

Abstract
The final, structure-determining step in the folding of membrane proteins involves the coalescence of preformed transmembrane helices to form the native tertiary structure. Here, we review recent studies on small peptide and protein systems that are providing quantitative data on the interactions that drive this process. Gel electrophoresis, analytical ultracentrifugation, and fluorescence resonance energy transfer (FRET) are useful methods for examining the assembly of homo-oligomeric transmembrane helical proteins. These methods have been used to study the assembly of the M2 proton channel from influenza A virus, glycophorin, phospholamban, and several designed membrane proteins—all of which have a single transmembrane helix that is sufficient for association into a transmembrane helical bundle. These systems are being studied to determine the relative thermodynamic contributions of van der Waals interactions, conformational entropy, and polar interactions in the stabilization of membrane proteins. Although the database of thermodynamic information is not yet large, a few generalities are beginning to emerge concerning the energetic differences between membrane and water-soluble proteins: the packing of apolar side chains in the interior of helical membrane proteins plays a smaller, but nevertheless significant, role in stabilizing their structure. Polar, hydrogen-bonded interactions occur less frequently, but, nevertheless, they often provide a strong driving force for folding helix–helix pairs in membrane proteins. These studies are laying the groundwork for the design of sequence motifs that dictate the association of membrane helices.
Keywords: Membrane protein, peptides, folding, helix association, FRET, analytical ultracentrifugation
Protein folding is a remarkable process in which a peptide chain folds into a well-defined structure with fine-tuned dynamic and functional properties. Over the last several decades, the kinetics and thermodynamics of the folding of water-soluble proteins have been extensively studied, and our present understanding of this subject has advanced significantly. In contrast, our understanding of the folding of membrane proteins remains in its infancy. For example, one can predict the energetic consequences of changing a buried nonpolar side chain in the interior of a water-soluble protein to a smaller side chain; a Leu-to-Ala mutation will “cost” ~2–5 kcal/mole (Pace 1992), depending on the extent of structural rearrangement accompanying the mutation (Xu et al. 1998). The corresponding range of values for membrane-soluble proteins is only now beginning to be measured by methods described in this review. In general, van der Waals interactions, electrostatic interactions, hydrogen bonding, and solvophobic effects will determine membrane protein stability, but the magnitude and relative importance of each of these forces remains to be determined. Fortunately, in recent years a number of simple transmembrane helical bundles have been developed that should allow these questions to be addressed quantitatively. In this review, we discuss several natural and designed systems and methods used to study the energetics of their association in membrane-like environments.
An understanding of membrane protein folding requires a consideration of the nature of the unfolded state, which is markedly different from the unfolded state of a water-soluble protein. In water-soluble proteins the unfolded state is largely devoid of secondary structure. In contrast, the unfolded state of helical membrane proteins retains considerable helical content. A two-stage model to account for the assembly of membrane proteins has been proposed by Popot and Engelman (1990) and further elaborated by White and coworkers (White and Wimley 1999) to describe the in vitro folding of membrane proteins. In the first stage of this process, the protein is inserted into a membrane. The driving force for insertion derives primarily from the transfer of hydrophobic side chains from water to the apolar region of the bilayer. Aromatic and positively charged side chains near the bilayer headgroup regions contribute to the appropriate orientation in biological membranes (Engelman and Steitz 1981; Popot and Engelman 1990, 2000; Liu and Deber 1998a; Deber et al. 1999; White and Wimley 1999). The helical conformation is stabilized in a membrane, because the backbone amide-carbonyl hydrogen bonds are satisfied within the helix and sequestered away from the nonpolar environment of the bilayer (Engelman and Steitz 1981; Popot and Engelman 1990, 2000; White and Wimley 1999). Once inserted in the bilayer, the protein is then able to fold via the coalescence of the helices to form the native tertiary structure.
Clearly, the above models represent an oversimplification of the actual kinetic process of protein insertion and folding as it occurs in vivo, in which chaparonins and translocon complexes come into play. Thus, these models provide valuable conceptual frameworks for understanding the overall thermodynamic features required to maintain the inserted, folded state of a protein. The insertion of transmembrane helices has been extensively studied, and is the topic of several reviews (Engelman and Steitz 1981; Popot and Engelman 1990, 2000; von Heijne 1999; White and Wimley 1999). We therefore will focus on the second step, in which the protein attains its native tertiary structure. Several problems have impeded the understanding of the thermodynamics of membrane protein folding. Membrane proteins are difficult to express and purify in high yields, although this obstacle can often be overcome for small membrane proteins. Furthermore, until recently, there have been few membrane proteins whose three-dimensional structures are known. However, the largest obstacle has been the difficulty in devising experimental systems in which membrane proteins can be shown to fold in a thermodynamically reversible manner. Among native membrane proteins, bacteriorhodopsin (Chen and Gouaux 1999) and diacylglycerol kinase (Y. Zhou et al. 2001) have been shown to fold reversibly under some conditions. A mutant of bacteriorhodopsin, which increased the hydrophobicity of the detergent-bound molecular surface, was shown to be less resistant to unfolding under equilibrium conditions (Chen and Gouaux 1999), although the effects have not been expressed in terms of differences in the free energy of folding of the protein. Here, we focus on more recently developed model systems comprised of homo-oligomeric transmembrane helices.
Homo-oligomeric transmembrane proteins provide attractive systems for the study of membrane protein folding because of their symmetry and relative simplicity. Here, we consider systems in which a single transmembrane helix associates to form a transmembrane helical bundle (Fig. 1 ▶). In many regards, these membrane proteins resemble water-soluble coiled coils, which have contributed significantly to our understanding of folding of water-soluble helical proteins (Cohen and Parry 1990; Harbury et al. 1993; Hodges 1996; Oakley and Kim 1998; Zhu et al. 2000). In both classes of structures, the formation of native structure is thermodynamically linked with the association step, providing a relatively easy and straightforward way to measure the thermodynamics of the process. The structures and properties of the monomeric helical peptides also can be studied in detail, allowing a rigorous characterization of the unfolded state of the protein. Furthermore, the noncovalent assembly of membrane peptides is likely to mimic the folding of larger native proteins; many membrane proteins have been cleaved into multiple fragments that nevertheless associate in membranes to adopt native, functional proteins (Popot and Engelman 2000). Thus, the folding process appears to be similar, irrespective of whether it occurs in an inter- or intramolecular manner.
Figure 1.

Diagram of the two-state model of membrane protein folding as described above. In the first stage, the preformed helix inserts into the lipid bilayer, and in the second stage, helices associate to form the folded structure.
In the following sections, we discuss methods to monitor the reversible folding of oligomeric membrane proteins. We then discuss the application of these methods to the study of glycophorin (GpA), the M2 proton channel from influenza A virus (M2), and phospholamban (PL), which, respectively, form 2-, 4-, and 5-helix bundle motifs (Fig. 2 ▶). Several designed systems are also discussed. Finally, we present a view of the features that stabilize the folding of membrane proteins as assessed from these experimental studies. The reader whose interest is primarily in the features stabilizing the structures of membrane proteins (rather than analytical methods and the details of individual membrane proteins) may wish to skip directly to this section.
Figure 2.
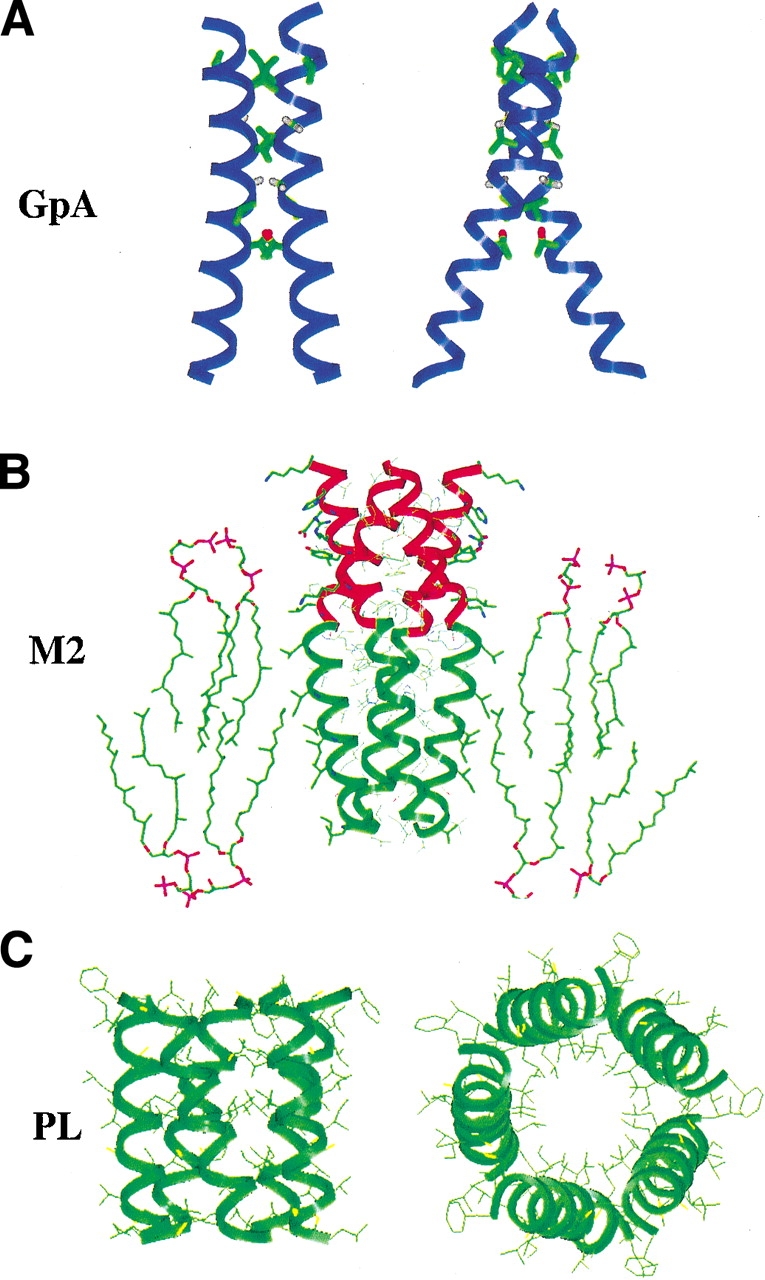
Homo-oligomeric membrane proteins. (A) The structure of GpA in ribbon representation showing residues (LIXXGVXXGVXXT) that are highly sensitive to mutation. (B) “Helical extension” model of the protease-resistant core of M2. Shown is a model of the TM peptide (green ribbon) with the water-soluble helical extension (residues 45–60, red ribbon) in a lipid bilayer. (C) Model of the transmembrane region of PL by Thomas and coworkers (Karim et al. 1998).
We also note, but do not discuss in this relatively brief review, several other interesting transmembrane (TM) helical systems that have been characterized experimentally. These include the phage M13 coat protein (Weiner et al. 1989; Deber et al. 1993, 1999; Peled and Shai 1993, 1994; Smith et al. 1996; Soekarjo et al. 1996; Gazit et al. 1997; Liu and Deber 1998a; Wang and Deber 2000; Constantinescu et al. 2001; Eisenhawer et al. 2001; Melnyk et al. 2001; Therien et al. 2001), a designed TM peptide (Liu and Deber 1998a), the Escherichia coli aspartate receptor TM peptide (Melnyk et al. 2001), helix–loop–helix TM segments of the CFTR (Therien et al. 2001), synthetic helices A and C of the Bacillus thuringiensis toxin (Gazit et al. 1997), S-2 and H-5 segments of the Shaker K+ channel (Peled and Shai 1993, 1994), the erythropoietin receptor TM domain (Constantinescu et al. 2001), the TM sequence of the neu/erbB-2 receptor (Smith et al. 1996), neu and c-neu (Weiner et al. 1989), peptides from the paramyxovirus fusion protein (Lamb and Pinto 1997), the TM domains of the integrin αIIbβ3 (R. Li et al. 2001), and the HIV Vpu protein transmembrane domain (Schubert et al. 1996).
Methods to study oligomerization-linked folding of membrane proteins
The folding of a membrane protein can be followed by monitoring any spectroscopic or physical property that differs between the folded and the unfolded state. For example, the acquisition of native structure in bacteriorhodopsin occurs with a concomitant change in the absorbance maximum and intensity of its cofactor, retinal (Popot et al. 1986). Also, the folding of diacylglycerol kinase gives rise to changes in its absorbance (Lau and Bowie 1997) spectrum and the near-UV CD spectrum of its aromatic side chains (Lau and Bowie 1997; Salom et al. 2000). Unfortunately, because the magnitude of these differences is much smaller than for most helical water-soluble proteins, other methods are needed.
For a homo-oligomeric protein, folding and oligomerization are thermodynamically coupled. Thus, one can study any property that varies between the monomer and oligomer states. In particular, analytical ultracentrifugation, fluorescence resonant energy transfer (FRET), and gel electrophoresis have been widely used for these purposes. Other methods that are outside the scope of this review include electron paramagnetic resonance (Hubbell et al. 1996, 1998; Oh et al. 1996; Langen et al. 1998), Fourier transform infrared spectroscopy (Chung and Thompson 1996; Smith et al. 1996; Torres et al. 2000a), and small angle X-ray scattering (Bu and Engelman 1999). Typically, FRET, ultracentrifugation, and gel electrophoresis have been used with membrane proteins solubilized in micelles rather than in lipid bilayers. Although micelles are clearly different from bilayers, there is qualitative agreement between studies in detergent micelles and oligomerization studies in biological membranes by the ToxR (Brosig and Langosch 1998) and the TOXCAT (Russ and Engelman 1999; F.X. Zhou et al. 2000, 2001) assays. Therefore, studies in detergents appear to have relevance to transmembrane helix association in the more ordered bilayer systems.
