

Abstract
Native-state hydrogen exchange (HX) studies, used in conjunction with NMR spectroscopy, have been carried out on Escherichia coli thioredoxin (Trx) for characterizing two folding subdomains of the protein. The backbone amide protons of only the slowest-exchanging 24 amino acid residues, of a total of 108 amino acid residues, could be followed at pH 7. The free energy of the opening event that results in an amide hydrogen exchanging with solvent (ΔGop) was determined at each of the 24 amide hydrogen sites. The values of ΔGop for the amide hydrogens belonging to residues in the helices α1, α2, and α4 are consistent with them exchanging with the solvent only when the fully unfolded state is sampled transiently under native conditions. The denaturant-dependences of the values of ΔGop provide very little evidence that the protein samples partially unfolded forms, lower in energy than the unfolded state. The amide hydrogens belonging to the residues in the β strands, which form the core of the protein, appear to have higher values of ΔGop than amide hydrogens belonging to residues in the helices, suggesting that they might be more stable to exchange. This apparently higher stability to HX of the β strands might be either because they exchange out their amide hydrogens in a high energy intermediate preceding the globally unfolded state, or, more likely, because they form residual structure in the globally unfolded state. In either case, the central β strands—β3, β2, and β4—would appear to form a cooperatively folding subunit of the protein. The native-state HX methodology has made it possible to characterize the free energy landscape that Trx can sample under equilibrium native conditions.
Keywords: Native-state HX, NMR spectroscopy, thioredoxin, folding, unfolded state
It has been difficult to characterize, in structural terms, the energy landscape separating the unfolded and the native state of a protein, using traditional optical probes like fluorescence and circular dichroism, because these probes do not provide residue-specific information about protein structure. Experimental approaches, which are not only more sensitive toward the detection of partially folded conformations, but which also provide residue-specific structural information, are therefore required. Hydrogen exchange (HX; Englander et al. 1996, 1997; Rumbley et al. 2001; Juneja and Udgaonkar 2003) is one such technique, which, when coupled with NMR spectroscopy, has been applied quite effectively to a number of proteins to obtain residue-specific information on structures present in kinetic and equilibrium folding intermediates, in the unfolded forms of proteins, and in partially unfolded forms (PUFs) that coexist very transiently with the native state in native conditions.
The native-state HX (Mayo and Baldwin 1993; Bai et al. 1995; Englander 1998) methodology can detect the fully unfolded as well as the partially unfolded conformations that can be sampled by a protein, even if only transiently, under strongly native conditions. The relative populations of PUFs can be perturbed using marginally destabilizing denaturant concentrations, and the perturbation is reflected in the exchange rates of individual amide protons. The exchange rate observed at each amide hydrogen site provides an estimate of the free energy of the opening event that leads to the exchange. Hence, information about the cooperatively unfolding units of a protein can be obtained. The native-state HX methodology has been applied to many proteins including ribonuclease A (Mayo and Baldwin 1993; Neira et al. 1999; Juneja and Udgaonkar 2002), cytochrome c (Bai et al 1995), staphylococcal nuclease (Loh et al. 1993), RNase H (Chamberlain et al. 1996), barnase (Clarke and Fersht 1996), chymotrypsin inhibitor 2 (Itzhaki et al. 1997), protein L (Yi et al. 1997), barstar (Bhuyan and Udgaonkar 1998), T4 lysozyme (Llinas et al. 1999), β-lactoglobulin (Forge et al. 2000), human acidic fibroblast growth factor (Srimathi et al. 2002), csp A (Rodriguez et al. 2002), cytochrome b562 (Chu et al. 2002), and transthyretin (Liu et al. 2002), and has usually led to the identification of PUFs of these proteins, which are populated under equilibrium native conditions. Native-state HX experiments have also provided useful insights about the various interactions that stabilize the native state, helped in comparing the pathogenic variants of a disease-causing protein with the normal protein (Hoshino et al. 2002; Liu et al. 2002), allowed the characterization of residual structure in the unfolded state (Wrabl and Shortle 1999), helped in comparing observed equilibrium intermediates with kinetic intermediates (Chu et al. 2002; Juneja and Udgaonkar 2002; Rodriguez et al. 2002), and helped to distinguish whether discrete intermediates or a continuum of intermediates populate the free energy landscape of a protein under native conditions (Loh et al. 1993; Mayo and Baldwin 1993; Bai et al 1995; Chamberlain et al. 1996; Clarke and Fersht 1996; Itzhaki et al. 1997; Yi et al. 1997; Bhuyan and Udgaonkar 1998; Llinas et al. 1999; Neira et al. 1999; Forge et al. 2000; Chu et al. 2002; Juneja and Udgaonkar 2002; Liu et al. 2002; Rodriguez et al. 2002; Srimathi et al. 2002).
Escherichia coli thioredoxin (Trx) is an oxidoreductase, whose sequence of 108 amino acids folds into a native state with two structural subdomains: a large β1α1β3α2β2 domain and a small β4β5α4 domain (Fig. 1 ▶; Holmgren et al. 1975; Dyson et al. 1989; Katti et al. 1990). The two subdomains of this mixed αβ protein are joined by an 18 amino acid residue segment consisting of a single turn α helix and a 310 helix. The protein has a twisted, five-stranded β-sheet as its central core, which is flanked by the four helices (Holmgren et al. 1975; Dyson et al. 1989; Katti et al. 1990). The refolding and unfolding kinetics of Trx have been studied using multiple probes, including intrinsic tryptophan (Trp) fluorescence, far UV circular dichroism (CD), near UV-CD, 8-anilino-1-naphthalene sulfonic acid binding, and regain or loss of native Trx activity (Kelley and Stellwagen 1984; Kelley et al. 1986; Kelley and Richards 1987; Kelley et al. 1987; Georgescu et al. 1998; Bhutani and Udgaonkar 2001). The unfolding kinetics of Trx appear to be two-state, without the accumulation of any detectable intermediates. The refolding of Trx is multiphasic, and has been shown to occur via a burst phase ensemble of intermediates, rich in β-content, which fold to the native state via multiple routes (Georgescu et al. 1998; Bhutani and Udgaonkar 2001). Fragment complementation studies have indicated that the initial event that occurs during the folding of Trx is the zippering together of the β2 and β4 strands, which form part of the hydrophobic core of the protein (Tasayco and Chao 1995; Tasayco et al. 2000). Detailed structural information about the various intermediates that populate the folding and unfolding pathways, as well as about the unfolded form, is still lacking.
Figure 1.

