

Abstract
The genetic characterization of Taiwanese influenza A and B viruses on the basis of analyses of pairwise amino acid variations, genetic clustering, and phylogenetics was performed. A total of 548, 2,123, and 1,336 sequences of the HA1 genes of influenza A virus subtypes H1 and H3 and influenza B virus, respectively, collected during 2003 to 2006 from an island-wide surveillance network were determined. Influenza A virus H3 showed activity during all periods, although it was dominant only in the winters of 2002-2003 and 2003-2004. Instead, influenza B virus and influenza A virus H1 were dominant in the winters of 2004-2005 and 2005-2006, respectively. Additionally, two influenza A virus H3 peaks were found in the summers of 2004 and 2005. From clustering analysis, similar characteristics of high sequence diversity and short life spans for the influenza A virus H1 and H3 clusters were observed, despite their distinct seasonal patterns. In contrast, clusters with longer life spans and fewer but larger clusters were found among the influenza B viruses. We also noticed that more amino acid changes at antigenic sites, especially at sites B and D in the H3 viruses, were found in 2003 and 2004 than in the following 2 years. The only epidemic of the H1 viruses, which occurred in the winter of 2005-2006, was caused by two genetically distinct lineages, and neither of them showed apparent antigenic changes compared with the antigens of the vaccine strain. For the influenza B viruses, the multiple dominant lineages of Yamagata-like strains with large genetic variations observed reflected the evolutionary pressure caused by the Yamagata-like vaccine strain. On the other hand, only one dominant lineage of Victoria-like strains circulated from 2004 to 2006.
Influenza A virus subtypes H1 and H3 and influenza B viruses have been the three kinds of influenza viruses most commonly isolated from humans during the past 40 years. It has been estimated that 250,000 to 500,000 deaths are directly associated with influenza virus epidemics around the world every year (21). In addition, genetic mutations in its hemagglutinin (HA) protein, often referred as antigenic drift, are considered the major way in which influenza viruses escape host defense mechanisms and are thus able to continuously infect humans and other species. For example, five antigenic sites on the HA1 domain of the H3 subtype were identified in antibody-combining or receptor binding sites by structural analysis (22, 23). Significantly more nonsynonymous than synonymous nucleotide substitutions were observed at these sites (8). Similar antigenic sites were also proposed for the H1 subtype (4), but none has been identified for influenza B virus. Furthermore, 18 residues of the HA1 domain of H3 were believed to be undergoing positive selection, as determined by empirical studies of global sequences (2, 3). An obvious codon bias for the HA gene instead of other internal genes was also observed (16). Other studies have inspected the relationship between amino acid substitutions and the corresponding changes in antigenicity in natural virus isolates (13, 14).
Starting in 2003, the Centers for Disease Control (CDC) of Taiwan has been receiving influenza virus isolates from its 12 contract virology laboratories around the island and has sequenced the HA1 region of many of these isolates. By July 2006, more than 3,000 HA1 sequences were obtained from influenza A viruses H1 and H3 and influenza B virus. In this study we used these sequences to determine the evolutionary properties of these Taiwanese influenza viruses by integrating their genetic features with local epidemiological information. Distance-based sequence clustering and phylogenetic analysis were both used to reveal the evolutionary pattern and important amino acid variations between Taiwanese isolates and the corresponding vaccine strains or global strains found in databases in the public domain.
MATERIALS AND METHODS
Sample collection and sequencing.
Details about the virology laboratories and the specimen collection, virus isolation, RNA extraction, reverse transcription-PCR, and nucleotide sequencing methods used can be found in a previous report (18). In summary, 12 virology laboratories throughout the island of Taiwan collected clinical samples and sent them to the core sequencing laboratory at the CDC of Taiwan for reverse transcription-PCR and nucleotide sequencing. This surveillance network consists of about 750 sentinel physicians and spans 22 metropolitan cities or counties. Approximately 75% of the 352 basic administrative units of Taiwan (cities, townships, or districts) are covered. A total of 34,312 samples from patients who were suspected of having respiratory tract infections from 2003 to 2006 were collected for virus isolation and further analysis. In addition to the normal negative control for PCR, we also checked the sequencing quality monthly by resequencing some specimens. Furthermore, sequence assembly tasks were carried out with the commercial program Sequencher (Gene Code Inc., Ann Arbor, MI), and all results were inspected manually. The counts for the isolates and the positions of the sequences of each type of influenza virus tested are listed in Table 1.
TABLE 1.
Amino acid sequence clustering of influenza A and B viruses in Taiwan during 2003 to 2006
| Virus | Subtype or lineage | Isolation period (mo [dates]) | No. of positive isolates | No. of available sequences | Amino acid position range (HA1) | Total no. of clusters | No. of clusters/mo | Ratio of no. of sequences/no. of clusters | No. (%) of dominant clustersb | No. (%) of sequences in largest cluster | Lifetime (mo) for dominant clusters
|
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Avg | Longest | |||||||||||
| Influenza A virus | H1 | 19 (01/2005 to 07/2006) | 548 | 535 | 22-190c | 201 | 10.58 | 2.66 | 17 (8.4) | 53 (9.9) | 5.76 | 16 |
| Influenza A virus | H3 | 43 (01/2003 to 07/2006) | 2,123 | 1,428 | 44-305d | 492 | 11.44 | 2.9 | 39 (7.9) | 284 (19.8) | 7.07 | 17 |
| Influenza B virus | Victoria | 31 (01/2004 to 07/2006) | 1,336a | 767 | 52-271 | 87 | 2.81 | 8.82 | 6 (6.9) | 534 (71.5) | 14.2 | 29 |
| Influenza B virus | Yamagata | 31 (01/2004 to 07/2006) | 1,336a | 532 | 52-271 | 93 | 3 | 5.72 | 8 (8.6) | 115 (21.5) | 14 | 30 |
Total count for influenza B virus isolates, including those of both the Yamagata and the Victoria lineages.
