

Abstract
Molecular machines order and disorder polypeptides as they form and dissolve large intermolecular interfaces, but the biological significance of coupled ordering and binding has been established in few, if any, macromolecular systems. The ordering and binding of GroES co-chaperonin mobile loops accompany an ATP-dependent conformational change in the GroEL chaperonin that promotes client protein folding. Following ATP hydrolysis, disordering of the mobile loops accompanies co-chaperonin dissociation, reversal of the GroEL conformational change, and release of the client protein. “High-affinity” GroEL mutants were identified by their compatibility with “low-affinity” co-chaperonin mutants and incompatibility with high-affinity co-chaperonin mutants. Analysis of binding kinetics using the intrinsic fluorescence of tryptophan-containing co-chaperonin variants revealed that excessive affinity causes the chaperonin to stall in a conformation that forms in the presence of ATP. Destabilizing the β-hairpins formed by the mobile loops restores the normal rate of dissociation. Thus, the free energy of mobile-loop ordering and disordering acts like the inertia of an engine’s flywheel by modulating the speed of chaperonin conformational changes.
Keywords: nuclear magnetic resonance, surface plasmon resonance, ATP hydrolysis, folding funnel, allele-specific genetic interaction, intrinsically unstructured protein
Order–disorder transitions in protein–ligand binding sites govern the thermodynamics and potentially the kinetics of binding. A substantial fraction of proteins are disordered or contain disordered domains in the native state (Wright and Dyson 1999; Dunker et al. 2002; Papoian and Wolynes 2003). All of the large (>1000 Å2) transient protein–ligand binding sites so far characterized exhibit a conformational change (Nooren and Thornton 2003), and to the extent that structural information on the unbound proteins is available, approximately one-third of them involve ordering of poly-peptide backbone segments (Lo Conte et al. 1999). Many disordered binding sites have important regulatory functions (Dyson and Wright 2002; Iakoucheva et al. 2002). Disordered binding sites have been proposed to provide two kinds of advantage: (1) The disordered domain could rapidly sample conformational space and therefore accelerate binding of otherwise rigid molecules (Shoemaker et al. 2000), and (2) the coupling of ordering and binding effectively lowers the binding affinity compared with rigid structures with the same size of interface (Frankel and Kim 1991; Alber 1993). The advantage of a lower affinity can be realized in terms of at least three potential cellular defects associated with excessive binding affinity: (1) Binding produces a significant amount of incorrect complex (incorrect site or aberrant binding at the correct site); (2) binding produces too much of the correct complex or leaves too little of the free subunit; or (3) dissociation of the correct complex is too slow to support its function. Cellular defects because of excessive binding affinity in molecular machines and regulatory complexes have not been well characterized, although the defect of at least one excessively tight protein–DNA complex was said to be slow dissociation (Hurlburt and Yanofsky 1990).
The Escherichia coli chaperonin GroEL and its co-chaperonin GroES constitute a molecular machine that assists the folding of nascent and misfolded polypeptides. GroEL is composed of 14 identical 57-kDa subunits, which are organized into two heptameric rings that are stacked back to back, thus forming a double-ring structure (Braig et al. 1994). Each GroEL subunit has an equatorial domain that has ATPase activity and forms the interface between the two rings, an apical domain that binds client proteins and GroES (Fenton et al. 1994) and an intermediate domain that connects the equatorial and apical domains. GroES forms a single-ring structure consisting of seven identical 10-kDa subunits (Hunt et al. 1996). Each GroES subunit uses a mobile loop with a conserved hydrophobic tripeptide for interaction with GroEL (Landry et al. 1993). The mobile loops are ~16 amino acids in length and undergo a transition from disordered loops to β-hairpins concomitant with binding the apical domains of GroEL (Shewmaker et al. 2001).
The GroE machine has been described picturesquely as a two-cylinder reciprocating engine (Lorimer 1997). Each ring of GroEL is negatively cooperative to the opposite ring (Yifrach and Horovitz 1995). The cooperative binding of ATP and GroES to the trans ring facilitates the departure of ADP and GroES from the cis ring (Todd et al. 1994). However, cycling time depends largely on the rate and cooperativity of ATP hydrolysis in the cis ring (Fridmann et al. 2002). The client protein binds to the inner surface of one of the GroEL rings (Braig et al. 1993). The subsequent binding of ATP and GroES to the client-bound GroEL ring causes the apical domains of that ring to reorient and sequester the hydrophobic surfaces used for binding the client protein (Xu et al. 1997). The client protein is released into the “Anfinsen cage” and potentially achieves a native fold (Saibil et al. 1993). The machine is believed to assist protein folding by preventing aggregation (Buchner et al. 1991), limiting the client protein’s conformational choices (Coyle et al. 1999; Brinker et al. 2001), and/or unfolding misfolded client protein molecules (Shtilerman et al. 1999; Chen et al. 2001).
The timing of articulations and energy transfers in a molecular machine must be tuned to match physical constraints, such as the molecular dimensions, temperature, and function. There exists a stringent interdependence between various alleles of E. coli groEL, and E. coli groES or bacteriophage-encoded co-chaperonin genes. Scores of mutant alleles that block and/or restore a functional interaction have been identified (Ang et al. 2000). These allelic interactions have two important features: First, many mutations that block the functional interaction of the proteins affect residues that are outside of the binding interface, and second, most of the mutations can be grouped into one of two phenotypic classes.
Bacteriophages T4 and RB49 encode co-chaperonins (Gp31 and CocO, respectively) that are distantly related to E. coli GroES (Ang et al. 2001; Keppel et al. 2002) and are required for successful bacteriophage propagation. During a bacteriophage T4 infection of E. coli, Gp31 functions with the host GroEL in folding the T4 coat protein Gp23 (Andreadis and Black 1998). Presumably, the bacteriophage RB49-encoded CocO homolog operates in a similar capacity. Why GroES is insufficient for folding Gp23 is unknown; however, it has been postulated on structural grounds that Gp31 and CocO, in conjunction with GroEL, form a larger “Anfinsen cage” (Hunt et al. 1997).
In a previous study, Richardson and coworkers classified GroEL(E191G) as a “low-affinity” GroEL mutant because it was unable to bind Gp31 (Richardson et al. 1999). Binding was restored with a mutant co-chaperonin encoded by T4 mutant 31(L35I), which had been selected to propagate on E. coli mutant groEL(E191G). T4 31(L35I) could not propagate on groEL(A383T) until it acquired a second mutation, resulting in T4 31(L35I, T31A). We proposed that T4 31(L35I) was incompatible with groEL(A383T) because the protein complex was overstabilized and that T4 31(L35I, T31A) was compatible with groEL(A383T) because T31A compensates for L35I. The substitutions L35I and T31A are expected to stabilize and destabilize the GroEL-bound β-hairpin formed by the Gp31 mobile loop, respectively.