Structure of Escherichia coli thioredoxin. (A) The crystal structure of thioredoxin (Katti et al. 1990) at 1.68 Å reveals two distinct domains: a large βαβαβ domain and a small ββα domain, joined by a segment consisting of a single turn α helix and a 310 helix. The figure has been generated using RASMOL (Sayle and Milner-White 1995) from the protein crystal structure 2TRX in the PDB, deposited by Katti et al. (B) Schematic representation of the topology of Trx, viewed down the axis of the β-sheet (Dyson et al. 1989). Boxes represent β strands, octagons represent α-helices, curved thick lines represent loops above, and curved thin lines represent loops below the β-sheet, respectively. The two subdomains are packed together via the β2 and β4 strands. (C) Location of the β strands, α-helices, and β-turns (Dyson et al. 1989) in Trx. The table also shows the amino acid residues whose backbone amide protons could be followed in this study.
In this study, the native state HX profile of Trx has been investigated under equilibrium native conditions at pH 7. Two folding subdomains have been identified, which can be distinguished on the basis of their apparently different stabilities to HX. Three helices, α1, α2, and α4, flanking the central β sheet core, appear to unfold cooperatively when the protein transiently samples the globally unfolded state under native conditions. A folding subdomain is also formed by the interface of the two structural subdomains of the protein, that is, the central β strands, β3, β2, and β4. The residues in these β strands appear to be more stable to exchange than do residues in the flanking helices, and it is argued that this is likely to be due to these residues participating in residual structure in the otherwise globally unfolded state. There is very little evidence indicating that the protein might be sampling PUFs lower in energy than the globally unfolded form under the native conditions used in this study.
Results
Equilibrium or kinetic unfolding intermediates cannot be detected by CD and fluorescence
The fluorescence of Trp 28 and Trp 31 is strongly quenched in native Trx because of the proximity of the active site disulphide bond between Cys 32 and Cys 35 (Stryer et al. 1967). There is a release of Trp quenching on unfolding. The guanidinium chloride (GdmCl)-induced equilibrium unfolding transition of Trx was monitored by measurement of Trp fluorescence as well as of far-UV CD at 222 nm. The unfolding transitions are observed to be identical by the tertiary structure probe as well as by the secondary structure probe, as shown in Figure 2A ▶. Equilibrium unfolding of Trx therefore appears to be two-state, without the accumulation of any intermediates. The values of the free energy of unfolding, ΔGu (H2O), and the midpoint of the unfolding transition, Cm, were determined to be 9.6 kcal mole−1 and 2.5 M, respectively.
Figure 2.
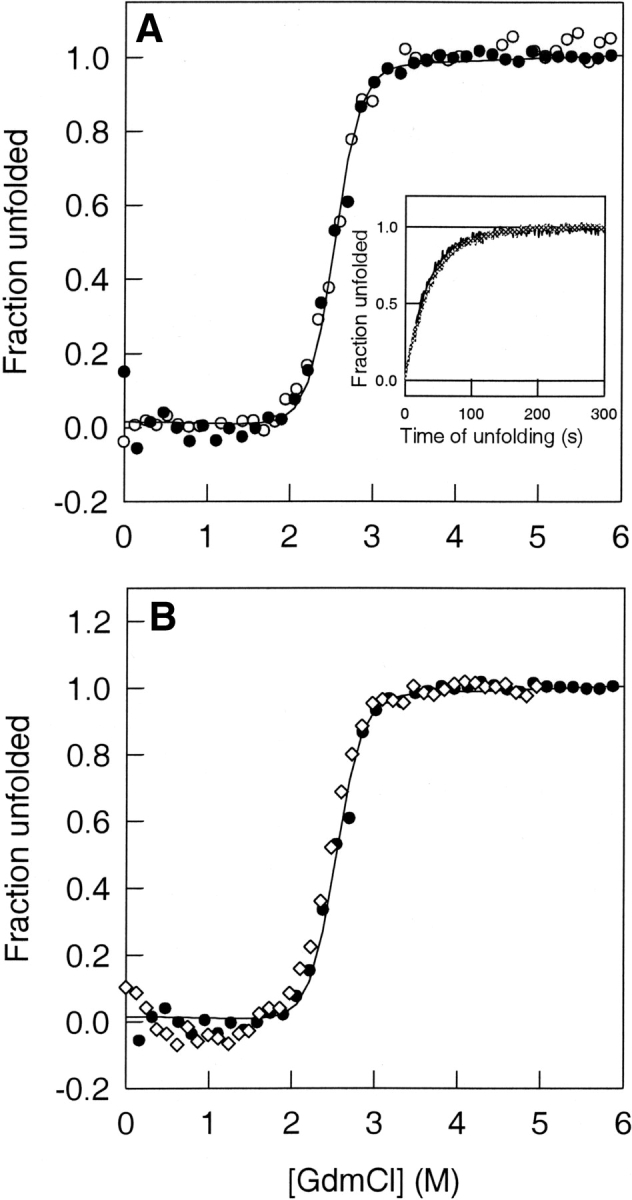
Global unfolding of thioredoxin. (A) The fraction of unfolded Trx molecules, as monitored by fluorescence (open circles) at 368 nm and CD (filled circles) at 222 nm, is plotted against the concentration of GdmCl in which Trx was equilibrated at pH 7. The solid line through the data is a fit of the data to a two-state N ⇌ U model (Agashe and Udgaonkar 1995) and yields a CM of 2.5 M. The inset to the figure shows the kinetics of unfolding of thioredoxin in 3.2 M GdmCl, followed by both fluorescence and CD. In either case, a fit (solid lines) to a single exponential yielded a rate of unfolding of 0.02 sec−1. In both cases, the data were normalized to a value of 0 for the native protein and to a value of 1 for the unfolded protein. (B) Equilibrium unfolding transitions of Trx in H2O (filled circles) and D2O (open diamonds) buffers at pH 7. The fraction of unfolded Trx molecules is plotted against the GdmCl concentration in which the protein is equilibrated. The equilibrium melts overlap, showing that there is no stabilization of Trx in D2O. The solid line through the data is a fit of the data to a two-state N ⇌ U model (Agashe and Udgaonkar 1995).
When Trx was unfolded at a final GdmCl concentration of 3.2 M, the increase in Trp fluorescence occurred in a single kinetic phase (Fig. 2A ▶, inset). The increase in the CD signal on unfolding in 3.2 M GdmCl took place at the same rate as that of the fluorescence change, that is, 0.02 sec−1, and the unfolding traces monitored by both CD and fluorescence overlapped, as shown in the inset to Figure 2A ▶. No kinetic unfolding intermediates had been detected in earlier studies (Kelley et al. 1986, 1987; Georgescu et al. 1998).
The equilibrium unfolding transition was monitored in H2O as well as in D2O solutions, using far-UV CD at 222 nm as the structural probe. Some proteins, like cytochrome c (Bai et al. 1994), have been shown to be more stable in D2O than in H2O. Figure 2B ▶ shows, however, that Trx, like ribonuclease A (Juneja and Udgaonkar 2002) and barstar (Bhuyan and Udgaonkar 1998) does not gain any extra stability in D2O. The equilibrium unfolding transition is identical in both D2O and H2O solutions, with the midpoint being at 2.5 M GdmCl.
