

Abstract
Changes in fluorescence were measured with an argon ion laser from the outer segments of isolated salamander rods containing the Ca2+-sensitive fluorescent dye fluo-5F. When the outer segments were exposed to a 0Ca2+/0Na+ solution designed to minimise surface membrane Ca2+ fluxes, exposure to intense light from the laser evoked a slow increase in fluorescence, reflecting a light-induced rise in outer segment [Ca2+]i. The time course of this slow fluorescence rise could be fitted with the sum of two asymptotic exponential functions of approximately equal amplitude, having time constants of approximately 200 ms and 5.7 s. When rods were exposed to saturating background light to reduce outer segment [Ca2+]i before laser illumination, the relative amplitude of the two exponentials was altered so as to reduce the contribution from the one with the shorter time constant. Examination of the initial time course of fluorescence when recording at high temporal resolution revealed a further rapid rise with a time constant of 1–2 ms, which could be observed even from rods in Ringer solution. This initial rapid rise could be abolished by pre-exposing the rod to bleaching illumination, whether the bleach was given in Ringer solution or in 0Ca2+/0Na+ solution. It would therefore appear that the rapid rise in fluorescence is generated in some way by the bleaching of the photopigment. Unlike the slower components of fluorescence increase, the rapid initial rise was virtually unaffected in waveform or amplitude when rods were pre-exposed in Ringer solution to light which was bright enough to suppress completely the circulating current but which bleached a negligible fraction of the photopigment. Furthermore, pre-incubation with the AM ester of the Ca2+ chelator BAPTA, although completely abolishing the slower components of fluorescence increase, had virtually no effect on the rapid rise. These results indicate that the rapid component, though triggered by rhodopsin bleaching, does not reflect an increase in outer segment [Ca2+]i. Neither the rapid nor the slower components of fluorescence increase were affected by exposure of the outer segment to 10 μm of the membrane-permeant compound N,N,N′,N′-tetrakis(2-pyridyl-methyl)ethylenediamine (TPEN), which chelates heavy metals such as Zn2+, or 100 μm 2-aminoethoxydiphenylborate (2-APB), a membrane-permeant blocker of IP3 receptors. These results appear to exclude a role for changes in heavy metal concentration or Ca2+ release via IP3 receptors in the light-induced increases in dye fluorescence. Estimates of absolute Ca2+ concentration and of rod buffering capacity suggest that the slower components of fluorescence increase represent the release of around 10–50 μmoles Ca2+ per litre cytoplasmic volume from bound or sequestered stores after bleaching.
The cytoplasmic free Ca2+ concentration ([Ca2+]i) within the outer segment is believed to play a crucial role in modulating the responses of vertebrate photoreceptors during light and dark adaptation (for recent reviews see Pugh et al. 1999; Fain et al. 2001). Ca2+ enters the outer segment through cyclic-nucleotide-gated channels (Yau & Nakatani, 1984a; Hodgkin et al. 1985) and leaves by means of a Na+/Ca2+-K+ exchange mechanism (Yau & Nakatani, 1984b; Hodgkin et al. 1987; Cervetto et al. 1989). The light-induced suppression of the outer segment conductance reduces Ca2+ influx and leads to a decrease in [Ca2+]i (Yau & Nakatani, 1985), which has been directly observed with the photoprotein aequorin (McNaughton et al. 1986), the ratiometric fluorescent dyes fura-2 (Ratto et al. 1988; McCarthy et al. 1994, 1996) and indo-1 (Gray-Keller & Detwiler, 1994), and the fluo family of dyes fluo-3, fluo-4, and fluo-5F (Sampath et al. 1998, 1999; Matthews & Fain, 2001a).
In addition to this reduction in [Ca2+]i, we have recently shown that light sufficiently intense to bleach a significant fraction of the photopigment can also evoke a release of Ca2+ from within the outer segment (Matthews & Fain, 2001a). This release of Ca2+ at the onset of bleaching illumination slows the decline in [Ca2+]i which accompanies the suppression of the circulating current in Ringer solution, and can be detected as an overt rise in [Ca2+]i when the outer segment is exposed to a 0Ca2+/0Na+ solution designed to minimise surface membrane Ca2+ fluxes (Matthews et al. 1988; Nakatani & Yau, 1988; Fain et al. 1989). We have now characterised the kinetics of this Ca2+ release by making fluorescence measurements over a wider bandwidth. Our experiments show that the release of Ca2+ can be characterised by two kinetic processes with time constants of approximately 200 ms and 5.7 s, whose amplitudes are differentially affected by prior light exposure and which may represent distinct sites for Ca2+ release. They also reveal a further much more rapid initial increase in fluorescence with a time constant of a few milliseconds, which is also evoked by the bleaching of rhodopsin but does not appear to reflect an increase in [Ca2+]i. Finally, estimates of absolute Ca2+ concentration and of rod buffering capacity suggest that of the order of 10–50 μmol Ca2+ per litre cytoplasmic volume are released from bound or sequestered stores after bleaching. Preliminary results of this study have been reported to the Association for Research in Vision and Ophthalmology (Matthews & Fain, 2001b).
Methods
Preparation
Aquatic tiger salamanders (Ambystoma tigrinum) purchased from Charles Sullivan (Nashville, TN, USA) were dark-adapted overnight. They were killed by decapitation and pithing under Home Office licence, their eyes removed, and the photoreceptors dissociated mechanically under infrared illumination (Matthews, 1996). Dissociated cells were incubated as described previously (Sampath et al. 1998) for 30 min with 10 μm fluo-5F acetoxymethyl ester (fluo-5F AM; Molecular Probes Europe, Leiden, Holland) in amphibian Ringer solution consisting of: 111 mm NaCl, 2.5 mm KCl, 1.6 mm MgCl2, 1.0 mm CaCl2, 10 μm EDTA, and 3 mm Hepes adjusted to pH 7.7-7.8 with NaOH. The Ca2+ chelator 1, 2-bis(2-aminophenoxy)-ethane-N,N,N‘,N‘-tetraacetic acid (BAPTA) was incorporated by including 50–100 μm BAPTA AM (Calbiochem, Nottingham, UK) together with 10 μm fluo-5F in the incubation medium. For the experiments of Fig. 6B, patch pipettes were filled with 100 μm free fluo-5F (Molecular Probes Europe) in a pseudo-intracellular solution containing 92 mm potassium aspartate, 7 mm NaCl, 5 mm MgCl2, 10 mm Hepes, 1 mm adenosine 5′-triphosphate (ATP), 1 mm guanosine 5′-triphosphate (GTP), and 20 μm BAPTA with no added CaCl2, pH 7.1. N,N,N‘,N‘-tetrakis(2-pyridyl-methyl)ethylenediamine (TPEN) and 2-aminoethoxydiphenyl borate (2-APB) were obtained from Sigma (Sigma-Aldrich Company Ltd, Poole, UK).
Figure 6. Effect of BAPTA and free fluo-5F on the magnitude of rapid and slow components of fluorescence increase in 0Ca2+/0Na+ solution.
