Fig. 3b ▶), and A/D-labeled CI2–Cys1′/Cys40 (at various concentrations of denaturant) (Fig. 3c–f ▶). One-dimensional (1D) histograms of the stoichiometric ALEX ratio S (shown in blue color to the right of each 2D E–S-histogram) are obtained by projection of the entire 2D S–E-histograms onto the vertical S-axis. Both D-only-labeled CI2–Cys40 and A-only-labeled CI2–Cys1′ exhibit the expected unimodal distribution of S with mean S values close to 1 (D-only) or 0 (A-only). The A/D-labeled CI2–Cys1′/Cys40 sample also shows a single subpopulation (>95% of histogram area) with an S value ≈ 0.5, which must arise from protein molecules that contain both D- and A-fluorophores. Notably, both D-only and A-only subpopulations are essentially absent in the A/D-labeled CI2 sample. It should be stressed that the lack of D-only and A-only species does not depend on the particular threshold burst size used (in this study, >80 photons per burst), as significantly lower values (10 photons per burst) resulted in similar S-histograms, but with larger width due to shot-noise (data not shown). This indicates that both labeling steps (steps 2 and 3 in Fig. 1 ▶) must have occurred essentially quantitatively, as any non-reacted Cys1′ would have been modified upon complex dissociation in the presence of a 10-fold stoichiometric access of A488, giving rise to a substantial D-only labeled subpopulation, which is not observed. The absence of any significant amounts of D-only and A-only subpopulations is also consistent with a 1:1 labeling deduced from MALDI-MS (see above).
Figure 3.

Probing labeling quality by alternating-laser excitation (ALEX) and fluorescence aided molecular sorting (FAMS). Sorting in ALEX-FAMS is achieved in 2D histograms using the FRETefficiency (E) and the ALEX-ratio (S) as independent variables. E sorts species according to FRET (interdye distance), S sorts on the D/A-stoichometry (extent of labeling). E-S histograms for D-only labeled CI2–Cys40 (a), A-only labeled CI2–Cys1′ (b), A/D-labeled CI2 Cys1′/Cys40 (c–f ) at various denaturant concentrations are shown.
One-dimensional (1D) histograms of the FRET-efficiency E for each sample are displayed at the top of each 2D E–S-histogram (histograms shown in purple). To construct these 1D E-histograms we exploited the fluorescence aided molecular sorting capabilities of ALEX and projected only the section of the 2D E–S-histogram onto the horizontal FRET-efficiency axis that contains A/D-labeled CI2 species (0.20<S-ratio< 0.60; selected area indicated by dashed purple box). This option enhances sorting even further by removing D-only and A-only species that contaminate the 1D E-histograms obtained with traditional single-laser excitation (Deniz et al. 1999, 2000; Schuler et al. 2002), due to partially labeled protein samples or samples with either photobleached D- or A-fluorophores in traditional single-laser excitation (Deniz et al. 1999, 2000; Schuler et al. 2002), and is thus particularly useful for the extraction of accurate mean E-values and subpopulations that significantly overlap with the D-only (E<0.2) or A-only (E>0.8) subpopulations. Under conditions where CI2 is folded ([GdnCl]<2.5 M), a unimodal, high-FRET distribution (E ≈ 0.95, S ≈ 0.45) is observed (Fig. 3c ▶). Higher denaturant concentrations lead to unfolding of CI2 and the coexistence of both high-FRET and low-FRET subpopulations (Fig. 3d,e ▶). At [GdnCl]>5 M, the unfolding process is complete and only the low-FRET subpopulation is visible (Fig. 3f ▶).
Effect of labeling on protein folding energetics
Protein labeling with large, aromatic extrinsic fluorophores is frequently accompanied by a decrease in protein stability (Ratner et al. 2002; Schuler et al. 2002), indicating a perturbation of free energy landscape of folding by the attached dye. To test the effect of fluorophore labeling on the folding thermodynamics of CI2 and to detect a possible dependence of protein stability on a particular dye positioning in the polypeptide chain (A/D-labeled CI2 vs. D/A-labeled CI2), protein stabilities of labeled and nonlabeled CI2 were measured at the ensemble level.
The free energies of folding of unlabeled CI2 is determined by following the increase in Trp-fluorescence upon unfolding (Fig. 4a ▶). Fitting the change in integrated Trp-fluorescence emission (310–420 nm) to a two-state model (Santoro and Bolen 1988) yields a folding free energy (ΔGN-U) of 27.4 ± 0.5 kJ mol−1 and an unfolding cooperativity (mG-value) of 7.6 ± 0.2 kJ mol−1 M−1.
Figure 4.