Native-state HX
Complete resonance assignments are available for the oxidized and reduced forms of Trx (Dyson et al. 1989). All experiments reported here have been carried out with oxidized Trx. The backbone amide proton peaks present in the fingerprint region of the 2D-NMR TOCSY spectra collected for oxidized Trx could be identified easily (Fig. 3 ▶) from the published NMR assignments. Only the slowest exchanging 24 backbone amide protons (Fig. 1C ▶) could be followed in this study, which was carried out at pH 7. The backbone amide protons of the residues present in all four β turns, in helix 3 and the 310 helix joining the two structural subdomains, as well as in the β strands 1 and 5, exchange fast and hence, could not be studied. Figure 3 ▶ shows how the peak intensities decrease with time in spectra collected at different time intervals for Trx in the absence of any GdmCl. For each peak, the decrease in intensity over time (Fig. 4 ▶) could be fitted to a single exponential (Equation 1).
Figure 3.

Fingerprint regions of TOCSY spectra of native thioredoxin in D2O at pD 7, 25°C, at (A) 0.5, (B) 4, (C) 48, and (D)185 h, after the change from H2O to D2O. Only the peaks in the first spectrum (A) have been labeled.
Figure 4.

Kinetics of HX. The proton-to-deuterium exchange kinetics of the backbone amide proton of Lys 36 (A) and Ala 46 (B), residues belonging to Class I, are shown at 0 (circles), 0.3 (triangles), 0.5 (diamonds), and 0.8 M (squares) GdmCl. The peak intensities were normalized with respect to the intensity in the first spectrum. The solid lines through the data are fits to Equation 1. (B) Kinetics of exchange of the amide protons of Leu 24 (C) and Leu 79 (D), residues belonging to Class II, are shown at 0 M (circles) and 0.3 M (triangles) GdmCl. The insets show the kinetics of exchange at 0.5 M (triangles) and 0.8 M (squares) GdmCl. The peak intensities were normalized with respect to the intensity in the first spectrum. The solid lines through the data are fits to Equation 1.
The exchange of the backbone amide protons was monitored as a function of GdmCl concentration in the concentration range 0 to 1.5 M. These concentrations of GdmCl correspond to native conditions as measured by optical probes like CD and fluorescence (Fig. 2A ▶). Figure 4 ▶ shows the kinetics of exchange of the amide protons of Lys 36, Ala 46, Leu 24, and Leu 79 at four different GdmCl concentrations. The kinetics of exchange of the amide protons of Lys 36 and Ala 46 have only a small dependence on the concentration of denaturant, implying that the exchange is dominated by local structural fluctuations in which there is no significant change in the surface area to which GdmCl can bind. For the much slower exchanging amide protons of Leu 24 and Leu 79, there is a significant increase in the rate of exchange with an increase in GdmCl concentration, signifying that exchange occurs through a structure-opening reaction in which there is a significant change in GdmCl-binding surface area.
HX isotherms defined by the GdmCl-dependence of exchange
The values of the free energy of opening to HX, ΔGop, were determined from the measured exchange rates, kex, as given by Equation 3. The dependence of ΔGop on GdmCl concentration, for each amide residue, is shown in Figures 5 ▶ and 6 ▶. The dependence defines the HX isotherm for each amide proton. Each HX isotherm was fitted to Equation 8. All fits, except those for Ile 41 and Leu 42, were constrained to the value of m that was obtained for global unfolding (Fig. 2 ▶). The straight solid lines in all panels of Figures 5 ▶ and 6 ▶ show the linear dependence of ΔGu on increasing GdmCl concentrations. This dependence of ΔGu values on GdmCl concentration was obtained from the global unfolding transition of Trx monitored by CD and fluorescence (Fig. 2 ▶). The dashed line shows the proline isomerization-corrected values of ΔGu at the different GdmCl concentrations, with 1.4 kcal mole−1 having been added to each of the values of ΔGu (see Discussion).
Figure 5.

HX isotherms of amide protons belonging to residues in helices. The GdmCl concentration-dependence of the free energy of opening to HX is shown for each of the amide protons from the helices that were used as probes. The solid line in each panel represents the free energy of global unfolding of Trx, as a function of GdmCl concentration, which was obtained from the equilibrium unfolding data of Figure 2 ▶. The dashed line shows the global unfolding free energy at each indicated GdmCl concentration, after correction for proline isomerization (see text). The solid lines through the data points are fits to Equation 8. All fits, except those for Ile 41 and Leu 42, were constrained to the value of m that was obtained for global unfolding (Fig. 2 ▶).
Figure 6.

HX isotherms of amide protons belonging to residues in the β-sheet. The GdmCl concentration-dependence of the free energy of opening to HX is shown for each of the amide protons from the β strands that were used as probes. The solid line in each panel represents the free energy of global unfolding of Trx, as a function of GdmCl concentration, which was obtained from the equilibrium unfolding data of Figure 2 ▶. The dashed line shows the global unfolding free energy at each indicated GdmCl concentration after correction for proline isomerization (see text). The solid lines through the data points are fits to Equation 8. All fits were constrained to the value of m that was obtained for global unfolding (Fig. 2 ▶).
Figure 5 ▶ shows the exchange isotherms of helix 1 amide protons. All the isotherms were fitted to Equation 8, with the fits constrained to the values of m and KU (water) that were obtained from optically monitored equilibrium unfolding studies (Fig. 2 ▶). At low GdmCl concentrations, exchange of the amide protons of Phe 12, Val 16, and Lys 18 appears to be independent of the concentration of GdmCl. The unfolded state does not appear to be populated to any significant extent, and HX is dominated by local structural fluctuations under these subdenaturing conditions. The HX isotherms for Phe 12, Val 16, and Lys 18 appear to merge with the global unfolding curve at high denaturant concentrations, in the transition zone, as expected from Equation 8.
Helix 2 appears to have two different sets of amide protons, as shown in Figure 5 ▶. The first set, comprising Lys 36 and Ala 46, shows exchange dominated by local structural fluctuations at low GdmCl concentrations, whereas at higher GdmCl concentrations, exchange appears to be dominated by global unfolding. Unlike the exchange isotherms for the helix 1 residues, which appears to merge with the uncorrected global unfolding curve, the isotherms of Lys 36 and Ala 46 merge with the proline isomerization-corrected global unfolding isotherm. The second set comprises Ile 41 and Leu 42, whose HX isotherms exhibit a linear dependence on GdmCl concentrations, like the global unfolding isotherm. The values obtained for m and ΔGu were, however, −2.7 kcal mole−1M−1 and 5.9 kcal mole−1, respectively, much lower than the values for global unfolding. The m value is indicative of the nonpolar surface area exposed during the opening reaction that leads to exchange. A value similar to the m value for global unfolding would therefore indicate that the exchange takes place via the globally unfolded state. The isotherms with a lower m value therefore represent exchange following a partial unfolding (structure-opening) reaction that leads to the exposure of less nonpolar surface area than the global unfolding reaction. Both the helix 4 amide protons, Leu 103 and Asp 104 (Fig. 5 ▶), show exchange dominated by local fluctuations at lower GdmCl concentrations, and by global unfolding at higher GdmCl concentrations. The HX isotherms for the helix 4 amide protons merge with the proline isomerization-corrected global unfolding isotherm at high GdmCl concentrations.