A dominant cluster was defined as one with at least five sequences.
A total of 25 antigenic and 144 nonantigenic sites.
A total of 125 antigenic and 137 nonantigenic sites.
Sequence preprocessing and clustering.
All nucleotide sequences were first translated into amino acid sequences and prealigned with the sequences of the corresponding vaccine strains for establishment of the correct amino acid positions, as suggested by the World Health Organization, by using the BLAST2 program (1). The sequences were inspected manually and were removed if any erroneous residues were found. We then trimmed the sequences such that they covered most of the known antigenic sites of the HA1 genes of the H1 and H3 subtypes. Note that most of the available sequences contained only partial HA regions (Table 1) due to the original sequencing strategy for surveillance purposes. Finally, a 507-nucleotide (nt) segment for influenza A virus subtype H1 (genomic positions 147 to 653, based on strain A/Puerto Rico/8/34 [GenBank accession no. NC_002017]), a 786-nt segment for influenza A virus subtype H3 (genomic positions 178 to 963, based on strain A/Hong Kong/1/68 [GenBank accession no. AF348176]), and a 660-nt segment for influenza B virus (genomic positions 232 to 891, based on strain B/Lee/40 [GenBank accession no. NC_002207]) were each selected from the HA1 domain for sequence analysis.
The average number of amino acid differences for the antigenic sites and the nonantigenic sites for H1 and H3 were calculated as
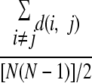 |
where d (i, j) is the number of amino acid differences from either antigenic or nonantigenic sites in each sequence pair and N represents the number of sequences used.
We subsequently performed protein sequence clustering for the Taiwanese influenza A virus subtypes H1 and H3 and influenza B viruses collected. Two sequences were assigned to a different cluster as long as there was one amino acid difference in the HA1 region analyzed. We then classified a cluster as dominant if it contained five or more sequences.
Statistical analysis.
The Student t test (two-tailed, two-sample test with unequal variance) was used to determine the significance in the average number of amino acid differences at antigenic sites and nonantigenic sites for influenza A virus subtypes H1 and H3 by use of SPSS software (version 13.0; SPSS Inc., Chicago, IL).
Phylogenetic construction.
Phylogenetic analysis of a partial region of HA1 (as shown in Table 1) of influenza A virus subtypes H1 and H3 and influenza B virus was performed on the basis of the nucleotide sequences. The MEGA program (11) was used for tree building by use of the neighbor-joining method and the Kimura two-parameter distance matrix. The number of bootstrap replications was set to 1,000, and bootstrap values were labeled on major tree branches for reference. Note that clustering based on nucleotides was first performed for the Taiwanese strains, and only dominant clusters were used to infer phylogenetic relationships. Similar to the clustering scheme used for the amino acid sequences, two strains with any observed nucleotide difference were assigned to different clusters, and a dominant cluster contained at least five sequences. The cluster counts and the lifetimes of the dominant clusters for nucleotide-based and amino acid-based clustering can be different due to synonymous substitutions at the nucleotide level. Aside from the definition of a dominant cluster for the purpose of selecting representative strains for phylogenetic analysis, we further defined a strongly dominant cluster as one that contained 20 or more sequences, for the purpose of the later discussion.
Nucleotide sequence accession numbers.
All sequences newly reported in this study have been deposited in the GenBank database under accession numbers EU068114 to EU068198.
RESULTS
Epidemiological distributions versus sequence diversities.
Clinical isolates collected by 12 virology laboratories in the influenza virus surveillance network were used to represent the influenza virus activities in Taiwan. Figure 1A shows the monthly distribution of positive virus counts from January 2003 to July 2006. The total numbers of isolates of influenza A virus subtypes H3 and H1 and influenza B virus were 2,123, 548, and 1,336, respectively. It is apparent that the influenza A virus H3 showed extensive activity in Taiwan from 2003 to 2006. Although the peak activities of subtype H3 were consistently found in each regular influenza season (the winter months from November-December to March-April of the next year), it is interesting to observe two large summer peaks of subtype H3 in August 2004 and June 2005, each of which was followed by a much smaller winter peak. No influenza A virus H1 activity was found until the 2004-2005 winter season, followed by a significant peak in 2005-2006. For the influenza B viruses, there was only one obvious peak, which occurred in the winter of 2004-2005. Note that the illnesses in these four winters were dominated by types H3, H3, B, and H1, respectively. Interestingly, the two most recent winters in which the peaks for H3 were much smaller were each dominated by influenza B virus and influenza A virus H1, respectively, and each of them followed a summer in which H3 viruses prevailed.
FIG. 1.

