

Abstract
Chemerin is a chemotactic protein that binds to the G protein–coupled receptor, ChemR23. We demonstrate that murine chemerin possesses potent antiinflammatory properties that are absolutely dependent on proteolytic processing. A series of peptides was designed, and only those identical to specific C-terminal chemerin sequences exerted antiinflammatory effects at picomolar concentrations in vitro. One of these, chemerin15 (C15; A140-A154), inhibited macrophage (MΦ) activation to a similar extent as proteolyzed chemerin, but exhibited reduced activity as a MΦ chemoattractant. Intraperitoneal administration of C15 (0.32 ng/kg) to mice before zymosan challenge conferred significant protection against zymosan-induced peritonitis, suppressing neutrophil (63%) and monocyte (62%) recruitment with a concomitant reduction in proinflammatory mediator expression. Importantly, C15 was unable to ameliorate zymosan-induced peritonitis in ChemR23−/− mice, demonstrating that C15's antiinflammatory effects are entirely ChemR23 dependent. In addition, administration of neutralizing anti-chemerin antibody before zymosan challenge resulted in a significant exacerbation of peritoneal inflammation (up to 170%), suggesting an important endogenous antiinflammatory role for chemerin-derived species. Collectively, these results show that chemerin-derived peptides may represent a novel therapeutic strategy for the treatment of inflammatory diseases through ChemR23.
Macrophages (MΦs) are key participants in innate immunity and contribute to the inflammatory response in part through the secretion of chemokines and cytokines that recruit and activate other immune cells. Despite the beneficial role of inflammatory mediators in host defense, their sustained production can lead to serious, chronic pathological conditions, such as atherosclerosis and rheumatoid arthritis. Therefore, although necessary for the elimination of pathogens, excessive and prolonged MΦ activation leads to serious deleterious effects in the host (1, 2).
Chemerin is a chemoattractant protein present in a range of human inflammatory exudates, including ascitic and synovial fluid (3, 4). Human prochemerin is secreted as a 142–amino acid (aa) precursor (145-aa Mus musculus), which undergoes C-terminal truncation to generate a 136-aa chemoattractant, chemerin (139-aa Mus musculus) (3, 5–7). Chemerin was identified as a natural ligand for the orphan heptahelical receptor ChemR23 through G protein–coupled receptor (GPCR) screening assays.
ChemR23 is a Gαi-linked receptor expressed primarily by monocytes, MΦs, and plasmacytoid dendritic cells (7, 8). ChemR23 shares phylogenetic homology with other chemoattractant receptors, including those for the antiinflammatory mediator lipoxin A4 and the neutrophil chemotaxins C5a and C3a (3, 7, 9). Extracellular terminal proteolysis is characteristic of ligands for some of these related receptors and appears to be used to modulate ligand bioactivites (3, 7, 9). At the time of writing, another receptor for chemerin, the GPCR GPR1, has been identified (10).
In addition to chemerin, ChemR23 binds an eicosapentenoic acid–derived lipid termed resolvin E1 (RvE1) (11). RvE1 is thought to exert antiinflammatory effects through activation of ChemR23 and has recently been found to be a ligand for the related receptor BLT1 (12). RvE1 suppresses inflammation in animal models, including sulfonic acid–induced colitis and zymosan-induced peritonitis (11, 13).
We show for the first time that chemerin undergoes proteolytic cleavage to generate potent antiinflammatory products that can exert profound antiinflammatory effects in vitro at low picomolar concentrations. A single injection of a synthetic chemerin-derived peptide significantly reduces zymosan-induced peritonitis, and injection of an anti-chemerin antibody (ChAb) increases neutrophil and monocyte recruitment by up to 170%.
RESULTS AND DISCUSSION
Chemerin exerts antiinflammatory effects on activated MΦs in a proteolysis-dependent manner
To investigate the potential antiinflammatory action of chemerin, we first evaluated the effect of murine chemerin on the production of inflammatory mediators by peritoneal MΦs (PMΦs) elicited by Bio-Gel polyacrylamide beads and stimulated with LPS and IFN-γ. PMΦs were pretreated with chemerin for 1 h followed by a 15-h challenge with LPS/IFN-γ to induce classical MΦ activation. Chemerin inhibited the production of the proinflammatory mediators TNF-α (70%), IL-1β (60%), IL-6 (42%), IL-12 p40 (54%), and RANTES (40%) by classically activated MΦs (Fig. 1 A). In addition, chemerin induced the expression of mRNA for the antiinflammatory cytokines TGF-β (twofold) and IL-10 (10-fold). These effects were dose dependent, with maximal responses observed at 1 pM (Fig. 1 A), and pertussis toxin (PTX) sensitive, indicating the involvement of a Gαi-linked GPCR (Fig. 1 B). In addition, antiinflammatory effects were observed at 4, 8, and 15 h after LPS/IFN-γ administration and were abrogated by PTX at all time points (Fig. 1 C).
Figure 1.

Antiinflammatory activity of chemerin on activated MΦs is proteolysis dependent. (A–D) PMΦs were pretreated with 1 pM chemerin (Chem), 1 pM chemerin plus protease inhibitor (leupeptin [Leu], E-64, pefabloc [Pef], pepstatin A [Pep A], calpeptin [Cal], cathepsin S inhibitor [Cath S], cathepsin L inhibitor [Cath L]), or 1 μM dexamethasone (Dexa) for 1 h and then stimulated with 100 ng/ml LPS and 20 ng/ml IFN-γ for 15 h. Cytokine expression in MΦ supernatants after 16 h was determined by Luminex assays. mRNA levels were quantified by quantitative RT-PCR (IL-10, TGF-β) and normalized to HPRT. (B) PMΦs were pretreated with 0.1–1 pM chemerin ± 200 ng/ml PTX before LPS/IFN-γ challenge. (C) PMΦs were pretreated with 1 pM chemerin for 1 h ± PTX and then stimulated with LPS/IFN-γ for 4, 8, or 15 h. ***, P < 0.001; **, P < 0.01; *, P < 0.05 relative to LPS/IFN-γ–treated samples; ###, P < 0.001; ##, P < 0.01; #, P < 0.05 relative to chemerin-treated samples. Graphs show mean values ± SEM from three to eight independent experiments. nd, below limit of detection for this assay. ns, not significant.