Figure 6 ▶ shows that, at high concentrations of GdmCl, the HX isotherms of all the amide protons of β strand 2, Ile 23, Leu 24, Asp 26, Phe 27, Trp28, and Ala 29, appear to merge with an isotherm that is higher in energy than the proline isomerization-corrected global unfolding curve by 1.3 kcal mole−1 at all GdmCl concentrations. At lower GdmCl concentrations, the exchange of all the amide protons of β strand 2 is dominated by local fluctuations, whereas at higher GdmCl concentrations, exchange appears to occur predominantly by global unfolding.
In β strand 3 (Fig. 6 ▶), the amide protons of Thr 54, Val 55, Ala 56, Leu 58, and Ile 60 appear to exchange via local fluctuations at lower GdmCl concentrations. At higher GdmCl concentrations, their HX isotherms, like those of the residues belonging to β strand 2, appear to merge with an isotherm that is higher in energy than the proline isomerization-corrected global unfolding isotherm by 1.3 kcal mole−1 at all GdmCl concentrations.
Figure 6 ▶ shows that the amide protons of Leu 78, Leu 79, Leu 80, and Phe 81 in β strand 4 exchange through local fluctuations at subdenaturing GdmCl concentrations. At higher GdmCl concentrations, the HX isotherms of all the amide protons merge with the high-energy isotherm defined by the residues of β strand 2 and β strand 3. This high-energy isotherm is higher in energy than the proline isomerization-corrected global unfolding isotherm by 1.3 kcal mole−1 at all GdmCl concentrations.
Two different classes of amide residues
On the basis of their HX isotherms, two different classes of amide residues could be broadly identified: Class I residues whose HX isotherms merge with the global unfolding isotherm, without or with the correction for proline isomerization, and Class II residues whose HX isotherms define an isotherm that is higher in energy than the proline isomerization-corrected global unfolding curve by 1.3 kcal mole−1 at all GdmCl concentrations. The residues belonging to Class I are Phe 12, Val 16, Lys 18, Lys 36, Ala 46, Leu 103, and Asp 104. The Class II residues are Ile 23, Leu 24, Asp 26, Phe 27, Trp 28, Ala 29, Thr 54, Val 55, Ala 56, Leu 58, Ile 60, Leu 78, Leu 79, Leu 80, and Phe 81 (Table 1). All the residues in Class I belong to helices, and all the residues belonging to Class II are present in β strands (Table 1). The range of the values of ΔGop in the absence of any denaturant, for the 24 observed amide protons is from 4.7 to 9.9 kcal mole−1. For Class I, ΔGop (water) is 6.3 ± 0.9 kcal mole−1, and for Class II, ΔGop (water) is 9.4 ± 0.4 kcal mole−1. Only residues Ile 41 and Leu 42 appear not to belong to either of these two classes. The HX isotherms of these two residues show a linear dependence on GdmCl concentrations even at subdenaturing concentrations (see earlier).
Table 1.
Spatial location and free energy of opening to HX, in the absence of denaturant, for the measured amide protons of Trx
| Residue | Structure | % Surface | H-bond | kint (h−1)a | ΔGop |
| Class I | |||||
| Phe 12 | helix 1 | 0 | 8 Thr O | 36165 | 6.4 |
| Val 16 | helix 1 | 4.4 | 12 Phe O | 4150 | 4.7 |
| Lys 18 | helix 1 | 15.7 | — | 30783 | 6.1 |
| Lys 36 | helix 2 | 25.2 | 33 Gly O | 177134 | 7.5 |
| Ala 46 | helix 2 | 7.1 | 42 leu O | 32223 | 7.0 |
| Leu 103 | helix 4 | 0 | 99 Leu O | 16531 | 8.8 |
| Asp 104 | helix 4 | 17.5 | 100 Lys O | 16916 | 7.1 |
| Class II | |||||
| Ile 23 | β strand 2 | 0 | 81 Phe O | 10191 | 9.1 |
| Leu 24 | β strand 2 | 0 | 54 Thr O | 8478 | 9.3 |
| Asp 26 | β strand 2 | 0.1 | 56 Ala O | 19875 | 7.9 |
| Phe 27 | β strand 2 | 0 | 77 Thr O | 20811 | 9.6 |
| Trp 28 | β strand 2 | 0 | 58 Leu O | 8478 | 9.2 |
| Ala 29 | β strand 2 | 0 | — | 42490 | 8.7 |
| Thr 54 | β strand 3 | 0 | — | 28724 | 7.8 |
| Val 55 | β strand 3 | 5.2 | — | 17308 | 9.8 |
| Ala 56 | β strand 3 | 0 | 24 Leu O | 39654 | 10.0 |
| Leu 58 | β strand 3 | 0 | 26 Asp O | 18979 | 6.6 |
| Ile 60 | β strand 3 | 16 | 28 Trp O | 21297 | 9.5 |
| Leu 78 | β strand 4 | 0 | 90 Lys O | 22820 | 7.3 |
| Leu 79 | β strand 4 | 0 | 25 Val O | 8877 | 9.0 |
| Leu 80 | β strand 4 | 0 | 88 Ala O | 8877 | 9.2 |
| Phe 81 | β strand 4 | 0.6 | 23 Ile O | 19422 | 9.8 |
| Ile 41 | helix 2 | 0 | — | 5864 | 5.6 |
| Leu 42 | helix 2 | 0 | 38 Ile O | 8478 | 6.1 |
a Values of kint were determined from model-peptide data, as described by Bai et al. 1993.
Figure 7 ▶ shows a comparison of the HX isotherms of the fast-exchanging helix 1 amide protons at two different pH values, pH 7 and at pH 5.7. The values of ΔGop for the residues Phe 12, Val 16, and Lys 18 were observed to be unchanged by the variation in pH, within experimental error, over different concentrations of GdmCl ranging from 0 M to 1 M.
Figure 7.

pH dependence of ΔGop. The GdmCl concentration-dependence of the free energy of opening to HX is shown for Phe 12, Val 16, and Lys 18, which are the fast-exchanging amide proton probes from helix 1, at pH 7 (open symbols) and at pH 5.7 (filled symbols), respectively. The solid and the dotted lines represent the free energies of global unfolding of Trx, as a function of GdmCl concentration, at pH 5.7 and at pH 7, respectively, and are given by the equations ΔGu = 10.7 − 3.5 [GdmCl] and ΔGu = 9.6 − 3.8 [GdmCl], respectively.