Monthly distribution of positive isolates of influenza A virus subtypes H3 and H1 and influenza B viruses in Taiwan from January 2003 to August 2006 (A). (B to D) Individual distributions incorporating the amino acid differences for subtypes H3, H1, and B, respectively. The amino acid differences for the antigenic and nonantigenic sites of subtypes H3 and H1 are graphed separately in panels B and C, respectively. While no antigenic site information was available for influenza B viruses, the amino acid differences for the Victoria and Yamagata lineages are graphed separately in panel D. The amino acid difference for a pair of sequences was computed by summing the number of aligned pairs that showed different amino acid residues. The average amino acid difference per month was then computed on the basis of all pairs of aligned sequences having their disease onset time within the same month. Months in which less than one pair of viruses was isolated and for which no amino acid difference could be produced are labeled with asterisks. a.a., amino acid.
To better understand the relationship between influenza virus activities and their genetic diversities, the average number of amino acid differences in the sequences from viruses causing disease onset in the same month along with the monthly isolation counts is shown separately in Fig. 1B to D for influenza A viruses H3 and H1 and influenza B virus, respectively. The numbers of amino acid differences for the antigenic and the nonantigenic sites were computed separately for the H3 virus subtype (an HA segment of 262 amino acids, including 125 antigenic and 137 nonantigenic sites) and the H1 virus subtype (169 amino acids, including 25 antigenic and 144 nonantigenic sites). Statistical analysis indicated that these differences were significant (Student t test, P < 0.05) for both H1 and H3. Three peaks of antigenic amino acid differences (shown as vertical black bars in Fig. 1B) of influenza A virus H3 over three flu seasons (winter of 2003-2004 winter, summer of 2004, and winter of 2004-2005) took place in October 2003, May-June 2004, and November 2004, respectively. Each of those peaks was followed by a swarming of influenza A H3 virus isolates, with a much larger virus count obtained if the diversity at the antigenic sites was dominant over that at the nonantigenic ones (as seen for the peaks of October 2003 and May-June 2004). All the antigenic sites showed differences equal to or greater than those at nonantigenic sites in all months except November 2005, implying that the viruses were under positive section pressure and were able to continuously cause epidemics in the population. The less apparent dominance of antigenic difference in October, November, and December 2005 might have been due to fewer available sequence counts (7, 2, and 5, respectively) and, thus, fewer pairs (21, 1, and 10, respectively) for use in the computation of diversity. Other low-sequence-count months included August 2003 (8 sequences), January 2006 (9 sequences), September 2003 and May 2006 (11 sequences each), and October 2003 (12 sequences).
It is apparent that from October 2003 to June 2004 the sequence diversity at the antigenic sites was higher than that during any other period. This observation is in line with laboratory hemagglutination inhibition (HI) test results (data not shown), which showed that a major antigenic drift from A/Panama/2007/99-like to A/Fujian/411/2002-like strains was first seen in the winter of 2003-2004, followed by detection of the first batch of local strains that were antigenically distinguishable from A/Fujian/411/2002 in the summer of 2004. Three subsequent smaller peaks of antigenic sites occurred in March 2005, September 2005, and May 2006, with only the one in March 2005 being followed by a major peak of H3.
We also calculated the average amino acid differences for five antigenic sites of H3 separately (data not shown) and found that antigenic sites B and D had higher degrees of diversity than the other three sites from 2003 to mid-2006, suggesting that these two sites were important hot spots when antigenic drift was seen in 2004 and 2005, when the Fujian-like strains were transformed into California-like strains. Finally, fewer amino acid differences were seen at the nonantigenic sites for H3 in 2003 and 2004 than in 2005 and 2006. The average numbers of differences at antigenic and nonantigenic sites in 2003 and 2004 were 2.76 and 0.43, respectively, while the average numbers of differences at antigenic and nonantigenic sites in 2005 and 2006 were 1.88 and 0.91, respectively. The less apparent gap between them in the two most recent years analyzed might suggest stabilization of the Fujian-like strains in the population, without further antigenic drift in the near future.
The corresponding activity of influenza A virus H1 in terms of the amino acid diversity at the antigenic and nonantigenic sites is shown in Fig. 1C. We analyzed only the sequences available in 2005 and 2006 because no apparent activity was detected in 2003 and 2004. Note that the HA1 region analyzed is shorter for the H1 virus (169 amino acids) than for the H3 virus (262 amino acids). The antigenic sites are also less abundant (25 of 169 for H1 and 125 of 262 for H3). In contrast to H3, on the other hand, the nonantigenic sites of H1 showed larger numbers of amino acid differences, on average, than the antigenic sites of viruses recovered during this time period. The average numbers of differences at antigenic and nonantigenic sites of H1 in 2005 and 2006 were 0.90 and 2.34, respectively, while the average numbers of differences at antigenic and nonantigenic sites of H3 from 2003 to 2006 were 2.63 and 0.68, respectively. This phenomenon corresponds well to the findings of a previous study that the HA1 gene of H1 undergoes neutral evolution rather than positive selection, as is observed in H3 (24). Moreover, we have observed that the overall average amino acid difference has steadily increased since February 2006. The average differences at antigenic and nonantigenic sites in 2005 were 0.39 and 0.67, respectively, while in the first 7 months of 2006 they were 0.98 and 2.93, respectively, which illustrates the trend for increasing H1 activity as well as for both antigenic and nonantigenic substitutions from 2005 to 2006.