Previous studies have demonstrated that serine proteases released by granulocytes upon degranulation cleave the C-terminal extremity of prochemerin and release its chemotactic potential (6). We therefore investigated the possibility that murine chemerin could undergo proteolytic processing by enzymes released upon murine MΦ activation. Coadministration of chemerin with leupeptin (a serine and cysteine protease inhibitor) or E-64 (a cysteine protease inhibitor) abolished its antiinflammatory effects (Fig. 1 D), whereas the acidic protease inhibitor pepstatin A and the serine protease inhibitor pefabloc exerted no effect on chemerin-mediated suppression of MΦ activation. These data demonstrate that chemerin exerts inhibitory effects on MΦ activation in a cysteine protease–dependent manner. A cathepsin L inhibitor (Z-FF-FMK), cathepsin S inhibitor (Z-FL-COCHO), and a calpain I and II inhibitor (calpeptin) were used to further probe the specific cysteine proteases involved in chemerin cleavage. It was found that chemerin's antiinflammatory effects were dependent on calpains and cathepsin S but were independent of cathepsin L. Collectively, our results demonstrate for the first time that classically activated MΦs are capable of converting chemerin into potent antiinflammatory peptides by cysteine protease–mediated cleavage of the parent molecule, most likely involving calpains and cathepsin S.
Design of chemerin peptides and assays for their bioactivity
Previous studies have indicated that the biological activity of chemerin depends upon C-terminal processing (6, 14). We therefore postulated that peptides formed upon C-terminal truncation could be responsible for the observed proteolysis-dependent effects of chemerin on MΦ activation. Conserved residues were identified by aligning the sequences of putative chemerin orthologs from several species using the ClustalW algorithm. A series of 11–20-aa peptides was designed and named C11 (P144-A154; PHGYFLPGQFA), C13 (P144-S156; PHGYFLPGQFAFS), C15 (A140-A154; AGEDPHGYFLPGQFA), C19 (A138-S156; AQAGEDPHGYFLPGQFAFS), N19 (E23-K41; ELSETQRRSLQVALEEFHK), and M20 (K86-K105; KPECTIKPNGRRRKCLACIK). C-terminal peptides C13 and C19 moderately suppressed LPS/IFN-γ–induced RANTES and TNF-α expression with an optimal dose of 100 pM (Table I). Chemerin15 (C15), however, retained the antiinflammatory activity shown by proteolyzed chemerin and inhibited cytokine expression with similar efficacy and potency as chemerin (optimal dose, 1 pM). In addition, C11, the N-terminal peptide (N19), midstream peptide (M20), and the control peptides (scrambled C15 [C15-S], GLFHDQAGPPAGYEF, and mutant C15 [C15-M], AGEDPHGYALPGQAA) were devoid of antiinflammatory activity in the MΦ activation assay. The 6-aa (R157-K162; RALRTK) and 8-aa (F155-K162; FSRALRTK) peptides removed during prochemerin cleavage by proteases during coagulation, allergy, and tissue injury (5), named C6 and C8, respectively, also possessed no detectable antiinflammatory activity in the MΦ activation assay.
Table I.
Antiinflammatory activity of chemerin-derived peptides
| Percentage inhibition of LPS/IFN-γ–induced inflammatory cytokine expression | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cytokine | Chemerin | C6 | C8 | C11 | C13 | C15 | C15-S | C15-M | C19 | N19 | M20 |
| TNF-α | 70 | 0 | 0 | 0 | 10 | 61 | 0 | 0 | 21 | 0 | 0 |
| RANTES | 40 | 0 | 0 | 0 | 32 | 47 | 0 | 0 | 41 | 0 | 0 |
| IL-1β | 60 | — | — | — | — | 54 | — | — | — | — | — |
| IL-12p40 | 54 | — | — | — | — | 47 | — | — | — | — | — |
| IL-6 | 42 | — | — | — | — | 43 | — | — | — | — | — |
PMΦs were challenged with 100 ng/ml LPS and 20 ng/ml IFN-γ for 15 h with or without pretreatment with peptides (0.1 pM–100 nM) for 1 h. Where peptides exhibited antiinflammatory properties, percentage inhibition of LPS/IFN-γ–induced MΦ activation represents effect with optimal dose (1 pM chemerin and C15 or 100 pM C13 and C19). Peptide sequences are as follows: C6 (R157-K162; RALRTK), C8 (F155-K162; FSRALRTK), C11 (P144-A154; PHGYFLPGQFA), C13 (P144-S156; PHGYFLPGQFAFS), C15 (A140-A154; AGEDPHGYFLPGQFA), C19 (A138-S156; AQAGEDPHGYFLPGQFAFS), N19 (E23-K41; ELSETQRRSLQVALEEFHK), and M20 (K86-K105; KPECTIKPNGRRRKCLACIK). Control peptides: C15-S (GLFHDQAGPPAGYEF) and C15-M (AGEDPHGYALPGQAA; F148A and F153A). Data represent mean percentage inhibition of cytokine production by classically activated MΦs from four to eight independent experiments as determined by ELISAs and Luminex assays.