Analytical ultracentrifugation
Analytical ultracentrifugation sedimentation equilibrium experiments have been extensively used to characterize water-soluble proteins (Laue et al. 1992; Teller 1973), and this method has also been adapted for membrane proteins solubilized in detergent micelles (Tanford et al. 1974; Tanford and Reynolds 1976; Fleming 2000). Analytical ultracentrifugation is used to determine the buoyant molecular weight (Mb) of a protein in solution. For a single species with molecular weight Mw with a partial specific volume ν̄ p in a solvent of density ρ, the buoyant molecular weight can be calculated as Mb = (1 − ν̄p ρ)Mw. However, for a membrane protein in a micelle, the detergent also contributes to the observed molecular weight. The method of Tanford and Reynolds (Tanford et al. 1974; Tanford and Reynolds 1976) resolves this issue. It is assumed that the partial specific volume of the protein–detergent complex ν̄c, with δd the weight ratio of detergent, can be approximated as the weighted average of the partial specific volumes of the detergent (ν̄d) and the protein (ν̄p) according to equation 1:
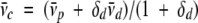 |
1 |
Therefore, the equation for the protein molecular weight becomes:
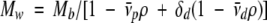 |
2 |
By adding D2O to the solution, the solvent density can be matched to the detergent density (ρ = 1√vd) such that the term from the detergent vanishes. Under these conditions, the molecular weight of the protein can be determined as though the detergent were not present.
Analytical ultracentrifugation data can be fit to a monomer–n-mer equilibrium with an aggregation number n. For a homogeneous species, the following equation applies:
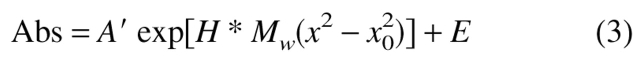 |
3 |
in which Abs is the absorbance at radius position x, A′ is the absorbance at x0, E is the baseline absorbance, H = (1 − ν̄pρ)ω2/2RT, and ω is the angular velocity in radians per second. To determine the degree of association (n) describing a monomer–n-mer equilibrium, one considers the contributions of the monomer and oligomer using the equilibrium constant according to equation 4:
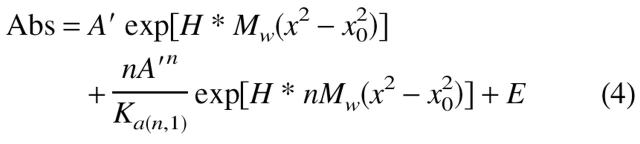 |
4 |
where Ka(n,1) is the disssociation constant for the interaction expressed in absorbance units. The appropriate extinction coefficient and path length can, of course, be used to convert Ka(n,1) to other units.
Equation 4 allows the determination of both the oligomerization state and the free energy of association for a monomer–n-mer species. However, it should be realized that this equation has several adjustable parameters, and it is not always trivial to obtain a unique set of values from a single run alone, particularly when the protein is only partially associated under the experimental conditions. Therefore, it is essential to analyze the sensitivity of the curve-fit to the assumptions of the model being used (Gratkowski et al. 2002). Global analysis of multiple curves obtained at different rotor speeds and protein concentrations is generally helpful in reducing both model and parameter uncertainty.
Figure 3 ▶ illustrates this approach for a peptide representing the influenza A virus M2 transmembrane segment, which is known from independent experiments to exist in a monomer–tetramer equilibrium near neutral pH (Salom et al. 2000). Figure 3A ▶ illustrates data collected for the peptide under conditions in which it is approximately half-tetrameric. The fitted curves are shown for either a pure dimer model (a monomer–dimer model gave no improvement) or a monomer–tetramer model. As can be seen both from the residuals (top panels) and the small differences in the dotted (dimer) versus solid (monomer–tetramer) fit lines, it would be difficult to argue for one model over the other. However, inclusion of two additional experiments (Fig. 3B ▶) shows the dimer model to consistently underpredict the absorbance at large radius, giving a less random set of residuals that strongly supports the argument against the dimer model. Inclusion of even more data covering a wider range of peptide/detergent ratios (Fig. 3C ▶) shows further that the residuals are clearly more random for a monomer–tetramer (Fig. 3C ▶, bottom panel) than either a dimer (Fig. 3C ▶, top) or a monomer–trimer (Fig. 3C ▶, middle panel) fit. Therefore, it is essential to perform a global analysis of multiple curves obtained at different rotor speeds and different protein concentrations. In cases in which concentrations are accurately known, material balance considerations can sometimes be invoked to distinguish between models giving otherwise equivalent fits (Gratkowski et al. 2002). Independent methods such as FRET and SDS-PAGE (see below) can also help distinguish among different models.
Figure 3.

Analytical ultracentrifugation data of a peptide from the transmembrane region of influenza A virus M2 proton channel in DPC micelles. A subset of these data was published in Kochendoerfer et al. (1999). Detergent-solubilized peptide at a peptide/detergent ratio of 1/284 (53 μM peptide, 15 mM DPC) in 50 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 0.5 mM TCEP was centrifuged to equilibrium at 42 krpm, and optical density at 280 nm versus radius data (bottom panel) was fit to functions describing sedimentation equilibrium of a dimer (dashed line) and a monomer in equilibrium with a tetramer (solid line). (A, top two panels) Fit residuals for dimer and monomer–tetramer models. (B) As in A, except data were obtained at two additional speeds (36 and 48 krpm). Fits were done using a single dissociation constant and baseline per model for all data sets. Residuals of all three data sets were concatenated for each of the upper two panels. (C) Concatenated residuals for, respectively, dimer, monomer–trimer, and monomer–tetramer model fits to data obtained as in A and B, but with samples prepared at two additional peptide/detergent ratios (1/126 and 1/554). Fits were done using a single dissociation constant per model for all data sets and a single baseline for each peptide/detergent ratio.
The association of a detergent-solubilized protein generally occurs in the micellar phase. Thus, the pertinent concentration variable is the mole fraction of the protein relative to the detergent, rather than the bulk concentration of the protein in aqueous solution. Experimental studies have shown this to be the case for MS1 (Choma et al. 2000) and GpA (Fisher et al. 1999). One possible exception is a fusion protein that contains the transmembrane helix from glycophorin attached to the water-soluble protein staphylococcal nuclease (Fleming et al. 1997; Fleming and Engelman 2001b).
Fluorescence resonance energy transfer (FRET)
It is important to consider multiple methods for determining the aggregation state and dissociation constant for a given system, because more than one model can often be fit to the data from a single method, such as analytical ultracentrifugation. In these cases, a second method can often decrease the ambiguity of the interpretation. One such method that has been extensively applied to the analysis of membrane proteins is fluorescence resonance energy transfer (FRET). A great advantage of FRET is that it can be performed in vesicles as well as micelles. In early work, this method was used to analyze the oligomerization state of gramicidin A in lipid vesicles (Veatch and Stryer 1977) and of bacteriorhodopsin in micelles (London and Khorana 1982). It has since been used extensively to examine the association of peptides in micelles and bilayers (Peled and Shai 1994; Shai 1995; Gazit et al. 1997; Eisenhawer et al. 2001). When applied to study homo-oligomers, this method uses two derivatives of a peptide: one bearing a fluorescent label and the other bearing a quencher. If the peptides associate, the emission of the fluorophore will be quenched by the presence of a sufficiently close quencher in the complex. The aggregation number can be determined by an experiment in which the fluorophore, quencher, and unlabeled derivatives of the peptide are mixed in lipid vesicles in different molar ratios. In the extreme case in which only the two labeled peptides (F and Q) are used, the observed fluorescence intensity, I, is related to the aggregation number, n, according to the equation:
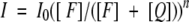 |
5 |
in which [F] and [Q] are the concentrations of fluorophore and quencher, respectively, and I0 is the fluorescence intensity measured in the absence of quencher.
Equation 5 is based on the assumption that the peptide or protein of interest exists in a single aggregation state. However, it is often the case that proteins exist in equilibrium between monomers and other aggregation states. To test for this possibility, it is important to conduct FRET quenching curves at multiple protein concentrations and protein/detergent or protein/phospholipid ratios. If the protein adopts a single aggregation state under the experimental conditions, the FRET curves should be superimposable within experimental error. However, if the protein populates more than a single aggregation state, and equilibration is sufficiently rapid, a family of curves will be obtained. In these cases, the experimental data are fit to a monomer–n-mer equilibrium by expressing the intensity as a sum of contributions from monomers (unquenched) and n-mers (fully quenched). The dissociation constant for the monomer–n-mer equilibrium is deduced by globally fitting to a function incorporating relevant equilibria. Experimental (Yano et al. 2002) or theoretical corrections for adventitious occurrences of quencher–fluorophore pairs in close proximity owing to high concentrations of the peptide in the detergent phase can be applied. These corrections, similar in spirit to those described for bilayers (Wolber and Hudson 1979), are kept small by adjustment of experimental conditions. It is also possible to consider situations in which the fluorophore–quencher distances are not uniform (Li et al. 1999) to obtain expressions for fluorescence lifetime distributions.
FRET experiments have been useful for determining the association equilibrium of MS1 (Choma et al. 2000; Fig. 3 ▶). Fluorescence quenching was monitored as a function of the donor-to-acceptor ratio at two different peptide–detergent ratios. Only a monomer–trimer equilibrium model adequately described the data. FRET has also been used to determine dissociation constants for the glycophorin dimer in micelles (Fisher et al. 1999).
SDS-PAGE
SDS electrophoresis has proved to be a quick and qualitative method for determining association states of many transmembrane proteins. Although SDS denatures some membrane proteins (London and Khorana 1982; Lau and Bowie 1997; Chen and Gouaux 1999), many small membrane proteins maintain their oligomeric structure in the presence of SDS (Fujii et al. 1989; Lemmon et al. 1991, 1994; Vorherr et al. 1993; Simmerman et al. 1996; Arkin et al. 1997; Karim et al. 1998; Choma et al. 2000; Splitt et al. 2000; F.X. Zhou et al. 2000, 2001; Gratkowski et al. 2001). SDS electrophoresis is useful for estimating molecular weights and screening of mutants, but it is not a quantitative method for determining exact association states or dissociation constants. However, a reasonable correlation has been demonstrated between the results of SDS gel electrophoresis and differences in free energies of association determined by analytical ultracentrifugation (Fleming and Engelman 2001b).
SDS-PAGE is particularly useful in conjunction with scanning or saturation mutagenesis. In this approach, each residue in the transmembrane sequence is mutated to one or more residues, and the extent of oligomer formation in the mutants is measured by gel electrophoresis. In this way, the residues that energetically contribute to the oligomerization surface can be determined. Furthermore, by examining mathematical and physical models of the results, one can obtain an approximation of the interhelical crossing angles. Thus, it was demonstrated that the dimers in GpA (Lemmon and Engelman 1994) and synaptobrevin (Fleming and Engelman 2001a; Laage and Langosch 1997; Laage et al. 2000) have a right-handed helical crossing angle, whereas the crossing angle of the transmembrane helices in phospholamban (Simmerman et al. 1996) is likely to be left-handed.
Homo-oligomeric membrane proteins
Glycophorin A, a paradigm for close interhelical packing in membrane proteins
Glycophorin A (GpA) is a small transmembrane protein that is found in erythrocyte membranes (Tomita et al. 1978). Although the function of this protein remains undefined, the molecular basis for its ability to form dimers has been extensively studied (Lemmon and Engelman 1994). It was discovered early that the dimeric form of this protein is stable to SDS, and subsequent studies showed that the transmembrane region of the protein was sufficient to mediate this dimerization (Bormann et al. 1989). The dimerization interface of GpA (Fig. 2A ▶) has been extensively studied by various methods in bilayers, native membranes, and detergent micelles. Site-directed mutagenesis has revealed that the residues involved in the association are LIXXGVXXGVXXT (where X represents any amino acid; Lemmon et al. 1992; Mingarro et al. 1996).
Computational modeling (Treutlein et al. 1992; Adams et al. 1996) indicated that the four glycines (two from each helix) line the dimer interface. The Gly residues at positions i and (i + 4) form a relatively flat surface that allows the polar backbone of the two helices to bind closely. The essential Val residues form a ridge, which extends the interaction interface, and the conserved Leu and Thr side chains further extend this ridge and pack against side chains in symmetry-related monomers. When the helices are docked to maximize their interaction along this surface, their interhelical crossing angle is right-handed with a value of ~30°–40°. Interestingly, Fleishman and Ben-Tal (2002) have developed a very simple potential function for docking transmembrane helices, which is based on the preference for small and β-branched amino acids to lie at the helix–helix interfaces of transmembrane helices. This potential function has been used to predict the conformation of the glycophorin dimer, which resulted in a good fit with the experimental structure.
The NMR structure of the transmembrane region of GpA in DPC (dodecylphosphocholine) micelles (MacKenzie et al. 1997) is in excellent agreement with a previously predicted model (Adams et al. 1996; rmsd = 0.8 Å for residues 74–91). Recently, the structure of GpA has been determined in lipid bilayers (Smith et al. 2001) by solid-state NMR, and the crossing angle was shown to be ~25°. Whether the differences are a result of more accurate distance estimates in the latter structure or a fundamental difference between micelles versus bilayers remains to be determined. The smaller crossing angle in the structure of the protein in bilayers allows a more extensive packing interface. Also, a Thr side chain that is known to stabilize the dimer appears to form interhelical hydrogen bonds in the bilayer structure (Smith et al. 2001, 2002), but not the micelle structure. Thus, further experiments are required to resolve the origins of these differences.
Irrespective of the precise details of the structure, the overall findings have revealed several important features of general relevance to the stabilization of membrane proteins (MacKenzie et al. 1997). Based on conformational free energy scales developed for water-soluble helices (e.g., Bryson et al. 1995; Munoz and Serrano 1995; Myers et al. 1997a, b), the GpA transmembrane sequence should have a very low helical potential because of the presence of numerous β-branched residues and Gly. However, this does not appear to present an energetic difficulty because the low dielectric environment of the membrane strongly favors helix formation (Liu and Deber 1998b). Also, the interaction interface contains two Val residues, an amino acid that has only a single low-energy rotamer in a helical conformation (for a review, see Doig and Sternberg 1995). Thus, dimerization of this helix does not require an unfavorable change in the side-chain entropy of these side chains.