Here, we used genetic and biochemical approaches to test two hypotheses, namely, (1) whether excessive co-chaperonin binding affinity can effectively block chaperonin function, and (2) whether destabilizing the mobile loop β-hairpin can reverse the excessive affinity and restore a functional interaction with a “high-affinity” GroEL mutant.
Isolation of high-affinity GroEL mutants
Putative high-affinity GroEL mutants were selected by demanding that groES(G24D) suppressors grow at high temperature (43°C) and simultaneously block bacteriophage T4. The E. coli mutant groES(G24D) is temperature-sensitive for growth (Landry et al. 1993), and the G24D substitution in the mobile loop has been suggested to disrupt formation of the GroEL-bound β-hairpin (Shewmaker et al. 2001). The rationale for believing the selected GroEL mutants would exhibit an higher affinity for GroES is twofold: First, they would be able to restore some of the interaction strength disrupted by the GroES(G24D) substitution, and second, they would be able to block bacteriophage T4 and RB49 propagation because of an excessive affinity for Gp31 and CocO, respectively. Following preliminary tests, we concentrated our efforts on two putative high-affinity GroEL mutant proteins, GroEL(V174F) (Zeilstra-Ryalls et al. 1994) and GroEL(E178K) (this work).
Because these suppressor mutants were isolated in the context of a groES mutant allele, it was of interest to determine whether the putative high-affinity GroEL mutations are detrimental for E. coli growth. Tests for bacteriophage P1 cotransduction and for overexpression toxicity indicated that neither the groEL(V174F) nor the groEL(E178K) allele is compatible with the wild-type groES+ allele (Supplemental Material). Thus, we conclude that the GroES–GroEL(V174F) and GroES–GroEL(E178K) combinations cannot sustain E. coli viability.
Various derivatives of T4 bacteriophage, differing only in gene 31, were plated on E. coli strains with various groEL backgrounds to determine if the putative high-affinity GroEL mutants were compatible with wild-type Gp31, high-affinity Gp31(L35I), low-affinity Gp31(T31A), or the double mutant Gp31(T31A,L35I) (Richardson et al. 1999). Only T4 31(T31A) propagated on a bacterial host encoding either GroEL(V174F) or GroEL(E178K) at any temperature (Table 1). This behavior is the exact opposite to the phenotype observed for the low-affinity strain encoding GroEL(E191G), on which only T4 31(L35I) propagated (Table 1).
Table 1.
T4 and RB49 bacteriophage propagation E. coli strains carrying various groEL alleles at 37°C
| E. coli groEL allele | ||||
| groEL(E191G) | groEL+ | groEL(V174F) | groEL(E178K) | |
| Phage T4 31 allele | ||||
| 31(wt) | − | + | − | − |
| 31(L35I) | + | + | − | − |
| 31(T31A) | − | + | + | + |
| 31(T31A,L35I) | − | + | − | − |
| Phage RB49 cocO allele | ||||
| cocO(wt) | − | + | − | − |
| cocO(Q36R) | + | − | − | − |
| cocO(S28G) | − | + | + | + |
| cocO(S28G,Q36R) | − | + | − | − |
The ability of each bacteriophage strain to form plaques on a given E. coli strain is indicated by either a plus sign or a minus sign; the plus sign indicates a plating efficiency of ~1.0; a minus sign indicates a plating efficiency of <1 × 10−6. Identical T4 plating phenotypes were also observed at 30°C with one notable exception: T4 (wt) forms small plaques on E. coli groEL(E191G).
Virtually the same pattern of phenotypes was observed using bacteriophage RB49, which encode a co-chaperonin with 34% amino acid sequence identity to bacteriophage T4 Gp31. A mutant bacteriophage RB49 was isolated by its ability to form plaques on E. coli groEL(E191G) mutant bacteria, and the mutation results in an amino-acid substitution in the co-chaperonin mobile loop (Q36R) (Ang et al. 2001). On the basis of the new positive charge encoded by this allele and the phenotypes of other cocO alleles affecting charges (data not shown), we speculate that Q36R increases the affinity for GroEL by an electrostatic mechanism. The increase in GroEL affinity caused by Q36R appears to be somewhat greater than that caused by L35I in Gp31 because the mutant bacteriophage RB49 cannot form plaques on groEL+ E. coli (Table 1).
We constructed a low-affinity RB49 co-chaperonin by incorporating a glycine-for-serine substitution (S28G) at the position homologous to the site of the T31A substitution in bacteriophage T4 Gp31. S28G is expected to strongly disfavor β-sheet formation (Minor and Kim 1994). The mutation encoding S28G was first introduced in a plasmid-borne gene and then recombined into the RB49 bacteriophage genome. As anticipated, the phenotype of RB49 cocO(S28G) was very similar to that of T4 31(T31A) (Table 1). When the S28G and Q36R substitutions were combined to produce RB49 cocO(S28G,Q36R), as for T4 31(T31A, L35I), the phenotypes of the two substitutions canceled each other out (Table 1).
Tryptophan fluorescence enhancement in WGp31–GroEL complexes
The binding of selected Gp31 and GroEL pairs was characterized by measuring the fluorescence emission of a Gp31 variant, designated WGp31, in which a tryptophan was substituted for isoleucine at position 36 (I36W) in the mobile loop. Isoleucine 36 is the second residue in the conserved hydrophobic tripeptide that contacts GroEL (Landry et al. 1993; Xu et al. 1997). Binding of WGp31 and GroEL can be monitored in vitro by the enhancement and blueshift in the tryptophan fluorescence (Richardson et al. 1999). The phenotypes of wild-type and mutant T4 bacteriophage encoding the I36W substitution are nearly identical to the phenotypes of the corresponding T4 bacteriophage without the I36W substitution (Supplementary Fig. 1 ▶). Thus, fluorescence-binding assays using the various purified WGp31 proteins could explain the phenotypes of mutations that affect chap-eronin function.
Figure 1.