MΦ chemoattractant properties of chemerin and its C-terminal–derived peptides
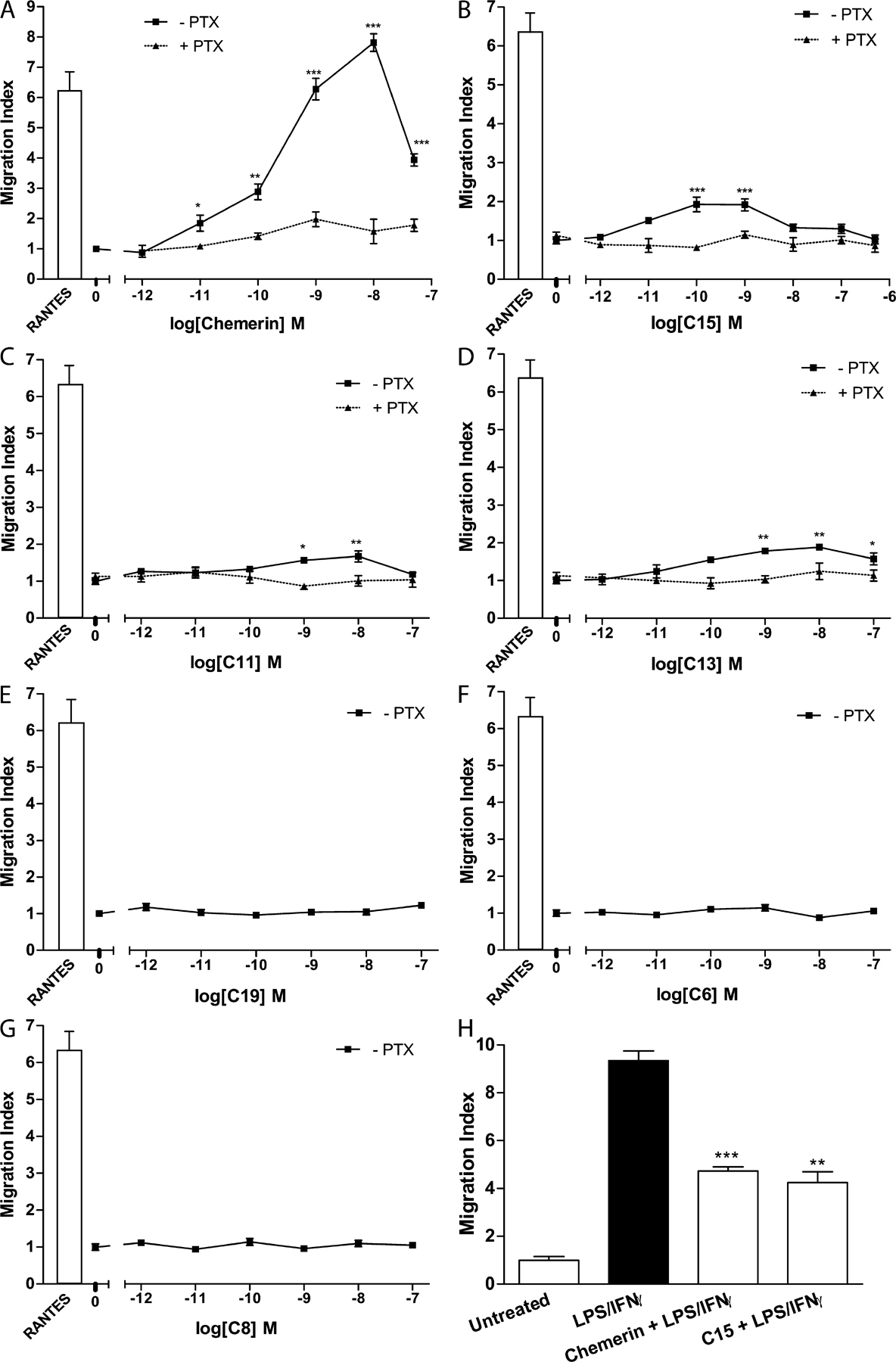
We confirmed previous reports that murine chemerin is a MΦ chemoattractant in modified Boyden chamber assays over a dose range of 10 pM to 50 nM (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20071601/DC1). C11, C13, and C15 (1 pM–100 nM; Fig. S1, B–D) possessed little MΦ chemotactic activity compared with chemerin or a positive control, the CC chemokine RANTES (25 ng/ml; 3 nM). Maximal MΦ migration was observed with 100 pM C15, 10 nM C13, and 10 nM C11 and was PTX sensitive. C19, however, displayed no chemotactic activity at all concentrations tested (0.1 pM–500 nM; Fig. S1 E). Thus, we were able to identify a chemerin-derived peptide that retains antiinflammatory activity but exhibits no chemotactic activity for MΦs, indicating the existence of distinct function-specific components of chemerin that could be exploited therapeutically. The prochemerin-derived peptides C6 and C8, which were found to be devoid of MΦ antiinflammatory activity, were also incapable of inducing MΦ migration at all concentrations tested (0.1 pM–500 nM; Fig. S1, F and G).
Ablation of the ChemR23 gene impairs antiinflammatory and chemotactic effects of chemerin and C15
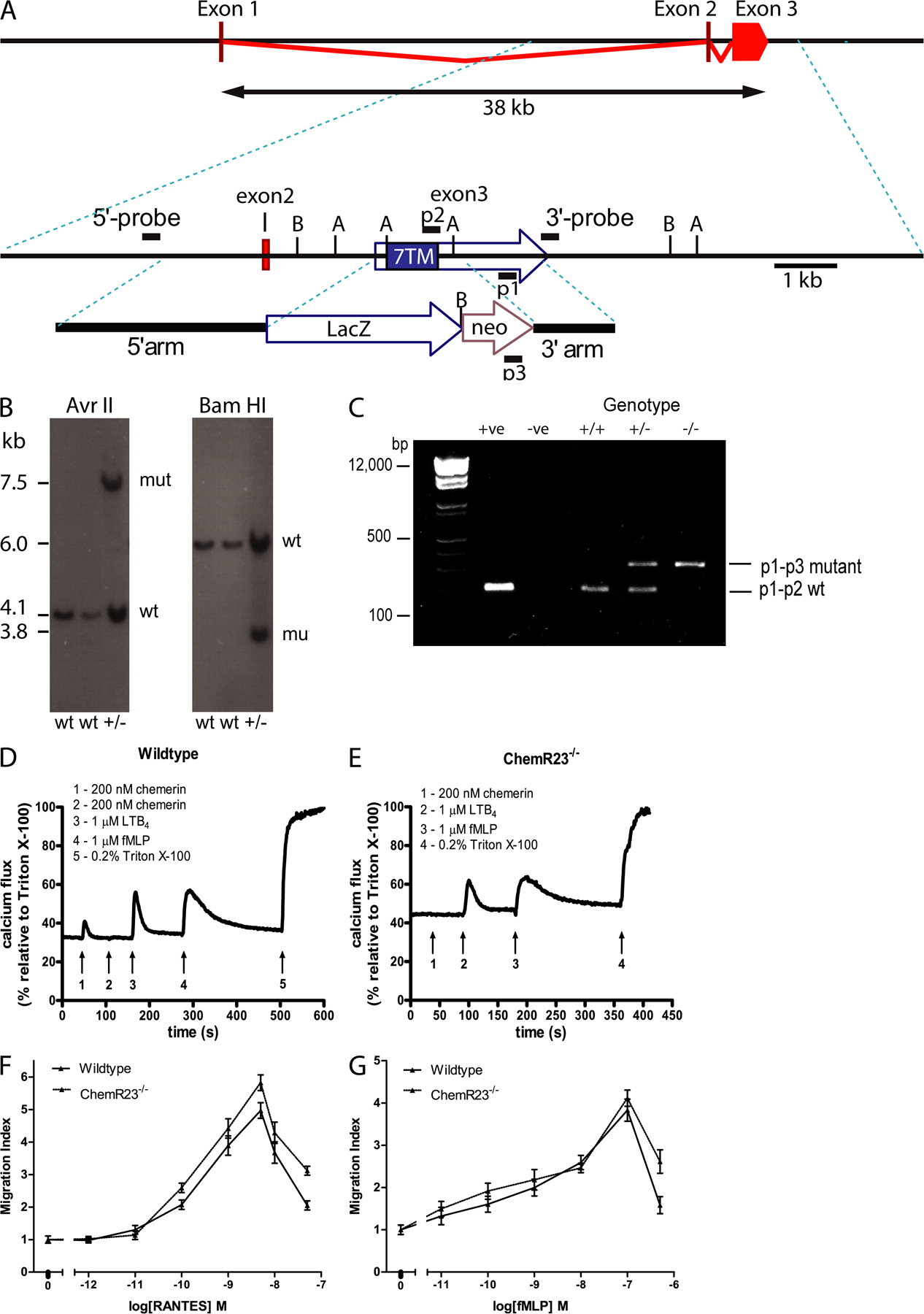
We next investigated whether the effects of chemerin and C15 in our in vitro models of MΦ activation and chemotaxis were mediated via the chemerin receptor ChemR23 through the use of KO mice. PCR and Southern blot analysis confirmed the presence of a disrupted ChemR23 allele in heterozygote and homozygote KO mice and the presence of a wild-type ChemR23 allele in heterozygote and wild-type animals (Fig. S2, A–C, available at http://www.jem.org/cgi/content/full/jem.20071601/DC1). Furthermore, chemerin was found to induce calcium flux in wild-type Bio-Gel–elicited peritoneal cells, but failed to do so in cells from ChemR23 KO mice (Fig. S2, D and E).
The chemotactic effects of chemerin and C15 were completely ablated in ChemR23 KO MΦs, indicating that induction of MΦ chemotaxis by chemerin and C15 is entirely ChemR23 dependent (Fig. 2, A and B). MΦ migration toward prototypical chemoattractants RANTES and fMLP was unaffected in MΦs from ChemR23−/− mice (Fig. S2, F and G), demonstrating that ChemR23−/− MΦs exhibited selective impairment of chemerin and C15-mediated chemotaxis.
Figure 2.

Effects of chemerin and C15 peptide are impaired in MΦs from ChemR23−/− mice. (A and B) PMΦs (0.4 × 106 cells) from wild-type and ChemR23−/− mice were allowed to migrate toward chemoattractant (chemerin or C15) in the bottom well of a modified Boyden chamber over 4 h. Filters were fixed in 4% formalin, and migrated cell nuclei were stained with DAPI, visualized, and quantified. Serum-free medium was used as a negative control and 25 ng/ml RANTES (migration index, 6 ± 0.59) as a positive control. Graphs indicate mean migration index ± SEM for each treatment group (n = 4 independent experiments). ***, P < 0.001; **, P < 0.01; *, P < 0.05 relative to migration induced by the same concentration of chemoattractant in ChemR23−/− PMΦs. (C and D) PMΦs from wild-type and ChemR23−/− mice were pretreated with 0.1–1 pM C15 (C) or 0.1–1 pM chemerin (D) for 1 h and then stimulated with 100 ng/ml LPS and 20 ng/ml IFN-γ for 15 h. Mean expression of cytokines ± SEM in MΦ supernatants after 16 h was determined by ELISA (n = 4 independent experiments). **, P < 0.01; *, P < 0.05 relative to LPS/IFN-γ–treated samples. ns, not significant.