It was reported that both the Victoria and the Yamagata lineages of influenza B viruses have cocirculated in Taiwan in recent years. Reassortants from these two lineages were detected in as early as 2002 and became dominant in the winter of 2004-2005 (5, 12, 19). Classification in either the Yamagata or the Victoria lineage was based on a BLAST search of their HA1 gene regions and comparison with the sequences in the nucleotide database of the National Center for Biotechnology Information. The Yamagata-lineage strains steadily showed a greater average amino acid difference (3.93) prior to May 2005, after which they became obsolete (Fig. 1D). On the other hand, the Victoria-lineage strains had a much lower level of amino acid diversity (0.94) compared with their sequences after December 2004, when they became prevalent, although they were apparently the dominant lineage during the course of their cocirculation with Yamagata viruses. It is known that B/Shanghai/361/2002 (a Yamagata-like strain) was selected as the influenza B virus vaccine strain for the Northern Hemisphere in the winters of 2004-2005 and 2005-2006. We found that the circulating Victoria viruses had lower levels of amino acid diversity, yet their sequences appeared to better fit those of the population of circulating viruses, probably due to a mismatch of vaccine strains. On the other hand, the greater diversity found among those Yamagata viruses might have been driven by the selection of this vaccine strain in the general population.
General features of sequence clusters.
The statistics for HA1 sequence clustering are summarized in Table 1. Each cluster, according to our definition that sequences are placed in separate clusters whenever a single amino acid difference exists between an aligned pair, has an HA1 region amino acid composition that is unique. The numbers of cluster (i.e., genetic variants) in influenza A viruses H1 and H3, the influenza B virus Victoria lineage, and the influenza B virus Yamagata lineage were 201, 497, 87, and 96, respectively. After normalization for their time spans, it is clear that the cluster counts per month for influenza A viruses H1 and H3 are comparable. They were, however, more prevalent than influenza B viruses, suggesting that influenza A viruses were more likely to evolve over the time period investigated. The influenza B virus Victoria lineage was found to be the largest cluster of all four subtypes, with the sequences of isolates of this lineage comprising 71.5% (534 of 767) of the sequence counts. On the other hand, the largest cluster of influenza A virus H1 sequences contained only 53 (9.9%) of 535 sequences, suggesting the presence of a dominant strain (from a genetic diversity point of view) among the influenza B virus Victoria-like strains, while such dominance was the least apparent for influenza A virus H1. This was also reflected by the cluster density, which was computed by dividing the total number of sequences by the total number of clusters, from which it was clear that influenza A virus isolates are far less dense (2.66 and 2.88 for H1 and H3, respectively) than influenza B viruses (8.59 and 5.57 for the Victoria and Yamagata lineages, respectively). In other words, more genetic variants of influenza A virus than genetic variants of influenza B virus circulated during 2003 to 2006 in Taiwan. The small percentage of dominant clusters (6.9 to 8.4%), in which each cluster contained five or more sequences, indicates that the prevalence of many of those clusters (over 90%) was sparse and the isolates failed to prevail in the general population. Among those clusters that dominated, influenza B virus Victoria strains seem to have aggregated as a limited number of genetic variants.
We also defined the duration or the lifetime of a dominant cluster as the time that elapsed (measured in months) from the earliest to the latest time of onset of sequences within that cluster. The lifetimes were found to be similar for the two influenza A virus subtypes according to either the average or the longest duration. However, for influenza B viruses, the lifetimes were double those for the influenza A viruses. This observation is in line with the findings of cluster analysis, mentioned above, that the influenza B viruses circulating in Taiwan over the past few years showed better genetic coherence into a number of major strains than the influenza A viruses did and thus were able to survive and cause disease for longer durations.
The monthly compositions of the cluster counts for each subtype were also computed and are shown in Fig. 2. Regardless of how many subtypes of influenza viruses were present or which subtype dominated in a specific month, there appears to be a good correlation between the sum of the cluster counts and the total virus counts. In other words, more circulating genetic variants appeared to contribute directly to sizable epidemics, as represented by the peaks in Fig. 2.
FIG. 2.

Monthly compositions of clusters and total numbers of influenza A virus subtypes H1 and H3 and influenza B virus from January 2003 to July 2006.
Phylogenetic analysis.