WGp31-GroEL binding monitored by tryptophan fluorescence. (A–D) Each protein pair was incubated at 25°C for 2 min prior to the addition of Mg2+ and K+. After an additional 30 sec, the protein interaction was triggered by the addition of 1 mM ATP. After 2 min, a 7.5-fold excess of His-GroES was added as competitor. All experiments were performed in 100 mM Tris-HCl (pH 7.5), 2 mM DTT, 5 mM MgCl2, 5 mM KCl with 2 μM GroEL, 2 μM WGp31, and 15 μM His-GroES proteins (protomer concentrations).
The fluorescence enhancements of the various WGp31 proteins on formation of GroEL complexes with ATP were 1.6- to 2.1-fold larger than for the complexes formed with ADP (Supplementary Fig. 2 ▶). The difference between ATP and ADP complexes cannot be attributed to differences in degree of complex formation. When the fluorescence enhancements were analyzed as a function of ADP concentration, the plots revealed highly cooperative transitions, and the signals for all four WGp31 proteins achieved >95% of their maximum values at 1 mM ADP. Thus, the difference in enhancement for ATP and ADP complexes must be intrinsic to the complexes formed.
Figure 2.

GroEL-GroES binding monitored by surface plasmon resonance. His-GroES was immobilized (~95 RU) on a chelating NTA sensor chip (Biacore, Inc.) in a BIAcore 2000 instrument, and the analysis was performed essentially as described (Hayer-Hartl et al. 1995). The buffer was 20 mM MOPS-KOH (pH 7.4), 100 mM KCl, 5 mM MgCl2, and 2 mM ATP. Association was performed with buffer containing 200 nM GroEL for 480 sec, followed by dissociation in buffer only.
Delayed dissociation of high-affinity WGp31–GroEL complexes
The dissociation of the various WGp31–GroEL complexes was monitored by the decay of the fluorescence enhancement after addition of excess His-tagged GroES as a competitor (Fig. 1A ▶). Dissociation rates for the various WGp31 proteins were almost indistinguishable from each other (Fig. 1A ▶, inset) and equal to the rate expected if dissociation was limited by the rate of ATP hydrolysis (one turnover every 10 to 12 sec; Todd et al. 1994), which is consistent with the ability of all of the mutant T4 bacteriophage to propagate on wild-type E. coli (Table 1).
As expected, the low-affinity chaperonin GroEL(E191G) bound weakly to WGp31(T31A,L35I) and not at all to WGp31(T31A) (Fig. 1B ▶), which is consistent with the failure of T4 31(T31A,L35I) and T4 31(T31A) to propagate on the E. coli groEL(E191G) mutant host (Table 1).
The binding kinetics of high-affinity GroEL(E178K) and GroEL(V174F) revealed a defect associated with excessive co-chaperonin affinity. The dissociation rates for the various WGp31 proteins differed greatly (Fig. 1C,D ▶), whereas association rates were indistinguishable from each other (Fig. 1 ▶; data not shown). Only WGp31(T31A) dissociated from the high-affinity chaperonins with wild-type rates, which is consistent with the unique ability of T4 31(T31A) to propagate on E. coli mutant strains that express the high-affinity chaperonins (Table 1). Dissociation rates for the other WGp31 proteins were much slower. The rank order of dissociation rates was WGp31(T31A) > WGp31(T31A, L35I) > WGp31 > WGp31(L35I), with decrements of ~10-fold from one WGp31 protein to the next. This ranking is consistent with the hypothesis that the corresponding co-chaperonin mutations were selected on the basis of restoring optimum binding affinity; for example, T4 31(L35I) was selected in the presence of GroEL(E191G) because L35I strengthened Gp31 binding, and T4 31(L35I, T31A) was selected in the presence of GroEL(A383T) because T31A weakened Gp31(L35I) binding.
Delayed dissociation of high-affinity GroES–GroEL complexes
The inability of high-affinity GroEL proteins to support E. coli growth can be explained by their slow dissociation from GroES. To evaluate this further, we immobilized His-tagged GroES on an NTA chip (BIAcore Inc.), and wild-type or mutant GroEL was allowed to bind in the presence of ATP. The association rates for all of the GroEL proteins were within an order of magnitude of 10 (±5) × 104 M−1 sec−1 (Fig. 2 ▶). The mutants GroEL(V174F) and GroEL(E178K) dissociated with very slow rates, 2.5 (±1) × 10−4 sec−1 and 0.4 (±0.2) × 10−4 sec−1, respectively, which are ~100-fold slower than the dissociation rate for wild-type GroEL (Hayer-Hartl et al. 1995). GroEL(E191G), a low-affinity mutant, showed a dissociation rate that was similar to that of wild-type GroEL (Fig. 2 ▶).
Slower cycling of high-affinity WGp31–GroEL complexes
Steady-state ATP hydrolysis in the various chaperonin complexes correlated with the rate of WGp31 release. In the absence of co-chaperonin, all of the GroEL proteins hydrolyzed ATP at essentially the same rate (0.15 ± 0.02 sec−1). In the presence of WGp31, the ATPase rate of GroEL was approximately half the rate determined for GroEL alone (Fig. 3 ▶). This is consistent with previous studies finding that GroES inhibits the steady-state ATPase rate of GroEL by 50% in similar conditions (Todd et al. 1993). At this “high” potassium ion concentration (5 mM), GroES binding to one ring of GroEL prevents nucleotide exchange in that ring until ATP hydrolysis is complete (Todd et al. 1994). A new cycle begins with binding of ATP and GroES to the trans ring and release of GroES and ADP from the cis ring. At a low potassium ion concentration (1 mM), GroES blocks ADP/ATP exchange in the trans ring and therefore completely inhibits chaperonin cycling. In the presence of WGp31 and 5 mM potassium ion, the ATPase rate for the low-affinity GroEL(E191G) was inhibited by <50%, consistent with weak binding of WGp31 to GroEL. In contrast, the ATPase rates for high-affinity GroEL(V174F) and GroEL(E178K) were inhibited by >50%, suggesting that the stronger binding of WGp31 delays the cycling of the high-affinity GroEL mutants, analogous to the delay in cycling caused by GroES at intermediate potassium ion concentrations.
Figure 3.

Inhibition by WGp31 of steady-state ATP hydrolysis in GroEL. GroEL or a GroEL mutant, with or without an equal concentration of WGp31 (2-, 3-, or 4-μM protomer concentrations), was incubated in 100 mM Tris-HCl (pH 7.5), 1 mM DTT, 1 mM ATP, 5 mM MgCl2, and 5 mM KCl for 30 min at 23°C. At this saturating concentration of ATP, the steady-state ATPase rate is determined by the rate of coordinated dissociation from GroEL of WGp31 and nucleotide. WGp31 inhibits the ATPase of the “low-affinity” GroEL mutant less than it inhibits the ATPase of wild-type GroEL, whereas WGp31 inhibits the ATPase of the “high-affinity” GroEL mutants more than it inhibits the ATPase of wild-type GroEL.