C15 was unable to suppress proinflammatory mediator production by ChemR23 KO MΦs, indicating that its antiinflammatory effects in this model are completely mediated through ChemR23 (Fig. 2 C). Interestingly, chemerin displayed a reduced but not completely abrogated antiinflammatory activity on ChemR23−/− MΦs (Fig. 2 D). These data are consistent with a model in which chemerin is cleaved by MΦ-derived proteases to generate antiinflammatory peptides that act through multiple GPCRs, including but not exclusively ChemR23. At the time of writing, a second receptor for chemerin, the GPCR GPR1, has been identified (10).
C15 ameliorates zymosan-induced peritonitis in a ChemR23-dependent manner
The zymosan–peritonitis model (for review see reference 15) has been used to demonstrate the antiinflammatory properties of dexamethasone (16), annexin peptide (17), and the lipid ChemR23 agonist RvE1 (13), which typically suppress both inflammatory cell recruitment into the peritoneal cavity and proinflammatory cytokine production.
Given the high chemotactic potential of chemerin and inherent requirement for proteolysis, we turned to the C-terminal synthetic peptide C15 for in vivo characterization of antiinflammatory effects in the sterile peritonitis model because C15 is largely devoid of chemotactic activity (Fig. S1 B) yet exerts comparable antiinflammatory effects to those of proteolyzed chemerin (Table I). To determine the antiinflammatory properties of C15 in vivo, a time-course experiment was performed extending over 48 h. Neutrophil (7/4high, Ly-6Ghigh) and monocyte (7/4high, Ly-6Glow) populations in peritoneal lavage fluid were determined by FACS analysis according to established protocols (18, 19). Administration of zymosan i.p. produced a time-dependent extravasation of inflammatory cells into the peritoneal cavity, which followed the typical profile of an acute inflammatory response (Fig. 3, A and B, solid line). Pretreatment with C15 (8 pg/mouse; ∼0.32 ng/kg) 1 h before zymosan challenge brought the peak neutrophilia forward from 4 h post-zymosan administration to 2 h with ∼50% the magnitude of that of zymosan-challenged mice (reduced from 1.25 × 106 to 0.62 × 106 cells; Fig. 3 A, dotted line). Suppression of neutrophil infiltration by C15 was seen at 2, (50%), 4 (66%), and 24 h (50%). A single dose of C15 peptide was also effective in reducing monocyte levels in inflamed cavities at all time points, with >60% suppression seen at 4 (63%), 8 (61%), and 48 h (64%; Fig. 3 B, dotted line). The rate of monocyte infiltration was highest 2–4 h after zymosan injection (0.51 × 106 cells/h), and C15 administration reduced the rate of influx (0.18 × 106 cells/h). A single dose of C15 peptide before zymosan challenge therefore provided significant protection against zymosan-induced peritoneal inflammation over 48 h.