The close approach of the two helices in GpA appears to be a feature that occurs frequently in both membrane proteins and water-soluble proteins (Kleiger et al. 2001, 2002). Glycine and other small side chains occur with a high frequency in helices of transmembrane proteins, and are often found in the packing interface (Javadpour et al. 1999; Eilers et al. 2000; Adamian and Liang 2001). An interesting hypothesis for why this motif is so strongly favored in transmembrane proteins is that the Cα hydrogen of the Gly residue forms a hydrogen bond with the backbone carbonyl of the associated helix (Senes et al. 2001). These interhelical backbone hydrogen bonds should be quite favorable in an apolar membrane environment. The GXXXG motif is also commonly found in water-soluble proteins, where it mediates a helix–helix interaction with a geometrically similar hydrogen-bonding pattern (Kleiger et al. 2001, 2002). Sequence analysis indicates that Gly-rich motifs might also mediate left-handed crossovers (Lemmon and Engelman 1994), although this remains to be demonstrated experimentally.
The Gly-rich motif has also been shown to be important for the association of other hydrophobic transmembrane sequences in biological membranes (Langosch et al. 1996; Brosig and Langosch 1998). An analysis of transmembrane domains in the SWISS-PROT database by Engelman and coworkers has identified GXXXG as a common motif that occurs 30% more frequently than the random expectation (Senes et al. 2000). It should also be noted that this sequence was commonly found with β-branched residues at neighboring positions. Also of particular interest, libraries were used to randomly select transmembrane sequences capable of association using the right-handed crossing motif of glycophorin (Russ and Engelman 2000). Of the total isolates showing strong association, 80% contained the GXXXG motif.
The formation of GpA dimers in micelles has been probed by FRET (Fisher et al. 1999) and sedimentation equilibrium ultracentrifugation (Fleming et al. 1997; Fleming and Engelman 2001b). The FRET study of Fisher et al. (1997) used synthetic peptides spanning the TM region and showed that the peptides formed dimers in several different types of micelles, including SDS, DDMAB, and DPC. The degree of dimerization in this study showed the expected dependence on the peptide/detergent ratio for the detergent SDS. However, in zwitterionic detergents, a much lower rate of equilibration was observed, presumably reflecting a significantly tighter degree of association.
Fleming (2000) has studied a series of mutants of the GpA transmembrane domain fused to staphylococcal nuclease in C8E5 micelles by analytical ultracentrifugation. These mutants show the same rank order of dimerization efficiency in C8E5 micelles, SDS micelles, and biological membranes (Fleming and Engelman 2001b). Thus, although the magnitude of the free energy of interaction depends on the hydrophobic environment, experiments on a series of variants in any one environment will allow subunit associations to be placed on a relative scale of interaction. In a particularly important contribution, the effect of Ala substitutions on the energetics of dimerization was determined (Fleming and Engelman 2001b). Ala substitutions have a large effect only when they occur at the interacting surface of the GpA helices. The largest effect was for a Gly-to-Ala mutation at position 83, which destabilized the dimer by ~1.6 kcal/mole of monomer. The introduction of the methyl group of Ala presumably destabilized the structure by forming unfavorable van der Waals interactions, which might also disrupt the interhelical hydrogen bond between the Cα hydrogen and the backbone carbonyl of the associated helix. The effects of mutating large apolar side chains to smaller residues were also investigated by mutating Leu, Ile, Val, or Thr residues to the smaller Ala side chain. Mutation of Leu 75 and Ile 76 to Ala resulted in a destabilization of the structure by 0.7 and 0.9 kcal/mole of monomer, respectively. Smaller changes were observed for mutating Val 80, Val 84, and Thr 87, ranging from 0.2 to 0.9 kcal/mole of monomer. Interestingly, even the largest effect seen for large-to-small mutations was much smaller than those typically seen for large-to-small mutations of buried hydrophobic residues in water-soluble proteins (Pace 1992; Xu et al. 1998) or coiled coils (Wagschal et al. 1999; Tripet et al. 2000; Acharya et al. 2002).
M2, a tetrameric proton channel from influenza A virus
The M2 proton channel from influenza A virus was first identified as the molecular target of the anti-influenza drug amantadine (Hay et al. 1985). This protein may be a prototype for a family of similar proteins found in other viruses, including the Vpu protein of HIV (Lamb and Pinto 1997). M2 is essential for uncoating of the virion within acidified endosomal vesicles. The M2 protein provides a conduit for passage of protons into the interior of the virus, thereby promoting the dissociation of the RNA from its matrix protein. In some strains of influenza A virus, the M2 protein is also important for delaying the acidification of the Golgi apparatus, thus preventing a premature conformational change in the viral hemagluttinin (Ciampor et al. 1995). Early studies identified M2 as an excellent candidate for biophysical and chemical investigations. It is a homotetramer (Holsinger and Lamb 1991) consisting of small, 97-residue monomers with a single transmembrane helix. The protein also has Cys residues at positions 17 and 19 on the viral exterior, which form a mixture of covalent dimers and tetramers (Holsinger et al. 1995). These species are easily measured on nondenaturing SDS gels, and provide a convenient readout of the ability of the protein to form tetramers (Sakaguchi et al. 1997). Significantly, these disulfides are not essential for assembly into functional tetrameric channels, and mutants in which Cys 17 and Cys 19 are converted to Ser are fully active in oocytes. Finally, the determinants for assembly (Kochendoerfer et al. 1999) and channel formation (Duff and Ashley 1992) appear to lie within the single membrane-spanning segment of the protein.
Several methods have been used to predict the structure of the channel-forming transmembrane four-helix bundle of M2. Cysteine scanning was used to generate a series of mutants with successive substitutions in the transmembrane segment of the protein (Pinto et al. 1997). Three distinct and essentially independent functional properties of the mutants were measured: The reversal potential was measured at pH 7.5 to determine whether the mutants disrupted the oocyte membranes. Currents were measured at pH 6.2 versus pH 7.5 to assess the extent of pH-activated channel formation. Finally, the ability of amantadine to inhibit the channel was determined. The fractional change in each of these three parameters (relative to wild type) was averaged for each mutant, providing a perturbation index for each position along the transmembrane helix. The resulting distribution shows a periodicity of ~3.5 residues, indicative of a left-handed interhelical crossing angle of ~10°–15°. Detailed modeling based on these experimental restraints led to a family of structures consistent with the functional characteristics of the channel (Pinto et al. 1997) and with in vacuo molecular dynamics simulations (Forrest et al. 1998). The tilt of the helices (relative to the central bundle axis) in these structures is, however, significantly less than the tilt of ~25°–35° determined by IR dichroism (Kukol et al. 1999; Torres et al. 2000b) and solid-state NMR (Kovacs and Cross 1997; Bauer et al. 1999; Wang et al. 2001; Tian et al. 2002). Unrestrained molecular dynamics simulations in more realistic environments (Zhong et al. 1998, 2000; Forrest et al. 2000) showed larger and variable helix tilt angles and a dependence of structure on the number of protonated His residues. Despite their differences, all of the models have essentially the same pore-lining residues. Also, most models of the channel show a continuous channel that is large enough to accommodate a hydrogen-bonded chain of water molecules running down most of the center of the bundle. High-resolution crystal structures and NMR structures (based on multiple distance restraints) should be very useful for further exploring the structural features of M2 affecting its function.
The minimal requirements for assembly and function of M2 were probed using a peptide spanning only the transmembrane regions of M2 (tmM2; Duff and Ashley 1992), as well as the full protein. To address the thermodynamics of association of the full-length protein, a single-site mutant (C17S), previously shown to produce active M2 tetramers (Holsinger and Lamb 1991), was chosen to reduce potential problems with disulfide formation. Analytical ultracentrifugation demonstrated that both the tmM2 peptide and the full-length protein existed in a reversible monomer–tetramer equilibrium in DPC (dodecylphosphocholine) micelles (Kochendoerfer et al. 1999). The full-length protein tetramerized with a more favorable free energy than the corresponding tmM2 peptide (ΔΔG° for the free energy of tetramerization of the full-length vs. the tm construct was −7 ± 1 kcal/mole). Thus, although the TM region alone is able to form tetramers, other regions in the chain appear to help stabilize this assembly. CD spectroscopy showed that the full-length protein contained more residues in a helical conformation than would be expected for the transmembrane helix alone. Furthermore, a region (residues 44–60) immediately C-terminal to the TM helix was highly protected from cleavage by proteases. This segment also has a high potential to form an amphiphilic α-helix, indicating that it may form a helical extension of the transmembrane helix (Fig. 2B ▶). Alternatively, these amphiphilic helices might be oriented parallel to the bilayer surface, creating a “rosette” structure.
The pH dependence of the assembly and amantadine binding of the tmM2 peptide has also been investigated in detail (Salom et al. 2000). The CD spectrum of the monomeric form of this peptide is slightly different from the tetrameric form, providing a convenient method to follow the assembly. Tetramerization is most favorable at high pH, where a critical transmembrane His 37 side chain is not protonated. The pH dependence is well described by a model in which the first His to be protonated in the tetramer has a pKa of 6.7 and the pKa associated with protonation of the second His in the tetramer is 5.7. These data are consistent with and have been interpreted in terms of a mechanism in which the His side chain acts as a proton shuttle. Addition of amantadine stabilizes the tetramer in a concentration-dependent manner through the formation of a complex. Amantadine binds exclusively to the neutral form of the channel with its His side chains fully deprotonated, which is consistent with resonance Raman studies of this peptide (Okada et al. 2001).
To determine which residues contribute to the conformational stability of the transmembrane tetramer, a series of variants of the tmM2 peptide was prepared. Each residue that projects toward the central axis of the tetramer (Val 27, Ala 30, Gly 34, His 37, and Trp 41) was individually changed to Ala and Phe. The free energies of tetramerization of the variants were determined by analytical ultracentrifugation, and can be understood in terms of present models for the structure of the protein (Fig. 4 ▶). Val 27 lies at the end of the transmembrane helix, and hence shows few structural constraints. A mutation of Val 27 to Ala occurs in naturally occurring variants of the virus, and indeed is slightly stabilizing to the structure of the tetramer. The larger residue, Phe, has not been observed at this position in naturally occurring variants, and slightly destabilizes the tetramer by 0.7 kcal/mole.
Figure 4.
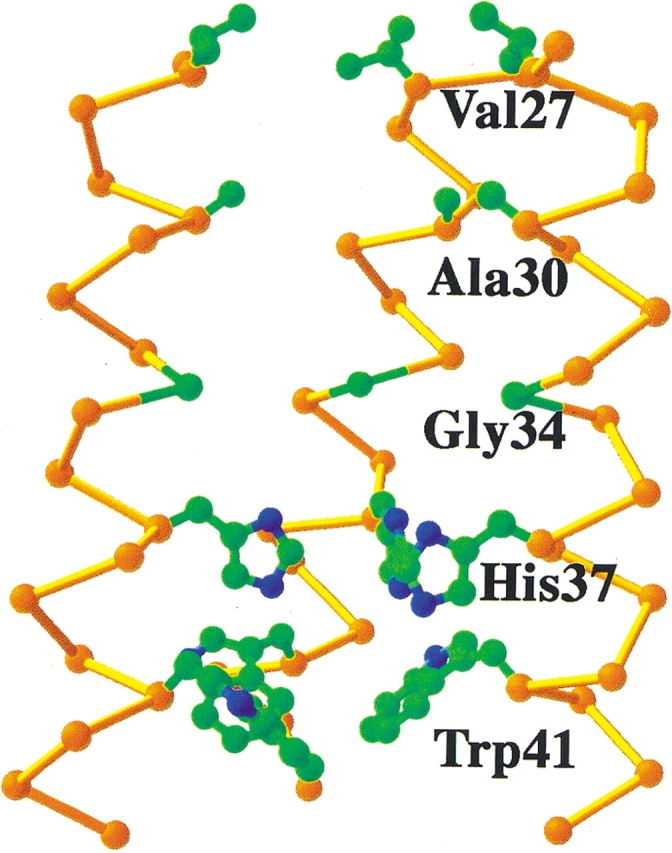
Predicted structure of the transmembrane region of the M2 proton channel in its “neutral” state, in which all four copies of His 37 are in the nonprotonated form. For clarity, only three monomers of the tetramer are shown. The side chains of the residues that were mutated are shown in ball-and-stick representations (carbon, green; nitrogen, blue). Note that the side chains pack in layers, with extensive van der Waals packing at the His 37 and Trp 41 layer. There is a large cavity associated with Gly 34 and Ala 30, allowing these side chains to be mutated to larger residues.
The pore widens substantially at Ala 30 and Gly 34, in part because of the small size of these side chains. Modeling indicated that the pore can accommodate residues as large as a Phe at position 30, and this mutation indeed results in a modest increase in the stability of the tetramer (ΔΔG = −1.2 kcal/mole). The less drastic mutation of Gly 34 to Ala has no significant effect on stability. These findings contrast with the effects of small-to-large mutations at the helix–helix interface of GP, in which a Gly-to-Ala mutation strongly destabilized the dimer, and is consistent with the expectation that Gly 34 lines a solvent-filled pore in the M2 tetramer.
Progressing through the channel from the outside of the virus toward the interior, one next encounters the side chains of His 37, which are in van der Waals contact in models of the neutral form of the channel. Mutation of this residue to Ala strongly destabilized the channel (ΔΔG = 3.1 kcal/mole), whereas the energetic penalty associated with mutating this residue is slightly less severe when it is instead mutated to the nearly isosteric but apolar residue, Phe (ΔΔG = 1.8 kcal/mole). The last side chain mutated was Trp 41, which appears to be essential for the gating of the channel (Tang et al. 2002). It is intriguing that changing W41 to either Ala or Phe is slightly stabilizing (ΔΔG = −1.2 kcal/mole). It is not uncommon for changes to functionally essential residues in soluble proteins to be stabilizing, because the structure in these regions is optimized for function rather than stability (Soichet et al. 1995). Furthermore, stability-enhancing mutations in membrane proteins are most prevalent near the edge of the bilayer (Bowie 2001).