Discussion
Validity of the EX2 mechanism
The data in Figures 5 ▶ and 6 ▶ have been analyzed according to the EX2 mechanism (see Materials and Methods), because a previous study of HX of oxidized and reduced forms of Trx (Jeng and Dyson 1995), in the absence of any denaturant, had indicated that the exchange of back-bone amide protons at pH 5.7 takes place via the EX2 mechanism. The validity of the assumption that the EX2 mechanism holds true over the entire range of the GdmCl concentration, 0–1.5 M, used in this study at pH 7, is indicated by the following observations.
1. The values of ΔGop for the fast-exchanging helix 1 residues, Phe 12, Val 16, and Lys 18, have been observed to be similar, within experimental error, at pH 5.7 and pH 7 in the 0–1 M range of GdmCl concentration (Fig. 7 ▶). The values of ΔGop have been determined using Equation 3 (see Materials and Methods). The observation that these values are similar at both pH 5.7 and pH 7, at all the concentrations of GdmCl between 0 M and 1 M, indicates that the values of kobs/kint for these residues are independent of pH over the entire range of GdmCl concentrations studied. Two different scenarios, for either the EX2 or EX1 mechanism being operative in such a case, can be considered:
(a) In the EX2 scenario, Kop = kobs/kint, and the observed pH independence of kobs/kint would imply that Kop is independent of pH. It is quite plausible that Kop, the equilibrium constant for the opening reaction leading to exchange, is affected only marginally by a change in pH from 5.7 to 7. In fact, the value of KU (water) for the N ⇌ U global unfolding transition for Trx at pH 5.7 is 0.2 × 10−7, whereas at pH 7 the value is 1 × 10−7, as obtained from optically monitored equilibrium unfolding transitions, indicating that at least the global unfolding reaction has only a small dependence on pH.
(b) In the EX1 scenario, kobs/kint = ko/kint, and the observed pH independence of kobs/kint would imply that the pH dependence of ko, the rate of the opening transition, would therefore have to be identical to the pH dependence of kint. It is very unlikely that the rate of any conformational change in a protein would have the very strong dependence on pH that characterizes the rate of an HX reaction.
Thus, the observations support the assumption that the mechanism of exchange remains EX2 over the entire range of GdmCl concentration that has been studied.
2. For the residues in the core β strands and helix 2, the dependence of the values of ΔGop at high GdmCl concentrations is consistent with the m value for global unfolding. For an EX2 mechanism of exchange, the values of ΔGop are expected to follow the dependence of ΔGu on the concentration of GdmCl at high concentrations of GdmCl where exchange is dominated by global unfolding (see Materials and Methods). This observation lends further support to the assumption that exchange takes place via the EX2 mechanism over the entire range of GdmCl concentrations that has been studied.
Presence of high free energy unfolded states
The HX isotherms for many amide protons are observed to be higher in energy than the global unfolding isotherm, which is defined by the dependence of ΔGu values on GdmCl concentrations. When the exchange-competent unfolded state is energetically and structurally identical to the globally unfolded state defined by optical probes, then ΔGop should be ≤ΔGu at all concentrations of GdmCl. HX is, however, a kinetic experiment, and if a transiently formed exchange-competent unfolded state is populated for a sufficient duration so that exchange takes place before it relaxes to its final equilibrium state, then ΔGop may be >ΔGu.
Equilibrium unfolded Trx is known to have at least three different unfolded states (Kelley and Stellwagen 1984; Georgescu et al. 1998), which have been identified on the basis of the differences in their rates of folding: UVR folds very rapidly, UR folds rapidly, and UM folds slowly. It is presumed that the presence of these multiple unfolded states is due to the five proline (Holmgren et al. 1975) residues at positions 34, 40, 64, 68, and 76, each of which can be in a cis or trans conformation in the unfolded state. Only Pro 76 is present in the cis conformation in the native state, whereas the rest of the Pro residues are present in trans conformations (Holmgren et al. 1975). Double-jump experiments have shown that native Trx unfolds rapidly to UVR, which must have all the proline residues in native conformations because it refolds very rapidly to the native state (Georgescu et al. 1998; Bhutani and Udgaonkar 2001). Under unfolding conditions, UVR has been found to relax slowly via two parallel processes to UR and UM (Georgescu et al. 1998; Bhutani and Udgaonkar 2001). Thus, UVR is a higher energy unfolded state that can equilibrate with two other lower energy unfolded states, with all three states expected to be exchange competent. In the context of HX, ΔGop for a particular amide hydrogen site is expected to be a measure of the energy difference between the native state and the high-energy unfolded state UVR. In contrast, ΔGu is the observed energy difference between the native state and the equilibrium mixture of UVR, UR, and UM.
Double-jump experiments as well as stopped-flow activity measurements have shown that UVR is populated to an extent of only 10% in the equilibrium distribution of UVR, UR, and UM (Georgescu et al. 1998; Bhutani and Udgaonkar 2001). The difference between ΔGop and ΔGu is therefore given by (Bhuyan and Udgaonkar 1998):
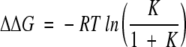 |
where K = [UVR]/[UR + UM] = 0.1/0.9 according to the equilibrium distribution of the three unfolded forms. The proline-isomerization related correction, that is, ΔΔG is therefore estimated to be 1.4 kcal mole−1. The addition of this ΔΔG to ΔGu at all concentrations of GdmCl yields the proline isomerization-corrected global isotherm, that is, the dashed line in Figures 5 ▶ and 6 ▶. In water, ΔGu from the proline isomerization-corrected global HX isotherm has a value of 9.6 kcal mole−1 + 1.4 kcal mole−1 = 11.0 kcal mole−1. The HX isotherms of the helix 2 residues Lys 36 and Ala 46, as well as the helix 4 residues Leu 103 and Asp 104 merge into the global unfolding isotherm having such a correction. The presence of multiple unfolded states, as a result of proline-isomerization after unfolding, has been observed for several proteins including ribonuclease A (Garel and Baldwin 1973; Schmid 1982), cytochrome c (Nall 1985), and barstar (Bhuyan and Udgaonkar 1998), and similar corrections were required to remove the discrepancy between the values of ΔGop and ΔGu observed for these proteins (Bai et al. 1995; Bhuyan and Udgaonkar 1998).
Folding subdomains of Trx
The amide protons of the β turns, helix 3, the 310-helix, β strand 1, and β strand 5 exchange too rapidly to be followed under the experimental conditions of this study. The amide protons whose exchange kinetics can be monitored have been classified on the basis of their HX isotherms: Class I comprises the residues whose HX isotherms merge with the global unfolding isotherm, without or with the correction for proline isomerization, and Class II comprises the residues whose HX isotherms define an isotherm, which is higher in energy than the proline isomerization-corrected global unfolding curve by 1.3 kcal mole−1 at all GdmCl concentrations. It appears that all the amide probes present in the helices, with the exception of Ile 41 and Leu 42 (see following) belong to Class I, and that the amide probes present in the β-sheet belong to Class II.