Figure 3.

C15 ameliorates zymosan-induced peritonitis in a ChemR23-dependent manner. C57Bl6/J mice were dosed i.p. with PBS or 0.32 ng/kg C15 followed by injection with PBS or 10 μg zymosan (∼2 × 106 particles per cavity) 1 h later. Peritoneal exudate cells (PECs) were harvested by peritoneal lavage at multiple time points (A and B; five to six mice/group) or after 4 h (C–E; 6–15 mice/group). Total PECs were quantified, and cellular composition (neutrophils vs. monocytes) was determined by FACS analysis. Cells were stained with Ly-6G-PE and 7/4-FITC. Gates were constructed around two populations, the neutrophils (7/4high, Ly-6Ghigh) and monocytes (7/4high, Ly-6Glow). (E) Representative FACS plots are shown for each treatment group at 4 h after zymosan injection. (F) Peritoneal lavage fluid was assayed for TNF-α, KC, IL-6, IL-1β, and JE by Luminex assay. (G) Mice (six to eight/treatment) were dosed i.p. with 10 μg zymosan and with 8 pg C15 or PBS either 1 h beforehand (C15 pretreatment) or 2 h later (C15 post-zymosan). Peritoneal lavage was performed 4 h after zymosan challenge. (H) Wild-type and ChemR23−/− mice on a 129SvEv background were dosed i.p. with PBS or 0.32 ng/kg C15 followed by injection with PBS or 10 μg zymosan 1 h later. PECs cells were harvested by peritoneal lavage 4 h after zymosan injection and characterized by FACS (six to eight mice/treatment). C15, chemerin15; Z, zymosan; ***, P < 0.001; **, P < 0.01 relative to zymosan-treated animals; nd, not detectable; ns, not significant.
The time-course experiment identified the 4-h post-zymosan time point as an appropriate point for validation of C15's antiinflammatory activity. In this study, a single dose of C15 produced a dose-dependent reduction in zymosan-elicited neutrophil and monocyte recruitment, which was maximal at 8 pg/mouse C15 (∼0.32 ng/kg; Fig. 3, C–E, and Fig. S3, which is available at http://www.jem.org/cgi/content/full/jem.20071601/DC1), although significant antiinflammatory effects were seen with a dose of 4 pg/mouse (∼0.16 ng/kg; Fig. S3). When C15 was administered 1 h before zymosan challenge, neutrophil numbers were reduced from 1.9 × 106 to 0.78 × 106 cells (63% decrease; Fig. 3 C) and monocyte levels from 0.69 × 106 to 0.30 × 106 cells (62% decrease; Fig. 3 D; representative FACS plots at the 4-h time point are shown in Fig. 3 E). C15 administration also diminished peritoneal expression of proinflammatory cytokines at 4 h, including TNF-α (51%), IL-1β (67%), IL-6 (67%), JE (59%), and KC (38%; Fig. 3 F). Control peptides C15-S and C15-M, which were devoid of in vitro antiinflammatory activity (Table I), were not protective in vivo when given at the same dose and time as C15, as judged by neutrophil and monocyte levels (Fig. 3, C and D). Suppression of neutrophil (1.9 × 106 to 0.83 × 106 cells; 60% decrease) and monocyte recruitment (0.69 × 106 to 0.42 × 106 cells; 42% decrease) was still seen 4 h after zymosan, when the same dose of C15 was given 2 h after zymosan injection (Fig. 3 G). This demonstrates that C15 can reduce neutrophil and monocyte recruitment in an established inflammatory setting, providing another indication that C15/C15 derivatives may represent attractive pharmacophores targeting inflammatory pathologies.
When the same experiments were performed in ChemR23−/− mice, C15 administration was not able to suppress zymosan-induced monocyte and neutrophil recruitment, indicating complete dependence of C15's antiinflammatory effects upon ChemR23 (Fig. 3 H). Furthermore, the inflammatory response of ChemR23−/− mice to zymosan was equivalent in magnitude to that of the wild-type controls in terms of neutrophil (+/+, 5.7 ± 0.37 vs. −/−, 5.5 ± 0.63 cells) and monocyte recruitment (+/+, 1.05 ± 0.09 vs. −/−, 0.96 ± 0.11 cells; Fig. 3 H).
Blockade of endogenous chemerin species exacerbates peritoneal inflammation
Given the apparent lack of ChemR23−/− phenotype in the zymosan-induced peritonitis model, we investigated a potential endogenous role for chemerin/chemerin-derived peptides by injecting mice i.p. with a neutralizing polyclonal ChAb or a control IgG 1 h before a 4- or 24-h zymosan challenge. We had previously shown that ChAb but not control IgG was capable of inhibiting C15 and chemerin-induced MΦ chemotaxis and antiinflammatory effects in vitro (Fig. 4, A and B). In vivo it was found that neutralization of endogenous chemerin species resulted in a rise in peritoneal neutrophil (63%) and monocyte (45%) levels at the 4-h time point relative to control IgG–treated mice and a 170 and 86% increase in peritoneal neutrophil and monocyte levels 24 h after zymosan injection (Fig. 4, C and D). This exacerbation of peritoneal inflammation over a 24-h period suggests an important antiinflammatory role for endogenous chemerin species in vivo in controlling the magnitude of the innate immune response. The absence of an exaggerated inflammatory phenotype in ChemR23−/− mice may be due to redundancy in the chemerin antiinflammatory system. In fact, an additional receptor for chemerin, the GPCR GPR1, has now been reported, which may represent a second target for chemerin-derived peptides (10). We therefore predict that ablation of the chemerin gene may result in mice with exaggerated inflammatory pathologies. An alternative hypothesis is that the ChemR23 inflammatory axis may be of particular importance only in certain inflammatory pathologies or that a ChemR23 KO phenotype may not manifest except in the context of chronic inflammation.
Figure 4.