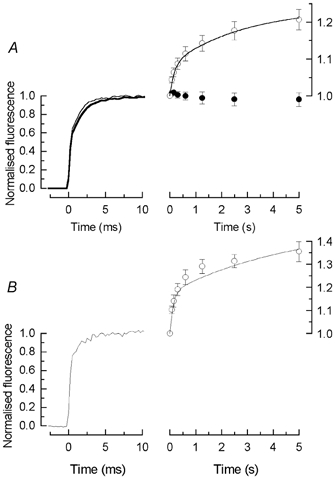
A, traces to the left compare the mean waveform for the initial 10 ms of the first laser flash, for rods incubated in Ringer solution containing 100 μm BAPTA AM together with 10 μm fluo-5F (thin trace), and for matched controls from the same retina incubated in 10 μm fluo-5F alone (thick trace). Responses have been normalised by setting the mean value of the fluorescence during the interval 15–19 ms equal to 1.0. Data to the right plot mean ± s.e.m. of the normalised photomultiplier current for each of 8 laser flashes for the same protocol as in Fig. 1. •, BAPTA AM; ○, matched controls (8 cells in each condition). Current amplitude has been measured as mean current during the interval 15–19 ms after beginning of laser flash and has been normalised to the amplitude of the first laser flash. Continuous curve is eqn (2) plotted with A1 / 0.08 ± 0.02, τ1= 0.17 ± 0.04 s, A2 = 0.16 ± 0.03, and τ2 = 2.9 ± 0.7 s determined as the means of the values obtained by fitting eqn (2) individually to the normalised data for each control cell (8 cells, mean ± s.e.m.). B, rapid and slow components of fluorescence increase for rods filled with free fluo-5F from a patch pipette (see Methods). Data to left give the mean initial time course of the fluorescence evoked by the first laser flash for 5 rods, normalised as in A. Data to the right plot mean ± s.e.m. of the photomultiplier current for each of 8 laser flashes for 3 of these cells using the same protocol as in Fig. 1, measured and normalised as in A; in the remaining 2 cells the individual fits to eqn (2) did not converge. Continuous curve is eqn (2) plotted with A1 = 0.18 ± 0.05, τ1 = 0.13 ± 0.04 s, A2 = 0.29 ± 0.10, and τ2 = 4.7 ± 3.6 s determined as the means of the values obtained by fitting eqn (2) individually to the normalised data for each cell (3 cells, mean ± s.e.m.).
An isolated rod was drawn inner segment first into a suction pipette so that the outer segment was exposed to the bathing solution. Rapid solution changes from Ringer solution to 0Ca2+/0Na+ solution were effected by laterally translating the microscope stage with a computer-controlled stepping motor (Matthews, 1996; Matthews & Fain, 2001a). The 0Ca2+/0Na+ solution was identical in composition to the ‘0 Ca2+, 0 Mg2+, 0 Na+ solution’ used previously (Matthews, 1996; Matthews & Fain, 2001a) and consisted of 111 mm choline chloride, 2.5 mm KCl, 2 mm EGTA, and 3.0 mm Hepes, adjusted to pH 7.7-7.8 with tetramethylammonium hydroxide.
Ca2+ measurement, recording and light stimulation
Free Ca2+ was measured as described previously (Matthews & Fain, 2000, 2001a). Dye fluorescence was excited by an air-cooled argon ion laser (Model 60, American Laser Corporation, Salt Lake City, UT, USA) tuned to 488 nm in light-control mode and collected with a 505 nm dichroic mirror and a 510 nm emission filter (Types 505DRLP and 510ALP, Omega Optical, Brattleboro, VT, USA). This excitation wavelength corresponds well to the maximum absorbance of fluo-5F, which is designed for maximum brightness with this argon line. The intensity of the laser was adjusted with reflective neutral density filters to 1.5-2 × 1011 photons μm−2 s−1, a factor of 10 higher than used previously (Matthews & Fain, 2001a) in order to reduce photon shot noise and hence to improve the signal-to-noise ratio over the wider bandwidth of our recordings. This increased laser intensity has the consequence that the overwhelming majority of the photopigment within the area of the laser spot will have been bleached by the first 20 ms laser flash. In the majority of experiments, noise due to laser intensity fluctuations was reduced with a beam stabiliser (Noise Eater, model CR-200A, Thorlabs, Newton, NJ, USA).
The intensity of fluorescence was measured with a photomultiplier tube having a Rb-Cs photocathode and antimony-caesium dynodes (Model 9124A, Electron Tubes Ltd, Ruislip, UK). This tube provided linearity of 1 % or better for anode currents of up to 20 μA when used with a resistive divider chain having 100 kΩ per stage. This gave a maximum working intensity an order of magnitude greater than for the photomultiplier used previously (Matthews & Fain, 2001a), though with a somewhat higher dark count. Such high anode currents were required to minimise the contribution of photon shot noise at the wide bandwidth used in these experiments. The signal from the photomultiplier was amplified as described previously (Matthews & Fain, 2001a) but filtered at 1 kHz and digitally sampled at 4 kHz. Multiple exponentials were fitted to the fluorescence data using the least squares algorithm of the plotting program Origin (OriginLab Corporation, Northampton, MA, USA).
Light stimuli were delivered via a dual-beam optical bench. The brightest intensity which could be delivered by the optical stimulator at 500 nm was of the order of 6–7 × 106 photons μm−2 s−1, which was typically attenuated with calibrated, neutral density filters. In some experiments, instead of 500 nm light the rods were stimulated with white light, whose absolute intensity had been estimated previously relative to 500 nm stimulation (Matthews & Fain, 2001a). Rhodopsin bleaching was calculated from the photosensitivity for a vitamin-A2-based pigment in solution (Dartnall, 1972), corrected for the difference in dichroism in free solution and disk membranes (Jones et al. 1993).
Results
We have previously shown that when the outer segment of a rod containing the fluorescent dye fluo-5F is rapidly exposed to 0Ca2+/0Na+ solution and then illuminated with intense laser light, there is an initial increase in fluorescence over approximately the first 5 s of laser illumination that can be suppressed by incorporation of the Ca2+ chelator BAPTA and reflects a light-induced release of Ca2+ within the rod outer segment (Matthews & Fain, 2001a). To characterise more quantitatively the time course of this release, we exposed the rod outer segment in 0Ca2+/0Na+ solution to a series of laser flashes as shown in Fig. 1A. Laser flashes 20 ms in duration were given at time intervals logarithmically spaced over a period of 5 s. The arrows in Fig. 1A indicate the amplitude of the photomultiplier current and hence the level of fluorescence for the first and last laser flashes (P1 and P8). During the sequence of laser flashes, the photomultiplier current increased from 10.5 to 13.1 μA, corresponding to a ratio P8/P1 of approximately 1.25.
Figure 1. Fluo-5F fluorescence recorded from a salamander rod in 0Ca2+/0Na+ solution in response to brief laser flashes.
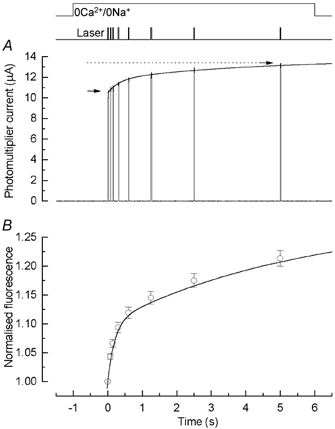
A, dark-adapted rod was stepped to 0Ca2+/0Na+ solution 1 s before a series of 8 laser flashes 20 ms in duration, spaced at approximately logarithmically equal intervals, beginning at the following times (ms): 0, 75, 150, 300, 600, 1250, 2500, and 5000 (i.e. 5 s). Arrows give magnitude of fluorescence for first and last laser flashes (P1 and P8). Continuous curve is eqn (2) fitted to the data (see text). B, summary of fluorescence amplitudes in 0Ca2+/0Na+ solution for laser flash protocol as in A, normalised individually for each cell to the amplitude of fluorescence evoked by the first laser flash. For each laser flash, photomultiplier current is given as the mean current during the interval 15–19 ms after the beginning of the fluorescence response. Data points are mean ± s.e.m. for 29 rods. Continuous curve is eqn (2) plotted with A1 / 0.10 ± 0.01, τ1 = 0.20 ± 0.02 s, A2 = 0.18 ± 0.02, and τ2 = 5.7 ± 0.7 s (mean ± s.e.m., 29 cells) determined as the means of the values obtained by fitting eqn (2) individually to the normalised data from each cell (see text).
The increase in fluorescence was poorly fitted by a single exponential asymptotic rise:
| (1) |
but could be adequately described by the sum of two exponentials:
| (2) |
This expression has been fitted to the raw photomultiplier current of Fig. 1A, yielding short and long time constants of 0.33 s and 4.2 s, respectively, with a value of 1.5 for the ratio of their amplitudes A2 /A1.