Figure 3A shows a ladder-like phylogenetic relationship of the HA genes of the Taiwanese influenza A virus H3N2 strains from 2003 to 2006, which is consistent with the relationship reported elsewhere (6, 7). Although only the dominant clusters (each of which contained at least five sequences) are included in Fig. 3, we had previously performed the analysis by including more nondominant clusters (see Figure S1 in the supplemental material) and found no apparent discrepancies in terms of the major evolutionary patterns and pathways. The largest two dominant clusters in 2004 and 2005, namely, H3-285 (91 sequences) and H3-507 (162 sequences), respectively, were found to be identical to A/Norway/807/2004 and A/Western Australia/2005, respectively (which are in clades H3a and H3b, respectively), for the HA1 region analyzed. Interestingly, they both first appeared in the early summer (June 2004) or late spring (April 2005), peaked in the summers (as seen in Fig. 1A), and carried their activities into the winters that followed. The peak activities of subtype H3 in these two summers can also be seen in Fig. 3A, which shows that other strains dominant in the same summer were positioned near these two strains, for example, H3-306 (July 2004 to October 2004; 11 sequences), H3-308 (July 2004 to August 2004; 10 sequences), and H3-314 (August 2004 to January 2005; 11 sequences) are close to H3-285 in clade H3a; and H3-597 (June 2005 to August 2005; 14 sequences), H3-567 (May 2005 to September 2005; 33 sequences), and H3-595 (June 2005 to September 2005; 14 sequences) are close to H3-507 in clade H3b. Between these two clades lies a group of strains from the early summer of 2005 (strains H3-535, H3-550, and H3-496) that connect the 2004 and the 2005-2006 epidemics. Further upstream in this ladder-like evolutionary pathway is another distinct clade resembling A/Ningbo/65/2004 (strains H3-263, H3-367, and H3-327) that bridges clades H3a and H3b to the earlier Fujian- or Wellington-like strains. Note that the amino acid changes at the major branches are largely located at the antigenic sites (one at site B and three at site D). This result is in line with the earlier observation in Fig. 1B that the amino acid changes found in these Taiwanese H3 strains were mostly antigenic.
FIG. 3.

Phylogenetic trees of the HA1 gene in influenza A virus H3N2 and H1N1 strains and influenza B virus strains. The nucleotide sequences of the H3N2 and H1N1 and the influenza B virus strains (positions 178 to 963, 147 to 653, and 232 to 891 of the HA gene, respectively, as described in Materials and Methods) were first aligned by use of the Clustal W program, and phylogenetic analysis was performed by use of the MEGA2 program and the neighbor-joining method. The percentages of bootstrap frequencies at the major branches are indicated. Strains A/Moscow/10/1999, A/Brevig Mission/1/1918, and B/Lee/40 were used as the outgroups for the H3N2 and H1N1 viruses and influenza B virus, respectively. The cluster identifier on each leaf node contains three sections: a unique cluster name, the earliest and latest onset month (month/year), and the size of the cluster. Solid triangles, vaccine strains selected by the World Health Organization; solid circles, strongly dominant clusters, each of which contains more than 20 sequences. The nucleotide sequences of the reference strains indicated in parentheses completely match the nucleotide sequences within that cluster. The amino acid changes at major branches are also labeled, with additional letters shown in parentheses if that position is a reported antigenic site.
Figure 3B shows the phylogenetic relationship of the HA genes of the H1N1 subtype, in which the 19 dominant clusters with isolates from 2005 to 2006 are separated into two major clades, clades H1a and H1b; the latter also includes A/New Caledonia/20/99, which has been a vaccine strain since 2000. Interestingly, one strongly dominant H1a cluster, namely, H1-61, with 48 sequences, together with another less dominant cluster, H1-54, actually more closely resembles a 2004 Canterbury strain, while all of the rest of the strains in cluster H1a resembled the 2006 reference strains. Viruses in half of the six clusters within clade H1b were isolated as early as January 2005 and produced the low levels of H1 activity seen in the first half of 2005 (as seen in Fig. 1A), while all except two isolates (which were recovered in December 2005) in all 13 clusters in the H1a lineage were recovered in 2006 and beyond, suggesting a shift in momentum for Taiwanese influenza A virus H1 away from clade H1b (which contains A/New Caledonia/20/99) to clade H1a beginning in 2006 (H1-26 to H1-61 to H1-273). We also noted that four H1 reference strains (Florida, England, Idaho, and South Australia) matched their tagged clusters in clade H1b 100%. The clusters in clade H1a, on the other hand, matched only one reference strain (strain Macau) found in the public databases, suggesting that these clusters are relatively unique in Taiwan and have yet to be reported. Upon comparison of these clusters at their amino acid level, we found that only 2 of the 13 amino acid changes in the major branches were antigenic (S69L at site Sa and V166A at site Ca2), which complies with the results in Fig. 1C that the number of amino acid differences at nonantigenic sites were, in general, higher than the number at antigenic sites.
Figure 3C shows the phylogenetic tree of the HA genes of the influenza B virus isolates, which is clearly divided into a Yamagata lineage and a Victoria lineage. The Yamagata-like strains showed a more diverse evolutionary pattern, which could be further grouped into three clades, namely, clades Ya, Yb, and Yc. The sequences of the isolates in clade Ya were similar to the sequence of B/Yamagata/1246/2003, while the sequences of the isolates in clades Yb and Yc were similar to the sequences of B/Georgia/9/2005 and B/New York/12/2005, respectively. For the Victoria-like lineage, the sequence of the largest cluster (V-2, with 376 sequences) completely matched the sequence of B/Nepal/1137/2005, which had the longest life span, from March 2004 to June 2006 (28 months). Although Yamagata-like strain B/Shanghai/361/2002 was chosen as the vaccine strain for the 2004-2005 and 2005-2006 seasons, the Taiwanese Victoria-like strains actually prevailed and outnumbered those Yamagata strains, and they were apparently less varied in terms of their HA1 sequences (fewer clusters, each with a relatively short tree branch). Although both influenza B virus lineages were dominant, in particular during the outbreak in the winter of 2004-2005, a Yamagata lineage containing Victoria clusters (and vice versa) was not observed, suggesting that no recombination took place in the HA1 domain analyzed.