Thermodynamic analysis of chemically unmodified and fluorophore- labeled CI2–Cys1′/Cys40. (a) Denaturation of unlabeled CI2– Cys1′/Cys40, followed by the change in Trp-fluorescence emission intensity. (b,c) Denaturation curve of A/D-labeled CI2–Cys1′/Cys40 (b) and D/A-labeled CI2–Cys1′/Cys40 (c). The change in the A-emission after D-excitation (FDA), normalized to A-emission (FA, excitation at 633 nm) is shown. Solid lines in a–c represent fits of the raw data to a two-state unfolding model. (d) Unfolding transitions shown in a–c after normalization to the fraction of folded protein (FN =1 − FU; FU defined by Equation 1b in Materials and Methods). Open circles, unlabeled CI2; open squares, A/D-labeled CI2; open rhombuses, D/A-labeled CI2.
To extract thermodynamic stabilities of A/D- and D/A-labeled CI2, the integrated change in fluorescence emission from 650 nm to 740 nm (F(DA)) of the A647 acceptor (A), after D-specific excitation at 470 nm (see Fig. 2d ▶), was monitored. To account for small variations in fluorescence intensity due to adsorption of the labeled protein to the quartz cuvette at the low protein concentrations employed (10 nM), we normalized F(DA) by the integrated fluorescence emission intensity of A after direct excitation at 630 nm (F(A)), and used the ratio F(DA)/F(A) as a progress coordinate for unfolding. Raw data obtained with D/A-labeled and A/D-labeled CI2–Cys1′/Cys40 are shown in Figure 4, b and c ▶, respectively. The decrease in F(DA)/F(A) is consistent with an increase in the interdye distance upon unfolding. Free energies of folding and cooperativity m-values of D/A-labeled and A/D-labeled CI2 are, within error, identical to those measured for the unmodified wt-CI2 reference (D/A-labeled CI2: ΔGN-U=27.2 ± 0.6 kJ mol−1, mG=7.5 ± 0.2 kJ mol−1 M−1; A/D-labeled CI2: ΔGN-U=28.7 ± 1.1 kJ mol−1, mG=8.0 ± 0.3 kJ mol−1 M−1). Unfolding transitions, normalized to the fraction of folded protein (see Materials and Methods for details), are superimposable within experimental error to the curve obtained with the unmodified CI2–Cys1′/Cys40 (Fig. 4d ▶). We conclude that labeling does not measurably perturb the folding free energy of CI2.
Discussion
We have introduced a novel approach to afford site-specific labeling of recombinant proteins for FRET-based single molecule studies. The new method, which we dub SLOPPI (for site-specific labeling of proteins using protein–protein interactions) in the following, invokes protein–protein interactions to selectively protect engineered cysteines that become buried in a binding interface upon protein–protein interaction against labeling in a multicysteine protein context. Unlike statistical two-step labeling (Sinev et al. 2000; Ratner et al. 2002; Schuler et al. 2002), SLOPPI can be performed in simple batch-mode and does not invoke time-consuming two-step chromatography to separate singly labeled protein from non- or doubly labeled side products required in conventional sequential labeling. SLOPPI may thus be useful for rapid optimization of dye pairs, for example, to optimize the FRET-efficiencies for protein folding studies (the FRET-efficiency E is most sensitive in the linear range where 0.3<E<0.8) or to minimize previously reported dye-induced destabilization of the labeled protein. SLOPPI may also be helpful in those cases where sequential labeling is difficult to achieve, for example, when neutral or zwitterionic dyes are employed and the singly labeled protein cannot be easily separated chromatographically from unlabeled or doubly labeled side products.
The suitability of SLOPPI as a tool for site-specific labeling of proteins was demonstrated for the binary CI2•Sbt complex as a model system. The same concept can be immediately applied to label several other proteins that serve as interesting model systems for protein folding studies, and that participate in high affinity binary or multisubunit complexes for which high-resolution structural information is available (Buckle et al. 1994; Welch et al. 1998; Lim et al. 2001; Fieulaine et al. 2002).
Because SLOPPI does not require N-terminal cysteines, it is compatible with and complementary to existing chemistries specific for N-terminal labeling (Shao and Tam 1995; Schuler and Pannell 2002). Figure 5 ▶ depicts how SLOPPI and specific N-terminal labeling could be combined to achieve three-color labeling of recombinantly expressed proteins for FRET-based single molecule protein folding studies. A hypothetical, recombinantly expressed triple-Cys protein (depicted as a four-helix bundle [red]) with Cys at the N terminus and at internal positions i and k along the chain (indicated by filled blue spheres) is first specifically modified at the N-terminal Cys with a thioester (COSR)-modified fluorophore (Schuler and Pannell 2002) F1 (green circle), followed by addition of an interacting protein to render Cys i solvent-inaccessible in a binding interface (step 1). The unlabeled Cys k is then conjugated with a second maleimide-functionalized fluorophore D2 (orange circle) (step 2). In the final step, the protecting protein masking Cys i is then removed by denaturation of the binary complex in high concentrations of chaotropes, followed by removal of the interacting protein via IMAC (only one of the interacting proteins carries a N- or C-terminal His6-tag) (this study) or by gel filtration (if the difference in molecular weights is sufficient) under strongly denaturing conditions. The unmasked Cys i in the doubly labeled hypothetical is then reacted with a third commercial maleimide-functionalized fluorophore D3 (red circle) yielding the site-specific, triply labeled protein of interest.