Nascent formation of a β-hairpin in the mobile loop correlates with GroEL binding affinity
Despite their dynamic flexibility, mobile loops of GroES, Gp31, and human Hsp10 form a nascent β-hairpin that resembles the conformation adopted by mobile-loop peptides bound to GroEL (Landry et al. 1993, 1996, 1997; Shewmaker et al. 2001). Thus, mutations could increase or decrease GroEL-binding affinity by increasing or decreasing the tendency to form the β-hairpin. As noted previously, the L35I and T31A substitutions are expected to increase and decrease the tendency to form β-sheet, respectively (Shewmaker et al. 2001). Nascent structures in the mobile loops of WGp31 and variants were compared by analysis of Hα chemical shifts (δHα) obtained from two-dimensional nuclear magnetic resonance (NMR) spectra. Polypeptide backbone resonances corresponding to the most dynamic region of the mobile loops, primarily in the N-terminal flank of the hairpin turn (Landry et al. 1997, 1999), were consistently observed in spectra for all of the variants.
The δHα for residues D27, E28, E29, and S33 were notably sensitive to the mobile-loop mutations (Fig. 4 ▶). In comparison to the δHα for wild-type WGp31, the δHα for all four residues in WGp31(L35I) moved downfield, whereas the δHα for these residues in WGp31(T31A) and WGp31(T31A,L35I) moved upfield. Downfield shifts indicate an increased tendency to adopt the β conformation, and upfield shifts indicate a decreased tendency to adopt the β conformation (Wishart et al. 1991). Thus, L35I favors the GroEL-bound β-hairpin conformation, and T31A disfavors the GroEL-bound β-hairpin conformation. The resulting tendency for the mobile loops to form the β-hairpin conformation follows the rank order, WGp31(T31A) < WGp31(T31A,L35I) < WGp31 < WGp31(L35I), which matches the rank order of dissociation rates, from fastest to slowest.
Figure 4.
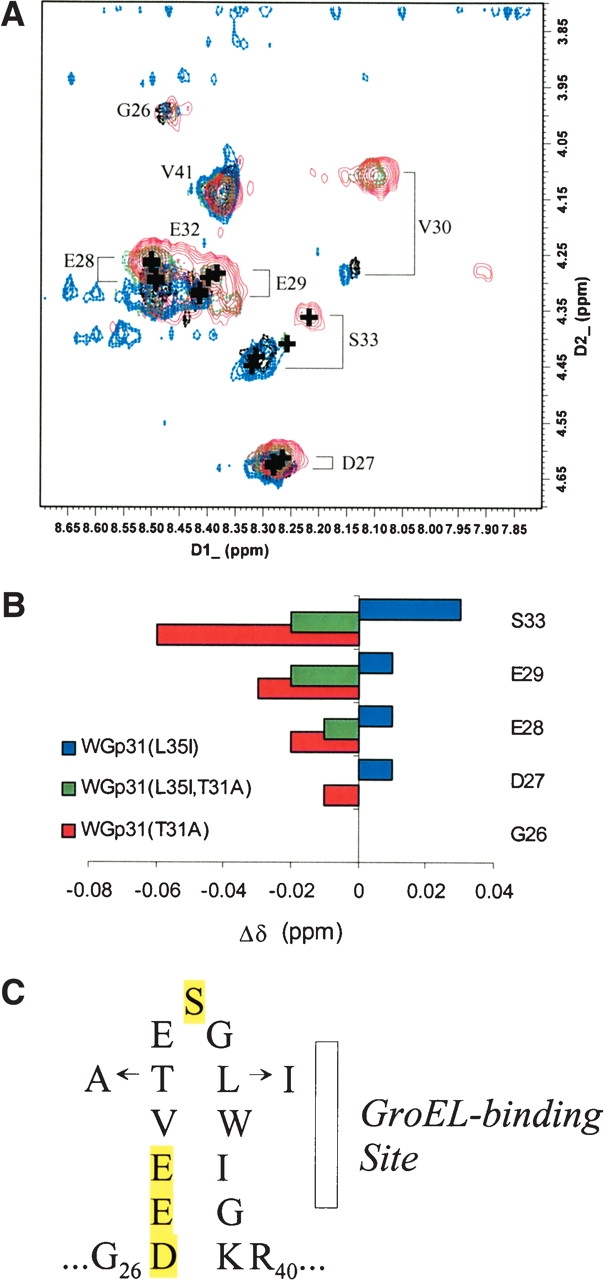
Affects of mutations on the tendency to form β-sheet in the most flexible region of the mobile loop. (A) The NH/Hα regions of total correlated (TOCSY) spectra of WGp31(L35I) (blue), WGp31 (black), WGp31(L35I,T31A) (green), and WGp31(T31A) (red). Backbone resonances for residues G34–K39 were not observed, most likely because of dipolar broadening caused by a microsecond–millisecond conformational fluctuation in proximity to the tryptophan side chain. (B) Changes in Hα chemical shift, indicating an increased tendency to adopt β-sheet (positive) or decreased tendency to adopt β-sheet (negative) in the mobile loops of mutant WGp31 proteins. Nearest-neighbor effects resulting from the T31A substitution confound interpretation of deviations for residues V30–E32. (C) The mobile-loop sequence that forms a nascent β-hairpin and binds to GroEL, highlighting the positions used to analyze conformational tendency.
Discussion
The high-affinity behavior of various GroEL and co-chaperonin proteins can be explained in terms of stabilization of the GroEL apical domain in the up conformation that binds co-chaperonin (Fig. 5 ▶). The amino-acid substitution in high-affinity GroEL(V174F) is likely to disrupt a hydrophobic cluster that stabilizes apical domains in the down conformation, therefore shifting the population of GroEL subunits toward the up conformation (Ang et al. 2000; Klein and Georgopoulos 2001). We propose a similar explanation for the high-affinity phenotype of GroEL(E178K) (Supplementary Fig. 3 ▶). When the apical domain is in the down conformation, E178 forms a salt bridge with R322. Disruption of the salt bridge would shift the population toward the up conformation.
Figure 5.