The observation that the HX isotherms of the amide probes present in the helix merge with the global unfolding isotherm, with or without the correction for proline isomerization, indicates that the helices unfold together when the globally unfolded state is sampled transiently by the protein in the native conditions used in this study. In that sense, helices α1, α2, and α4 form a folding subdomain. Similarly, a second folding subdomain appears to be formed by the core β strands, β2, β3, and β4.
Apparently high stability of β strand amide hydrogens to exchange
Two simple explanations for why the residues in the core β strands form a HX isotherm higher in energy than even the proline isomerization-corrected global unfolding isotherm are easily ruled out. The first possibility, that the protein is stabilized in D2O with respect to that in H2O, as seen for some proteins (Bai et al. 1994), is ruled out because Trx does not show any increase in stability in D2O (Fig. 2B ▶). The second possibility, that the value obtained for ΔGu by monitoring optical probes does not reflect the true value for the stability of the protein, is ruled out because the values obtained for ΔGu by monitoring optical probes and by using calorimetry are identical (Santoro and Bolen 1992).
A third possible explanation is that the β strands exchange out not in UVR but in a high-energy unfolding intermediate I, which has an even higher free energy than UVR. I would then be a form in which the core β strands have unfolded, but in which the α helices are intact. It seems unlikely that the core β strands can unfold fully without affecting the stability to exchange of the α helices that flank it. Because it is observed that the α helices exchange only in completely unfolded forms, it is unlikely that intermediate I is populated on the kinetic pathway of unfolding of N. Moreover, kinetic unfolding studies using optical probes would not miss detecting an intermediate with only partial secondary structure. Thus, this explanation can also be ruled out.
The most likely explanation is that residual structure is present in the unfolded state. Residual structure in the unfolded state would be expected to lower the intrinsic rates of HX of the amides involved. Because the intrinsic rates used in the calculation of the values of ΔGop are those estimated from random-coil peptide models (Bai et al. 1993), such rates would be much faster than the actual rates applicable to an unfolded protein with residual structure; consequently ΔGop would be overestimated. The presence of residual structure in the unfolded state leading to such an overestimation of the values of ΔGop has been previously observed for cytochrome c (Bai et al. 1995).
Thus, the native-state HX results for Trx indicate that the β strands, β2, β3, and β4, which form the core of the protein in the native state, are involved in residual structure in the unfolded state in native conditions at pH 7. Native residual secondary structure has also been observed in the cold denatured state of barstar (Wong et al. 1996), the acid unfolded state of apomyoglobin (Eliezer et al. 1998; Yao et al. 2001), and the denatured state of barnase (Freund et al. 1996; Bond et al. 1997), and native-state HX studies have been applied elegantly to probe the effect of single point mutations on the structure of the denatured state of Staphylococcus nuclease (Wrabl and Shortle 1999).
Structure in the unfolded state and the folding pathway of Trx
The unfolded states of many proteins contain specific residual structure, either secondary structure or localized hydrophobic clusters, in some cases in a native topology (Neri et al. 1992; Alexandrescu et al. 1994; Freund et al. 1996; Saab-Rincon et al. 1996; Wang and Shortle 1996; Wong et al. 1996; Bond et al. 1997; Gillespie and Shortle 1997; Schwalbe et al. 1997; Eliezer et al. 1998; Mok et al. 1999; Hodsdon and Frieden 2001;Yao et al. 2001; Klein-Seetharaman et al. 2002). Such residual structure is likely to determine the initial protein folding events for these proteins (Bond et al. 1997; Nolting et al. 1997). Theoretical studies have also indicated that such structure in the unfolded state facilitates folding by forming nucleation sites around which structure can be formed (Dill et al. 1993; Smith et al. 1996; Klimov and Thirumalai 1998).
Because the core β strands, β2, β3, and β4 appear to be present in the unfolded state itself, it is expected that folding studies of Trx will indicate that they are also present in the earliest folding intermediates. No detailed structural information about the unfolding and refolding pathways of Trx is, however, available at present. Nevertheless, studies using the fragment-complementation approach (Tasayco and Chao 1995; Tasayco et al. 2000) have provided indirect structural information on the early intermediate in Trx folding. Three complementary fragments of Trx were generated: the N (1–37 residues), the M fragment (38–73 residues), and the C fragment (74–108 residues) (Tasayco et al. 2000). Using size-exclusion chromatography, far-UV CD, and three-dimensional NMR spectroscopy, it was observed that the N and the C fragments were essential for native complex formation. 1H-15N HSQC spectra of the complex formed by the three fragments showed cross-peaks corresponding to almost all the native-like secondary structure elements, whereas the N/C complex showed cross-peaks corresponding mainly to the β2 and β4 strands. The N/M and M/C fragment complexes failed to show any cross-peaks corresponding to native-like secondary structures. It was therefore suggested that the initial folding event is governed by the interaction of the N and C fragments, and that folding is initiated by the zippering together of the β2 and β4 strands. These observations correlate well with the native-state HX results reported here, in which the core β strands, β2, β3, and β4 appear to be involved in residual structure in the unfolded state itself.
Absence of PUFs of Trx
In the case of many proteins, native-state HX studies (Loh et al. 1993; Mayo and Baldwin 1993; Bai et al 1995; Chamberlain et al. 1996; Llinas et al. 1999; Neira et al. 1999; Forge et al. 2000; Chu et al. 2002; Juneja and Udgaonkar 2002; Liu et al. 2002; Rodriguez et al. 2002; Srimathi et al. 2002) have led to the identification of PUFs that are sampled transiently by the protein under native conditions. PUFs represent structural segments of a protein with different stabilities, and especially in the case of cytochrome c (Bai et al. 1995; Hoang et al. 2002), they have been useful in dissecting out the unfolding pathway.
PUFs are usually identified by determining whether a group of amide hydrogens displays overlapping HX isotherms whose dependence on denaturant concentration is less than that of ΔGU, and by determining whether an extrapolation of the HX isotherm to zero denaturant yields a value for ΔGop that is less than that of ΔGU. In the case of Trx, the isotherms of Ile 41 and Leu 42 show a linear dependence even on low concentrations of GdmCl, having an m value of −2.7 kcal mole−1M−1 and a ΔGop value of 5.9 kcal mole−1. An m value lower than the value for global unfolding, as seen for the isotherms of Ile 41 and Leu 42, signifies a partial unfolding reaction, in which the exposure of nonpolar surface area is less than that which occurs during global unfolding. The partial unfolding reactions defined by the isotherms of Ile41 and Leu 42 can be used to define a PUF of Trx, in which these two residues are unfolded. It might appear surprising that helix 2 is not a cooperatively unfolding unit, but it should be noted that it is the longest helix in Trx (residues 35–49) and contains two Pro residues, Pro 34 and Pro 40, out of which Pro34 is accommodated within the helical turns, and Pro 40 causes a break in the helix (Katti et al. 1990). This break in the helix may account for the lower protection against HX of the two residues, Ile 41 and Leu 42, that follow in sequence. Thus, the HX isotherms of Ile 41 and Leu 42 may not reflect a real subglobal opening event.