A total of 31 rods were stimulated in 0Ca2+/0Na+ solution with the series of laser flashes used for the cell in Fig. 1A. For each cell the mean fluorescence was measured over the interval 15–19 ms from the beginning of each 20 ms laser flash, in order to avoid a contribution from an initial rapid fluorescence rise to be described subsequently (see Fig. 2). The fluorescence values for each cell were then normalised to the fluorescence evoked by the first flash. The mean increase in fluorescence, given by the ratio P8/P1, was 1.21 ± 0.01 but varied in individual cells from 1.06 to 1.38. In 29 of these 31 cells the data were fitted individually for each cell with the double exponential asymptotic rise of eqn (2) with the value of i0 constrained to unity, after normalising the photomultiplier current for each cell individually according to the current amplitude evoked by the first flash. In the remaining two cells the fitting procedure did not converge, and they have been omitted from the mean data of Fig. 1B. The mean values of the free parameters obtained individually from each of the 29 cells were: A1 / 0.10 ± 0.01, τ1 = 0.20 ± 0.02 s, A2 = 0.18 ± 0.02, and τ2 = 5.7 ± 0.7 s (mean ± s.e.m., 29 cells). These data are summarised in Fig. 1B, which superimposes upon the mean values of normalised fluorescence evoked by each laser flash the continuous curve given by eqn (2) plotted using these mean parameter values. This function provides a good description of the mean data, indicating the presence of two distinct kinetic processes of Ca2+ release. It is of some interest that the values of the two time constants differed by more than an order of magnitude (see Discussion).
Figure 2. Rapid component of fluorescence response.
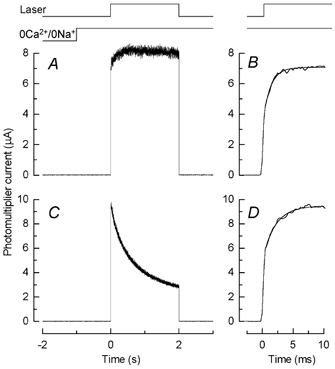
A, dark-adapted rod was stepped to 0Ca2+/0Na+ solution 1 s before beginning of a 2 s laser exposure. Monitor traces (top) indicate timing of laser exposure and solution change. Note rapid rising phase of fluorescence increase. B, initial time course of fluorescence increase for cell in A plotted on a more rapid time base. Photomultiplier current has been fitted with eqn (1) with a best-fitting value for the time constant τ0 of 1.2 ms. C and D, fluorescence measurements from a different rod with the same protocol as for A and B except that recordings were made in Ringer solution instead of in 0Ca2+/0Na+ solution. Rapid component of fluorescence increase in D fitted with eqn (1) but with τ0 / 2.0 ms.
An additional component of rapid fluorescence increase
When the waveform of the photomultiplier current was examined at higher temporal resolution, an even faster rise in fluorescence was revealed (Fig. 2). In Fig. 2A is shown the response of a dark-adapted rod in 0Ca2+/0Na+ solution to a 2 s laser exposure. The initial rounding of the current waveform, which was only present for the first laser flash in the series, is plotted on a more rapid time base in Fig. 2B, where the current has been fitted with a single exponential asymptotic rise (eqn (1)). The best fitting value for the time constant τ0 for this cell was 1.2 ms, but the mean of this parameter from 48 cells exposed to 0Ca2+/0Na+ solution was somewhat larger, with a value of 1.8 ± 0.15 ms (mean ± s.e.m.). Note that this is two orders of magnitude shorter than the time constant for the faster of the two kinetic components of Ca2+ release, which thus makes a negligible contribution over this time scale.
It is difficult to estimate accurately the amplitude of this rapid component because of the limited frequency response of the fluorescent dye itself (Escobar et al. 1997). Nevertheless, for the purposes of comparison, the current at zero time (i0) was estimated by fitting eqn (1) individually to each cell and back-extrapolating to time zero. The fractional contribution of the fluorescence response to the first laser flash contributed by the rapid component (frapid) was then calculated for each cell according to:
| (3) |
When calculated in this way, frapid was 0.56 of the total response for the cell of Fig. 2B and 0.55 ± 0.02 (mean ± s.e.m.) of the response for the 48 cells exposed to laser light in 0Ca2+/0Na+ solution.
This calculation shows that the initial component of fluorescence increase accounts for a large fraction of the total amplitude of the signal in Fig. 2B. It should also be sufficiently rapid to be recorded even from cells in Ringer solution. Previous measurements in Ringer solution have been made with recordings of lower bandwidth and have not revealed a rapid increase in fluorescence like that shown in Fig. 2B. Instead, as we and others have shown, the signal from a fluorescent or luminescent Ca2+ indicator measured from cells in Ringer solution shows a prominent decrease following the rapid suppression of the dark current upon bright light exposure (as in Fig. 2C) as Ca2+ is removed from the cell by Na+/Ca2+-K+ exchange (McNaughton et al. 1986; Ratto et al. 1988; Gray-Keller & Detwiler, 1994; McCarthy et al. 1994, 1996; Sampath et al. 1998, 1999). This decrease has been described as the sum of at least two exponential functions of the form:
| (4) |
where A3 and A4 are approximately equal in amplitude and sum to give the apparent value of the current at the beginning of the laser exposure. Our previous measurements of the decline in [Ca2+]i which accompanies laser exposure in Ringer solution yield values for τ3 and τ4 of 0.8-0.9 s and 7–8 s, respectively (Matthews & Fain, 2001a). In contrast, the rapid increase in fluorescence is mostly complete within the first 10 ms (Fig. 2B) within which interval eqn (4) would predict that Ca2+ extrusion would cause the fluorescence to decrease by less than 1 % of its apparent initial value. It is therefore not surprising that cells in Ringer solution recorded over a wide bandwidth also show an initial component of fluorescence increase. This is illustrated in Fig. 2D, where the photomultiplier current has again been fitted with a single exponential with a time constant in this case of 2.0 ms. For 28 cells from which similar measurements were made in Ringer solution, the mean time constant fitted individually to the fluorescence increase in each cell was 1.8 ± 0.1 ms (s.e.m.), a value not significantly different from the time constant in 0Ca2+/0Na+ solution; frapid as calculated from eqn (3) was 0.45 ± 0.02, a value somewhat smaller than for the responses in 0Ca2+/0Na+ solution.
Effect of bleaching light on the rapid and slower components of fluorescence increase
When responses to a series of laser flashes were recorded as in Fig. 1A, the initial rapid component of fluorescence was only apparent for the first flash in the series. Since this first flash at the intensity used in the experiment of Fig. 1 was sufficiently bright to have bleached essentially all of the rhodopsin within the area of the laser spot, the absence of a rapid increase for the second laser flash suggested that this component of fluorescence might depend in some way on rhodopsin bleaching.
This possibility was investigated further in the experiments illustrated in Fig. 3. In Fig. 3A, a rod was stepped into 0Ca2+/0Na+ solution in darkness and kept there for 30 s. Then, still in 0Ca2+/0Na+ solution, the outer segment was exposed to the same series of laser flashes used in Fig. 1, which evoked a clear increase in fluorescence. The responses to the first two laser flashes are shown for this cell at higher temporal resolution in Fig. 3B. The first laser flash (trace 1) evoked a rapid rise in fluorescence, and has been fitted with a single exponential with a time constant 1.4 ms. This is in contrast to the second laser flash (trace 2), for which no rise could be detected. The value of frapid was 0.48, much as for rods exposed to 0Ca2+/0Na+ solution for a much briefer period in darkness (see Fig. 2B). Mean values from individually normalised fits to all of the rods for which this protocol was employed are given in Table 1.
Figure 3. Effect of bleaching light on rapid and slow components of fluorescence increase.