Conformational and energetic events during reciprocation of the chaperonin machine. Seven ATP molecules stabilize the cis ring of subunits with apical domains up, and co-chaperonin is bound with mobile loops folded into the GroEL-bound hairpin conformation. ATP hydrolysis destabilizes the cis-ring complex with the co-chaperonin (indicated by the reshaped folding funnel). Mobile loop unfolding and co-chaperonin dissociation from the cis ring, coupled with ATP and co-chaperonin binding to the trans ring, cause the chaperonin to reciprocate to the conformation with trans subunits up and cis subunits down. The structures at the beginning and the end of the cycle are indistinguishable. For wild-type GroEL, the ADP-bound intermediate (bracketed) decays rapidly and steadily because mobile loop dissociation/unfolding (cis) and association/folding (trans) encounter no significant enthalpic barriers. Thus, the free energy of mobile-loop unfolding/folding acts like a flywheel to prevent the chaperonin machine from stalling, and the overall rate of reciprocation is controlled by the rate of ATP hydrolysis. “High-affinity” GroEL mutants stall in the ADP-bound intermediate. “Low-affinity” co-chaperonins can restore reciprocation of high-affinity GroEL mutants.
The L35I substitution in Gp31 intensifies the high-affinity phenotype by stabilizing the GroEL-bound β-hairpins, which further stabilizes the GroEL apical domains in the up conformation. The T31A substitution compensates by destabilizing the GroEL-bound β-hairpins. The stabilizing effect of L35I is not as strong as the destabilizing effect of T31A. The upfield changes in Hα chemical shift due to T31A were larger than the downfield changes due to L35I, and Hα chemical shifts for the double mutant were upfield from those of wild-type Gp31. The changes in β-hairpin stability correlate with expected changes in intrinsic β-sheet propensity from host–guest studies (Minor and Kim 1994). L35I is expected to stabilize the β conformation by 0.49 kcal mole−1 when the affected position is in the hydrophobic center of a β-sheet, an environment that may be similar to the β-hairpin strand that is buried against GroEL (Fig. 4C ▶). T31A is expected to destabilize the β conformation by 0.83 kcal mole−1 when the affected position is at the solvent-exposed edge of a β-sheet, an environment that may be similar to the β-hairpin strand opposite to the GroEL-bound strand. Thus, T31A overcompensates for L35I, and this is evident in the faster dissociation from high-affinity GroEL proteins of Gp31(L35I,T31A) compared with wild-type Gp31.
Excessive affinity causes the chaperonin machine to stall in a complex that forms in the presence of ATP but not ADP. ADP complexes exhibit generally lower fluorescence intensity than the corresponding ATP complexes (Supplementary Fig. 2 ▶), suggesting that the ADP complexes have different structures, possibly with fewer or less buried tryptophans. Moreover, in contrast to expectation, the ADP complexes with high-affinity GroEL proteins dissociate faster than the ADP complexes with wild-type GroEL (Supplementary Fig. 4 ▶). These results can be explained by a failure of the ADP complexes to achieve the conformational state that exhibits high-affinity behavior.
The difference between ATP and ADP complexes could be related to the number of mobile loops that are ordered in the complex. The crystallographic structure of the GroES–GroEL–ADP complex, including a mimic of the ATP γ-phosphate, aluminum fluoride (AlFx), appeared to be identical to the ADP complex without AlFx, despite the fact that the complex with AlFx was much more stable (Chaudhry et al. 2003). However, poor resolution of the mobile loops and symmetry averaging of the electron density may have obscured differences in the ordering of individual GroES mobile loops. It is possible that the stability of the complex depends on the number of tightly bound mobile loops. The complex formed in ATP may have a greater number of tightly bound mobile loops than the complex formed in ADP. Likewise, high-affinity GroEL may have a greater number of tightly bound mobile loops than wild-type GroEL has in the complexes formed in ATP. This model implies that fewer than seven tightly bound mobile loops are necessary to stabilize the wild-type complex. In experiments with GroEL containing mixtures of wild-type and mutant apical domains, as few as one or two wild-type apical domains were sufficient for stable binding of GroES (Farr et al. 2000).
Ordering of the mobile loops creates a mechanism for the co-chaperonin to promote transitions between the two predominant GroEL conformations, analogous to the action of a flywheel in a reciprocating engine (Fig. 5 ▶). On binding of ATP, formation of the β-hairpin and binding of the mobile loops drive GroEL subunits into the up conformation. After ATP hydrolysis, dissociation and unfolding of the mobile loops drive GroEL subunits into the down conformation. As for any two-state protein-folding transition, the pathway for ordering and disordering the mobile loop can be described by a funnel in which there are no significant enthalpic barriers, and thus the kinetics of ordering and disordering are determined by only the time required to search conformational space (Papoian and Wolynes 2003). Mobile-loop ordering and disordering can provide a smooth and gradual driving force for GroEL conformational changes as well as co-chaperonin binding and dissociation.
The intrinsic kinetics of mobile-loop disordering are not likely to limit the rate of co-chaperonin dissociation. NMR studies indicated that the β-hairpin forms and dissolves on a timescale of no longer than milliseconds (Shewmaker et al. 2001), whereas wild-type co-chaperonin dissociation occurs on a timescale of seconds.
The thermodynamics of the co-chaperonin mobile-loop order–disorder transition govern the kinetics of the GroEL conformational change. The fact that the effects of amino acid substitutions in Gp31 and GroEL are additive suggests that they modulate a single kinetic step. If all seven subunits participate in binding, the difference in dissociation rates for WGp31 and WGp31(T31A) corresponds to a reduction in the kinetic barrier to dissociation of 0.4 kcal mole−1 per subunit (Supplementary Fig. 5 ▶). Because a T-for-A substitution can destabilize a β-sheet by more than twice this amount, destabilization of the GroEL-bound β-hairpin by T31A can explain the restoration of normal dissociation rates and chaperonin function with high-affinity GroEL proteins.
The paradigm of the co-chaperonin mobile loop suggests that the ordering of binding domains could be widely used to sustain appropriate kinetics in macromolecular machines and other processes that involve reversible multivalent protein–protein interactions. Because domain ordering depends on the contribution of many intramolecular and intermolecular bonds, the kinetics are robust and can be finely tuned.