Figure 5.

Combining selective labeling of interacting proteins and specific labeling of N-terminal Cys to afford site-specific three-color labeling of a hypothetical recombinant protein with Cys at the N terminus, and internal positions i and k. See main text for details.
By using recombinant single-chain fragments (scFv) or Fab-fragments of antibodies as interacting domains and Cys-protecting tools, we envision that SLOPPI can be developed into a general method for site-specific protein labeling. The affinity of antibody fragments directed against protein antigens is typically in the nanomolar range (Hawkins et al. 1993; Kelley and O’Connell 1993; Chakrabarti and Janin 2002), high enough to afford essentially irreversible binding at the micromolar protein concentrations typically employed in labeling experiments. The buried interfaces in these antibody–antigen complexes range from 1250 Å2 to 2320 Å2 (Li et al. 1997; Ding et al. 2000; Chakrabarti and Janin 2002), providing enough flexibility in the positioning of unique interfacial cysteines. Advanced mass spectrometric methods in combination with hydrogen–deuterium exchange (Mandell et al. 1998; Hughes et al. 2001; Anand et al. 2003; Chu et al. 2004) and thiol-accessibility assays similar to those described in the text can be utilized to map binding interfaces in these antibody–antigen complexes, allowing the engineering of Cys into the binding surface even if no high-resolution structural data are available. Moreover, directed evolution of antibodies against the protein of interest (Feldhaus et al. 2003), together with epitope mapping, could produce several different monoclonal antibodies against different epitopes of the same target. Cys residues could be engineered into each of the epitope interfaces. Such an approach could, in principle, afford the labeling of n engineered Cys in a single chain. In the first labeling step, n − 1 monoclonal antibodies will protect n − 1 Cys, and only the single-exposed Cys will be labeled. After denaturation and separation, n − 2 monoclonal antibodies will be used to protect the remaining Cys except for the second one to be labeled. After n − 1 similar steps, a single chain could be site-specifically labeled with n fluorophores.
Materials and methods
Materials
Alexa Fluor 488 maleimide (A488) and Alexa Fluor 647 maleimide (A647) were purchased from Molecular Probes. Guanidinium thiocyanate (GdnSCN) was from Sigma; Guanidinium chloride (GdnCl, sequanal grade) was from Pierce. A plasmid for recombinant expression of a truncated 64-residue double mutant (Glu26Ala/Lys53Arg) of CI2, obtained by deletion of the first, unstructured 19 amino acid residues and replacement of Leu20 with a new starting Met was a gift from Dr. Daniel Koshland (University of California at Berkeley). This truncated and mutated CI2 variant has been shown to retain the structure and function of full-length CI2 and is referred to as wild type hereafter (Radisky and Koshland 2002, 2003). Three additional CI2 variants are described in the text. The first mutant is a single-Cys variant in which Met40 in the inhibitory loop was replaced by a Cys (dubbed CI2–Cys40 hereafter). This mutant is used as a control to test site-specific labeling of a double-Cys variant of CI2 in the CI2•Sbt complex. The second variant carries a unique Cys engineered between the initiation methionine (Met1) and lysine at position 2. Electrospray-ionization mass spectrometry indicates that Met1 is not proteolytically removed in vivo (data not shown). In order to retain the amino acid numbering scheme of wt-CI2, the inserted Cys was dubbed Cys1′, and the corresponding CI2-variant called CI2–Cys1′. The third variant (dubbed CI2–Cys1′/Cys40 hereafter) contains both Cys1′and Cys40. The CI2–Cys1′/Cys40 variant is used for labeling with a unique D/A-FRET pair.
A plasmid for the expression of a catalytically inactive and stability-engineered variant of subtilisin BPN′ (dubbed wt Sbt hereafter) (Bryan et al. 1995) was kindly provided by Dr. Bryan (CARB, University of Maryland Biotechnology Institute). A hexa-His tag was added to the C terminus of the protease to facilitate separation of Sbt from labeled CI2 under denaturating conditions using batch-mode IMAC.