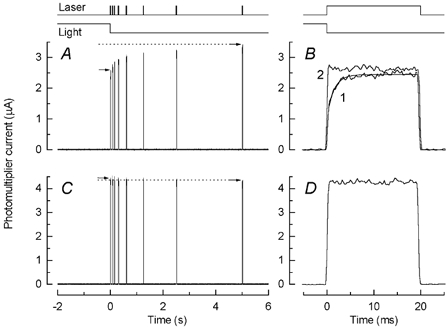
A, a dark-adapted rod was stepped into 0Ca2+/0Na+ solution and kept there for 30 s. The rod, still in 0Ca2+/0Na+ solution, was exposed to the same series of 20 ms laser flashes used in Fig. 1. A, rod maintained in darkness until first laser flash. The response to the first (trace 1) and second (trace 2) laser exposures for this cell are shown at higher temporal resolution in B; trace 1 has been fitted with eqn (1) with τ0 / 1.4 ms. C, protocol as for A except that rod was moved into 0Ca2+/0Na+ solution and then 1 s later exposed for 3 s to a white light of equivalent intensity of 2.4 × 108 photons μm−2 s−1(at 500 nm), sufficiently bright to have bleached 100 % of the rhodopsin in the outer segment. Arrows in A and C give magnitude of fluorescence for first and last laser flashes (P1 and P8). Note elimination of slow rise in fluorescence. D, response to first laser flash for cell in C. Note absence of rapid component of fluorescence increase.
Table 1.
Effect of prior illumination on rapid and slower components of fluorescence increase
| Prior illumination | frapid | τ (ms) | P8/P1 | n |
|---|---|---|---|---|
| None (darkness) | 0.55 ± 0.03 | 1.7 ± 0.3 | 1.32 ± 0.03 | 8 |
| 0.03% | 0.50 ± 0.04 | 1.7 ± 0.2 | 1.28 ± 0.01 | 8 |
| 100% | — | — | 0.98 ± 0.01* | 8 |
Prior illumination for 30 s in 0Ca2+/0Na+ solution, as in Fig. 3. Intensity of prior illumination given as percentage of rhodopsin bleached by 30 s light exposure (see text). Values given as means ± s.e.m., values for frapid and τ obtained from eqn (3) fitted individually to each cell.
Value of P8/P1 for 100% bleach significantly different at 5% level (Student's t test) both from value for 0.03% bleach and from value for darkness. For 100% bleach, the rapid initial fluorescence increase was too small for frapidand τ to be determined.
In Fig. 3C another dark-adapted rod was stepped into 0Ca2+/0Na+ solution but was then exposed for 30 s to a light sufficiently bright to have bleached 100 % of the rhodopsin in the outer segment. Since the light exposure took place in 0Ca2+/0Na+ solution, it would have produced only a relatively modest decrease in [Ca2+]i during that period (see Matthews & Fain, 2001a). Nevertheless, prior bleaching of the visual pigment eliminated the rapid component of the fluorescence increase (Fig. 3D), yielding a fluorescence trace indistinguishable from that obtained under the same recording conditions from a film of fluo-5F in agar. Any fractional contribution of this rapid rise, frapid, was too small to be measured under these conditions. Bleaching of the visual pigment also eliminated the slower increase in fluorescence to a series of laser flashes in 0Ca2+/0Na+ solution (Fig. 3C and Table 1, see also Matthews & Fain, 2001a).
The suppression of the fast component of fluorescence increase in Fig. 3D was produced by bleaching of rhodopsin and not by light exposure per se, since if the intensity of the 30 s exposure was reduced to 1.9 × 103 photons μm−2 s−1, sufficiently bright to suppress completely the circulating current but to bleach only 0.03 % of the visual pigment, the mean amplitude and time constant of the fast component were not significantly different (Student's t test, 5 % level) from those for rods kept in darkness during this 30 s period (see Table 1).
Effect of sub-bleaching light on the rapid and slower components of fluorescence increase
The abolition of the initial component by prior exposure to intense light suggested that this rapid fluorescence increase was triggered in some way by rhodopsin bleaching. It was therefore important to determine whether it represents an increase in [Ca2+]i, as is the case for the later components of fluorescence increase (Matthews & Fain, 2001a).
One approach to this question, illustrated in Fig. 4, involves pre-exposure of the rod to light; in contrast to Fig. 3, however, the period of illumination was only 3 s in duration, was initially delivered in Ringer solution, and was chosen to be of such an intensity as to suppress completely the circulating current but not to bleach a significant fraction of the photopigment. We have shown previously that just-saturating light gradually depletes the pool of Ca2+ available to be released by bleaching, presumably as a secondary consequence of the drop in outer segment [Ca2+]i (Matthews & Fain, 2001a). The depletion of the pool has a time constant of around 4 s and so occurs more slowly than the faster of the two exponential components (τ / 0.8-0.9 s) of the decrease in [Ca2+]i following the suppression of the circulating current in Ringer solution (see Fig. 2C and Matthews & Fain, 2001a). This has the consequence that a brief pre-exposure to saturating illumination evokes a relatively small decrease in the releasable pool but a larger decrease in [Ca2+]i, so that the relative rise in [Ca2+]i produced by a series of laser flashes in 0Ca2+/0Na+ solution is larger after a brief light exposure in Ringer solution than if no previous light had been given (Matthews & Fain, 2001a).
Figure 4. Effect of prior illumination in Ringer solution on the magnitude of rapid and slow components of fluorescence increase in 0Ca2+/0Na+ solution.
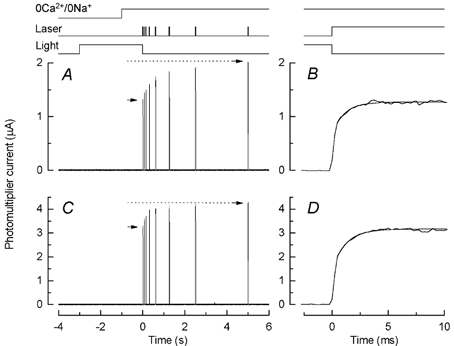
A, fluorescence responses evoked by a series of 20 ms laser flashes in 0Ca2+/0Na+ solution as in Fig. 1A. Arrows give magnitude of fluorescence for first and last laser flashes (P1 and P8). Rod was exposed to 3 s illumination of intensity 1.9 × 103 photons μm−2 s−1, bright enough to suppress completely the dark current but to bleach only 0.003 % of the rhodopsin in the outer segment over the duration of the exposure. Timing of illumination given by uppermost traces. B, response of rod in A to first laser flash on a more rapid time base. Rapid component of fluorescence increase has been fitted with eqn (1) with τ0 / 1.07 ms. C, same protocol as for A but with no light exposure before the first laser flash. Note smaller proportional increase in fluorescence between first and last laser exposure (arrows). D, response of rod in C to first laser flash on more rapid time base. Rapid component of fluorescence increase has been fitted with eqn (1) with τ0 = 1.21 ms.
This effect can be seen by comparing Fig. 4A with Fig. 4C. For the rod of Fig. 4A, which was pre-exposed for a brief period in Ringer solution to just-saturating light, P8/P1 was 1.56, whereas for the matched control of Fig. 4C, in which the rod was maintained in darkness, it was 1.28, a value similar to that in Fig. 1. The rise in [Ca2+]i after brief light exposure was therefore proportionately larger, although its absolute value would of course have diminished since some decrease of the releasable pool would have accompanied the light exposure before the first laser flash was delivered (Matthews & Fain, 2001a).
If the rapid increase in fluorescence also reflected a release of Ca2+ from some store within the outer segment, it might similarly be expected to become a larger fraction of the total response after pre-illumination in Ringer solution. Comparison of the responses in Fig. 4B and D shows, however, that this is not the case. For the cell exposed to just-saturating light in Fig. 4B, τ0 was 1.07 ms and frapid was 0.48; whereas for the cell maintained in darkness in Fig. 4D, τ0 was 1.21 ms and frapid was 0.54. Exposure to the background appeared not to augment the fraction of the fluorescence increase attributable to the rapid component but, if anything, to decrease it slightly.