Materials and methods
Construction of recombinant bacteriophage
First, the desired change in gene 31 was introduced by site-directed mutagenesis of pALEX36 (Richardson et al. 1999), which encodes WGp31. Second, a specific E. coli strain was transformed with the recombinant plasmid. Third, under permissive conditions, a specific bacteriophage T4 strain was used to infect the cells carrying the plasmid in order to create a lysate. Fourth, the lysate was spotted onto an E. coli strain known to be nonpermissive to the initial T4 strain. Fifth, gene 31 from the bacteriophage DNA of individual plaques was sequenced to determine if the desired change was picked up. For the production of W-T4 31(L35I), E. coli strain B178 carrying pALEX36(L35I) was infected with wild-type T4 and the desired bacteriophage was subsequently selected on groEL(E191G) (Richardson et al. 1999). For the production of W-T4 31(T31A), DH5 carrying pALEX(T31A) was infected with T4 31(L35I) and the desired bacteriophage was subsequently selected on strain JZ661 (Zeilstra-Ryalls et al. 1994). For the production of W- T4 31(T31A,L35I), B178 carrying pALEX36(T31A, L35I) was infected with T4 31(L35I) and the desired bacteriophage was subsequently selected on groEL(A383T) (Richardson et al. 1999).
Protein preparations
Wild-type GroEL was expressed in E. coli B178 from a pBAD22 derivative containing the groE operon between the EcoRI and HindIII sites (kind gift of France Keppel, University of Geneva). Mutations causing single amino-acid substitutions were introduced by standard site-directed mutagenesis protocols. Mutant GroEL proteins were expressed in B178-derived strains that possessed identical chromosomal mutations, thus preventing the formation of mixed oligomers. Purification of GroEL and its mutants was as described previously (Shewmaker et al. 2001).
Expression of wild-type and mutant WGp31 proteins was performed in E. coli MC1009, and the purification was a modification of the protocol previously described (van der Vies et al. 1994). Three liters of preheated LB medium (50 μg/mL ampicillin) was inoculated with an overnight culture (1:20 dilution) and incubated at 37°C with constant shaking. At OD600 = 0.6, protein expression was induced by the addition of 0.02% l-arabinose. Cells were harvested after 5 h of induction and resuspended in 34 mL of 10 mM Tris-HCl (pH 7.7), 100 mM NaCl, 1 mM EDTA, and 1 mM β-mercaptoethanol. After resuspension, 1 mL of Sigma Protease Inhibitor Cocktail (cat. no. P-8465) was added, and the cells were lysed by passage through a French Pressure Cell three times.
The cell lysate was diluted to four times its volume and centrifuged for 30 min at 23,000g to remove debris. The supernatant was adjusted to 5% w/v with streptomycin sulfate and slowly mixed for 15–20 min at 4°C. The precipitated nucleic acid was removed by centrifugation at 23,000g for 15 min. The supernatant was brought to 36% ammonium-sulfate saturation at 4°C by slow addition of 100% saturated ammonium sulfate while stirring.
The precipitated protein was recovered by centrifugation for 30 min at 23,000g and then resuspended in 25 mL of Buffer Q (20 mM Tris-HCl at pH 7.7, 1 mM EDTA, 1 mM β-mercaptoethanol). The protein solution was dialyzed against 2 L of the same buffer for ~4 h at 4°C and then centrifuged for 10 min at 33,000g to remove precipitated contaminants. The protein was loaded on Q-Sepharose (Pharmacia) equilibrated with Buffer Q. The column was washed with one bed volume of Buffer Q, and then the protein was eluted with a gradient of 0–0.8 M NaCl in Buffer Q. The Gp31-containing fractions were dialyzed overnight against 3 L Buffer H (20 mM sodium phosphate at pH 7.4, 1 mM EDTA, 2 mM β-mercapto-ethanol). The protein was loaded onto a hydroxy-apatite column equilibrated with Buffer H. The column was washed with one bed volume of Buffer H, and the protein was eluted with a 0.02–0.275 M sodium-phosphate gradient (pH 7.4, 1 mM EDTA, 2 mM β-mercaptoethanol). The Gp31-containing fractions were pooled and stored in 70% saturated ammonium sulfate.
Fluorescence spectroscopy
All fluorescence experiments were performed on a Photon Technologies Quanta Master luminescence spectrometer with a gloved cuvette holder attached to a water bath for temperature maintenance. Excitation was at 293 nm, and emission was recorded at 334 nm. All kinetic experiments were performed under constant stirring in a total volume of at least 1.5 mL. Component concentrations for individual experiments are given in figure and table legends. The proteins used for all experiments were recovered by sedimentation and resuspension after storage under 70% saturated ammonium sulfate. Thus, small amounts of residual NH4+ ions (submillimolar) are part of the experimental conditions.
Steady-state ATPase activity
The GroEL proteins were separated from the bulk of the solution by using centrifugal filter columns (Microcon YM-50, Millipore; cat. no. 42415). The liberated inorganic phosphate from the hydrolysis of ATP was then quantitated using the Enzchek Phosphate Assay Kit (Molecular Probes; cat. no. E6646). In brief, this assay allows for the quantitation of inorganic phosphate produced from the hydrolysis of ATP. The assay is based on the enzyme purine nucleoside phosphorylase, which can convert the substrate 2-amino-6-mercapto-7-methylpurine riboside into ribose 1-phosphate and 2-amino-6-mercapto-7-methyl-purine. This enzymatic conversion causes a spectrophotometric shift in maximum absorbance to 360 nm for the enzymatic product from 330 nm for the substrate. The rates of inorganic phosphate production were determined at several chaperonin concentrations, first without and then with equal concentrations of co-chaperonin. The plots of ATPase rate versus chaperonin concentration were linear, suggesting that differences in ATPase rate were not due to differences in chaperonin assembly. After linear regression of these plots, the slopes were taken as the ATPase rate constants.
NMR spectroscopy
Samples were prepared in 550 μL of 50 mM potassium phosphate buffer (pH 6.0), and then 60 μL of D2O, 5 μL 20% (w/v) 3,3,3-(trimethylsilyl)propionate (TSP) in D2O, and 1 μL of 80 mM NaN3 were added. The final concentration of wild-type or variant WGp31 was 0.5 mM. All NMR spectra were recorded on a Bruker DRX 500 equipped with a 5-mm Bruker TXI probe. Proton chemical shifts were referenced to TSP (0 ppm). The NOESY experiment (homonuclear correlation via dipolar coupling) used a NOESY mixing time of 200 msec; and phase-sensitive data were acquired using the States-TPPI method (Jeener et al. 1979). The TOCSY experiment (homonuclear Hartman-Hahn transfer) used the MLEV17 sequence for mixing (60 msec) and two power levels for excitation and spin lock; phase-sensitive data were acquired using the States-TPPI method (Bax and Davis 1985). In both pulse sequences, water was suppressed by using the 3-9-19 pulse sequence with gradients (Piotto et al. 1992; Sklenar et al. 1993). Spectra of width 6000 Hz were acquired with 2048 points in t2 and 512 increments in t1 and then processed into a frequency-domain matrix of size 2048 × 2048.