Protein expression and purification
Protein expression and purification was performed as described (Bryan et al. 1995; Radisky and Koshland 2002, 2003). Purified wt-CI2 and Sbt were dialyzed against buffer A (20 mM sodium phosphate [pH 7.0], 100 mM sodium chloride) and stored at 4°C until further use. The Cys variants of CI2 were stored in buffer B (20 mMsodium phosphate, 100 mM sodium chloride [pH 7.0], 10 mM dithiotreitol [DTT]) at 4°C until used in labeling experiments.
Labeling specificity in the CI2•Sbt complex
A quantity of reduced CI2–Cys40 or CI2–Cys1′/CysC40 (10 μM, in buffer B) was passed through a PD10 column (Pharmacia), equilibrated in buffer A, and mixed with a twofold excess of Sbt (in buffer A) and incubated for 30 min to allow complex formation. A 10-fold excess of A647 (or A488) was added and the solution was incubated for 4 h at 25°C in the dark. Excess dye was removed by repeated concentration/dilution of the protein solution in a Centricon YM3 concentration device until the absorbance of the flow-through fraction at 647 nm (or 488 nm) was negligible. The washed protein solution was transferred to a 1.5 mL Eppendorf tube. The volume of the solution was adjusted to 1 mL and transferred to a 10-mm quartz cuvette.Absorbance spectra were recorded from 220nmto 760 nm in a Perkin-Elmer model Lambda 25 UV/Vis spectrophotometer (Perkin-Elmer). The extent of Cys labeling was judged from the sample absorbance at 647 nm (absorbance maximum of A647) or 488 nm (absorbance maximum of A488).
Protein labeling
For the labeling of CI2–Cys1′ and CI2–Cys40, a quantity of freshly purified protein (in buffer B) was passed through a PD10 column, equilibrated in buffer A. A 10-fold excess of maleimide-fluorophore (in buffer A) was added to the solution and incubated for 4 h at 25°C. The protein solution was washed extensively on a Centricon YM3 centrifugal filter membrane (Millipore Corp.) to remove excess unreacted fluorophore. Singly labeled protein solutions were stored at 4°C in the dark until further use.
Two doubly labeled CI2 variants are described in the text. In the first variant, Cys 1′ is labeled with A647 (A) and Cys40 is labeled with A488 (D) (A/D-labeled CI2 hereafter). In variant 2, the dye positions are permutated and Cys 1′ is labeled with A488, while Cys 40 is modified with A647 (D/A-labeled CI2 hereafter).
To prepare A/D-labeled CI2, a quantity of purified and reduced CI2–Cys1′/Cys40 (10 μM in buffer B) was passed through a PD10 column, equilibrated in buffer A, mixed with a twofold excess of Sbt (in buffer A) and incubated for 30 min at room temperature to allow complex formation. A 10-fold excess of A647 (in buffer A) was added to the protein solution and incubated for 4 h at 25°C in the dark. Unreacted dye was removed by extensively washing the protein solution with buffer A on a Centricon YM-3 centrifugal filter. To release CI2–Cys1′/Cys40 (singly labeled with A647 at Cys1′) from the CI2•Sbt complex, the protein solution was mixed with a stock solution of GdnSCN (in buffer A) to give a final concentration of 4 M denaturant. After incubation for 2 h at 25°C in the dark, a 10-fold excess of A488 (in buffer A) was added to the solution and incubated for another 4 h at 25°C in the dark. To separate the hexa-His-tagged Sbt from A/D-labeled CI2, 1 mL of a slurry of IMAC resin (Pharmacia), equilibrated in buffer A, was added to the protein solution. After incubation and gentle agitation of the mixture for 1 h at ambient temperature in the dark, the slurry was centrifuged for 10 min in a benchtop centrifuge and the supernatant fraction containing A/D-labeled CI2 recovered. To remove excess dye, the protein solution was then exchanged into buffer C (20 mM sodium phosphate [pH 6.3], 100 mM sodium chloride) (PD10 column) and stored at 4°C in the dark until used. D/Alabeled CI2 was prepared accordingly by changing the order of the added dyes. A/D- and D/A-labeled CI2 was monomeric, as confirmed by injecting small aliquots of the protein solutions onto a HighLoad 16/60 Superdex 75 size-exclusion column (Pharmacia), equilibrated in buffer C (data not shown).