Phospholamban, a possible left-handed transmembrane coiled coil
Although GpA is an excellent model for close helix–helix packing interactions in membrane proteins, helices in membrane proteins pack with a range of interhelical distances highly overlapping those of water-soluble proteins (Bowie 1997; Eilers et al. 2000). Indeed, many helix–helix interfaces in membrane proteins (Langosch and Heringa 1998; North et al. 2001) show interaction patterns that are typical of the ubiquitous coiled coils found in water-soluble proteins (Cohen and Parry 1990; Seo and Cohen 1993; Brown et al. 1996). Phospholamban (PL) appears to provide an excellent example of this type of transmembrane helical interface.
PL is a small transmembrane protein of 52 amino acids. It is a component of the sarcoplasmic reticulum that is involved in the regulation of Ca2+ ATPase. PL inhibits ATPase, and this function can be reversed by phosphorylation of PL. PL has one transmembrane helix, which appears to form pentamers, based on SDS-PAGE (Arkin et al. 1994; Simmerman et al. 1996). Five bands are observed corresponding to monomers, dimers, trimers, tetramers, and pentamers, with monomers and pentamers predominating. When samples are boiled prior to electrophoresis, the breakdown into smaller oligomers is more prevalent. Phosphorylation of PL increases the stability of the pentamer formation (Cornea et al. 1997; Li et al. 1998).
IR of site-specifically labeled PL in oriented bilayers (Ludlam et al. 1996; Torres et al. 2000a) and CD in vesicles (Arkin et al. 1995) support models in which the transmembrane helix of PL is helical and perpendicular to the membrane bilayer. Two models have been proposed for the structure of PL. Engelman and coworkers proposed a structure based on a low-energy conformer from modeling studies (Adams et al. 1995). However, another model has been suggested by Jones and coworkers (Simmerman et al. 1996) in which the helices are rotated by 45°. This second model (Fig. 2C ▶) is based on extensive mutagenesis experiments by this group that have identified Leu and Ile side chains at i and (i + 3) with a 7-residue periodicity. This repeat is highly indicative of a left-handed coiled coil. Interestingly, Harbury et al. (1993) have introduced a Leu at each a and an Ile at each d position of a water-soluble, two-stranded coiled coil, GCN4 P1. This substitution switches the aggregation state of the peptide to that of a tetramer (Harbury et al. 1993), most probably because the Ile side chains at d positions destabilize the dimeric structure, and favor the formation of the higher-order aggregate. The features that cause PL to form a pentamer rather than a tetramer are not yet known. The structure of PL may be very similar to the recently solved structure of a five-stranded water-soluble coiled coil, COMP (Malashkevich et al. 1996).
FRET has also been performed on PL as discussed (Li et al. 1999; Reddy et al. 1999). EPR studies indicated that phosphorylation did increase the amount of higher-order aggregate (Cornea et al. 1997). In similar FRET experiments, PL was reconstituted in the presence of Ca2+ ATPase, and smaller oligomers were found than those observed with PL alone (Reddy et al. 1999), consistent with the idea that PL interacts with ATPase as a monomer. The energetics of oligomerization of PL have not yet been systematically investigated in the same detail as GpA.
It should also be noted that a smaller integral membrane protein, sarcolipin (SLN), has been identified, and it is homologous to PL (Odermatt et al. 1997). SLN does not have the extensive cytoplasmic domain that is present with PL, and SLN does not contain a highly conserved phosphorylation domain. SLN appears to be monomeric, by SDS electrophoresis (Hellstern et al. 2001). Analytical ultracentrifugation has shown that it does form oligomers, although it does not associate as strongly as PL (Hellstern et al. 2001).
Designed coiled-coil peptides
De novo design has proved to be a useful approach for testing the features required for the folding and function of water-soluble proteins (Beasley and Hecht 1997; Baltzer and Broo 1998; DeGrado et al. 1999); now this method is beginning to show its promise for the understanding of membrane protein folding. Peptides have been designed to delineate the features required for helix formation and insertion into membranes (for review, see Deber et al. 1999; Wimley and White 2000). Membrane proteins have also been designed to probe the minimal requirements for forming close Gly-mediated helix–helix pairings in the GXXXG motif (Lemmon et al. 1994). More recently, de novo peptide design has been applied to probe the role of polar side chains in driving the association of transmembrane helices in bilayers (Bowie 2000; Choma et al. 2000; F.X. Zhou et al. 2000, 2001; Gratkowski et al. 2001; Pasternak et al. 2001).
These studies were stimulated, in part, by the analysis of Rees and Eisenberg, who showed that the interiors of membrane-soluble proteins are similar in packing and polarity to those of water-soluble proteins (Rees et al. 1989). Although this analysis has been challenged (Stevens and Arkin 1999), more recent computational studies on known structures of membrane proteins have provided additional detail (Samatey et al. 1995; Bowie 1997; Eilers et al. 2000; Adamian and Liang 2001), and the essential conclusions of Rees and Eisenberg (2000) appear to be not only essentially correct but remarkably prescient. The solvent-inaccessible core of both types of proteins predominantly consist of well-packed apolar residues. Buried polar side chains occur less frequently, and generally appear to be important for function, conformational specificity, and thermodynamic stability. Therefore, it might be possible to design water-soluble analogs of membrane proteins by introducing highly polar residues on their surface (see, e.g., Frank et al. 2000; H. Li et al. 2001). More pertinent to this review, it has now been shown that a water-soluble protein can be converted into a membrane-soluble structure by increasing the hydrophobicity of its surface side chains.
Two groups independently designed a water-soluble, two-stranded coiled coil, GCN4-P1, to render it soluble in membranes (Choma et al. 2000; F.X. Zhou et al. 2000). The GCN4-P1 dimer is stabilized in aqueous solution by a series of hydrophobic interactions, predominantly from Val side chains at position a and Leu side chains at position d of the heptad repeat (Fig. 5 ▶). Only one polar interaction occurs between buried side chains, involving a hydrogen bond between the carboxamide groups of an Asn side chain on neighboring chains. Mutational studies indicate that this interaction is actually destabilizing relative to a hydrophobic interaction, although it is important for specifying a dimeric state, relative to other aggregation states that are observed when this Asn is changed to hydrophobic residues (Harbury et al. 1993, 1994; Zhou et al. 2000). Structures of several variants of GCN4-P1 producing dimeric, trimeric, and tetrameric aggregation states have been determined (O’Shea et al. 1991; Harbury et al. 1993, 1994; Gonzalez et al. 1996a,b,c; Akey et al. 2001). Thus, GCN4-P1 should provide an excellent system for comparing the effects of substitutions in water-soluble versus membrane-soluble proteins.
Figure 5.

Helical wheel representation of GCN4-P1 and MS1 sequences showing side chains that occupy a and d positions in both peptides.
The GCN4-P1 peptide was converted to a membrane-soluble peptide by changing its exterior, polar side chains to a combination of apolar side chains, while maintaining its buried side chains invariant. In the sequence designed in our laboratory (Choma et al. 2000), three heptads of solvent-exposed residues were replaced with randomly chosen aliphatic hydrophobic side chains, resulting in MS1 (membrane soluble 1; Fig. 5 ▶). MS1 associates in a monomer–trimer equilibrium as revealed by FRET and analytical ultracentrifugation. The Asn residue was essential for association. Independently, Engelman and coworkers (F.X. Zhou et al. 2000) replaced solvent-exposed residues of GCN4-P1 with leucine and fused this sequence with that of staphylococcal nuclease. This protein, characterized in SDS micelles by PAGE, formed dimers, and mutational analysis again showed the Asn side chain to be essential for association. Furthermore, a TOXCAT assay (Russ and Engelman 1999) was used to demonstrate that the transmembrane helix formed oligomers in the cytoplasmic membrane of E. coli.
The differences between the trimeric aggregation state observed in our work versus the dimer observed by Zhou et al. are related in part to the presence of the fused staphylococcal nuclease domain used in the latter study. This large protein domain appears to inhibit the formation of trimers, as shown in an experiment in which synthetic peptides with the membrane sequence alone were allowed to compete with the fusion protein (F.X. Zhou et al. 2000). Mixtures of monomers, dimers, and trimers were observed, indicating that both dimers and trimers are energetically reasonable species. To further examine the cooperativity of the association reaction, we reanalyzed the sedimentation equilibrium data of our peptide in zwitterionic C14-betaine micelles using a monomer–dimer–trimer equilibrium and found a slightly better (20% reduction in χ2) fit to the data. A more extensive analysis (Gratkowski et al. 2002) showed that up to approximately 20% dimer could be accommodated near the midpoint of the transition, without a serious erosion of the fit. Thus, the trimer formation of MS1 is reasonably cooperative in this detergent. However, in micelles of a different zwitterionic detergent (DPC), a much less cooperative process was observed. Analytical ultracentrifugation indicated that ~70% dimer is present near the midpoint of the transition. This was further examined by NMR spectroscopy using a sample of the MS1 peptide that was labeled at its critical Asn with 15N. Three peaks were observed in the HSQC spectrum, which could be assigned to the monomer, dimer, and trimer by comparing their intensities with those predicted for the monomer–dimer–trimer equilibrium in DPC (Gratkowski et al. 2002).
In conclusion, MS1 behaves very much like the GCN4-P1 system, on which it was based. Extensive studies of GCN4-P1 showed that it tended to form trimers unless some feature was introduced to destabilize this aggregation state in favor of a dimeric or tetrameric state (Harbury et al. 1993; Gonzalez et al. 1996a,b,c; Schneider et al. 1998). For MS1, we find that the dimer is significantly populated only within a limited concentration range, in which trimers and monomers are also present. At lower concentrations, monomers predominate, and at higher concentrations, the trimer will predominate. Thus, MS1 provides an attractive system for measuring the thermodynamics, kinetics, and cooperativity of interactions of membrane helices, in much the same way that GCN4-P1 has proved to be useful for studying the association of water-soluble helices.
Variants of MS1 and the related peptide sequence from the Engelman laboratory have been used to probe the importance of polar side chains in mediating the assembly of transmembrane helices. NMR studies indicate that the Asn side chains engage in hydrogen-bonded interaction (F.X. Zhou et al. 2000), implying that these interactions are formed as in the design, and that they might provide a strong driving force for oligomerization. Furthermore, when Asn was changed to Val, the peptides failed to associate appreciably in micelles or membranes (Choma et al. 2000; F.X. Zhou et al. 2000). Thus, this Asn plays a different energetic role in the folding of water-soluble versus membrane-soluble versions of GCN4-P1. The Asn is important for thermodynamic stability in MS1, whereas the same residue in GCN4 provides conformational specificity at the expense of thermodynamic stability. This finding indicates that interactions between well-packed side chains are relatively weak in membrane proteins relative to water-soluble proteins. To address this conclusion further, individual Val and Leu side chains at a and d positions of MS1 were mutated to Ala. These mutations had little effect on the free energy of oligomerization (Lear et al. 2001); thus, packing interactions between apolar side chains in MS1 are significantly less favorable than the corresponding mutations in water-soluble proteins. This conclusion is also consistent with the above-mentioned energetic analysis of the association of variants of GpA (Fleming and Engelman 2001b).
A second question that has been addressed, both by the Engelman group as well as our group, is the effect of varying the position and number of Asn side chains in a transmembrane peptide. A peptide, based on MS1, was designed with two Asn residues in adjacent a positions, one heptad apart. When the additional Asn was located in the apolar region of the transmembrane helix, it increased the stability of MS1 by at least −2.0 kcal/mole of monomer. Interestingly, when the additional Asn occurred at the interface between the polar and apolar regions of the transmembrane helix, the second Asn failed to contribute significantly to the free energy of association. Thus, the Asn side chain must occur in an apolar environment to provide a strong driving force for oligomerization. Engelman and coworkers have investigated the effect of a second Asn residue on the association of transmembrane helices in the inner membrane of E. coli. The introduction of an appropriately placed second Asn side chain into a Leu-rich, transmembrane peptide increased the extent of association, as tested by the TOXCAT assay (F.X. Zhou et al. 2000).
MS1 has also provided an excellent system for examining polar interactions between side chains other than Asn (Gratkowski et al. 2001). A series of mutants was prepared in which the critical Asn was changed to various other side chains, and the change in free energy of association was determined by analytical ultracentrifugation (Gratkowski et al. 2001). It was determined that two polar atoms on the side chain were essential to drive association of MS1. The variants consisted of Ala and Leu, representing small and large aliphatic side chains, respectively; Ser, Thr, and Lys, all of which have one side chain polar atom; and Asp, Asn, Gln, and Glu, which all have two polar atoms. Only Asp, Asn, Gln, and Glu showed an appreciable degree of association. The difference in free energy between peptides that had side chains with two polar atoms as opposed to those with one polar atom ranges between ~0.8 kcal/mole and 1.5 kcal/mole, although these are probably lower limits for the values of ΔΔG.
F.X. Zhou et al. (2001) also examined a series of variants, using the TOXCAT assay. Their results indicated that Asn, Asp, and Glu were capable of driving oligomerization, and Leu, Ser, and Thr only showed modest levels of aggregation. Two additional side chains were examined, His and Tyr. Whereas His associated, the Tyr variant showed small levels of association similar to Leu, Ser, and Thr. In a competition assay in SDS, chimeras were mixed with the purified parent peptide that had one Asn at an a position. Interestingly, Asp, Glu, and Gln variants heteroassociated, but His did not. Our results, together with those of the Engelman laboratory, demonstrate that hydrogen bonding can play a decisive role in determining the thermodynamics of association in a membrane environment.
Although a single Ser–Ser contact between transmembrane helices may not be sufficient to drive association, cooperative interactions between multiple Ser residues appear to provide adequate stability for assembly. Statistical pairwise contact potentials have shown that Ser residues have a high tendency to form interhelical interactions (Adamian and Liang 2001, 2002). Using the TOXCAT method, Engelman and coworkers showed that SLL(x)SSLLT motifs are able to drive association of transmembrane helices (Dawson et al. 2002). Interestingly, a similar sequence is embedded within a designed peptide that has been shown to assemble to form proton and ion channels (LSSLLSLLSSLLSLLSSLLSL; Lear et al. 1988, 1994; DeGrado et al. 1989). These findings are consistent with the recent work of Liang and coworkers, who recently described a “serine zipper” motif in which interacting Ser side chains are spaced at 7-residue increments in the crystal structures of transmembrane proteins (Adamian and Liang 2002).