DISCUSSION
We have reported on comprehensive influenza virus surveillance activities performed in Taiwan over the past four flu seasons (2003 to 2006). In addition to those four winters, during which strong influenza virus activities were observed, as in other countries or regions, two summer influenza virus subtype H3 peaks were found in 2004 and 2005 (Fig. 1A). Each of these two summer peaks of H3 was followed by significantly less H3 activity during the next winter (2004-2005 and 2005-2006) compared with that during the previous two winters (2002-2003 and 2003-2004). Instead, influenza B viruses and influenza A viruses H1 took over as the dominant influenza viruses in the winters of 2004-2005 and 2005-2006, respectively. The reason why the emergence of the influenza H3 viruses in those two summer led to changes in the dominant strains in the following winters remains unsolved. One hypothesis is that these two summer peaks of H3 were caused by imported strains rather than strains that had been circulating locally in Taiwan in the previous two winter seasons. A recent study (15) also made a similar conclusion that H3 viruses circulating in one region during one season were mainly derived from newly imported strains rather than from local isolates circulating in the previous season. If they were indeed imported (see the two largest H3 clusters in Fig. 3A), is it possible that they were equipped with a certain growth advantage over local strains? When the summer epidemic ended, under this scenario, the local strains were no longer able to prevail in the winter that followed, thus allowing the influenza B and H1 viruses to take over. In terms of the amino acid sequence changes computed from those circulating H3 strains, it was found that the diversity at the antigenic sites was much higher at the beginning of the winter of 2003-2004, when an H3 peak was observed, and declined accordingly over the next two winters, when little H3 activity was detected. The cross-immunity caused by those summer H3 strains may also explain the low levels of activity of H3 and the low antigenic sequence variations in the two following winters. Coincidently, no apparent H3 activities were detected in the summer of 2006, and one sharp but short epidemic was found in the winter of 2006-2007 (data not shown).
Another interesting observation for H3 is the relative amino acid sequence variations between antigenic sites and nonantigenic sites. From April 2003 to June 2004, the ratio of the number of antigenic site changes to the total number of variations was higher than that during any other month. This period corresponds to a time when certain antigenic changes and strain transitions from Moscow (or Panama) to Fujian strains and, following that, to California (or Wellington) strains were observed. It seems that Fujian strains were converted to California strains in a short period of time, and they were closer in terms of their evolutionary distance than the Moscow and Fujian strains. In contrast, from April 2005 to February 2006, the ratio of the number of antigenic changes to the total number of variations was the lowest among those that occurred during the period of time under investigation. Indeed, A/California/7/2004 was used as the vaccine strain in the winter of 2005 and had apparently relieved some positive selection pressure on the antigenic sites for H3 cases from the fall of 2005 and beyond.
In contrast to the circulating H3 viruses, for which several peaks were found during the 4 years of surveillance and the antigenic changes were overwhelmingly higher than the nonantigenic ones, only one H1 peak, which occurred in the winter of 2005-2006, was found, and the amino acid changes were mostly nonantigenic. Although the average number of amino acid differences at the H3 antigenic sites was greater than the number at the H1 antigenic sites in our studies (2.63 and 0.90, respectively), it should be noted that the average number of amino acid differences per residue at H1 antigenic sites (0.04) was larger than the average number at H3 antigenic sites (0.02). Nevertheless, it is our belief that the number of amino acid variations at all antigenic sites determined the overall antigenic variations in the HA1 region. Evidence for this is that most H1 strains isolated in Taiwan, based on their HI test data, still showed high titers against A/New Caledonia/20/99. Furthermore, the lower level of fitness of H3 during the winter mentioned above might have given H1 a chance to cause an epidemic, as we have described here. It seems that the H3 and H1 viruses were competing somewhat and were holding each other up over these 4 years. That is, there were almost no cases of H1 infection in Taiwan prior to the winter of 2005-2006, over which this long period of time H3 was dominant and revealed major strain transitions from Moscow to Fujian to California. On the other hand, H1 viruses took over in the winter of 2005-2006, when less H3 activity was found.
Although no antigenic site has been reported for influenza B viruses, higher levels of variation in the HA gene of the Yamagata lineage might indicate that this lineage had been under more evolutionary pressure than the Victoria lineage. The choice of vaccine strain could be the possible reason for this observation. In the winter of 2004-2005, the Shanghai strain (which is of the Yamagata lineage) was used as the vaccine strain and offered only limited protection against viruses of the Victoria lineage. In other words, the reassortment of these Victoria-like strains could describe another means by which they gained a better chance of survival and the competitive ability to cause epidemics.
The positive correlation found between the isolates and the number of clusters indicates that although one or some limited number of dominant strains contributed to one epidemic, other related strains also appeared during the epidemic, and the scenario seems to be similar to that for a “swarm” defined in a previous study (17). Different patterns of cluster distributions in these three subtypes also illustrated the disparity of the strategy of evolution for HA. For H1 and H3 viruses that had higher mutation rates, a shorter life span of the clusters is expected. Actually, no apparent difference between the cluster count per month, the average cluster size, the average lifetime, or the longest lifetime was found between the H1 and the H3 viruses (Table 1). Although the H1 viruses showed notable activity only in 2005-2006, the H1 and H3 subtypes revealed common characteristics by cluster analysis. For the influenza B viruses, on the other hand, very different cluster statistics were observed. In addition, the two lineages of influenza B viruses showed similar patterns in cluster statistics, although the Yamagata viruses revealed more sequence variations than the Victoria viruses.