Acknowledgments
We acknowledge France Keppel and Abdul Waheed for technical assistance. Supported by NSF grant MCB-9512711 (S.J.L.), Swiss National Foundation grant FN31–65403 (C.P.), the Canton of Geneva (C.P.), and Deutsche Forschungsgemeinschaft (SFB 594; M.H.-H.).
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Supplemental material: see www.proteinscience.org
Article published online ahead of print. Article and publication date are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04773204.
References
- Alber, T. 1993. How GCN4 binds DNA. Curr. Biol. 3 182–184. [DOI] [PubMed] [Google Scholar]
- Andreadis, J.D. and Black, L.W. 1998. Substrate mutations that bypass a specific Cpn10 chaperonin requirement for protein folding. J. Biol. Chem. 273 34075–34086. [DOI] [PubMed] [Google Scholar]
- Ang, D., Keppel, F., Klein, G., Richardson, A., and Georgopoulos, C. 2000. Genetic analysis of bacteriophage-encoded cochaperonins. Annu. Rev. Genet. 34 439–456. [DOI] [PubMed] [Google Scholar]
- Ang, D., Richardson, A., Mayer, M.P., Keppel, F., Krisch, H., and Georgopoulos, C. 2001. Pseudo-T-even bacteriophage RB49 encodes CocO, a co-chaperonin for GroEL, which can substitute for Escherichia coli’s GroES and bacteriophage T4’s Gp31. J. Biol. Chem. 276 8720–8726. [DOI] [PubMed] [Google Scholar]
- Bax, A. and Davis, D.G. 1985. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 65 355–360. [Google Scholar]
- Braig, K., Simon, M., Furuya, F., Hainfeld, J.F., and Horwich, A.L. 1993. A polypeptide bound by the chaperonin groEL is localized within a central cavity. Proc. Natl. Acad. Sci. 90 3978–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braig, K., Otwinowski, Z., Hegde, R., Boisvert, D.C., Joachimiak, A., Horwich, A.L., and Sigler, P.B. 1994. The crystal structure of the bacterial chaperonin GroEL at 2.8 Å. Nature 371 578–586. [DOI] [PubMed] [Google Scholar]
- Brinker, A., Pfeifer, G., Kerner, M.J., Naylor, D.J., Hartl, F.U., and Hayer-Hartl, M. 2001. Dual function of protein confinement in chaperonin-assisted protein folding. Cell 107 223–233. [DOI] [PubMed] [Google Scholar]
- Buchner, J., Schmidt, M., Fuchs, M., Jaenicke, R., Rudolph, R., Schmid, F.X., and Kiefhaber, T. 1991. GroE facilitates refolding of citrate synthase by suppressing aggregation. Biochemistry 30 1586–1591. [DOI] [PubMed] [Google Scholar]
- Chaudhry, C., Farr, G.W., Todd, M.J., Rye, H.S., Brunger, A.T., Adams, P.D., Horwich, A.L., and Sigler, P.B. 2003. Role of the γ-phosphate of ATP in triggering protein folding by GroEL-GroES: Function, structure and energetics. EMBO J. 22 4877–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J., Walter, S., Horwich, A.L., and Smith, D.L. 2001. Folding of malate dehydrogenase inside the GroEL-GroES cavity. Nat. Struct. Biol. 8 721–728. [DOI] [PubMed] [Google Scholar]
- Coyle, J.E., Texter, F.L., Ashcroft, A.E., Masselos, D., Robinson, C.V., and Radford, S.E. 1999. GroEL accelerates the refolding of hen lysozyme without changing its folding mechanism. Nat. Struct. Biol. 6 683–690. [DOI] [PubMed] [Google Scholar]
- Dunker, A.K., Brown, C.J., Lawson, J.D., Iakoucheva, L.M., and Obradovic, Z. 2002. Intrinsic disorder and protein function. Biochemistry 41 6573–6582. [DOI] [PubMed] [Google Scholar]
- Dyson, H.J. and Wright, P.E. 2002. Coupling of folding and binding for unstructured proteins. Curr. Opin. Struct. Biol. 12 54–60. [DOI] [PubMed] [Google Scholar]
- Farr, G.W., Furtak, K., Rowland, M.B., Ranson, N.A., Saibil, H.R., Kirchhausen, T., and Horwich, A.L. 2000. Multivalent binding of nonnative substrate proteins by the chaperonin GroEL. Cell 100 561–573. [DOI] [PubMed] [Google Scholar]
- Fenton, W.A., Kashi, Y., Furtak, K., and Horwich, A.L. 1994. Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371 614–619. [DOI] [PubMed] [Google Scholar]
- Frankel, A.D. and Kim, P.S. 1991. Modular structure of transcription factors: Implications for gene regulation. Cell 65 717–719. [DOI] [PubMed] [Google Scholar]
- Fridmann, Y., Kafri, G., Danziger, O., and Horovitz, A. 2002. Dissociation of the GroEL-GroES asymmetric complex is accelerated by increased cooperativity in ATP binding to the GroEL ring distal to GroES. Biochemistry 41 5938–5944. [DOI] [PubMed] [Google Scholar]
- Hayer-Hartl, M.K., Martin, J., and Hartl, F.U. 1995. Asymmetrical interaction of GroEL and GroES in the ATPase cycle of assisted protein folding. Science 269 836–841. [DOI] [PubMed] [Google Scholar]
- Hunt, J.F., Weaver, A.J., Landry, S.J., Gierasch, L., and Deisenhofer, J. 1996. The crystal structure of the GroES co-chaperonin at 2.8 Å resolution. Nature 379 37–45. [DOI] [PubMed] [Google Scholar]
- Hunt, J.F., van der Vies, S.M., Henry, L., and Deisenhofer, J. 1997. Structural adaptations in the specialized bacteriophage T4 co-chaperonin Gp31 expand the size of the Anfinsen cage. Cell 90 361–371. [DOI] [PubMed] [Google Scholar]
- Hurlburt, B.K. and Yanofsky, C. 1990. Enhanced operator binding by trp superrepressors of Escherichia coli. J. Biol. Chem. 265 7853–7858. [PubMed] [Google Scholar]
- Iakoucheva, L.M., Brown, C.J., Lawson, J.D., Obradovic, Z., and Dunker, A.K. 2002. Intrinsic disorder in cell-signaling and cancer-associated proteins. J. Mol. Biol. 323 573–584. [DOI] [PubMed] [Google Scholar]
- Jeener, J., Meier, B.H., Bachmann, P., and Ernst, R.R. 1979. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 71 4546–4553. [Google Scholar]