Determinants of folding in water-soluble versus membrane-soluble helical proteins
Helix formation and conformational entropy
Helix formation is an essential step in the folding of both membrane and water-soluble proteins. In water-soluble proteins the formation of isolated helices is intrinsically unfavorable, but can be driven by the formation of stabilizing interactions between residues from adjacent elements of secondary structure. Often, these interactions are hydrophobic in nature, and this effect is sufficient to stabilize the folding of peptides and proteins into molten globular or well-defined structures (DeGrado and Lear 1985; Bryson et al. 1995; Beasley and Hecht 1997; DeGrado et al. 1999; Baltzer et al. 2001). The hydrophobic effect plays a somewhat different role in stabilizing helices in membranes (Popot and Engelman 1990; Deber et al. 1999; White and Wimley 1999); this force is responsible for the transfer of hydrophobic peptides from water to the membrane surface, and ultimately to the membrane-spanning inserted state. Within a membrane, the exposure of the polar amide backbone to the lipid acyl chains is unfavorable relative to the formation of intramolecular helical hydrogen bonds. Thus, the α-helix is strongly stabilized, and folding of a helical membrane protein corresponds to the association of preformed helices to form a well-defined three-dimensional structure.
Helical membrane proteins appear to fold with a significantly smaller loss in conformational entropy than water-soluble proteins. First, the loss of backbone entropy is significantly decreased because the helices are preformed in the unfolded state. Furthermore, the fold of a membrane protein is highly directed by its biosynthetic insertion into a membrane, which forces the helices to lie with their axes roughly perpendicular to the plane of the membrane, and also defines the parallel versus antiparallel orientation of the helices relative to one another. Finally, Engelman et al. have pointed out that the loss in side-chain entropy is minimal for the Gly-rich interface of GpA (MacKenzie et al. 1997). This interface consists of two Gly residues, which lack side chains, and one Val residue, which adopts only a single low-energy rotamer in helices. The loss in conformational entropy of the remaining side chains in the interfacial region (Leu, Ile, and Thr) is expected to be relatively small: within a helix, Leu and Ile have similar conformational entropies (Leu has two low-energy χ1 rotamers and one χ2 rotamer, whereas Ile has one low-energy χ1 rotamer and two χ2 rotamers; Dunbrack and Karplus 1993), and the loss of entropy for Leu, Ile, and Thr at room temperature is expected to be less than 1 kcal/mole for each side chain, assuming that they adopt a single rotamer upon dimerization (Doig and Sternberg 1995). Similarly, three of the five residues that line the pore of the M2 proton channel (Gly, Ala, and Val) lose very little conformational entropy upon folding, although the nature of these residues presumably reflects the requirements for folding as well as function.
Burial and packing of apolar side chains
In water-soluble proteins, the burial of hydrophobic side chains in the interior of the protein provides a very strong driving force for folding into the native structure. For example, the mutation of a Leu to Ala will destabilize a protein or a coiled coil by 2 to 5 kcal/mole (Pace 1992), depending on the extent to which structural rearrangements accompany the mutation. The destabilization has two components: one arising from the free energy of transfer of an apolar side chain from water to a less polar environment. The second destabilizing component is the unfavorable free energy of forming a cavity in the protein. Thus, both van der Waals packing interactions as well as the classical hydrophobic effect contribute to stability. Although the hydrophobic effect alone is a potent driving force for folding, tight and regular packing also plays an important role. For example, proteins in which the interior side chains were maintained at approximately the same overall volume, but whose identities were changed, show marked reductions in thermodynamic stability (Baldwin and Matthews 1994; Gassner et al. 1996; Xu et al. 1998; Chen and Stites 2001a,b,c; Holder et al. 2001).
Within the acyl-chain region of the bilayer, the hydrophobic effect will not contribute to folding. Although related solvophobic contributions are possible, they presumably would contribute less to the overall driving force than in an aqueous environment. Furthermore, the free energy of cavitation is expected to be smaller in a membrane because the surface tension of hydrocarbons is considerably lower than that of water. The remaining force to consider is van der Waals packing. If folding results in overall better packing of the system (helices with helices, helices with lipid, and lipid with lipid), this force will favor folding. The analysis of crystallographic structures of membrane proteins indeed shows that the side chains of transmembrane helices are packed at least as well as in water-soluble proteins (Javadpour et al. 1999; Eilers et al. 2000; Adamian and Liang 2001, 2002; Chen and Stites 2001,b,c). Thus, one might expect that the packing of apolar side chains contributes to the folding of membrane proteins. Nevertheless, as compared with water-soluble proteins, their contribution to the free energy of folding would be strongly attenuated because of the lack of a hydrophobic driving force. The few quantitative tests of this prediction are consistent with this expectation. When large apolar residues along the GpA dimer interface are mutated to smaller Ala residues, they destabilize folding by only 0.2–0.9 kcal/mole of monomer. Conversely, the introduction of a Phe residue that appears to fill a pore in the M2 proton channel stabilizes the structure only slightly by −0.3 kcal/mole of monomer.
Although the burial of apolar side chains in membrane proteins appears to provide a relatively modest driving force for folding, it nevertheless appears to be sufficient to drive folding in some cases. For example, the synaptobrevin transmembrane helix forms dimers that are stable to SDS-PAGE (Laage et al. 2000). Mutagenesis has revealed that a collection of apolar side chains comprises its dimerization interface. The only critical polar residue in this case is a Cys, which appears to form an intra- rather than an interhelical hydrogen bond (Fleming and Engelman 2001a). Also, Gly-rich helical interfaces often are devoid of residues that can form conventional hydrogen bonds, although it has been hypothesized that a significant portion of their stability derives from relatively weak interhelical hydrogen bonds between CαH and carbonyl groups (Senes et al. 2001). In this context, it is important to remember that the interhelical interactions in membrane proteins need not be as favorable as in water-soluble proteins, because there is a considerably lower loss of conformational entropy associated with the folding of a membrane protein.
Polar interactions
Polar interactions are particularly important for function and conformational specificity in water-soluble proteins. Furthermore, site-directed mutagenesis has shown that the replacement of pairs of hydrogen-bonded side chains with Ala residues can destabilize a protein by 1–2 kcal/mole (Pace et al. 1996). However, in these experiments, it is difficult to deconvolute the effects of introducing a void in the protein (Xu et al. 1998) from the effect of deleting the hydrogen-bonded interaction. Calculations and experimental studies have indicated that hydrogen-bonded interactions are generally destabilizing relative to hydrophobic interactions between similarly sized residues, because the energetic penalty associated with the burial of the polar group is frequently larger than the compensating energy associated with hydrogen-bond formation (Hendsch and Tidor 1994; Honig and Yang 1995; Hendsch et al. 1996). In a recent experimental study particularly relevant to the present review, Hu and coworkers (Zhu et al. 2000) examined the energetic effects of placing Ile or Asn residues at four buried a positions in a two-stranded coiled coil. Ile was, indeed, favored over Asn by 1.3–2.4 kcal/mole of monomer, but this energetic range is less than that predicted by considering only the hydrophobic effect. Thus, interhelical hydrogen bonds between the side chains of the buried Asn residues appear to contribute to the conformational stability of the coiled-coil peptides.
The formation of a hydrogen bond in a membrane protein is expected to be significantly more stabilizing than in a water-soluble protein, because, within the apolar region of the bilayer, the formation of a hydrogen bond does not require dehydration of the two interacting groups. Thus, polar interactions are expected to provide a strong driving force for folding in membranes. Indeed, a recent study (Adamian and Liang 2002) clearly demonstrated that interhelical hydrogen bonds are particularly important for the stabilization of helix–helix interactions in membrane proteins; most transmembrane helices in polytopic membrane proteins have at least one interhelical hydrogen bond, and hydrogen-bonded transmembrane helices tend to interact more extensively than non-hydrogen-bonded helices. Similarly, experimental studies (described in detail above) with designed transmembrane helices have shown that a single polar residue, such as Asn, Asp, Glu, Gln, or His, is sufficient to mediate homo-oligomerization of the helix in biological membranes and micelles. Finally, as discussed in the section on glycophorin, hydrogen bonding between the Cα hydrogen of a Gly residue and a carbonyl from a neighboring helix might be particularly stabilizing in membrane proteins (Senes et al. 2001).
These experimental results are also consistent with measurements of the frequencies of occurrence of amino acids in the transmembrane helical portions of membrane proteins of known three-dimensional structure (Eilers et al. 2000). Two features contribute to these observed frequencies: first, a transmembrane helix must partition into the bilayer, which tends to maximize the number of hydrophobic side chains and minimize polar residues. However, the requirement for function and folding dictate the occasional need for polar side chains. This trade-off between partitioning versus folding and function is apparent in a comparison of the frequencies of occurrence of polar side chains in the population of type I single-span (nonoligomeric) membrane proteins (Landolt-Marticorena et al. 1993). Amino acids that mediate oligomerization of MS1 occur several-fold more frequently in multispan than in single-span helices in transmembrane proteins (Gratkowski et al. 2001). Thus, the requirements for folding and function lead to an increase in the number of these polar side chains in the transmembrane region. Indeed, when Asp, Asn, Glu, and Gln occur in multispan proteins, they are usually found in the head group region. When they are found in the interior of the protein (Gratkowski et al. 2001), they appear to be essential for folding, proton translocation activity, or other functional roles. In contrast, the presence of even a single Asn or Gln in a single-span membrane protein might potentially lead to deleterious oligomerization (F.X. Zhou et al. 2000, 2001).
Both statistical and experimental studies have shown that the location of polar residues within a transmembrane helix has an important influence on the stability of a given hydrogen bond. In a recent study (J.D. Lear, H. Gratkowski, and W.F. DeGrado, unpubl.), Val and Asn residues were placed singly and in pairs at three a positions within a model transmembrane helix, MS1. When placed near the middle of the transmembrane helix, folding into a trimeric bundle was stabilized by at least 2 kcal/mole of monomer. In comparison, when the Asn was placed at the interface between the hydrophobic and polar regions of the peptide, ΔΔG for Val versus Asn was 0.1 ± 0.5 kcal/mole, and a related Ile-for-Asn replacement in a two-stranded coiled coil is destabilizing by approximately 1.5 kcal/mole of monomer (Acharya et al. 2002). Thus, as the polarity of the environment of the Asn side chain (in the unfolded state) increases, the free energy of the hydrogen bond switches from favorable to unfavorable.
A similarly large dependence on the location of the polar residues was found in a statistical survey of helical transmembrane proteins in which the tendency of different types of residues to be buried in the interiors versus being exposed to lipids was analyzed (data not shown). Asn and Gln had a very strong tendency to be buried when they were located near the middle of a transmembrane helix. However, when placed near the ends of transmembrane helices, they showed little preference for the surface versus the interior of the protein.
Biological perspectives
The association, folding, and misfolding of transmembrane domains play important roles in physiological as well as pathophysiological processes. For example, the receptor tyrosine kinase encoded by the neu proto-oncogene is constitutively activated by a single Val-to-Glu substitution in the predicted membrane-spanning sequence of the receptor (Bargmann et al. 1986; Bargmann and Weinberg 1988). Mutations in the transmembrane domain of cystic fibrosis transmembrane conductance regulator also lead to accumulation of misfolded protein (Wigley et al. 1999, 2002; Corboy et al. 2002) or aberrant association of helices in mutant forms of the protein (Therien et al. 2001; Wigley et al. 2002). Recently, it has been proposed that Pro residues in TM helices can serve to suppress similar misfolded states (Wigley et al. 2002).
Transient associations of TM domains are also believed to be important for the regulation of a variety of proteins. For example, the transmembrane helices of the α and β chains of integrins have been proposed to associate in the inactive state of this highly regulated family of proteins (Woodside et al. 2001), but to move apart in the activated state (Lu et al. 2001; Shimaoka et al. 2001, 2002; Takagi et al. 2001). Furthermore, when released from cytoskeletal restraints, the cytoplasmic and TM domains of these proteins have a strong tendency to self-associate (R. Li et al. 2001), which may be an important step in the clustering of these integrins into focal adhesions.
The principles described in this review provide the means to identify and test the role of potential transmembrane associations in biology. For example, mutations can be introduced to either enhance or interrupt a potential transmembrane helical association, to test the role of such interactions in the regulation and activities of a variety of proteins.
Abbreviations
FRET, fluorescence resonance energy transfer
NBD, 7-nitrobenz-2-oxa-1,3-diazole
C-14 betaine, N-tetradecyl-N,N-dimethyl-3-ammonio-1-propanesulfonate
MF, mole fraction
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0236503.