The summer outbreaks caused by H3 were mainly caused by cluster H3-285 (which contained 91 sequences) in 2004 and by cluster H3-507 (which contained 162 sequences) and H3-567 (which contained 33 sequences) in 2005 (Fig. 1B and 3A). During the two winter seasons in 2002-2003 and 2003-2004 in which H3 caused the outbreaks, on the other hand, the virus strains generally dispersed into different clusters, and no single cluster could be considered strongly dominant (that is, the cluster contained 20 or more sequences). In other words, these winter outbreaks were caused by sporadic strains, while the outbreaks in the following summers were caused by the strains that were strongly dominant during this period. Furthermore, the ladder-like shape of the phylogenetic tree also indicates that only one major strain and other related minor strains of influenza A virus H3 circulated during a specific epidemic season, which supports well the proposed scenario (10, 20) from the epochal model of the evolution of influenza A virus.
Unlike the situation in which only one dominant strain of Taiwanese H3 viruses circulated in one season, two genetically distinct lineages of influenza A virus H1 cocirculated in Taiwan in the winter of 2005-2006. There was no apparent antigenic change in these H1 viruses, according to the results of the HI test (data not shown). From the analysis of the amino acid variations, we also saw a closer antigenic similarity of H1 to A/New Caledonia/20/99, which was used as the vaccine strain from 1999 to 2006. As A/Solomon Islands/3/2006 was selected as the 2007-2008 vaccine strain, it was indeed found to have a higher degree of sequence identity with the clade H1a viruses, which represented the dominant lineage in 2006.
Among the influenza B viruses, the dominant clusters in the Yamagata lineage were separated into three subclades, while in the Victoria lineage there was a major dominant cluster (cluster V-2) that contained 376 sequences and that was prevalent from March 2004 to June 2006. The observation that the dominant strains in the Yamagata lineage had shorter prevalence times than those in the Victoria lineage might have resulted from recently reported reassortants and might have involved their HA and neuraminidase genes (5, 12, 19). During the winter of 2006-2007, these reassortants caused one serious epidemic that was even larger than the other epidemics that occurred after 2000 (9). In contrast, the introduction of vaccine strain B/Shanghai/361/2002 during this time period, which set up greater evolutionary pressure for the Yamagata viruses, apparently drove them to evolve into more different subclades. Despite these genetic transitions, however, they seemed to have less of a competitive advantage than the Victoria strains in the winter of 2006-2007 that followed (data not shown).
In this work we performed the genetic characterization of Taiwanese influenza viruses on the basis of pairwise analysis of amino acid variations (at antigenic and nonantigenic sites), genetic clustering, and phylogenetic analyses. Although they have provided a good description of the evolutionary story for HA, some questions remain to be answered. One is the evolutionary relationship between these clusters. For example, we are interested in knowing whether the dominant clusters were located in the center of the sequence space for all clusters found in the same epidemic. Another important task is to find the most likely ancestor of these dominant clusters so that the transition between epidemics may be better described. In addition to clustering analysis, we measured the relationship of these clusters with the vaccine strain and other local strains that cocirculated, which may also provide more insight into the evolution of the HA gene in influenza A and B viruses.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Science Council (grants 94-0324-19-F-01-00-00-00-35, 95-0324-19-F-01-00-00-00-35, and 96-0324-19-F-01-00-00-00-35) and the CDC, Department of Health, Taiwan.
We thank the investigators of the CDC-Taiwan Contracted Virology Reference Laboratory Network for collecting and providing clinical samples: Chuan-Liang Kao, National Taiwan University Hospital, Taipei; Jang-Jih Lu, Tri-Service General Hospital, Taipei; Yu-Jiun Chan, Veterans General Hospital, Taipei; Kuo-Chien Tsao, Chang Gung Memorial Hospital, Linkou; Ming-Jer Ding, Veterans General Hospital, Taichung; Mu-Chin Shih, Chinese Medical University Hospital, Taichung; Jen-Shiou Lin, Changhua Christian Hospital, Changhua; Jen-Ren Wang, National Cheng Kung University Hospital, Tainan; Kuei-Hsiang Lin, Kaohsiung Medical University Hospital, Kaohsiung; Yung-Ching Liu, Veterans General Hospital, Kaohsiung; Hock-Liew Eng, Chang Gung Memorial Hospital, Kaohsiung; and Li-Kuang Chen, Tzuchi Hospital, Hualien.
Footnotes
Published ahead of print on 6 February 2008.