ChAb neutralizes chemerin species and exacerbates peritoneal inflammation. (A) PMΦs were used in MΦ chemotaxis assays (as detailed in Fig. 2) and allowed to migrate toward RANTES, chemerin, or C15 ± ChAb or control IgG. Graphs indicate mean migration index ± SEM (n = 4 independent experiments). ***, P < 0.001; **, P < 0.01 relative to chemoattractant. (B) PMΦs were pretreated with 1 pM C15 or 1 pM chemerin ± ChAb or control IgG for 1 h and then stimulated with 100 ng/ml LPS and 20 ng/ml IFN-γ for 15 h. Mean expression of RANTES ± SEM in MΦ supernatants after 16 h was determined by ELISA (n = 4 independent experiments). ***, P < 0.001 relative to LPS/IFN-γ–treated samples. (C and D) C57Bl6/J mice were dosed i.p. with PBS, 100 ng/mouse ChAb, or 100 ng/mouse control IgG followed by injection with PBS or 10 μg/cavity zymosan 1 h later. PECs were harvested 4 and 24 h after zymosan injection and processed as outlined in Fig. 3. Z, zymosan; ChAb, anti-chemerin antibody; **, P < 0.01 relative to zymosan-challenged mice; ns, not significant.
C-terminal chemerin peptides: novel antiinflammatory mediators
Many endogenous proinflammatory mediators have been described, several of which are active in the picomolar range, including TNF-α (10−11 M), fMLP (10−11 M), and IL-1 (10−14 M) (20, 21). To our knowledge, our synthetic chemerin peptides are the first antiinflammatory mediators shown to be active in vitro and in vivo in the picomolar range. Pharmaceutical preparations such as dexamethasone are commonly administered at concentrations in the micromolar range in vitro (22). Indeed, dexamethasone achieves a 50% suppression of monocyte and neutrophil influx in the zymosan-induced peritonitis model at 30 μg/mouse (1.2 mg/kg) (16), whereas C15 produces equivalent antiinflammatory effects at a dose of 8 pg/mouse (0.32 ng/kg). C15 is also more potent than the lipid ligand for ChemR23, RvE1, which reduces PMN numbers in peritoneal exudates by up to 25% when administered at 300 ng/mouse i.p. (12 μg/kg) (23). The in vitro and in vivo concentrations of chemerin used in our experiments are ∼500-fold lower than concentrations of chemerin found in murine plasma and serum (8). This could mean that little endogenous chemerin may need to be present at sites of inflammation for antiinflammatory effects to become evident in vivo.
Here, we have demonstrated that chemerin exhibits antiinflammatory effects on MΦs in vitro in a proteolysis-dependent fashion. We found that C-terminal chemerin peptides, in particular C15, exhibit potent antiinflammatory properties in in vitro and in vivo models of inflammation when administered in the picogram range that are mediated through ChemR23. These findings suggest that C15/C15 derivatives may represent a novel therapeutic strategy for the treatment of inflammatory pathologies via ChemR23.
MATERIALS AND METHODS
Animals.
All animal studies were conducted with ethical approval from the Dunn School of Pathology Local Ethical Review Committee and in accordance with the UK Home Office regulations (Guidance on the Operation of Animals, Scientific Procedures Act, 1986).
MΦ activation assays.
Bio-Gel P100 polyacrylamide beads (1 ml of 2% wt/vol in PBS; Bio-Rad Laboratories) were injected into the peritoneal cavity of 8–12-wk-old C57BL/6 mice. 4 d later, mice were killed and peritoneal exudates collected in PBS plus 2 mM EDTA. Harvested cells were centrifuged and resuspended in OptiMEM medium (Invitrogen) supplemented with 2 mM glutamine (Invitrogen), 50 U/ml penicillin, and 50 μg/ml streptomycin (Invitrogen). Cells (1.5 × 106/well) were plated in six-well tissue culture plates (35-mm diameter; Costar) and allowed to adhere for 2 h at 37°C in a humidified atmosphere containing 5% CO2 to isolate MΦ populations by adherence. Nonadherent cells were removed by washing with PBS after 2 h. Adherent cells were judged to be >95% MΦs by morphological criteria. MΦs were preincubated with 10−12–10−8 M rmChemerin (R&D Systems), 10−12–10−8 M chemerin peptides (Bio-Synthesis Inc.), or 1 μM dexamethasone (Sigma-Aldrich) for 1 h and then challenged with 100 ng/ml LPS (Sigma-Aldrich) and 20 ng/ml IFN-γ (PeproTech) for 15 h. Protease inhibitors (Sigma-Aldrich and EMD) were administered with chemerin to determine dependency upon proteolysis. MΦ media were harvested and stored at −80°C until use in ELISAs (R&D Systems) and Luminex assays (Bio-Rad Laboratories). Protease inhibitors were used at the following concentrations: leupeptin, 15 μg/ml; E-64, 5 μM; pefabloc, 10 μM; pepstatin A, 1 μM; calpeptin, 5 μM; cathepsin S inhibitor, 250 nM; cathepsin L inhibitor, 1 μM. Chemerin peptides were prepared by Bio-Synthesis Inc., and purity was assessed to be >95% as assessed by HPLC and mass spectrometry (data provided by the manufacturer). Chemerin and chemerin-derived peptides were not found to affect cell viability adversely as determined using the MTS assay (Promega) and had no detectable endotoxin as judged by Limulus amebocyte lysate assay (Cambrex).
Detection of secreted protein by ELISAs and Luminex.
RANTES, TNF-α, and KC concentrations in cell supernatants and peritoneal fluid were assessed by ELISA (R&D Systems). IL-12 p40, IL-1β, TNF-α, JE, and IL-6 levels were determined by Luminex multiplex bead assay (Bio-Rad 6 plex assay). Detection limits were 100 pg/ml for ELISAs and 10–50 pg/ml for Luminex assays depending on cytokine.
RNA preparation and RT-PCR.
Total RNA was extracted using QIAGEN RNeasy kits, reverse transcribed, and subjected to quantitative RT-PCR using the Sybr-Green method. Data were analyzed using the 2−ΔΔCT method (24).
Chemotaxis assay.