Thermodynamic analysis
Stability measurements were performed by mixing CI2 with increasing amounts of chaotrope (0–6 M GdnCl). Denaturant concentrations were determined refractometrically (Pace 1986). The thermodynamic stability of unlabeled wt-CI2 was determined by monitoring the increase in fluorescence emission of the single tryptophan (Trp5) upon unfolding. Protein concentrations of 5 μM in buffer D (20 mM sodium phosphate [pH 6.3]) were employed. After incubation for 4 h at 25°C, fluorescence emission spectra were recorded from 310 nm to 420 nm (excitation at 295 nm). The stability of the D/A-labeled CI2– Cys1′/Cys40 variant (or the A/D-permutant thereof) was determined in buffer D (containing 100 μg/mL bovine serum albumine (BSA) to minimize adsorption of the protein to the cuvette wall) by recording acceptor fluorescence emission spectra from 650 nm to 740 nm after excitation at 488 nm (A-emission due to FRET) and 630 nm (direct excitation of A) as a function of denaturant concentration. A protein concentration of 10 nM was used. Changes in free energy of folding (ΔGN-U) were estimated by a six-parameter least-squares fit, assuming a two-state unfolding model (Santoro and Bolen 1988):
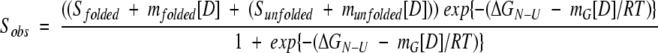 |
(1a) |
Sobs is either the observed integrated fluorescence emission between 310 nm and 420 nm (unlabeled CI2) or the integrated fluorescence intensity of A647 after excitation of the donor at 488 nm, normalized by the integrated fluorescence intensity of A647 after direct excitation (630 nm) at denaturant concentration [D] (labeled protein). Sfolded, Sunfolded, mfolded, and munfolded represent intercepts and slopes of native and unfolded baselines, respectively; mG is a cooperativity parameter related to the change in exposure of hydrophobic surface area upon unfolding (Myers et al. 1995); R is the gas constant; and T is the absolute temperature. Unfolding transitions were normalized to the fraction of unfolded protein, FU (Santoro and Bolen 1988):
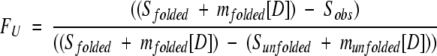 |
(1b) |
Fluorescence aided single molecule sorting (FAMS)
Single-molecule measurements were performed with an inverted fluorescence microscope (Zeiss Axiovert 100, 100 × 1.4 NA oil-immersion objective, 100 μm pinhole), modified to allow alternating laser excitation (ALEX) using a two-laser excitation source (488 nm Ar+-laser, 638 nm diode laser). A detailed description of ALEX is given elsewhere (Kapanidis et al. 2004). Briefly, alternation of the two lasers with a period of 100 μsec, a timescale faster than the residence time in the confocal spot (~600 μsec for CI2) allows simultaneous, direct probing of both A488 (D) and A647 (A) in the diffusing CI2 molecule. ALEX allows the fluorescence-aided molecular sorting of species that differ in their emission profiles using two ratiometric expressions, the traditional FRET-efficiency E and the stoichiometry ratio S. The FRET-efficiency E is defined as:
 |
(2a) |
FDemDexc is the background-corrected D-excitation-based D-emission, FAemDexc is the D-excitation-based A-emission, and γ is a detection and quantum yield correction factor. The novel stoichiometric ALEX-ratio S is defined as:
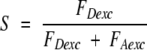 |
(2b) |
FDexc is the sum of D-excitation-based emissions, FAexc is the sum of A-excitation-based emissions. For a D-only molecule (e.g., single D-fluorophore or bleached A-fluorophore), FAexc is negligible and S is ≈ 1. For an A-only molecule, both FDexc and S values are around 0. The S-ratio of a D/A-labeled molecule depends on the excitation power used. If the excitation is adjusted such that FDexc ≈ FAexc (this study), S ≈ 0.5.
All measurements were carried out in buffer D (containing 100 μg/mL BSA). The concentration of the protein was 100 pM and was calculated from the absorbance of A at 647 nm. D- and A-excitation power was adjusted to 50 μW. Fluorescence bursts indicating the presence of a molecule in the laser confocal spot were detected by binning the recorded photons in 500 μsec time bins and defining the beginning and end of a burst using a threshold (D+A photons) set such as to reject most of the background (Dahan et al. 1999; Deniz et al. 2001). Only bursts containing >80 photons were retained for further analysis. E- and S-ratios were calculated for each burst and represented as E- or S-histograms. Denaturation studies were carried out in buffer D with GdnCl concentrations varying from 0 M to 6 M. GdnCl from Pierce (sequanal grade) was found to be sufficiently background-free for these single molecule experiments. Data acquisition was for 25 min, and histograms were calculated and analyzed using in-house written LabView software.
Acknowledgments
We thank Prof. Daniel Koshland (Chemistry Department, UC Berkeley) and Dr. Phil Bryan (CARB, University of Maryland Biotechnology Institute, Rockville, MD) for providing expression plasmids for CI2 and Sbt, respectively. This work was funded by NIH grant no. 1R01-GM65382 (to S.W.).