Figure 5 summarises the results for all of the cells which were exposed to sub-bleaching illumination in Ringer solution. The cells in Fig. 5A were treated as in Fig. 4: the light was present for a total of 3 s, the first 2 s in Ringer solution followed by 1 s in 0Ca2+/0Na+ solution. In Fig. 5B, the light was present for a total of 30 s, the first 29 s in Ringer solution then 1 s in 0Ca2+/0Na+ solution. The traces to the left show on a fast time scale the mean normalised increase in fluorescence during the first laser flash for cells kept in darkness (thick trace) and for cells pre-exposed to light (thin trace). The plots to the right give the response to the sequence of laser flashes after prior light exposure (○) and for matched controls in darkness (•), normalised to the response to the first flash (P1). The ratio P8/P1 was 1.71 ± 0.06 (n = 8) for the cells exposed to light for 3 s but fell to 1.35 ± 0.04 (n = 7) for the cells exposed for 30 s. This decrease reflects the gradual decline in the pool of Ca2+ available to be released (Matthews & Fain, 2001a). As in Fig. 1B, the peak fluorescence evoked by each laser flash was fitted for each cell with eqn (2). It is of some interest that the ratio A1 /A2 of the mean amplitudes of the fast and the slow increases in fluorescence are smaller after light exposure that in darkness, decreasing for both the 3 s and the 30 s light exposures, in the former case from 1.1 ± 0.3 to 0.48 ± 0.05 (mean ± s.e.m., n = 8), and in the latter from 0.59 ± 0.15 to 0.29 ± 0.07 (mean ± s.e.m., n = 7). The reduction in this ratio may reflect preferential release during light exposure from a pool of Ca2+ which is bound less tightly to sites within the outer segment (see Discussion).
Figure 5. Summary of the fluorescence increase in 0Ca2+/0Na+ solution after prior illumination in Ringer solution.
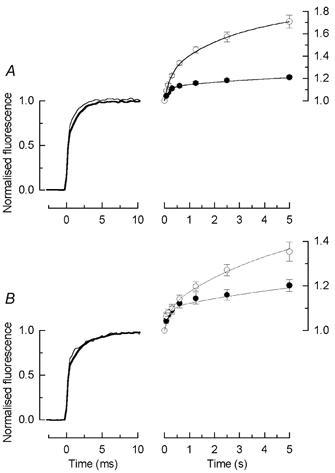
Experimental protocol as for Fig. 4: before presentation of first laser flash, rods were exposed to light of intensity 1.9 × 103 photons μm−2 s−1, bright enough to suppress completely the circulating current but not to produce significant bleaching of rhodopsin. A, duration of illumination was 3 s (2 s in Ringer solution, 1 s in 0Ca2+/0Na+ solution), as in Fig. 4A; B, duration of illumination was 30 s (29 s in Ringer solution, 1 s in 0Ca2+/0Na+ solution). In both A and B, traces to the left compare the mean waveform during the initial 10 ms of the first laser flash (8 cells in each condition). Thin traces: rods exposed to prior illumination; thick traces, matched controls from the same retina treated identically but kept in darkness until the first laser flash. Data to the right plot mean ± s.e.m. of fluorescence evoked by each of the 8 laser flashes as in Fig. 4A and C; ○, after prior light exposure; •, matched controls in darkness. Amplitude has been measured as mean current during the interval 15–19 ms after the beginning of the laser flash and has been normalised for each cell to the amplitude of the first laser flash. Continuous curves in the right hand panels plotted according to eqn (2) with parameters set to the mean values obtained by fitting eqn (2) individually to the normalised data from each cell. A, 3 s prior illumination (○, mean ± s.e.m., 8 cells): A1 / 0.25 ± 0.02, τ1 = 0.25 ± 0.02 s, A2 = 0.56 ± 0.06, τ2 = 3.0 ± 0.3 s; dark control (•, mean ± s.e.m., 8 cells): A1 = 0.12 ± 0.01, τ1 = 0.18 ± 0.02 s, A2 = 0.13 ± 0.02, τ2 = 5.2 ± 1.5 s. B, 30 s prior illumination (○, mean ± s.e.m., 7 cells; fit did not converge for remaining cell): A1 = 0.10 ± 0.02, τ1 = 0.23 ± 0.08 s, A2 = 0.43 ± 0.10, and τ2 = 5.3 ± 1 s; dark control (•, mean ± s.e.m., 7 cells; fit did not converge for remaining cell): A1 = 0.10 ± 0.02, τ1 = 0.18 ± 0.04 s, A2 = 0.20 ± 0.03, τ2 = 8.2 ± 1.8 s.
The experiments of Fig. 4 and Fig. 5 showed that pre-illumination dramatically altered the relative amplitudes of the slower kinetic components reflecting the release of Ca2+ within the outer segment but had relatively little effect on the amplitude or time constant of the rapid component of the fluorescence increase. The results in Fig. 5B are especially noteworthy. During a 29 s exposure to saturating light in Ringer solution, the Ca2+ concentration in the outer segment would be expected to decline from a resting level of several hundred nanomolar to of the order of 10 nm (H. R. Matthews & G. L. Fain, unpublished data). Nevertheless, the rapid component of the fluorescence increase remained nearly the same fraction of the total signal as in darkness. These observations would only be consistent with the rapid fluorescence increase representing a release of Ca2+ if the pool from which it originated were depleted by steady light to exactly the same degree as was free Ca2+ concentration, as would only be the case for release from a binding site of extremely low affinity.
Effect of BAPTA incorporation
In order to exclude the possibility that the rapid increase in fluorescence might result from the release of Ca2+ from a site of very low affinity, the Ca2+ chelator BAPTA was incorporated into the rod as its AM ester along with the fluorescent dye (see Methods).
The results of these experiments are presented in Fig. 6A. For eight cells without BAPTA incorporation (○), P8/P1 was 1.21 ± 0.03 (mean ± s.e.m.); whereas for eight cells from the same retina pre-incubated with BAPTA/AM, P8/P1 was 0.99 ± 0.02. These data confirm our previous observation (Matthews & Fain, 2001a) that BAPTA can suppress the slower fluorescence increase, indicating that it results from an actual release of Ca2+ within the outer segment.
To the left are plotted for these same groups of cells the mean normalised wave form of the rapid fluorescence increase, with the thick trace for control cells and the thin trace for the cells pre-incubated with BAPTA/AM. There is almost no difference between these two: τ0 and frapid for the BAPTA-incorporated cells were 1.64 ± 0.03 ms and 0.53 ± 0.04 (mean ± s.e.m., fitted individually to each cell), while for the controls without BAPTA the corresponding values were 1.99 ± 0.33 ms and 0.55 ± 0.04. We therefore conclude that the rapid component, although induced by the bleaching of rhodopsin, does not reflect an actual increase of [Ca2+]i in the outer segment.
Further evidence for this view is given in Fig. 6B, for which patch pipettes containing 100 μm of the free form of the dye fluo-5F were sealed onto the inner segment of a rod held by a suction pipette. After establishing a whole-cell recording, the dye was allowed to diffuse into the cytoplasm for 1 min, and the patch pipette was withdrawn so that the membrane could reseal. The inner segment of the cell was then drawn entirely into the suction pipette and the outer segment rapidly stepped into 0Ca2+/0Na+ solution and exposed to the same sequence of 20 ms laser flashes as in Fig. 1.
The trace on the left represents the average of the normalised fluorescence evoked by the first laser flash for five rods filled with free fluo-5F. The slower components of fluorescence increase are shown to the right for the three of these cells for which the double exponential fit to eqn (2) converged. A comparison of the amplitude of the rapid component can be made by viewing the thin trace to the left in Fig. 6A, the mean initial fluorescence increase for eight control cells filled with fluo-5F AM, with the corresponding trace in Fig. 6B for the five cells filled with free fluo-5F. Although the time constants were not significantly different, frapid was clearly smaller, averaging 0.37 ± 0.03 for the cells filled with free fluo-5F and 0.55 ± 0.04 for the eight control cells in Fig. 6A (or 0.55 ± 0.02 for the entire sample of 48 cells from which recordings were made). This difference in frapid was significant at the 0.01 level (Student's t test). Although our sample of cells filled with free fluo-5F was small, it would appear that the amplitude of the rapid component depended upon the way the dye was loaded into the cell (Zhao et al. 1997), more consistent with the notion that this component reflects some change in the property of the dye rather than an increase in [Ca2+]i.