4 × 105 Bio-Gel-elicited MΦs were harvested and placed on 96-well Neuroprobe membranes (6-mm diameter, 8-μm pore size; ChemoTX) in RPMI supplemented with 25 mM Hepes (Invitrogen) and 0.1% BSA (Sigma-Aldrich). Cells were allowed to migrate toward 1 pM–100 nM chemerin peptides, 1 pM–50 nM chemerin, 25 ng/ml RANTES (PeproTech), or serum-free media for 4 h. Signal transduction via Gαi was blocked by preincubating cells with 200 ng/ml PTX (Sigma-Aldrich) for 30 min before chemotaxis assays. Migrated cells on the underside of membranes were fixed (3% formalin) and stained with DAPI. Migration was quantified as total pixel count of DAPI (Sigma-Aldrich) -stained nuclei under the fluorescence microscope (two photos/membrane and a minimum of three replicate wells per treatment). Images were analyzed using Metamorph software to determine percentage threshold areas (TA) occupied by migrated cells. Migration indices were obtained by dividing treatment TA by serum-free media TA.
Murine peritonitis.
C57BL6/J mice were administered 500 μl C15 (0.32 ng/kg) or vehicle (PBS) i.p. 1 h before administration of 500 μl of 10 μg zymosan A (Sigma-Aldrich) i.p. After 2, 4, 8, 16, 24, and 48 h and after mice were killed, peritoneal exudates were collected by peritoneal lavage with 5 ml of sterile PBS-3 mM EDTA. Total cell counts were determined, and the cellular composition (neutrophils vs. monocytes) of peritoneal lavage fluid were obtained by FACS analysis. In brief, 105 cells were blocked with anti–mouse 2.4G2 FcγII/III (0.5 μg/0.105 cells; Serotech) for 10 min and stained for 10 min with FITC-conjugated anti–mouse 7/4 and PE-conjugated anti–mouse Ly-6G (0.5 μg/0.5 × 106 cells; clones rmC5-3 and RB6-8C5, respectively; BD Biosciences). Gates were constructed around two populations, the neutrophils (7/4high, Ly-6Ghigh) and inflammatory monocytes (7/4high, Ly-6Glow). The percentage of total events in each population was obtained. In addition, cell-free lavage fluid was collected for use in ELISAs and Luminex assays.
Generation and characterization of ChemR23−/− mice.
ChemR23 is encoded by the Cmklr1 gene (Ensembl Gene ID, ENSMUSG00000042190). The gene-targeting vector was designed to delete the majority of the coding sequence from 33 bp after the initiating methionine to beyond the translational stop codon, thereby removing all but the first 12 of the 371 protein residues and replacing them with an internal ribosome entry site LacZ reporter gene. Targeted embryonic stem cell clones were identified by PCR and confirmed by Southern blot analysis. Correctly targeted clones were used to generate chimeras that transmitted the ChemR23 KO allele at the expected Mendelian frequency. Heterozygous mice were viable and fertile as were the homozygous KO mice. PCR was performed with the following primers: P1, 5′-CCCAGTCACAGGAGCTTCACCAAGATG-3′; p2, 5′-AATGGCCGCTTTTCTGGATTCATCGAC-3′; and p3, 5′-ATGAGCCGCCTTGTGCAAAATTCAGTG-3′. DNA fragments were analyzed by agarose gel electrophoresis. A product of 378 bp (neomycin insert) corresponded to the −/− genotype, a 215 bp product (ChemR23) corresponded to the +/+ genotype, and the presence of both the 215- and 378-bp products indicated the −/+ genotype.
Calcium flux.
4-d Bio-Gel–elicited cells were harvested and resuspended in HBSS with CaCl2 and MgCl supplemented with 0.5% BSA and 10 mM Hepes (HBSS/BSA) at room temperature to a cell density of 107 cells/ml. The cell suspension was incubated with 2 μM Fura-PE3/AM (EMD) for 45 min at room temperature. The cells were then washed twice and resuspended in HBSS/BSA (3 × 107 cells/ml). 100 μl of cell suspension (3 × 106 cells) was added to a cuvette containing 400 μl of prewarmed (37°C) HBSS/BSA and placed in a luminescence spectrometer (LS55; PerkinElmer). After a period of at least 60 s, fluorescence measurements were recorded at excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm. 10 μl of agonist was added under constant stirring at appropriate intervals. At the end of each experiment, 10 μl 10% Triton X-100 was added to determine maximum calcium flux.
Statistics.
Student's t test and one-way ANOVA (with Tukeys post-hoc test) were performed using GraphPad Prism software.
Online supplemental material.
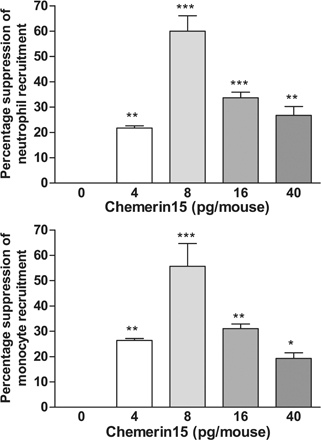
Fig. S1 shows MΦ chemotaxis induced by chemerin and chemerin-derived peptides. Fig. S2 provides detailed methods for the generation and characterization of ChemR23−/− mice, including Southern blots and targeting constructs, Ca2+ flux, and chemotaxis data. Fig. S3 shows the dose-dependent antiinflammatory effects of C15 in zymosan-induced peritonitis.
Supplemental Material
Acknowledgments
We thank Phil Taylor for advice and Linda Randall and Denise Jelfs for technical assistance. D.R. Greaves thanks Siamon Gordon for continuing support and encouragement. J.L. Cash thanks Gemma White and Eileen McNeil for advice.
J.L. Cash and D.R. Greaves planned the project, designed and performed the experiments, and wrote the manuscript. R. Hart did Ca2+ flux experiments. A. Russ, J.P.C. Dixon, A.G. Hendrick, J. Doran, and M.B.L. Carlton generated ChemR23−/− mice.
Work in the laboratory of D.R. Greaves is funded by the British Heart Foundation.
D.R. Greaves has received fees from Chemocentryx and Takeda Cambridge. The authors have no other conflicting financial interests.