References
- Acharya, A., Rubinov, S.B., Gal, J., Moll, J.R., and Vinson, C. 2002. A heterodimerizing leucine zipper coiled coil system for examining the specificity of a position interactions: Amino acids I, V, L, N, A, and K. Biochemistry 41 14122–14131. [DOI] [PubMed] [Google Scholar]
- Adamian, L. and Liang, J. 2001. Helix–helix packing and interfacial pairwise interactions of residues in membrane proteins. J. Mol. Biol. 311 891–907. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Interhelical hydrogen bonds and spatial motifs in membrane proteins: Polar clamps and serine zippers. Proteins 47 209–218. [DOI] [PubMed] [Google Scholar]
- Adams, P.D., Arkin, I.T., Engelman, D.M., and Brünger, A.T. 1995. Computational searching and mutagenesis suggest a structure for the pentameric transmembrane domain of phospholamban. Nat. Struct. Biol. 2 154–162. [DOI] [PubMed] [Google Scholar]
- Adams, P.D., Engelman, D.M., and Brünger, A.T. 1996. Improved prediction for the structure of the dimeric transmembrane domain of glycophorin obtained through global searching. Proteins 26 257–261. [DOI] [PubMed] [Google Scholar]
- Akey, D.L., Malashkevich, V.N., and Kim, P.S. 2001. Buried polar residues in coiled-coil interfaces. Biochemistry 40 6352–6360. [DOI] [PubMed] [Google Scholar]
- Arkin, I.T., Adams, P.D., MacKenzie, K.R., Lemmon, M.A., Brünger, A.T., and Engelman, D.M. 1994. Structural organization of the pentameric transmembrane α-helices of phospholamban, a cardiac ion channel. EMBO J. 13 4757–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkin, I.T., Rothman, M., Ludlam, C.F.C., Aimoto, S., Engelman, D.M., Rothschild, K.J., and Smith, S.O. 1995. Structural model of the phospholamban ion channel complex in phosholipid membranes. J. Mol. Biol. 248 824–834. [DOI] [PubMed] [Google Scholar]
- Arkin, I.T., Adams, P.D., Brünger, A.T., Aimoto, S., Engelman, D.M., and Smith, S.O. 1997. Structure of the transmembrane cysteine residues in phospholamban. J. Membrane Biol. 155 199–206. [DOI] [PubMed] [Google Scholar]
- Baldwin, E.P. and Matthews, B.W. 1994. Core-packing constraints, hydrophobicity and protein design. Curr. Opin. Biotechnol. 5 396–402. [DOI] [PubMed] [Google Scholar]
- Baltzer, L. and Broo, K.S. 1998. De novo designed polypeptide catalysts with adopted folded structures. Biopolymers 47 31–40. [Google Scholar]
- Baltzer, L., Nilsson, H., and Nilsson, J. 2001. De novo design of proteins—What are the rules? Chem. Rev. 101 3153–3163. [DOI] [PubMed] [Google Scholar]
- Bargmann, C.I. and Weinberg, R.A. 1988. Increased tyrosine kinase activity associated with the protein encoded by the activated neu oncogene. Proc. Natl. Acad. Sci. 85 2371–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann, C.I., Hung, M.C., and Weinberg, R.A. 1986. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell 45 649–657. [DOI] [PubMed] [Google Scholar]
- Bauer, C.M., Pinto, L.H., Cross, T.A., and Lamb, R.A. 1999. The influenza virus M2 ion channel protein: Probing the structure of the transmembrane domain in intact cells by using engineered disulfide cross-linking. Virology 254 196–209. [DOI] [PubMed] [Google Scholar]
- Beasley, J.R. and Hecht, M.H. 1997. Protein design: The choice of de novo sequences. J. Biol. Chem. 272 2031–2034. [DOI] [PubMed] [Google Scholar]
- Bormann, B.-J., Knowles, W.J., and Marchesi, V.T. 1989. Synthetic peptides mimic the assembly of transmembrane glycoproteins. J. Biol. Chem. 264 4033–4037. [PubMed] [Google Scholar]
- Bowie, J.U. 1997. Helix packing in membrane proteins. J. Mol. Biol. 272 780–789. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Understanding membrane protein structure by design. Nat. Struct. Biol. 7 91–94. [DOI] [PubMed] [Google Scholar]
- ———. 2001. Stabilizing membrane proteins. Curr. Opin. Struct. Biol. 11 397–402. [DOI] [PubMed] [Google Scholar]
- Brosig, B. and Langosch, D. 1998. The dimerization motif of the glycophorin A transmembrane segment in membranes: Importance of glycine residues. Protein Sci. 7 1052–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, J.H., Cohen, C., and Parry, D.A. 1996. Heptad breaks in α-helical coiled coils: Stutters and stammers. Proteins 26 134–145. [DOI] [PubMed] [Google Scholar]
- Bryson, J.W., Betz, S.F., Lu, H.S., Suich, D.J., Zhou, H.X., O’Neil, K.T., and DeGrado, W.F. 1995. Protein design: A hierarchic approach. Science 270 935–941. [DOI] [PubMed] [Google Scholar]
- Bu, Z. and Engelman, D.M. 1999. A method for determining transmembrane helix association and orientation in detergent micelles using small angle X-ray scattering. Biophys. J. 77 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, G.Q. and Gouaux, E. 1999. Probing the folding and unfolding of wild-type and mutant forms of bacteriorhodopsin in micellar solutions: Evaluation of reversible unfolding conditions. Biochemistry 38 15380–15387. [DOI] [PubMed] [Google Scholar]
- Chen, J. and Stites, W.E. 2001a. Energetics of side chain packing in staphylococcal nuclease assessed by systematic double mutant cycles. Biochemistry 40 14004–14011. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Higher-order packing interactions in triple and quadruple mutants of staphylococcal nuclease. Biochemistry 40 14012–14019. [DOI] [PubMed] [Google Scholar]
- ———. 2001c. Packing is a key selection factor in the evolution of protein hydrophobic cores. Biochemistry 40 15280–15289. [DOI] [PubMed] [Google Scholar]
- Choma, C., Gratkowski, H., Lear, J.D., and DeGrado, W.F. 2000. Asparagine-mediated self-association of a model transmembrane helix. Nat. Struct. Biol. 7 161–166. [DOI] [PubMed] [Google Scholar]
- Chung, L.A. and Thompson, T.E. 1996. Design of membrane-inserting peptides: Spectroscopic characterization with and without lipid bilayers. Biochemistry 35 11343–11354. [DOI] [PubMed] [Google Scholar]
- Ciampor, F., Cmarko, D., Cmarkova, J., and Zavodska, E. 1995. Influenza-virus M2 protein and hemagglutinin conformation changes during intracellular-transport. Acta Virol. 39 171–181. [PubMed] [Google Scholar]
- Cohen, C. and Parry, D.A.D. 1990. α-Helical coiled coils and bundles: How to design an α-helical protein. Proteins 7 1–15. [DOI] [PubMed] [Google Scholar]
- Constantinescu, S.N., Keren, T., Socolovsky, M., Nam, H., Henis, Y.I., and Lodish, H.F. 2001. Ligand-independent oligomerization of cell-surface erythropoietin receptor is mediated by the transmembrane domain. Proc. Natl. Acad. Sci. 98 4379–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corboy, M.J., Thomas, P.J., and Wigley, W.C. 2002. CFTR degradation and aggregation. Methods Mol. Med. 70 277–294. [DOI] [PubMed] [Google Scholar]
- Cornea, R.L., Jones, L.R., Autry, J.M., and Thomas, D.D. 1997. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry 36 2960–2967. [DOI] [PubMed] [Google Scholar]
- Dawson, J.P., Weinger, J.S., Engelman, D.M., and Sturgis, J.N. 2002. Motifs of serine and threonine can drive association of transmembrane helices. J. Mol. Biol. 316 799–805. [DOI] [PubMed] [Google Scholar]
- Deber, C.M., Khan, A.R., Li, Z., Joensson, C., Glibowicka, M., and Wang, J. 1993. Val to Ala mutations selectively alter helix–helix packing in the transmembrane segment of phage M13 coat protein. Proc. Natl. Acad. Sci. 90 11648–11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deber, C.M., Liu, L.P., and Wang, C. 1999. Perspective: Peptides as mimics of transmembrane segments in proteins. J. Pept. Res. 54 200–205. [DOI] [PubMed] [Google Scholar]
- DeGrado, W.F. and Lear, J.D. 1985. Induction of peptide conformation at apolar/water interfaces: A study with peptides of defined hydrophobic periodicity. J. Am. Chem. Soc. 107 76–84. [Google Scholar]
- DeGrado, W.F., Wasserman, Z.R., and Lear, J.D. 1989. Protein design, a minimalist approach. Science 243 622–628. [DOI] [PubMed] [Google Scholar]
- DeGrado, W.F., Summa, C.M., Pavone, V., Nastri, F., and Lombardi, A. 1999. De novo design and structural characterization of proteins and metalloproteins. Ann. Rev. Biochem. 68 779–819. [DOI] [PubMed] [Google Scholar]
- Doig, A.J. and Sternberg, M.J. 1995. Side-chain conformational entropy in protein folding. Protein Sci. 4 2247–2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff, K.C. and Ashley, R.H. 1992. The transmembrane domain of influenza A M2 protein forms amantadine-sensitive proton channels in planar lipid bilayers. Virology 190 485–489. [DOI] [PubMed] [Google Scholar]
- Dunbrack, Jr., R.L. and Karplus, M. 1993. Backbone-dependent rotamer library for proteins. Application to side-chain prediction. J. Mol. Biol. 230 543–574. [DOI] [PubMed] [Google Scholar]
- Eilers, M., Shekar, S.C., Shieh, T., Smith, S.O., and Fleming, P.J. 2000. Internal packing of helical membrane proteins. Proc. Natl. Acad. Sci. 97 5796–5801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhawer, M., Cattarinussi, S., Kuhn, A., and Vogel, H. 2001. Fluorescence resonance energy transfer shows a close helix–helix distance in the transmembrane M13 procoat protein. Biochemistry 40 12321–12328. [DOI] [PubMed] [Google Scholar]
- Engelman, D.M. and Steitz, T.A. 1981. The spontaneous insertion of proteins into and across membranes: The helical hairpin hypothesis. Cell 23 411–422. [DOI] [PubMed] [Google Scholar]
- Fisher, L.E., Engelman, D.M., and Sturgis, J.N. 1999. Detergents modulate dimerization, but not helicity, of the glycophorin A transmembrane domain. J. Mol. Biol. 293 639–651. [DOI] [PubMed] [Google Scholar]
- Fleishman, S.J. and Ben-Tal, N. 2002. A novel scoring function for predicting the conformations of tightly packed pairs of transmembrane α-helices. J. Mol. Biol. 321 363–378. [DOI] [PubMed] [Google Scholar]
- Fleming, K.G. 2000. Probing stability of helical transmembrane proteins. Methods Enzymol. 323 63–77. [DOI] [PubMed] [Google Scholar]
- Fleming, K.G. and Engelman, D.M. 2001a. Computation and mutagenesis suggest a right-handed structure for the synaptobrevin transmembrane dimer. Proteins 45 313–317. [DOI] [PubMed] [Google Scholar]
- ———. 2001b. Specificity in transmembrane helix–helix interactions can define a hierarchy of stability for sequence variants. Proc. Natl. Acad. Sci. 98 14340–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, K.G., Ackerman, A.L., and Engelman, D.M. 1997. The effect of point mutations on the free energy of transmembrane α-helix dimerization. J. Mol. Biol. 272 266–275. [DOI] [PubMed] [Google Scholar]
- Forrest, L.R., DeGrado, W.F., Dieckmann, G.R., and Sansom, M.S. 1998. Two models of the influenza A M2 channel domain: Verification by comparison. Fold Des. 3 443–448. [DOI] [PubMed] [Google Scholar]
- Forrest, L.R., Kukol, A., Arkin, I.T., Tieleman, D.P., and Sansom, M.S. 2000. Exploring models of the influenza A M2 channel: MD simulations in a phospholipid bilayer. Biophys. J. 78 55–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, S., Kammerer, R.A., Hellstern, S., Pegoraro, S., Stetefeld, J., Lustig, A., Moroder, L., and Engel, J. 2000. Toward a high-resolution structure of phospholamban: Design of soluble transmembrane domain mutants. Biochemistry 39 6825–6831. [DOI] [PubMed] [Google Scholar]
- Fujii, J., Maruyama, K., Tada, M., and MacLennan, D.H. 1989. Expression and site-specific mutagenesis of phospholamban. Studies of residues involved in phosphorylation and pentamer formation. J. Biol. Chem. 264 12950–12955. [PubMed] [Google Scholar]
- Gassner, N.C., Baase, W.A., and Matthews, B.W. 1996. A test of the “jigsaw puzzle” model for protein folding by multiple methionine substitutions within the core of T4 lysozyme. Proc. Natl. Acad. Sci. 93 12155–12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazit, E., Burshtein, N., Ellar, D.J., Sawyer, T., and Shai, Y. 1997. Bacillus thuringiensis cytolytic toxin associates specifically with its synthetic helices A and C in the membrane bound state. Implications for the assembly of oligomeric transmembrane pores. Biochemistry 36 15546–15554. [DOI] [PubMed] [Google Scholar]
- Gonzalez, Jr., L., Brown, R.A., Richardson, D., and Alber, T. 1996a. Crystal structures of a single coiled-coil peptide in two oligomeric states reveal the basis for structural polymorphism. Nat. Struct. Biol. 3 1002–1009. [DOI] [PubMed] [Google Scholar]
- Gonzalez, Jr., L., Plecs, J.J., and Alber, T. 1996b. An engineered allosteric switch in leucine-zipper oligomerization. Nat. Struct. Biol. 3 510–515. [DOI] [PubMed] [Google Scholar]
- Gonzalez, Jr., L., Woolfson, D.N., and Alber, T. 1996c. Buried polar residues and structural specificity in the GCN4 leucine zipper. Nat. Struct. Biol. 3 1011–1018. [DOI] [PubMed] [Google Scholar]
- Gratkowski, H., Lear, J.D., and DeGrado, W.F. 2001. Polar sidechains drive the association of model, transmembrane peptides. Proc. Natl. Acad. Sci. 98 880–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratkowski, H., Dai, Q.H., Wand, A.J., DeGrado, W.F., and Lear, J.D. 2002. Cooperativity and specificity of association of a designed transmembrane peptide. Biophys. J. 83 1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbury, P.B., Zhang, T., Kim, P.S., and Alber, T. 1993. A switch between two-, three-, and four-stranded coiled coils. Science 262 1401–1407. [DOI] [PubMed] [Google Scholar]