TPEN and 2-APB
It seemed conceivable that the rapid fluorescence increase might be produced by a change in the concentration of some heavy metal, such as Zn2+, for which there is some evidence for a role in photoreceptor function (He et al. 2000; Ugarte & Osborne, 2001). This possibility was addressed by exposing the photoreceptor to the lipid-soluble heavy metal chelator N,N,N‘,N‘-tetrakis(2-pyridyl-methyl)ethylenediamine (TPEN), which has been shown to permeate cell membranes and has a reported affinity for Zn2+ of greater than 1015m−1 (Arslan et al. 1985). No effect of 10 μm TPEN was detected on either the rapid or slower components of fluorescence increase (see Table 2). This observation appears to exclude the possibility that either the rapid or slower components can be attributed to the release of heavy metal ions within the outer segment following bleaching, and reinforce our conclusion that the slower components of fluorescence increase represent an actual release of Ca2+ ions. At higher concentrations, TPEN appeared to block the cGMP-gated channels, but this effect was not examined in detail.
Table 2.
Effect of TPEN and 2-APB on rapid and slower components of fluorescence increase
| Drug | frapid | τ(ms) | P8/P1 | n |
|---|---|---|---|---|
| None (control) | 0.55 ± 0.02 | 2.0 ± 0.2 | 1.21 ± 0.01* | 31 |
| 10 μm TPEN | 0.50 ± 0.03 | 2.1 ± 0.4 | 1.18 ± 0.03 | 8 |
| 100 μm 2-APB | 0.49 ± 0.01 | 2.2 ± 0.3 | 1.29 ± 0.02* | 8 |
Values given as means ± s.e.m.
Values of P8/P1 for control rods and rods exposed to 2-APB were significantly different at 5% level, Student's t test. TPEN is N,N,N′,N′-tetrakis(2-pyridyl-methyl)-ethylenediamine, 2-APB is 2-aminoethoxydiphenylborate.
We also investigated a possible role for IP3-gated release of Ca2+ from intracellular stores in the slower components of fluorescence increase, since photoreceptors have been reported to contain both a PLCβ (Peng et al. 1997) and IP3 receptors (Wang et al. 1999). The membrane-permeant compound 2-aminoethoxydiphenylborate (2-APB) has been reported to block IP3-gated release in several cell types (see for example Ma et al. 2000; Chorna-Ornan et al. 2001). However for salamander rods, 100 μm 2-APB had little effect on either of the components of fluorescence increase (Table 2). The value for P8/P1 was actually significantly larger in 2-APB than for control rods (5 % level, Student's t test), but fell within the range observed in different animals and may not reflect a specific effect of the drug. These results appear to exclude a role for Ca2+ release via IP3 receptors in the light-induced increases in dye fluorescence.
Discussion
Our experiments show that when the rod outer segment is exposed to 0Ca2+/0Na+ solution to minimise fluxes of Ca2+ across the plasma membrane, fluo-5F fluorescence increases with multiple exponential kinetics, first very rapidly with a time constant of about 2 ms, then more slowly with time constants of about 200 ms and 5.7 ms. Neither the rapid nor the slower fluorescence increase was observed if the rod was first exposed to bleaching light. We therefore infer that both components must somehow be triggered by the photoisomerisation of rhodopsin by the intense light from the laser, which can be calculated to bleach the overwhelming majority of the photopigment within the area of the laser spot during the first laser flash.
Previous experiments have demonstrated that the slower increase in fluorescence is produced by an increase in [Ca2+]i in the outer segment (Matthews & Fain, 2001a). This seems not to be the case for the more rapid fluorescence increase, however, since the experiments of Fig. 4 and Fig. 5 show that the rapid increase is unaffected by treatments which greatly alter the properties of Ca2+ release. Furthermore, incorporation of the Ca2+ chelator BAPTA into the rod suppresses the slow increase in fluorescence but produces almost no change in the rapid increase.
Origin of rapid component of fluorescence increase
Recordings of fluo-5F fluorescence from mouse rod outer segments in Ringer solution (Woodruff et al. 2002) also show a rapid early component of fluorescence increase, which is suppressed by prior exposure to bleaching illumination and unaffected by BAPTA incorporation. The rapid component in mouse is unaltered by knockout of the G-protein transducin and has a more rapid rate of rise when the rate of rhodopsin bleaching is increased by increasing the intensity of the laser light. These observations, taken together with the present experiments on salamander rods, indicate that the initial component of fluorescence increase may reflect some change in the properties of the fluorescent dye induced by the activation of rhodopsin.
The time course of the rapid component of fluorescence increase is of the same order as the rate of the formation of the rhodopsin bleaching intermediate metarhodopsin II (Baumann, 1976). The formation of meta II produces a net uptake of protons with a time course similar to that of the initial fluorescence increase (Szundi et al. 1998; Meyer & Hofmann, 2000), which would cause the outer segment pH to rise. Although this change in pH is unlikely to affect the properties of the fluorescent dye (Grynkiewicz et al. 1985), it might cause some fraction of dye, previously bound and inaccessible, to be released into the outer segment cytoplasm.
A change in pH (or the bleaching of rhodopsin directly) might alter the extent of binding of dye to protein in the outer segment. There is considerable evidence for many fluorescent indicators that a fraction of dye can be bound to protein within the cell, and that the Kd of the dye is thereby altered, increasing with increased binding (see for example Hove-Madsen & Bers, 1992; Bassani et al. 1995; Baylor & Hollingworth, 2000). If alteration of pH or disk membrane surface charge by rhodopsin bleaching increased the binding of protein to the disk membrane, it might reduce the concentration of soluble protein in the outer segment, diminish binding of protein to the dye, decrease dye Kd, and thereby increase the amplitude of the fluorescence signal. Measurements of the diffusion of fluo-3 in bleached rods indicate that there is little or no binding of the dye to protein (Nakatani et al. 2002), but comparable measurements do not exist for dark-adapted rods.
It seems unlikely that the rapid component of fluorescence increase is produced by the movement of transducin, which is thought to come out of the cytoplasm and bind to the disks following photon absorption (Kuhn et al. 1981). Transducin binding occurs more slowly than the rapid rise in fluorescence we have observed, and a rapid fluorescence increase similar to the one we have seen in salamander rods is present in mouse rods lacking transducin (Woodruff et al. 2002). A similar argument would apply for other proteins later in the transduction cascade, such as for example the phosphodiesterase (Heck & Hofmann, 1993).
Whatever its cause, this initial rise in fluorescence is sufficiently rapid that it does not obscure either the slower increase in fluorescence in 0Ca2+/0Na+ solution, produced by light-dependent calcium release, or the decrease in fluorescence in Ringer solution, generated by Ca2+ efflux via the Na+/Ca2+-K+ exchanger. Indeed, this rapid component is so fast that it was not previously detected in earlier recordings over a lower bandwidth (Gray-Keller & Detwiler, 1994; Sampath et al. 1998; Matthews & Fain, 2001a). We cannot exclude the possibility that, in addition to the initial rise, there are also slower changes in dye properties that affect the time course or amplitude of the fluorescence changes we have recorded, but our experiments incorporating BAPTA into the rods suggest that these effects are likely to be small (see Fig. 6A and Matthews & Fain, 2001a).
The time course and magnitude of light-induced Ca2+ release
The slower increase in fluorescence rises in most cells with two distinct time constants, differing by more than an order of magnitude. The relative amplitudes of these two exponentials are differentially affected by light exposure (Fig. 5), suggesting that the more rapid of the two components (with a time constant of about 200 ms) is selectively depleted as [Ca2+]i is lowered. This observation is consistent with the notion that this component represents a fraction of Ca2+ binding within the outer segment to a site of relatively low affinity. In contrast, the slower time constant of [Ca2+]i increase might reflect a release of Ca2+ either from a binding site of higher affinity or from a sequestered pool such as the disks (Schröder & Fain, 1984; Fain & Schröder, 1990).
In order to estimate the quantity of Ca2+ released on exposure to bleaching light it is first necessary to translate the fluorescence increase into a rise in absolute Ca2+ concentration. This can most simply be done by expressing the rise in [Ca2+]i evoked by bleaching light as a fraction of the dark resting [Ca2+]i, for which several groups have reported measurements (McNaughton et al. 1986; Ratto et al. 1988; Gray-Keller & Detwiler, 1994; McCarthy et al. 1994, 1996; Sampath et al. 1998), yielding a value for [Ca2+]i in darkness of around 500–700 nm. Our demonstration of an initial rapid change in fluorescence at the onset of dye excitation does, however, have two potential implications for these measurements.