References
- 1.Taylor, P.R., L. Martinez-Pomares, M. Stacey, H.H. Lin, G.D. Brown, and S. Gordon. 2005. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 23:901–944. [DOI] [PubMed] [Google Scholar]
- 2.Han, J., and R.J. Ulevitch. 2005. Limiting inflammatory responses during activation of innate immunity. Nat. Immunol. 6:1198–1205. [DOI] [PubMed] [Google Scholar]
- 3.Wittamer, V., J.D. Franssen, M. Vulcano, J.F. Mirjolet, E. Le Poul, I. Migeotte, S. Brezillon, R. Tyldesley, C. Blanpain, M. Detheux, et al. 2003. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 198:977–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meder, W., M. Wendland, A. Busmann, C. Kutzleb, N. Spodsberg, H. John, R. Richter, D. Schleuder, M. Meyer, and W.G. Forssmann. 2003. Characterization of human circulating TIG2 as a ligand for the orphan receptor ChemR23. FEBS Lett. 555:495–499. [DOI] [PubMed] [Google Scholar]
- 5.Zabel, B.A., S.J. Allen, P. Kulig, J.A. Allen, J. Cichy, T.M. Handel, and E.C. Butcher. 2005. Chemerin activation by serine proteases of the coagulation, fibrinolytic, and inflammatory cascades. J. Biol. Chem. 280:34661–34666. [DOI] [PubMed] [Google Scholar]
- 6.Wittamer, V., B. Bondue, A. Guillabert, G. Vassart, M. Parmentier, and D. Communi. 2005. Neutrophil-mediated maturation of chemerin: a link between innate and adaptive immunity. J. Immunol. 175:487–493. [DOI] [PubMed] [Google Scholar]
- 7.Samson, M., A.L. Edinger, P. Stordeur, J. Rucker, V. Verhasselt, M. Sharron, C. Govaerts, C. Mollereau, G. Vassart, R.W. Doms, and M. Parmentier. 1998. ChemR23, a putative chemoattractant receptor, is expressed in monocyte-derived dendritic cells and macrophages and is a coreceptor for SIV and some primary HIV-1 strains. Eur. J. Immunol. 28:1689–1700. [DOI] [PubMed] [Google Scholar]
- 8.Zabel, B.A., T. Ohyama, L. Zuniga, J.Y. Kim, B. Johnston, S.J. Allen, D.G. Guido, T.M. Handel, and E.C. Butcher. 2006. Chemokine-like receptor 1 expression by macrophages in vivo: regulation by TGF-beta and TLR ligands. Exp. Hematol. 34:1106–1114. [DOI] [PubMed] [Google Scholar]
- 9.Zabel, B.A., L. Zuniga, T. Ohyama, S.J. Allen, J. Cichy, T.M. Handel, and E.C. Butcher. 2006. Chemoattractants, extracellular proteases, and the integrated host defense response. Exp. Hematol. 34:1021–1032. [DOI] [PubMed] [Google Scholar]
- 10.Barnea, G., W. Strapps, G. Herrada, Y. Berman, J. Ong, B. Kloss, R. Axel, and K.J. Lee. 2008. From the cover: the genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. USA. 105:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arita, M., F. Bianchini, J. Aliberti, A. Sher, N. Chiang, S. Hong, R. Yang, N.A. Petasis, and C.N. Serhan. 2005. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 201:713–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arita, M., T. Ohira, Y.P. Sun, S. Elangovan, N. Chiang, and C.N. Serhan. 2007. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 178:3912–3917. [DOI] [PubMed] [Google Scholar]
- 13.Arita, M., M. Yoshida, S. Hong, E. Tjonahen, J.N. Glickman, N.A. Petasis, R.S. Blumberg, and C.N. Serhan. 2005. Resolvin E1, an endogenous lipid mediator derived from omega-3 eicosapentaenoic acid, protects against 2,4,6-trinitrobenzene sulfonic acid-induced colitis. Proc. Natl. Acad. Sci. USA. 102:7671–7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wittamer, V., F. Gregoire, P. Robberecht, G. Vassart, D. Communi, and M. Parmentier. 2004. The C-terminal nonapeptide of mature chemerin activates the chemerin receptor with low nanomolar potency. J. Biol. Chem. 279:9956–9962. [DOI] [PubMed] [Google Scholar]
- 15.Lawrence, T., D.A. Willoughby, and D.W. Gilroy. 2002. Anti-inflammatory lipid mediators and insights into the resolution of inflammation. Nat. Rev. Immunol. 2:787–795. [DOI] [PubMed] [Google Scholar]
- 16.Getting, S.J., R.J. Flower, and M. Perretti. 1997. Inhibition of neutrophil and monocyte recruitment by endogenous and exogenous lipocortin 1. Br. J. Pharmacol. 120:1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Damazo, A.S., S. Yona, R.J. Flower, M. Perretti, and S.M. Oliani. 2006. Spatial and temporal profiles for anti-inflammatory gene expression in leukocytes during a resolving model of peritonitis. J. Immunol. 176:4410–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henderson, R.B., J.A.R. Hobbs, M. Mathies, and N. Hogg. 2003. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 102:328–335. [DOI] [PubMed] [Google Scholar]
- 19.Taylor, P.R., D.M. Reid, S.E. Heinsbroek, G.D. Brown, S. Gordon, and S.Y. Wong. 2005. Dectin-2 is predominantly myeloid restricted and exhibits unique activation-dependent expression on maturing inflammatory monocytes elicited in vivo. Eur. J. Immunol. 35:2163–2174. [DOI] [PubMed] [Google Scholar]
- 20.Movat, H.Z., and M.I. Cybulsky. 1987. Neutrophil emigration and microvascular injury. Role of chemotaxins, endotoxin, interleukin-1 and tumor necrosis factor alpha. Pathol. Immunopathol. Res. 6:153–176. [DOI] [PubMed] [Google Scholar]
- 21.Movat, H.Z., M.I. Cybulsky, I.G. Colditz, M.K. Chan, and C.A. Dinarello. 1987. Acute inflammation in gram-negative infection: endotoxin, interleukin 1, tumor necrosis factor, and neutrophils. Fed. Proc. 46:97–104. [PubMed] [Google Scholar]
- 22.Choi, J.-H., E. Brummer, Y.J. Kang, P.P. Jones, and D.A. Stevens. 2006. Inhibitor [kappa]B and nuclear factor [kappa]B in granulocyte-macrophage colony-stimulating factor antagonism of dexamethasone suppression of the macrophage response to Aspergillus fumigatus conidia. J. Infect. Dis. 193:1023–1028. [DOI] [PubMed] [Google Scholar]
- 23.Schwab, J.M., N. Chiang, M. Arita, and C.N. Serhan. 2007. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature. 447:869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak, K.J., and T.D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}