It therefore seems that discrete well-defined PUFs are not sampled by Trx under the native conditions used in this study. In the case of barnase (Clarke and Fersht 1996), chymotrypsin inhibitor 2 (Itzhaki et al. 1997), and protein L (Yi et al. 1997), discrete PUFs have not been detected. The absence of PUFs for chymotrypsin inhibitor 2 (Itzhaki et al. 1997) and protein L (Yi et al. 1997) is not surprising because these proteins fold without the accumulation of folding intermediates (Jackson and Fersht 1991; Scaley et al. 1997). The failure to detect PUFs for Trx is surprising because Trx is known to fold via multiple folding intermediates (Georgescu et al. 1998; Bhutani and Udgaonkar 2001). Barnase, too, is known to fold via a stable folding intermediate (Bycroft et al. 1990; Matouschek et al. 1990; Oliveberg and Fersht 1996). More recent, kinetic pulse-labeling HX experiments have, however, failed to detect this intermediate of barnase (Chu et al. 1999; Takei et al. 2000), and on the contrary, the observed kinetics have been interpreted to indicate that the folding intermediate is not populated on the folding pathway (Chu et al. 1999; Takei et al. 2000). It is expected that kinetic pulse-labeling HX experiments on Trx, which are currently in progress, will allow a better understanding of why Trx does not appear to sample discrete PUFs in the native conditions studied.
Materials and methods
Buffers and protein purification
D2O, GdmCl, and buffer components used were ultra pure grade reagents from Sigma. Values of the pH reported for the D2O buffers correspond to the pH meter readings uncorrected for isotope effects. GdmCl was deuterated by dissolving it in D2O and lyophilizing the solution; this was repeated three times. Trx was deuterated by dissolving it in D2O at pH 7, and heating to 80°C for 30 min. It was then refolded on ice and lyophilized. The procedure was repeated three times to ensure complete deuteration. Complete deuteration was checked by an electrospray ionization mass spectrum of the deuterated protein, with the deuterated protein showing an increase in mass of 130 D over the protonated protein, which showed a mass of 11,673 D.
The procedure for purification of Trx has been described previously (Bhutani and Udgaonkar 2001). Trx concentrations were determined using a molar extinction coefficient of 13,700 M−1cm−1.The activity of Trx was measured by using its ability to catalyze the DTT-mediated reduction of insulin, and monitoring the subsequent aggregation of the insulin B chain (Holmgren 1979).
Equilibrium unfolding experiments
GdmCl-induced equilibrium unfolding was monitored by CD at 222 nm and by fluorescence. Fluorescence intensities at 368 nm were measured on a SPEX Fluorimeter with excitation at 295 nm. Protein concentrations used were typically 2 –4 μM. A slit width of 0.37 nm was used for excitation, and a slit width of 10 nm for emission. For CD measurements, a Jasco J720 spectropolarimeter was used with the bandwidth set to 1 nm. The final protein concentrations used were 20–25 μM for equilibrium unfolding in H2O as well as D2O solutions. Deuterated protein was used for equilibrium unfolding in D2O. The buffer used was 30 mM sodium phosphate, 0.1 M KCl (pH 7.0) without any reducing agent in order to maintain Trx in its oxidized form under all conditions. All experiments were carried out at 25°C.
Unfolding kinetics
Manual mixing experiments were carried out to observe the unfolding kinetics of Trx by CD (222 nm) as well as by fluorescence. The mixing dead time was ~10 sec for both sets of experiments. Native Trx was unfolded to a final GdmCl concentration of 3.2 M, with the protein concentration being 20 μM for CD and 4 μM for fluorescence. For fluorescence, the excitation was at 295 nm, and the emission was collected at 368 nm.
Native-state HX experiments
Equilibrium hydrogen-deuterium exchange experiments were carried out at different concentrations of GdnDCl (0–1.5 M). The final protein concentrations used typically for NMR were 1–2 mM. To initiate the H→D exchange, a 5-mL gel filtration column packed with G-25 was used. The column was equilibrated with the deuterated buffer containing 30 mM sodium phosphate and 0.1 M KCl (pH 7), and containing different GdnDCl concentrations. Exchange was initiated as soon as the protein in the aqueous buffer was loaded on the column. The protein eluted in the void volume was collected, and the typical run time for the column was 2 min.
After initiating the exchange, the minimum time after which the NMR spectrum was recorded for any of the samples was 20–25 min, and this dead time was taken into account for each of the samples. 2D 1H TOCSY spectra were recorded on a 600-MHz (Varian Unity Plus) NMR spectrometer with a spectral width of 7500 Hz and at a temperature of 25°C. Each TOCSY spectra was recorded with a mixing time of 80 msec, and 128 data points were collected along the t1 domain and 2048 data points along the t2 domain. A total of eight scans were collected at each point along the t1 domain. Residual water in the samples was suppressed by presaturation. Before Fourier transformation, sine-bell squared window functions, using phase shifts of 90° and 30° were applied to the data in the D1 and D2 dimensions, and the data were also zero-filled to 2048 and 4096 points.
Each TOCSY spectrum took 1 h to record, and spectra were collected back to back over 8 to 24 h for samples in different GdnDCl concentrations. For the GdnDCl concentrations for which the exchange was observed over several days, the samples were maintained at 25°C in an incubator while they were kept outside the spectrometer. The cross-peaks were assigned according to the published proton chemical shifts of oxidized Trx (Dyson et al. 1989). Cross-peak intensities were analyzed using the Felix 97 software. The intensities of the nonexchangeable aromatic ring protons of Trp 28 and Tyr 49 were used as the internal references for the TOCSY spectra. The intensity of each CαH-NH cross-peak was divided by the sum of the intensities of the reference cross-peaks. The decrease in the normalized peak intensities of each amide proton was measured as a function of time, and fitted to single-exponential kinetics to obtain the exchange rate according to the following equation:
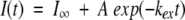 |
(1) |
where I∞ is the intensity at infinite time, A is the total change in intensity, and kex is the observed rate of HX.
Exchange for the slow-exchanging amide protons could be followed only over a period of 10–12 d, where the decay in their peak intensities is small. For the fits of the decreases in intensities of such slow-exchanging amide protons, I∞ was constrained to a value that was 0.5%–2% of the intensity of the corresponding peak in the earliest spectrum recorded. This was done because the experimentally observed values of I∞ for the fast exchanging Class I amide protons, whose complete exchange could be observed, were 0.5%–2% of the intensities in the earliest spectrum recorded. It was observed for different residues that constraining I∞ in this way did not alter the observed rates by more than 1%–2%. Even if I∞ was constrained to 10% of the intensity of the corresponding peak in the earliest spectrum recorded, the variation in the observed rates was <10%.