The first is the uncertainty as to whether existing measurements of the resting [Ca2+]i in darkness might have been compromised by a failure to resolve a rapid component of fluorescence increase. This seems to us to be readily dismissed, since the initial value of the rapid exponential component would correspond to an extrapolated value for [Ca2+]i in darkness only if the rapid component of fluorescence increase reflected an actual increase in [Ca2+]i. Our experiments show, however, that the rapid component is instead more likely to reflect a change in the accessibility or Kd of the fluorescent dye. Consequently, the best estimate of the dark resting [Ca2+]i will be provided by the level of fluorescence attained once the rapid component of fluorescence increase is complete, as would be obtained in measurements of lower bandwidth.
The second problem, as to whether the Kd value obtained in vitro can be used reliably in calculating [Ca2+]i from the fluorescence signal is not so readily resolved. As discussed above, the rapid component of fluorescence increase may reflect a change in the Kd of the dye induced by rhodopsin bleaching. If the latter were the case, then Kd values determined in vitro would not be appropriate. However, recent measurements indicate that binding of fluo-3 to intracellular components is minimal in bleached photoreceptors (Nakatani et al. 2002), suggesting that an effect of bleaching on dye Kd may make only a modest contribution in practice.
If the dark resting free-calcium concentration in a rod outer segment were 500 nm, and if light produces an increase in dye fluorescence of 6–38 % (mean 21 %, see Fig. 1B), then [Ca2+]i increases by 30–190 nm (mean 105 nm) if the modest non-linearity of fluo-5F fluorescence with Ca2+ concentration below the dye Kd is disregarded. In order to translate this increase in [Ca2+]i into the total quantity of released Ca2+, a value is required for the buffering capacity of the outer segment. In the detached gecko rod outer segment, the buffering capacity for Ca2+ has been estimated as 500 (Gray-Keller & Detwiler, 1994), implying that this increase in [Ca2+]i represents a total release of Ca2+ of 15–95 μmol per litre cytoplasmic volume (mean of about 50 μmol per litre cytoplasmic volume). In the intact salamander rod, two components of Ca2+ buffering have been detected (Lagnado et al. 1992), the first of low capacity and high affinity, binding 74 out of every 75 Ca2+ ions; the second of high capacity and low affinity, binding 15 out of every 16 Ca2+ ions. Since the value of [Ca2+]i in darkness is below the ≈0.7 μmKd of the high affinity, low capacity buffer, these measurements imply an effective buffering capacity of approaching one-fifth of that in gecko, corresponding to a net Ca2+ release of around 10 μmol per litre cytoplasmic volume. These numbers would be underestimated, however, if the Kd values for the indicator dyes were larger in the cytosol of the outer segment than in vitro in solution, as has been demonstrated for calcium-sensitive indicators in several other cell types (see for example Hove-Madsen & Bers, 1992; Bassani et al. 1995). These considerations suggest that the amount of Ca2+ released by light in the outer segment is unlikely to be much less than 10 μmol but probably not more than 100 μmol per litre cytoplasmic volume.
This estimate is at least an order of magnitude smaller than that from measurements of total outer segment calcium with enery dispersive X-ray microanalysis (EDX) and laser micromass analysis (Schröder & Fain, 1984; Fain & Schröder, 1990). The reason for this discrepancy is unclear. It may simply reflect some error in the calibration either of the free or total calcium in the rod. On the other hand, if some substantial fraction of total calcium were contained within the disks (Schnetkamp, 1979; Fain & Schröder, 1985), it is possible that some of this calcium could leave the outer segment without causing an increase in the fluorescence of dye in the cytosol. It also seems possible that there might be mechanisms of calcium homeostasis in the rod whose effects on the transport and free concentration of Ca2+ we still do not understand. It might be possible to resolve some of these uncertainties if the free concentration of Ca2+ within the disks could be measured. Recent measurements of disk pH and Ca2+ with fluorometric dyes (Chen et al. 2002) give hope that this may eventually prove possible.
Acknowledgments
This work was supported by a grant from the Wellcome Trust (to H.R.M.) and by a grant from the National Eye Institute of the National Institutes of Health (EY-01844 to G.L.F.).
References
- Arslan P, Di Virgilio F, Beltrame M, Tsien RY, PoANZZ T. Cytosolic Ca2+ homeostasis in Ehrlich and Yoshida carcinomas. A new, membrane-permeant chelator of heavy metals reveals that these ascites tumor cell lines have normal cytosolic free Ca2+ Journal of Biological Chemistry. 1985;260:2719–2727. [PubMed] [Google Scholar]
- Bassani JW, Bassani RA, Bers DM. Calibration of indo-1 and resting intracellular [Ca]i in intact rabbit cardiac myocytes. Biophysical Journal. 1995;68:1453–1460. doi: 10.1016/S0006-3495(95)80318-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann C. The formation of metarhodopsin380 in the retinal rods of the frog. Journal of Physiology. 1976;259:357–366. doi: 10.1113/jphysiol.1976.sp011470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor SM, Hollingworth S. Measurement and interpretation of cytoplasmic [Ca2+] signals from calcium-indicator dyes. News in Physiological Science. 2000;15:19–26. [PubMed] [Google Scholar]
- Cervetto L, Lagnado L, Perry RJ, Robinson DW, McNaughton PA. Extrusion of calcium from rod outer segments is driven by both sodium and potassium gradients. Nature. 1989;337:740–743. doi: 10.1038/337740a0. [DOI] [PubMed] [Google Scholar]
- Chen C, Jiang Y, Koutalos Y. Dynamic behaviour of rod photoreceptor disks. Biophysical Journal. 2002;83 doi: 10.1016/S0006-3495(02)73911-8. (in the Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorna-Ornan I, Joel-Almagor T, Ben-Ami HC, Frechter S, Gillo B, Selinger Z, Gill DL, Minke B. A common mechanism underlies vertebrate calcium signaling and Drosophila phototransduction. Journal of Neuroscience. 2001;21:2622–2629. doi: 10.1523/JNEUROSCI.21-08-02622.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dartnall HJA. Photosensitivity. In: Dartnall HJA, editor. Handbook of Sensory Physiology. 1. VII. Berlin: Springer-Verlag; 1972. pp. 122–145. [Google Scholar]
- Escobar AL, VeleZ P, Kim AM, Cifuentes F, Fill M, Vergara JL. Kinetic properties of DM-nitrophen and calcium indicators: rapid transient response to flash photolysis. Pflügers Archiv. 1997;434:615–631. doi: 10.1007/s004240050444. [DOI] [PubMed] [Google Scholar]
- Fain GL, Lamb TD, Matthews HR, Murphy RLW. Cytoplasmic calcium concentration as the messenger for light adaptation in salamader rods. Journal of Physiology. 1989;416:215–243. doi: 10.1113/jphysiol.1989.sp017757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Adaptation in vertebrate photoreceptors. Physiological Reviews. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- Fain GL, Schröder WH. Calcium content and calcium exchange in dark-adapted toad rods. Journal of Physiology. 1985;368:641–665. doi: 10.1113/jphysiol.1985.sp015881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fain GL, Schröder WH. Light-induced calcium release and re-uptake in toad rods. Journal of Neuroscience. 1990;10:2238–2249. doi: 10.1523/JNEUROSCI.10-07-02238.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray-Keller MP, Detwiler PB. The calcium feedback signal in the phototransduction cascade of vertebrate rods. Neuron. 1994;13:849–861. doi: 10.1016/0896-6273(94)90251-8. [DOI] [PubMed] [Google Scholar]