- Keppel, F., Rychner, M., and Georgopoulos, C. 2002. Bacteriophage-encoded cochaperonins can substitute for Escherichia coli’s essential GroES protein. EMBO Rep. 3 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, G. and Georgopoulos, C. 2001. Identification of important amino acid residues that modulate binding of Escherichia coli GroEL to its various cochaperones. Genetics 158 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry, S.J., Zeilstra-Ryalls, J., Fayet, O., Georgopoulos, C., and Gierasch, L.M. 1993. Characterization of a functionally important mobile domain of GroES. Nature 364 255–258. [DOI] [PubMed] [Google Scholar]
- Landry, S.J., Taher, A., Georgopoulos, C., and van der Vies, S.M. 1996. Interplay of structure and disorder in co-chaperonin mobile loops. Proc. Natl. Acad. Sci. 93 11622–11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry, S.J., Steede, N.K., and Maskos, K. 1997. Temperature dependence of backbone dynamics in loops of human mitochondrial heat shock protein 10. Biochemistry 36 10975–10986. [DOI] [PubMed] [Google Scholar]
- Landry, S.J., Steede, N.K., Garaudy, A.M., Maskos, K., and Viitanen, P.V. 1999. Chaperonin function depends on structure and disorder in co-chaperonin mobile loops. In acific Symposium on Biocomputing ‘99 (eds. R.B. Altman et al.), pp. 520–531. World Scientific, Singapore. [DOI] [PubMed]
- Lo Conte, L., Chothia, C., and Janin, J. 1999. The atomic structure of protein-protein recognition sites. J. Mol. Biol. 285 2177–2198. [DOI] [PubMed] [Google Scholar]
- Lorimer, G. 1997. Protein folding. Folding with a two-stroke motor. Nature 388 720. [DOI] [PubMed] [Google Scholar]
- Minor, D.L. and Kim, P.S. 1994. Context is a major determinant of β-sheet propensity. Nature 371 264–267. [DOI] [PubMed] [Google Scholar]
- Nooren, I.M. and Thornton, J.M. 2003. Structural characterisation and functional significance of transient protein-protein interactions. J. Mol. Biol. 325 991–1018. [DOI] [PubMed] [Google Scholar]
- Papoian, G.A. and Wolynes, P.G. 2003. The physics and bioinformatics of binding and folding—an energy landscape perspective. Biopolymers 68 333–349. [DOI] [PubMed] [Google Scholar]
- Piotto, M., Saudek, V., and Sklenar, V. 1992. Gradient-tailored excitation for single-quantum NMR-spectroscopy of aqueous-solutions. J. Biomol. NMR 2 661–665. [DOI] [PubMed] [Google Scholar]
- Richardson, A., van der Vies, S.M., Keppel, F., Taher, A., Landry, S.J., and Georgopoulos, C. 1999. Compensatory changes in GroEL/Gp31 affinity as a mechanism for allele-specific genetic interaction. J. Biol. Chem. 274 52–58. [DOI] [PubMed] [Google Scholar]
- Saibil, H.R., Zheng, D., Roseman, A.M., Hunter, A.S., Watson, G.M.F., Chen, S., auf der Mauer, A., O’Hara, B.P., Wood, S.P., Mann, N.H., et al. 1993. ATP induces large quaternary rearrangements in a cage-like chaperonin structure. Curr. Biol. 3 265–273. [DOI] [PubMed] [Google Scholar]
- Shewmaker, F., Maskos, K., Simmerling, C., and Landry, S.J. 2001. The disordered mobile loop of GroES folds into a defined β-hairpin upon binding GroEL. J. Biol. Chem. 276 31257–31264. [DOI] [PubMed] [Google Scholar]
- Shoemaker, B.A., Portman, J.J., and Wolynes, P.G. 2000. Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. Proc. Natl. Acad. Sci. 97 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtilerman, M., Lorimer, G.H., and Englander, S.W. 1999. Chaperonin function: Folding by forced unfolding. Science 284 822–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sklenar, V., Piotto, M., Leppik, R., and Saudek, V. 1993. Gradient-tailored water suppression for H-1-N-15 Hsqc experiments optimized to retain full sensitivity. J. Magn. Reson. A 102 241–245. [Google Scholar]
- Todd, M.J., Viitanen, P.V., and Lorimer, G.H. 1993. Hydrolysis of adenosine 5′-triphosphate by Escherichia coli GroEL—Effects of GroES and potassium ion. Biochemistry 32 8560–8567. [DOI] [PubMed] [Google Scholar]
- ———. 1994. Dynamics of the chaperonin ATPase cycle: Implications for facilitated protein folding. Science 265 659–666. [DOI] [PubMed] [Google Scholar]
- van der Vies, S.M., Gatenby, A.A., and Georgopoulos, C. 1994. Bacteriophage-T4 encodes a co-chaperonin that can substitute for Escherichia coli GroES in protein folding. Nature 368 654–656. [DOI] [PubMed] [Google Scholar]
- Wishart, D.S., Sykes, B.D., and Richards, F.M. 1991. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J. Mol. Biol. 222 311–333. [DOI] [PubMed] [Google Scholar]
- Wright, P.E. and Dyson, H.J. 1999. Intrinsically unstructured proteins: Reassessing the protein structure-function paradigm. J. Mol. Biol. 293 321–331. [DOI] [PubMed] [Google Scholar]
- Xu, Z.H., Horwich, A.L., and Sigler, P.B. 1997. The crystal structure of the asymmetric GroEL-GroES-(ADP)(7) chaperonin complex. Nature 388 741–750. [DOI] [PubMed] [Google Scholar]
- Yifrach, O. and Horovitz, A. 1995. Nested cooperativity in the ATPase activity of the oligomeric chaperonin GroEL. Biochemistry 34 5303–5308. [DOI] [PubMed] [Google Scholar]
- Zeilstra-Ryalls, J., Fayet, O., and Georgopoulos, C. 1994. Two classes of extragenic suppressor mutations identify functionally distinct regions of the GroEL chaperone of Escherichia coli. J. Bacteriol. 176 6558–6565. [DOI] [PMC free article] [PubMed] [Google Scholar]