Supplemental material for this article may be found at http://jcm.asm.org/.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 253389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bush, R. M., C. A. Bender, K. Subbarao, N. J. Cox, and W. M. Fitch. 1999. Predicting the evolution of human influenza A. Science 2861921-1925. [DOI] [PubMed] [Google Scholar]
- 3.Bush, R. M., W. M. Fitch, C. A. Bender, and N. J. Cox. 1999. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol. Biol. Evol. 161457-1465. [DOI] [PubMed] [Google Scholar]
- 4.Caton, A. J., G. G. Brownlee, J. W. Yewdell, and W. Gerhard. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31417-427. [DOI] [PubMed] [Google Scholar]
- 5.Chen, G. W., S. R. Shih, M. R. Hsiao, S. C. Chang, S. H. Lin, C. F. Sun, and K. C. Tsao. 2007. Multiple genotypes of influenza B viruses cocirculated in Taiwan in 2004 and 2005. J. Clin. Microbiol. 451515-1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Earn, D. J. D., J. Dushoff, and S. A. Levin. 2002. Ecology and evolution of the flu. Trends Ecol. Evol. 17334-340. [Google Scholar]
- 7.Ferguson, N. M., A. P. Galvani, and R. M. Bush. 2003. Ecological and immunological determinants of influenza evolution. Nature 422428-433. [DOI] [PubMed] [Google Scholar]
- 8.Fitch, W. M., R. M. Bush, C. A. Bender, and N. J. Cox. 1997. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc. Natl. Acad. Sci. USA 947712-7718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jian, J. W., C. T. Lai, C. Y. Kuo, S. H. Kuo, L. C. Hsu, P. J. Chen, H. S. Wu, and M. T. Liu. 2007. Genetic analysis and evaluation of the reassortment of influenza B viruses isolated in Taiwan during the 2004-2005 and 2006-2007 epidemics. Virus Res. 131243-249. [DOI] [PubMed] [Google Scholar]
- 10.Koelle, K., S. Cobey, B. Grenfell, and M. Pascual. 2006. Epochal evolution shapes the phylodynamics of interpandemic influenza A (H3N2) in humans. Science 3141898-1903. [DOI] [PubMed] [Google Scholar]
- 11.Kumar, S., K. Tamura, and M. Nei. 2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5150-163. [DOI] [PubMed] [Google Scholar]
- 12.Lin, J. H., S. C. Chiu, M. W. Shaw, Y. C. Lin, C. H. Lee, H. Y. Chen, and A. Klimov. 2006. Characterization of the epidemic influenza B viruses isolated during 2004-2005 season in Taiwan. Virus Res. 124204-211. [DOI] [PubMed] [Google Scholar]
- 13.Nakajima, K., E. Nobusawa, A. Nagy, and S. Nakajima. 2005. Accumulation of amino acid substitutions promotes irreversible structural changes in the hemagglutinin of human influenza AH3 virus during evolution. J. Virol. 796472-6477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakajima, K., E. Nobusawa, K. Tonegawa, and S. Nakajima. 2003. Restriction of amino acid change in influenza A virus H3HA: comparison of amino acid changes observed in nature and in vitro. J. Virol. 7710088-10098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson, M. I., L. Simonsen, C. Viboud, M. A. Miller, J. Taylor, K. S. George, S. B. Griesemer, E. Ghedi, N. A. Sengamalay, D. J. Spiro, I. Volkov, B. T. Grenfell, D. J. Lipman, J. K. Taubenberger, and E. C. Holmes. 2006. Stochastic processes are key determinants of short-term evolution in influenza A virus. PLoS Pathog. 2e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Plotkin, J. B., and J. Dushoff. 2003. Codon bias and frequency-dependent selection on the hemagglutinin epitopes of influenza A virus. Proc. Natl. Acad. Sci. USA 1007152-7157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plotkin, J. B., J. Dushoff, and S. A. Levin. 2002. Hemagglutinin sequence clusters and the antigenic evolution of influenza A virus. Proc. Natl. Acad. Sci. USA 996263-6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shih, S. R., G. W. Chen, C. C. Yang, W. Z. Yang, D. P. Liu, J. H. Lin, S. C. Chiu, H. Y. Chen, K. C. Tsao, C. G. Huang, Y. L. Huang, C. K. Mok, C. J. Chen, T. Y. Lin, J. R. Wang, C. L. Kao, K. H. Lin, L. K. Chen, H. L. Eng, Y. C. Liu, P. Y. Chen, J. S. Lin, J. H. Wang, C. W. Lin, Y. J. Chan, J. J. Lu, C. A. Hsiung, P. J. Chen, and I. J. Su. 2005. Laboratory-based surveillance and molecular epidemiology of influenza virus in Taiwan. J. Clin. Microbiol. 431651-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai, H. P., H. C. Wang, D. Kiang, S. W. Huang, P. H. Kuo, C. C. Liu, I. J. Su, and J. R. Wang. 2006. Increasing appearance of reassortant influenza B virus in Taiwan from 2002 to 2005. J. Clin. Microbiol. 442705-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Nimwegen, E. 2006. Epidemiology. Influenza escapes immunity along neutral networks. Science 3141884-1886. [DOI] [PubMed] [Google Scholar]
- 21.WHO. 2002. Influenza vaccines. Wkly. Epidemiol. Rec. 28230-239. [PubMed] [Google Scholar]
- 22.Wiley, D. C., I. A. Wilson, and J. J. Skehel. 1981. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 289373-378. [DOI] [PubMed] [Google Scholar]
- 23.Wilson, I. A., and N. J. Cox. 1990. Structural basis of immune recognition of influenza virus hemagglutinin. Annu. Rev. Immunol. 8737-771. [DOI] [PubMed] [Google Scholar]
- 24.Wolf, Y. I., C. Viboud, E. C. Holmes, E. V. Koonin, and D. J. Lipman. 2006. Long intervals of stasis punctuated by bursts of positive selection in the seasonal evolution of influenza A virus. Biol. Direct. 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.