- Harbury, P.B., Kim, P.S., and Alber, T. 1994. Crystal structure of an isoleucine-zipper trimer. Nature 371 80–83. [DOI] [PubMed] [Google Scholar]
- Hay, A.J., Wolstenholme, A.J., Skehel, J.J., and Smith, M.H. 1985. The molecular basis of the specific anti-influenza action of amantadine. EMBO J. 4 3021–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstern, S., Pegoraro, S., Karim, C.B., Lustig, A., Thomas, D.D., Moroder, L., and Engel, J. 2001. Sarcolipin, the shorter homologue of phospholamban, forms oligomeric structures in detergent micelles and in liposomes. J. Biol. Chem. 276 30845–30852. [DOI] [PubMed] [Google Scholar]
- Hendsch, Z.S. and Tidor, B. 1994. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 3 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendsch, Z.S., Jonsson, T., Sauer, R.T., and Tidor, B. 1996. Protein stabilization by removal of unsatisfied polar groups: Computational approaches and experimental tests. Biochemistry 35 7621–7625. [DOI] [PubMed] [Google Scholar]
- Hodges, R.S. 1996. Boehringer Mannheim award lecture 1995. La conference Boehringer Mannheim 1995. De novo design of α-helical proteins: Basic research to medical applications. Biochem. Cell Biol. 74 133–154. [DOI] [PubMed] [Google Scholar]
- Holder, J.B., Bennett, A.F., Chen, J., Spencer, D.S., Byrne, M.P., and Stites, W.E. 2001. Energetics of side chain packing in staphylococcal nuclease assessed by exchange of valines, isoleucines, and leucines. Biochemistry 40 13998–14003. [DOI] [PubMed] [Google Scholar]
- Holsinger, L.J. and Lamb, R.A. 1991. Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology 183 32–43. [DOI] [PubMed] [Google Scholar]
- Holsinger, L.J., Shaughnessy, M.A., Micko, A., Pinto, L.H., and Lamb, R.A. 1995. Analysis of the posttranslational modifications of the influenza-virus M(2) protein. J. Virology 69 1219–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig, B. and Yang, A.-S. 1995. Free energy balance in protein folding. Adv. Prot. Chem. 46 27–58. [DOI] [PubMed] [Google Scholar]
- Hubbell, W.L., McHaourab, H.S., Altenbach, C., and Lietzow, M.A. 1996. Watching proteins move using site-directed spin labeling. Structure 4 779–783. [DOI] [PubMed] [Google Scholar]
- Hubbell, W.L., Gross, A., Langen, R., and Lietzow, M.A. 1998. Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8 649–656. [DOI] [PubMed] [Google Scholar]
- Javadpour, M.M., Eilers, M., Groesbeek, M., and Smith, S.O. 1999. Helix packing in polytopic membrane proteins: Role of glycine in transmembrane helix association. Biophys. J. 77 1609–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim, C.B., Stamm, J.D., Karim, J., Jones, L.R., and Thomas, D.D. 1998. Cysteine reactivity and oligomeric structures of phospholamban and its mutants. Biochemistry 37 12074–12081. [DOI] [PubMed] [Google Scholar]
- Kleiger, G., Perry, J., and Eisenberg, D. 2001. 3D structure and significance of the GPhiXXG helix packing motif in tetramers of the E1β subunit of pyruvate dehydrogenase from the archeon Pyrobaculum aerophilum. Biochemistry 40 14484–14492. [DOI] [PubMed] [Google Scholar]
- Kleiger, G., Grothe, R., Mallick, P., and Eisenberg, D. 2002. GXXXG and AXXXA: Common α-helical interaction motifs in proteins, particularly in extremophiles. Biochemistry 41 5990–5997. [DOI] [PubMed] [Google Scholar]
- Kochendoerfer, G.G., Salom, D., Lear, J.D., Wilk-Orescan, R., Kent, S.B., and DeGrado, W.F. 1999. Total chemical synthesis of the integral membrane protein influenza A virus M2: Role of its C-terminal domain in tetramer assembly. Biochemistry 38 11905–11913. [DOI] [PubMed] [Google Scholar]
- Kovacs, F.A. and Cross, T.A. 1997. Transmembrane four-helix bundle of influenza A M2 protein channel: Structural implications from helix tilt and orientation. Biophys. J. 73 2511–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukol, A., Adams, P.D., Rice, L.M., Brünger, A.T., and Arkin, I.T. 1999. Experimentally based orientational refinement of membrane protein models: A structure for the influenza A M2 H+ channel. J. Mol. Biol. 286 951–962. [DOI] [PubMed] [Google Scholar]
- Laage, R. and Langosch, D. 1997. Dimerization of the synaptic vesicle protein synaptobrevin (vesicle-associated membrane protein) II depends on specific residues within the transmembrane segment. Eur. J. Biochem. 249 540–546. [DOI] [PubMed] [Google Scholar]
- Laage, R., Rohde, J., Brosig, B., and Langosch, D. 2000. A conserved membrane-spanning amino acid motif drives homomeric and supports heteromeric assembly of presynaptic SNARE proteins. J. Biol. Chem. 275 17481–17487. [DOI] [PubMed] [Google Scholar]
- Lamb, R.A. and Pinto, L.H. 1997. Do Vpu and Vpr of human immunodeficiency virus type 1 and NB of influenza B virus have ion channel activities in the viral life cycles? Virology 229 1–11. [DOI] [PubMed] [Google Scholar]
- Landolt-Marticorena, C., Williams, K.A., Deber, C.M., and Reithmeier, R.A. 1993. Non-random distribution of amino acids in the transmembrane segments of human type I single span membrane proteins. J. Mol. Biol. 229 602–608. [DOI] [PubMed] [Google Scholar]
- Langen, R., Isas, J.M., Luecke, H., Haigler, H.T., and Hubbell, W.L. 1998. Membrane-mediated assembly of annexins studied by site-directed spin labeling. J. Biol. Chem. 273 22453–22457. [DOI] [PubMed] [Google Scholar]
- Langosch, D. and Heringa, J. 1998. Interaction of transmembrane helices by a knobs-into-holes packing characteristic of soluble coiled coils. Proteins 31 150–159. [DOI] [PubMed] [Google Scholar]
- Langosch, D., Brosig, B., Kolmar, H., and Fritz, H.J. 1996. Dimerisation of the glycophorin A transmembrane segment in membranes probed with the ToxR transcription activator. J. Mol. Biol. 263 525–530. [DOI] [PubMed] [Google Scholar]
- Lau, F.W. and Bowie, J.U. 1997. A method for assessing the stability of a membrane protein. Biochemistry 36 5884–5892. [DOI] [PubMed] [Google Scholar]
- Laue, T., Shaw, B.D., Ridgeway, T.M., and Pelletier, S.L. 1992. Analytical ultracentrifugation in biochemistry and polymer science. In Analytical ultracentrifugation in biochemistry and polymer science (eds. S.E. Harding et al.), pp. 90–125. The Royal Society of Chemistry, Cambridge, UK.
- Lear, J.D., Wasserman, Z.R., and DeGrado, W.F. 1988. Synthetic amphiphilic peptide modes for protein ion channels. Science 240 1177–1181. [DOI] [PubMed] [Google Scholar]
- ———. 1994. The use of synthetic peptides for the study of membrane protein structure. In Membrane protein structure (ed. S.H. White), pp. 335–354. Oxford University Press, New York.
- Lemmon, M.A. and Engelman, D.M. 1994. Specificity and promiscuity in membrane helix interactions. Quart. Rev. Biophys. 27 157–218. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A., Flanagan, J.M., Hunt, J.F., Adair, B.D., Bormann, B.-J., Dempsey, C.E., and Engelman, D.M. 1991. Glycophorin A dimerization is driven by specific interactions between transmembrane α helices. J. Biol. Chem. 267 7683–7689. [PubMed] [Google Scholar]
- Lemmon, M.A., Flanagan, J.M., Treutlein, H.R., Zhang, J., and Engelman, D.M. 1992. Sequence specificity in the dimerization of transmembrane α helices. Biochemistry 31 12719–12725. [DOI] [PubMed] [Google Scholar]
- Lemmon, M.A., Treutlein, H.R., Adams, P.D., Brünger, A.T., and Engelman, D.M. 1994. A dimerization motif for transmembrane α helices. Nat. Struct. Biol. 1 157–163. [DOI] [PubMed] [Google Scholar]
- Li, H., Cocco, M.J., Steitz, T.A., and Engelman, D.M. 2001. Conversion of phospholamban into a soluble pentameric helical bundle. Biochemistry 40 6636–6645. [DOI] [PubMed] [Google Scholar]
- Li, M., Cornea, R.L., Autry, J.M., Jones, L.R., and Thomas, D.D. 1998. Phosphorylation-induced structural change in phospholamban and its mutants, detected by intrinsic fluorescence. Biochemistry 37 7869–7877. [DOI] [PubMed] [Google Scholar]
- Li, M., Reddy, L.G., Bennett, R., Silva, Jr., N.D., Jones, L.R., and Thomas, D.D. 1999. A fluorescence energy transfer method for analyzing protein oligomeric structure: Application to phospholamban. Biophys. J. 76 2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, R., Babu, C.R., Lear, J.D., Wand, A.J., Bennett, J.S., and DeGrado, W.F. 2001. Oligomerization of the integrin αIIbβ3: Roles of the transmembrane and cytoplasmic domains. Proc. Natl. Acad. Sci. 98 12462–12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L.P. and Deber, C.M. 1998a. Guidelines for membrane protein engineering derived from de novo designed model peptides. Biopolymers 47 41–62. [DOI] [PubMed] [Google Scholar]
- ———. 1998b. Uncoupling hydrophobicity and helicity in transmembrane segments. α-Helical propensities of the amino acids in non-polar environments. J. Biol. Chem. 273 23645–23648. [DOI] [PubMed] [Google Scholar]
- London, E. and Khorana, H.G. 1982. Denaturation and renaturation of bacteriorhodopsin. J. Biol. Chem. 257 7003–7011. [PubMed] [Google Scholar]
- Lu, C., Takagi, J., and Springer, T.A. 2001. Association of the membrane proximal regions of the α and β subunit cytoplasmic domains constrains an integrin in the inactive state. J. Biol. Chem. 276 14642–14648. [DOI] [PubMed] [Google Scholar]
- Ludlam, C.F.C., Arkin, I.T., Liu, X.-M., Rothman, M.S., Rath, P., Aimoto, S., Smith, S.O., Engelman, D.M., and Rothschild, K.J. 1996. Fourier transform infrared spectroscopy and site-directed isotope labeling as a probe of local secondary structure in the transmembrane domain of phospholamban. Biophys. J. 70 1728–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie, K.R., Prestegard, J.H., and Engelman, D.M. 1997. A transmembrane helix dimer: Structure and implications. Science 276 131–133. [DOI] [PubMed] [Google Scholar]
- Malashkevich, V.N., Kammerer, R.A., Efimov, V.P., Schulthess, T., and Engel, J. 1996. The crystal structure of a five-stranded coiled coil in COMP: A prototype ion channel? Science 274 761–765. [DOI] [PubMed] [Google Scholar]
- Melnyk, R.A., Partridge, A.W., and Deber, C.M. 2001. Retention of native-like oligomerization states in transmembrane segment peptides: Application to the Escherichia coli aspartate receptor. Biochemistry 40 11106–11113. [DOI] [PubMed] [Google Scholar]
- Mingarro, I., Whitley, P., Lemmon, M.A., and von Heijne, G. 1996. Ala-insertion scanning mutagenesis of the glycophorin A transmembrane helix: A rapid way to map helix–helix interactions in integral membrane proteins. Protein Sci. 5 1339–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz, V. and Serrano, L. 1995. Helix design, prediction and stability. Curr. Opin. Biotechnol. 6 382–386. [DOI] [PubMed] [Google Scholar]
- Myers, J.K., Pace, C.N., and Scholtz, J.M. 1997a. A direct comparison of helix propensity in proteins and peptides. Proc. Natl. Acad. Sci. 94 2833–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1997b. Helix propensities are identical in proteins and peptides. Biochemistry 36 10923–10929. [DOI] [PubMed] [Google Scholar]
- North, B., Summa, C.M., Ghirlanda, G., and DeGrado, W.F. 2001. D(n)-symmetrical tertiary templates for the design of tubular proteins. J. Mol. Biol. 311 1081–1090. [DOI] [PubMed] [Google Scholar]
- Oakley, M.G. and Kim, P.S. 1998. A buried polar interaction can direct the relative orientation of helices in a coiled coil. Biochemistry 37 12603–12610. [DOI] [PubMed] [Google Scholar]
- Odermatt, A., Taschner, P.E., Scherer, S.W., Beatty, B., Khanna, V.K., Cornblath, D.R., Chaudhry, V., Yee, W.C., Schrank, B., Karpati, G., et al. 1997. Characterization of the gene encoding human sarcolipin (SLN), a proteolipid associated with SERCA1: Absence of structural mutations in five patients with Brody disease. Genomics 45 541–553. [DOI] [PubMed] [Google Scholar]
- Oh, K., Zhan, H., Cui, C., Hideg, K., Collier, R., and Hubbell, W. 1996. Organization of diphtheria toxin T domain in bilayers: A site-directed spin labeling study. Science 273 810–812. [DOI] [PubMed] [Google Scholar]
- Okada, A., Miura, T., and Takeuchi, H. 2001. Protonation of histidine and histidine–tryptophan interaction in the activation of the M2 ion channel from influenza A virus. Biochemistry 40 6053–6060. [DOI] [PubMed] [Google Scholar]
- O’Shea, E.K., Klemm, J.D., Kim, P.S., and Alber, T.A. 1991. X-ray structure of the GCN4 leucine zipper, a two-stranded coiled coil. Science 254 539–544. [DOI] [PubMed] [Google Scholar]
- Pace, C.N. 1992. Contribution of the hydrophobic effect to globular protein stability. J. Mol. Biol. 226 29–35. [DOI] [PubMed] [Google Scholar]
- Pace, C.N., Shirley, B.A., McNutt, M., and Gajiwala, K. 1996. Forces contributing to the conformational stability of proteins. FASEB J. 10 75–83. [DOI] [PubMed] [Google Scholar]
- Pasternak, A., Kaplan, J., Lear, J.D., and Degrado, W.F. 2001. Proton and metal ion-dependent assembly of a model diiron protein. Protein Sci. 10 958–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peled, H. and Shai, Y. 1993. Membrane interaction and self-assembly within phospholipid membranes of synthetic segments corresponding to the H-5 region of the shaker K+ channel. Biochemistry 32 7879–7885. [DOI] [PubMed] [Google Scholar]