Abbreviations
A, FRET-acceptor
A488, Alexa Fluor 488
A647, Alexa Fluor 647
ALEX, alternating laser excitation, a novel single-molecule spectroscopic technique
CI2, chymotrypsin inhibitor 2
Cys, cysteine
D, FRET-donor
DTT, dithiotreitol
E, FRET-efficiency
FAMS, fluorescence-aided molecular sorting
FRET, fluorescence resonance energy transfer
FU, fraction of unfolded protein
GdnCl, guanidinium chloride
GdnSCN, guanidinium thiocyanate
KD, dissociation constant
R0, Förster radius
S, ALEX-ratio S, accounting for D/A-stoichometry in D/A-labeled biomolecules
Sbt BPN′, subtilisin BPN′, a serine protease
SLOPPI, site-specific labeling of proteins using protein–protein interactions
spFRET, single-pair FRET
wt, wild type.
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.051384705.
References
- Anand, G.S., Law, D., Mandell, J.G., Snead, A.S., Tsigelny, I., Taylor, S.S., Ten Eyck, L.F., and Komives, E.A. 2003. Identification of the protein kinase A regulatory R′ α-catalytic subunit interface by amide H/D-exchange and protein docking. Proc. Natl. Acad. Sci. 100 13264– 13269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan, A.A. and Thorn, K.S. 1998. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 280 1–9. [DOI] [PubMed] [Google Scholar]
- Brasselet, S. and Moerner, W.E. 2000. Fluorescence behaviour of singlemolecule pH-sensors. Single Molecules 1 17–23. [Google Scholar]
- Bryan, P., Wang, L., Hoskins, J., Ruvinov, S., Strausberg, S., Alexander, P., Almog, O., Gilliland, G., and Gallagher, T. 1995. Catalysis of a protein folding reaction: Mechanistic implications of the 2.0 Å structure of the subtilisin-prodomain complex. Biochemistry 34 10310–10318. [DOI] [PubMed] [Google Scholar]
- Buckle, A.M., Schreiber, G., and Fersht, A.R. 1994. Protein–protein recognition: Crystal structural analysis of a barnase-barstar complex at 2.0 Å resolution. Biochemistry 33 8878–8889. [DOI] [PubMed] [Google Scholar]
- Chakrabarti, P. and Janin, J. 2002. Dissecting protein–protein recognition sites. Proteins 47 334–343. [DOI] [PubMed] [Google Scholar]
- Chelius, D. and Shaler, T.A. 2003. Capture of peptides with N-terminal serine and threonine: A sequence-specific chemical method for peptide mixture simplification. Bioconjugate Chem. 14 205–211. [DOI] [PubMed] [Google Scholar]
- Chu, F., Shan, S., Moustakas, D.T., Alber, F., Egea, P.F., Stroud, R.M., Walter, P., and Burlingame, A.A. 2004. Unraveling the interface of signal recognition particle and its receptor by using chemical crosslinking and tandem mass spectrometry. Proc. Natl. Acad. Sci. 101 16454–16459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg, R.M. 1992. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 211 353–388. [DOI] [PubMed] [Google Scholar]
- Dahan, M., Deniz, A.A., Ha, T., Chemla, D.S., Schultz, P.G., and Weiss, S. 1999. Ratimetric measurement and identification of single diffusing molecules. Chem. Phys. 247 85–106. [Google Scholar]
- Dawson, P.E., Muir, T.W., Clark-Lewis, I., and Kent, S.B. 1994. Synthesis of proteins by native chemical ligation. Science 266 776–779. [DOI] [PubMed] [Google Scholar]
- Deniz, A.A., Dahan, M., Grunwell, J.R., Ha, T., Faulhaber, A.E., Chemla, D.S., Weiss, S., and Schultz, P.G. 1999. Single-pair fluorescence energy transfer on freely diffusing molecules: Observation of Foerster distance dependence and subpopulations. Proc. Natl. Acad. Sci. 96 3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A.A., Laurence, T.A., Beligere, G.S., Dahan, M., Martin, A.B., Chemla, D.S., Dawson, P.E., Schultz, P.G., and Weiss, S. 2000. Singlemolecule protein folding: Diffusion fluorescence energy transfer studies of the denaturation of chymotrypsin inhibitor 2. Proc. Natl. Acad. Sci. 97 5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz, A.A., Laurence, T.A., Dahan, M., Chemla, D.S., Schultz, P.G., and Weiss, S. 2001. Ratiometric single-molecule studies of freely diffusing biomolecules. Annu. Rev. Phys. Chem. 52 233–253. [DOI] [PubMed] [Google Scholar]
- Ding, W., Huang, X., Yang, X., Dunn, J.J., Luft, B.J., Koide, S., and Lawson, C.L. 2000. Structural identification of a key protective B-cell epitope in Lyme disease antigen OspA. J. Mol. Biol. 302 1153–1164. [DOI] [PubMed] [Google Scholar]