- GrynkiewicZ G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- He F, Seryshev AB, Cowan CW, Wensel TG. Multiple zinc binding sites in retinal rod cGMP phosphodiesterase, PDE6alpha beta. Journal of Biological Chemistry. 2000;275:20572–20577. doi: 10.1074/jbc.M000440200. [DOI] [PubMed] [Google Scholar]
- Heck M, Hofmann KP. G-protein-effector coupling: a real-time light-scattering assay for transducin-phosphodiesterase interaction. Biochemistry. 1993;32:8220–8227. doi: 10.1021/bi00083a024. [DOI] [PubMed] [Google Scholar]
- Hodgkin AL, McNaughton PA, Nunn BJ. The ionic selectivity and calcium dependence of the light-sensitive pathway in toad rods. Journal of Physiology. 1985;358:447–468. doi: 10.1113/jphysiol.1985.sp015561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, McNaughton PA, Nunn BJ. Measurement of sodium-calcium exchange in salamander rods. Journal of Physiology. 1987;391:347–370. doi: 10.1113/jphysiol.1987.sp016742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Madsen L, Bers DM. Indo-1 binding to protein in permeabilized ventricular myocytes alters its spectral and Ca binding properties. Biophysical Journal. 1992;63:83–97. doi: 10.1016/S0006-3495(92)81597-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GJ, Fein A, MacNichol EF, Jr, Cornwall MC. Visual pigment bleaching in isolated salamander retinal cones. Microspectrophotometry and light adaptation. Journal of General Physiology. 1993;102:483–502. doi: 10.1085/jgp.102.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Bennett N, Michel-VillaZ M, Chabre M. Interactions between photoexcited rhodopsin and GTP-binding protein: kinetic and stoichiometric analyses from light-scattering changes. Proceedings of the National Academy of Sciences of the USA. 1981;78:6873–6877. doi: 10.1073/pnas.78.11.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagnado L, Cervetto L, McNaughton PA. Calcium homeostasis in the outer segments of retinal rods from the tiger salamander. Journal of Physiology. 1992;455:111–142. doi: 10.1113/jphysiol.1992.sp019293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Patterson RL, van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol trisphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- McCarthy ST, Younger JP, Owen WG. Free calcium concentrations in bullfrog rods determined in the presence of multiple forms of Fura-2. Biophysical Journal. 1994;67:2076–2089. doi: 10.1016/S0006-3495(94)80691-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy ST, Younger JP, Owen WG. Dynamic, spatially nonuniform calcium regulation in frog rods exposed to light. Journal of Neurophysiology. 1996;76:1991–2004. doi: 10.1152/jn.1996.76.3.1991. [DOI] [PubMed] [Google Scholar]
- McNaughton PA, Cervetto L, Nunn BJ. Measurement of the intracellular free calcium concentration in salamander rods. Nature. 1986;322:261–263. doi: 10.1038/322261a0. [DOI] [PubMed] [Google Scholar]
- Matthews HR. Static and dynamic actions of cytoplasmic Ca2+ in the adaptation of responses to saturating flashes in salamander rods. Journal of Physiology. 1996;490:1–15. doi: 10.1113/jphysiol.1996.sp021123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR, Fain GL. Laser spot confocal technique to measure cytoplasmic calcium concentration in photoreceptors. Methods in Enzymology. 2000;316:146–163. doi: 10.1016/s0076-6879(00)16722-9. [DOI] [PubMed] [Google Scholar]
- Matthews HR, Fain GL. A light-dependent increase in free Ca2+ concentration in the salamander rod outer segment. Journal of Physiology. 2001a;532:305–321. doi: 10.1111/j.1469-7793.2001.0305f.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews HR, Fain GL. Time course of light-induced increase in Ca2+ in salamander rod outer segments. Investigative Ophthalmology and Visual Science. 2001b;42:S119. [Google Scholar]
- Matthews HR, Murphy RLW, Fain GL, Lamb TD. Photoreceptor light adaptation is mediated by cytoplasmic calcium concentration. Nature. 1988;334:67–69. doi: 10.1038/334067a0. [DOI] [PubMed] [Google Scholar]
- Meyer CK, Hofmann KP. Monitoring proton uptake from aqueous phase during rhodopsin activation. Methods in Enzymology. 2000;315:377–387. doi: 10.1016/s0076-6879(00)15855-0. [DOI] [PubMed] [Google Scholar]
- Nakatani K, Chen C, Koutalos Y. Calcium diffusion coefficient in rod photoreceptor outer segments. Biophysical Journal. 2002;82:728–739. doi: 10.1016/S0006-3495(02)75435-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani K, Yau K-W. Calcium and light adaptation in retinal rods and cones. Nature. 1988;334:69–71. doi: 10.1038/334069a0. [DOI] [PubMed] [Google Scholar]
- Peng YW, Rhee SG, Yu WP, Ho YK, Schoen T, Chader GJ, Yau KW. Identification of components of a phosphoinositide signaling pathway in retinal rod outer segments. Proceedings of the National Academy of Sciences of the USA. 1997;94:1995–2000. doi: 10.1073/pnas.94.5.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh EN, Jr, Nikonov S, Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaptation. Current Opinion in Neurobiology. 1999;9:410–418. doi: 10.1016/S0959-4388(99)80062-2. [DOI] [PubMed] [Google Scholar]
- Ratto GM, Payne R, Owen WG, Tsien RY. The concentration of cytosolic free calcium in vertebrate rod outer segments measured with fura-2. Journal of Neuroscience. 1988;8:3240–3246. doi: 10.1523/JNEUROSCI.08-09-03240.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath AP, Matthews HR, Cornwall MC, Bandarchi J, Fain GL. Light-dependent changes in outer segment free Ca2+ concentration in salamander cone photoreceptors. Journal of General Physiology. 1999;113:267–277. doi: 10.1085/jgp.113.2.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampath AP, Matthews HR, Cornwall MC, Fain GL. Bleached pigment produces a maintained decrease in outer segment Ca2+ in salamander rods. Journal of General Physiology. 1998;111:53–64. doi: 10.1085/jgp.111.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnetkamp PPM. Calcium translocation and storage of isolated intact cattle rod outer segments in darkness. Biochimica et Biophysica Acta. 1979;554:441–459. doi: 10.1016/0005-2736(79)90383-3. [DOI] [PubMed] [Google Scholar]
- SchrÖder WH, Fain GL. Light-dependent calcium release from photoreceptors measured by laser micro-mass analysis. Nature. 1984;309:268–270. doi: 10.1038/309268a0. [DOI] [PubMed] [Google Scholar]
- Szundi I, Mah TL, Lewis JW, Jager S, Ernst OP, Hofmann KP, Kliger DS. Proton transfer reactions linked to rhodopsin activation. Biochemistry. 1998;37:14237–14244. doi: 10.1021/bi981249k. [DOI] [PubMed] [Google Scholar]
- Ugarte M, Osborne NN. Zinc in the retina. Progress in Neurobiology. 2001;64:219–249. doi: 10.1016/s0301-0082(00)00057-5. [DOI] [PubMed] [Google Scholar]
- Wang TL, Sterling P, Vardi N. Localization of type I inositol 1, 4, 5-triphosphate receptor in the outer segments of mammalian cones. Journal of Neuroscience. 1999;19:4221–4228. doi: 10.1523/JNEUROSCI.19-11-04221.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff ML, Sampath AP, Matthews HR, Krasnoperova NV, Lem J, Fain GL. Measurement of cytoplasmic calcium concentration in the rods of wild-type and transducin knock-out mice. Journal of Physiology. 2002;542:843–854. doi: 10.1113/jphysiol.2001.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau K-W, Nakatani K. Cation selectivity of light-sensitive conductance in retinal rods. Nature. 1984a;309:352–354. doi: 10.1038/309352a0. [DOI] [PubMed] [Google Scholar]
- Yau K-W, Nakatani K. Electrogenic Na-Ca exchange in retinal rod outer segment. Nature. 1984b;311:661–663. doi: 10.1038/311661a0. [DOI] [PubMed] [Google Scholar]
- Yau K-W, Nakatani K. Light-induced reduction of cytoplasmic free calcium in retinal rod outer segment. Nature. 1985;313:579–582. doi: 10.1038/313579a0. [DOI] [PubMed] [Google Scholar]
- Zhao M, Hollingworth S, Baylor SM. AM-loading of fluorescent Ca2+ indicators into intact single fibers of frog muscle. Biophysical Journal. 1997;72:2736–2747. doi: 10.1016/S0006-3495(97)78916-1. [DOI] [PMC free article] [PubMed] [Google Scholar]