- ———. 1994. Synthetic S-2 and H-5 segments of the Shaker K+ channel: Secondary structure, membrane interaction, and assembly within phospholipid membranes. Biochemistry 33 7211–7219. [DOI] [PubMed] [Google Scholar]
- Pinto, L.H., Dieckmann, G.R., Gandhi, C.S., Papworth, C.G., Braman, J., Shaughnessy, M.A., Lear, J.D., Lamb, R.A., and DeGrado, W.F. 1997. A functionally defined model for the M2 proton channel of influenza A virus suggests a mechanism for its ion selectivity. Proc. Natl. Acad. Sci. 94 11301–11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popot, J.-L. and Engelman, D.M. 1990. Membrane Protein folding and oligomerization: The two-stage model. Biochemistry 29 4031–4037. [DOI] [PubMed] [Google Scholar]
- ———. 2000. Helical membrane protein folding, stability, and evolution. Annu. Rev. Biochem. 69 881–922. [DOI] [PubMed] [Google Scholar]
- Popot, J.L., Trewhella, J., and Engelman, D.M. 1986. Reformation of crystalline purple membrane from purified bacteriorhodopsin fragments. EMBO J. 5 3039–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy, L.G., Jones, L.R., and Thomas, D.D. 1999. Depolymerization of phospholamban in the presence of calcium pump: A fluorescence energy transfer study. Biochemistry 38 3954–3962. [DOI] [PubMed] [Google Scholar]
- Rees, D.C. and Eisenberg, D. 2000. Turning a reference inside-out: Commentary on an article by Stevens and Arkin entitled: “Are membrane proteins ‘inside-out’ proteins?” Proteins 38 121–122. [PubMed] [Google Scholar]
- Rees, D.C., DeAntonio, L., and Eisenberg, D. 1989. Hydrophobic organization of membrane proteins. Science 245 510–513. [DOI] [PubMed] [Google Scholar]
- Russ, W.P. and Engelman, D.M. 1999. TOXCAT: A measure of transmembrane helix association in a biological membrane. Proc. Natl. Acad. Sci. 96 863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2000. The GxxxG motif: A framework for transmembrane helix–helix association. J. Mol. Biol. 296 911–919. [DOI] [PubMed] [Google Scholar]
- Sakaguchi, T., Qiang, T., Pinto, L.H., and Lamb, R.A. 1997. The active oligomeric state of the minimalist influenza virus M2 ion channel is a tetramer. Proc. Natl. Acad. Sci. 94 5000–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salom, D., Hill, B.R., Lear, J.D., and DeGrado, W.F. 2000. pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry 39 14160–14170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samatey, F.A., Xu, C., and Popot, J.-L. 1995. On the distribution of amino acid residues in transmembrane α helix bundles. Proc. Natl. Acad. Sci. 92 4577–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, J.P., Lombardi, A., and DeGrado, W.F. 1998. Analysis and design of three-stranded coiled coils and three-helix bundles. Fold Des. 3 R29–R40. [DOI] [PubMed] [Google Scholar]
- Schubert, U., Ferrer-Montiel, A.V., Oblatt-Montal, M., Henklein, P., Strebel, K., and Montal, M. 1996. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett. 398 12–18. [DOI] [PubMed] [Google Scholar]
- Senes, A., Gerstein, M., and Engelman, D.M. 2000. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J. Mol. Biol. 296 921–936. [DOI] [PubMed] [Google Scholar]
- Senes, A., Ubarretxena-Belandia, I., and Engelman, D.M. 2001. The Cα—H…O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. 98 9056–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo, J. and Cohen, C. 1993. Pitch diversity in α-helical coiled coils. Proteins 15 223–234. [DOI] [PubMed] [Google Scholar]
- Shai, Y. 1995. Molecular recognition between membrane-spanning polypeptides. Trends Biochem. Sci. 20 460–464. [DOI] [PubMed] [Google Scholar]
- Shimaoka, M., Lu, C., Palframan, R.T., von Andrian, U.H., McCormack, A., Takagi, J., and Springer, T.A. 2001. Reversibly locking a protein fold in an active conformation with a disulfide bond: Integrin αL I domains with high affinity and antagonist activity in vivo. Proc. Natl. Acad. Sci. 98 6009–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimaoka, M., Takagi, J., and Springer, T.A. 2002. Conformational regulation of integrin structure and function. Annu. Rev. Biophys. Biomol. Struct. 31 485–516. [DOI] [PubMed] [Google Scholar]
- Simmerman, H.K., Kobayashi, Y.M., Autry, J.M., and Jones, L.R. 1996. A leucine zipper stabilizes the pentameric membrane domain of phospholamban and forms a coiled-coil pore structure. J. Biol. Chem. 271 5941–5946. [DOI] [PubMed] [Google Scholar]
- Smith, S.O., Smith, C.S., and Bormann, B.J. 1996. Strong hydrogen bonding interactions involving a buried glutamic acid in the transmembrane sequence of the neu/erbB-2 receptor. Nat. Struct. Biol. 3 252–258. [DOI] [PubMed] [Google Scholar]
- Smith, S.O., Song, D., Shekar, S., Groesbeek, M., Ziliox, M., and Aimoto, S. 2001. Structure of the transmembrane dimer interface of glycophorin A in membrane bilayers. Biochemistry 40 6553–6558. [DOI] [PubMed] [Google Scholar]
- Smith, S.O., Eilers, M., Song, D., Crocker, E., Ying, W., Groesbeek, M., Metz, G., Ziliox, M., and Aimoto, S. 2002. Implications of threonine hydrogen bonding in the glycophorin A transmembrane helix dimer. Biophys. J. 82 2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soekarjo, M., Eisenhawer, M., Kuhn, A., and Vogel, H. 1996. Thermodynamics of the membrane insertion process of the M13 procoat protein, a lipid bilayer traversing protein containing a leader sequence. Biochemistry 35 1232–1241. [DOI] [PubMed] [Google Scholar]
- Soichet, B.K., Baase, W.A., Kuroki, R., and Matthews, B.W. 1995. A relationship between protein stability and protein function. Proc. Natl. Acad. Sci. 92 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splitt, H., Meuser, D., Borovok, I., Betzler, M., and Schrempf, H. 2000. Pore mutations affecting tetrameric assembly and functioning of the potassium channel KcsA from Streptomyces lividans. FEBS Lett. 472 83–87. [DOI] [PubMed] [Google Scholar]
- Stevens, T.J. and Arkin, I.T. 1999. Are membrane proteins “inside-out” proteins? Proteins 36 135–143. [DOI] [PubMed] [Google Scholar]
- Takagi, J., Erickson, H.P., and Springer, T.A. 2001. C-Terminal opening mimics “inside-out” activation of integrin α5β1. Nat. Struct. Biol. 8 412–416. [DOI] [PubMed] [Google Scholar]
- Tanford, C. and Reynolds, J.A. 1976. Characterization of membrane proteins in detergent solutions. Biochim. Biophys. Acta 457 133–170. [DOI] [PubMed] [Google Scholar]
- Tanford, C., Nozaki, Y., Reynolds, J.A., and Makino, S. 1974. Molecular characterization of proteins in detergent solutions. Biochemistry 13 2369–2376. [DOI] [PubMed] [Google Scholar]
- Tang, Y., Zaitseva, F., Lamb, R.A., and Pinto, L.H. 2002. The gate of the influenza virus M2 proton channel is formed by a single tryptophan residue. J. Biol. Chem. 277 39880–39886. [DOI] [PubMed] [Google Scholar]
- Teller, D.C. 1973. Characterization of proteins by sedimentation equilibrium in the analytical ultracentrifuge. Methods Enzymol. 27 346–441. [DOI] [PubMed] [Google Scholar]
- Therien, A.G., Grant, F.E., and Deber, C.M. 2001. Interhelical hydrogen bonds in the CFTR membrane domain. Nat. Struct. Biol. 8 597–601. [DOI] [PubMed] [Google Scholar]
- Tian, C., Tobler, K., Lamb, R.A., Pinto, L.H., and Cross, T.A. 2002. Expression and initial structural insights from solid-state NMR of the M2 proton channel from influenza A virus. Biochemistry 41 11294–11300. [DOI] [PubMed] [Google Scholar]
- Tomita, M., Furthmayr, H., and Marchesi, V.T. 1978. Primary structure of human erythrocyte glycophorin A. Isolation and characterization of peptides and complete amino acid sequence. Biochemistry 17 4756–4770. [DOI] [PubMed] [Google Scholar]
- Torres, J., Adams, P.D., and Arkin, I.T. 2000a. Use of a new label, (13)=(18)O, in the determination of a structural model of phospholamban in a lipid bilayer. Spatial restraints resolve the ambiguity arising from interpretations of mutagenesis data. J. Mol. Biol. 300 677–685. [DOI] [PubMed] [Google Scholar]
- Torres, J., Kukol, A., and Arkin, I.T. 2000b. Use of a single glycine residue to determine the tilt and orientation of a transmembrane helix. A new structural label for infrared spectroscopy. Biophys. J. 79 3139–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein, H.R., Lemmon, M.A., Engelman, D.M., and Brünger, A.T. 1992. The glycophorin A transmembrane domain dimer: Sequence-specific propensity for a right-handed supercoil of helices. Biochemistry 31 12726–12733. [DOI] [PubMed] [Google Scholar]
- Tripet, B., Wagschal, K., Lavigne, P., Mant, C.T., and Hodges, R.S. 2000. Effects of side-chain characteristics on stability and oligomerization state of a de novo-designed model coiled-coil: 20 amino acid substitutions in position “a” and “d”. J. Mol. Biol. 300 377–402. [DOI] [PubMed] [Google Scholar]
- Veatch, W. and Stryer, L. 1977. The dimeric nature of the gramicidin A transmembrane channel: Conductance and fluorescence energy transfer studies of hybrid channels. J. Mol. Biol. 113 89–102. [DOI] [PubMed] [Google Scholar]
- von Heijne, G. 1999. Recent advances in the understanding of membrane protein assembly and structure. Quart. Rev. Biophys. 32 285–307. [DOI] [PubMed] [Google Scholar]
- Vorherr, T., Wrzosek, A., Chiesi, M., and Carafoli, E. 1993. Total synthesis and functional properties of the membrane-intrinsic protein phospholamban. Protein Sci. 2 339–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagschal, K., Tripet, B., Lavigne, P., Mant, C., and Hodges, R.S. 1999. The role of position a in determining the stability and oligomerization state of α-helical coiled coils: 20 amino acid stability coefficients in the hydrophobic core of proteins. Protein Sci. 8 2312–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C. and Deber, C.M. 2000. Peptide mimics of the M13 coat protein transmembrane segment. Retention of helix–helix interaction motifs. J. Biol. Chem. 275 16155–16159. [DOI] [PubMed] [Google Scholar]
- Wang, J., Kim, S., Kovacs, F., and Cross, T.A. 2001. Structure of the transmembrane region of the M2 protein H+ channel. Protein Sci. 10 2241–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, D.B., Liu, J., Cohen, J.A., Williams, W.V., and Greene, M.I. 1989. A point mutation in the neu oncogene mimics ligand induction of receptor aggregation. Nature 339 230–231. [DOI] [PubMed] [Google Scholar]
- White, S.H. and Wimley, W.C. 1999. Membrane protein folding and stability: Physical principles. Annu. Rev. Biophys. Biomol. Struct. 28 319–365. [DOI] [PubMed] [Google Scholar]
- Wigley, W.C., Fabunmi, R.P., Lee, M.G., Marino, C.R., Muallem, S., DeMartino, G.N., and Thomas, P.J. 1999. Dynamic association of proteasomal machinery with the centrosome. J. Cell Biol. 145 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigley, W.C., Corboy, M.J., Cutler, T.D., Thibodeau, P.H., Oldan, J., Lee, M.G., Rizo, J., Hunt, J.F., and Thomas, P.J. 2002. A protein sequence that can encode native structure by disfavoring alternate conformations. Nat. Struct. Biol. 9 381–388. [DOI] [PubMed] [Google Scholar]
- Wimley, W.C. and White, S.H. 2000. Designing transmembrane α-helices that insert spontaneously. Biochemistry 39 4432–4442. [DOI] [PubMed] [Google Scholar]
- Wolber, P.K. and Hudson, B.S. 1979. An analytic solution to the Forster energy transfer problem in two dimensions. Biophys. J. 28 197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodside, D.G., Liu, S., and Ginsberg, M.H. 2001. Integrin activation. Thromb. Haemost. 86 316–323. [PubMed] [Google Scholar]
- Xu, J., Baase, W.A., Baldwin, E., and Matthews, B.W. 1998. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 7 158–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano, Y., Takemoto, T., Kobayashi, S., Yasui, H., Sakurai, H., Ohashi, W., Niwa, M., Futaki, S., Sugiura, Y., and Matsuzaki, K. 2002. Topological stability and self-association of a completely hydrophobic model transmembrane helix in lipid bilayers. Biochemistry 41 3073–3080. [DOI] [PubMed] [Google Scholar]
- Zhong, Q., Husslein, T., Moore, P.B., Newns, D.M., Pattnaik, P., and Klein, M.L. 1998. The M2 channel of influenza A virus: A molecular dynamics study. FEBS Lett. 434 265–271. [DOI] [PubMed] [Google Scholar]
- Zhong, Q., Newns, D.M., Pattnaik, P., Lear, J.D., and Klein, M.L. 2000. Two possible conducting states of the influenza A virus M2 ion channel. FEBS Lett. 473 195–198. [DOI] [PubMed] [Google Scholar]
- Zhou, F.X., Cocco, M.J., Russ, W.P., Brüger, A.T., and Engelman, D.M. 2000. Interhelical hydrogen bonding drives strong interactions in membrane proteins. Nat. Struct. Biol. 7 154–160. [DOI] [PubMed] [Google Scholar]
- Zhou, F.X., Merianos, H.J., Brüger, A.T., and Engelman, D.M. 2001. Polar residues drive association of polyleucine transmembrane helices. Proc. Natl. Acad. Sci. 98 2250–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y., Lau, F.W., Nauli, S., Yang, D., and Bowie, J.U. 2001. Inactivation mechanism of the membrane protein diacylglycerol kinase in detergent solution. Protein Sci. 10 378–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, H., Celinski, S.A., Scholtz, J.M., and Hu, J.C. 2000. The contribution of buried polar groups to the conformational stability of the GCN4 coiled coil. J. Mol. Biol. 300 1377–1387. [DOI] [PubMed] [Google Scholar]