- Feldhaus, M.J., Siegel, R.W., Opresko, L.K., Coleman, J.R., Feldhaus, J.M., Yeung, Y.A., Cochran, J.R., Heinzelman, P., Colby, D., Swers, J., et al. 2003. Flow-cytometric isolation of human antibodies from a non. Nat. Biotechnol. 21 163–170. [DOI] [PubMed] [Google Scholar]
- Fieulaine, S., Morera, S., Poncet, S., Mijakovic, I., Galinier, A., Janin, J., Deutscher, J., and Nessler, S. 2002. X-ray structure of a bifunctional protein kinase in complex with its protein substrate HPr. Proc. Natl. Acad. Sci. 99 13437–13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan, K.F. and Stroh, J.G. 1992. Site-directed conjugation of nonpeptide groups to peptides and proteins via perjodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjugate Chem. 3 138–146. [DOI] [PubMed] [Google Scholar]
- Guillaumie, F., Thomas, O.R.T., and Jensen, K.J. 2002. Immobilization of pectin fragments on solid supports: Novel coupling by thiazolidine formation. Bioconjugate Chem. 13 285–294. [DOI] [PubMed] [Google Scholar]
- Ha, T. 2001. Single-molecule fluorescence energy transfer. Methods 25 78–86. [DOI] [PubMed] [Google Scholar]
- ———. 2004. Structural dynamics and processing of nucleic acids revealed by single-molecule spectroscopy. Biochemistry 43 4055–4063. [DOI] [PubMed] [Google Scholar]
- Hawkins, R.E., Russell, S.J., Baier, M., and Winter, G. 1993. The contribution of contact and non-contact residues of antibody in the affinity of binding to antigen. J. Mol. Biol. 234 958–964. [DOI] [PubMed] [Google Scholar]
- Hohng, S., Joo, C., and Ha, T. 2004. Single-molecule three-color FRET. Biophys. J. 87 1328–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, C.A., Mandell, J.G., Anand, G.S., Stock, A.M., and Komives, E.A. 2001. Phosphorylation causes subtle changes in the solvent accessibility at the interdomain interface of methylesterase CheB. J. Mol. Biol. 307 967–976. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E., and Fersht, A.R. 1991. Folding of Chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 30 10428–10435. [DOI] [PubMed] [Google Scholar]
- ———. 1994. Contribution of residues in the reactive loop of chymotrypsin inhibitor 2 to protein stability and activity. Biochemistry 33 13880– 13887. [DOI] [PubMed] [Google Scholar]
- Jia, Y., Talaga, D.S., Lau, W.L., Lu, H.S.M., DeGrado, W.F., and Hochstrasser, R.M. 1999. Folding dynamics of single GCN-4 peptides by fluorescence resonant energy transfer confocal microscopy. Chem. Phys. 247 69–83. [Google Scholar]
- Kapanidis, A.N., Lee, N.K., Laurence, T.A., Doose, S., Margeat, E., and Weiss, S. 2004. Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser-excitation of single molecules. Proc. Natl. Acad. Sci. 101 8936–8941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley, R.F. and O’Connell, M.P. 1993. Thermodynamic analysis of an antibody functional epitope. Biochemistry 32 6828–6835. [DOI] [PubMed] [Google Scholar]
- Li, H., Dunn, J.J., Luft, B.J., and Lawson, C.L. 1997. Crystal structure of Lyme disease antigen outer surface protein complexed with an Fab. Proc. Natl. Acad. Sci. 94 3584–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, D., Park, H.U., De Castro, L., Kang, S.G., Lee, H.S., Jensen, S., Lee, K.L., and Strynadka, N.C.J. 2001. Crystal structure and kinetic analysis of β-lactamase inhibitor protein-II in complex with TEM-1 β-lactamase. Nat. Struct. Biol. 8 848–852. [DOI] [PubMed] [Google Scholar]
- Liu, J. and Lu, Y. 2002. FRET study of a trifluorophore-labeled DNAzyme. J. Am. Chem. Soc. 124 15208–15216. [DOI] [PubMed] [Google Scholar]
- Lo Conte, L., Chothia, C., and Janin, J. 1999. The atomic structure of protein–protein recognition sites. J. Mol. Biol. 285 2177–2198. [DOI] [PubMed] [Google Scholar]
- Mandell, J.G., Falick, A.M., and Komives, E.A. 1998. Identification of protein–protein interfaces by decreased amide proton solvent accessibility. Proc. Natl. Acad. Sci. 95 14705–14710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moerner, W.E. and Orrit, M. 1999. Illuminating single molecules in condensed matter. Science 283 1670–1676. [DOI] [PubMed] [Google Scholar]
- Muir, T.W., Sondhi, D., and Cole, P.A. 1998. Expressed protein ligation: A general method for protein engineering. Proc. Natl. Acad. Sci. 95 6705–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1995. Denaturant m-values and heat capacity changes: Relation to changes in accessible surface areas of protein unfolding. Protein Sci. 4 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace, C.N. 1986. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 131 266–280. [DOI] [PubMed] [Google Scholar]
- Radisky, E.S. and Koshland Jr., D.E. 2002. A clogged gutter mechanism for protease inhibitors. Proc. Natl. Acad. Sci. 99 10316–10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003. The role of the protein core in the inhibitory power of the classic serine protease inhibitor, chymotrypsin inhibitor 2. Biochemistry 42 6484–6492. [DOI] [PubMed] [Google Scholar]
- Radisky, E.S., Kwan, G., Lu, K., and Koshland Jr., D.E. 2004. Binding, proteolytic, and crystallographic analyses of mutations in the proteaseinhibitor interface of the subtilisin BPN′/chymotrypsin inhibitor 2 complex. Biochemistry 43 13648–13656. [DOI] [PubMed] [Google Scholar]
- Ratner, V., Kahana, E., Eichler, M., and Haas, E. 2002. A general strategy for site-specific double labeling of globular proteins for kinetic FRET studies. Bioconjugate Chem. 13 1163–1170. [DOI] [PubMed] [Google Scholar]
- Rhoades, E., Gussakovsky, E., and Haran, G. 2003. Watching proteins fold one molecule at a time. Proc. Natl. Acad. Sci. 100 3197–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1988. Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27 8063–8068. [DOI] [PubMed] [Google Scholar]
- Schuler, B. and Pannell, L.K. 2002. Specific labeling of polypeptides at amino-terminal cysteine residues using Cy5-benzyl thioester. Bioconjugate Chem. 13 1039–1043. [DOI] [PubMed] [Google Scholar]
- Schuler, B., Lipman, E.A., and Eaton, W.A. 2002. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature 419 743–747. [DOI] [PubMed] [Google Scholar]
- Selvin, P.R. 2000. The reaissance of fluorescence resonance energy transfer. Nat. Struct. Biol. 7 730–734. [DOI] [PubMed] [Google Scholar]
- Shao, J. and Tam, J.P. 1995. Unprotected peptides as building blocks for the synthesis of peptide dentrimers with oxime, hydrazone and thiazolidine linkages. J. Am. Chem. Soc. 117 3893–3898. [Google Scholar]
- Sinev, M., Landsmann, P., Sineva, E., Ittah, V., and Haas, E. 2000. Design consideration and probes for fluorescence resonance energy transfer studies. Bioconjugate Chem. 11 352–362. [DOI] [PubMed] [Google Scholar]
- Stryer, L. and Haugland, R.P. 1967. Energy transfer: A spectroscopic ruler. Proc. Natl. Acad. Sci. 58 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talaga, D.S., Lau, W.L., Roder, H., Tang, J., Jia, Y., DeGrado, W.F., and Hochstrasser, R.M. 2000. Dynamics and folding of single two-stranded coiled-coil peptides studied by fluorescent energy transfer confocal microscopy. Proc. Natl. Acad. Sci. 97 13021–13026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolbert, T.J. and Wong, C.H. 2002. New methods for proteomic research: Preparation of proteins with N-terminal cysteines for labeling and conjugation. Angew. Chem. Int. Ed. 41 2171–2174. [DOI] [PubMed] [Google Scholar]
- Watrob, H.M., Pan, C., and Barkley, M.D. 2003. Two-step FRET as a structural tool. J. Am. Chem. Soc. 125 7336–7343. [DOI] [PubMed] [Google Scholar]
- Weiss, S. 2000. Measuring conformational dynamics of biomolecules by single molecule fluorescence spectroscopy. Nat. Struct. Biol. 7 724–729. [DOI] [PubMed] [Google Scholar]
- Welch, M., Chinardet, N., Mourey, L., Birck, C., and Samama, J.P. 1998. Structure of the CheY-binding domain of histidine kinase CheA in complex with CheY. Nat. Struct. Biol. 5 25–29. [DOI] [PubMed] [Google Scholar]
- Zhang, L. and Tam, J.P. 1996. Thiazolidine formation as a general and site-specific conjugation method for synthetic peptides and proteins. Anal. Biochem. 233 87–93. [DOI] [PubMed] [Google Scholar]