Data analysis
Data were analyzed according to a “two process model” (Woodward and Rosenberg 1971; Woodward and Hilton 1980), in which the exchangeable amide protons, protected as a result of hydrogen bonding or burial, can exchange with the solvent deuterons via global as well as local unfolding reactions, as represented in the following equation:
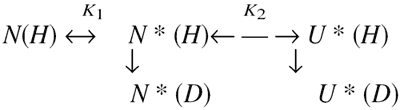 |
where N is the exchange-incompetent or closed native state, N* is the locally unfolded native state that is exchange competent or open for a particular backbone amide, U* is the globally unfolded state that is exchange competent or open for all amide residues, and K1 (=N*/N) and K2 (=U*/N*) are the two structure-opening equilibria. The closed-to-open reaction for HX according to the Linderstrøm-Lang is represented by Hvidt and Nielsen (1966):
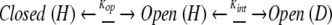 |
where N is identified with the closed state, N* and U* are identified with the open state, Kop = kop/kcl is the equilibrium constant for the opening reaction, and kint is the intrinsic rate constant of the amide proton as calculated from unfolded peptide models (Bai et al. 1993). The model makes the assumption that the intrinsic rates of exchange at the globally and locally open sites are the same (Bhuyan and Udgaonkar 1998).
Under the EX2 limit, where the rate of the closing reaction, kcl, is faster than both the rate of the opening reaction, kop, and the intrinsic rate of exchange, kint, the observed exchange rate, kex, is given by:
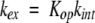 |
(2) |
The free energy of production of exchange-competent states (N* + U*) from the exchange-incompetent N, is then given by:
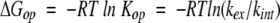 |
(3) |
where R is the gas constant and T is the absolute temperature.
Because two exchange competent forms are present,
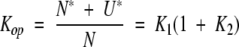 |
(4) |
If U* is taken to be equivalent to the equilibrium unfolded state U, then the equilibrium constant defining the global unfolding of N and N* is given by:
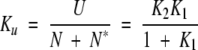 |
(5) |
Combining Equations 3, 4, and 5 yields Equation 6,
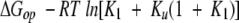 |
(6) |
It is assumed that K1 has no significant dependence on GdmCl concentration and that the denaturant dependence of Ku is as obtained from the linear-free energy model of global unfolding (Santoro and Bolen 1992).
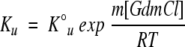 |
(7) |
where 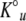 is the equilibrium constant for the N ⇌ U reaction in water, and m is related to the denaturant-binding surface area exposed on unfolding. Combining Equations 6 and 7 gives the following equation for the dependence of ΔGop on the denaturant concentration:
is the equilibrium constant for the N ⇌ U reaction in water, and m is related to the denaturant-binding surface area exposed on unfolding. Combining Equations 6 and 7 gives the following equation for the dependence of ΔGop on the denaturant concentration:
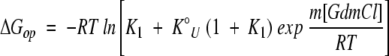 |
(8) |
Acknowledgments
All NMR spectra were recorded at the NMR facility at TIFR, Mumbai. We thank N. S. Bhavesh for useful discussions and help with the spectrometer, and M.K Mathew and R. Varadarajan for discussions. This work was funded by the Tata Institute of Fundamental Research, the Wellcome Trust, and the Department of Science and Technology, Government of India. J.B.U. is the recipient of a Swarnajayanti Fellowship from the Government of India.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.0239503.
References
- Agashe, V.R. and Udgaonkar, J.B. 1995. Thermodynamics of denaturation of barstar: Evidence for cold denaturation and evaluation of the interaction with guanidinium chloride. Biochemistry 34 3286–3299. [DOI] [PubMed] [Google Scholar]
- Alexandrescu, A.T., Abeygunawardana, C., and Shortle, D. 1994. Structure and dynamics of a denatured 131-residue fragment of staphylococcal nuclease: A heteronuclear NMR study. Biochemistry 3 1063–1072. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Milne, J.S., Mayne, L., and Englander, S.W. 1993. Primary structure effects on peptide group hydrogen exchange. Proteins 17 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 1994. Protein stability parameters measured by hydrogen exchange. Proteins 20 4–14. [DOI] [PubMed] [Google Scholar]
- Bai, Y., Sosnick, T.R., Mayne, L., and Englander, S.W. 1995. Protein folding intermediates: Native-state hydrogen exchange. Science 269192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani, N. and Udgaonkar, J.B. 2001. GroEL channels the folding of thioredoxin along one kinetic route. J. Mol. Biol. 314 1167–1179. [DOI] [PubMed] [Google Scholar]
- Bhuyan, A.K. and Udgaonkar, J.B. 1998. Two structural subdomains of barstar detected by rapid mixing NMR measurement of amide hydrogen exchange. Proteins 30 295–308. [PubMed] [Google Scholar]
- Bond, C.J., Wong, K.B., Clarke, J., Fersht, A.R., and Daggett, V. 1997. Characterization of residual structure in the thermally denatured state of barnase by simulation and experiment: Description of the folding pathway. Proc. Natl. Acad. Sci. 94 13409–13413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft, M., Matouschek, A., Kellis Jr., J.T., Serrano, L., and Fersht, A.R. 1990. Detection and characterization of a folding intermediate in barnase by NMR. Nature 346 488–490. [DOI] [PubMed] [Google Scholar]
- Chamberlain, A.K., Handel, T.M., and Marqusee, S. 1996. Detection of rare partially folded molecules in equilibrium with the native conformation of RNaseH. Nat. Struct. Biol. 3 782–787. [DOI] [PubMed] [Google Scholar]
- Chu, R.A., Takei, J., Barchi Jr., J.J., and Bai, Y. 1999. Relationship between the native-state hydrogen exchange and the folding pathways of barnase. Biochemistry 38 14119–14124. [DOI] [PubMed] [Google Scholar]
- Chu, R., Pei, W., Takei, J., and Bai, Y. 2002. Relationship between the native-state hydrogen exchange and folding pathways of a four-helix bundle protein. Biochemistry 41 7998–8003. [DOI] [PubMed] [Google Scholar]
- Clarke, J. and Fersht, A.R. 1996. An evaluation of the use of hydrogen exchange at equilibrium to probe intermediates on the protein folding pathway. Fold. Des. 1 243–254. [DOI] [PubMed] [Google Scholar]
- Dill, K.A., Fiebig, K.M., and Chan, H.S. 1993.Modeling compact denatured states of proteins. Proc. Natl. Acad. Sci. 90 1942–1946.7680482 [Google Scholar]
- Dyson, H.J., Holmgren, A., and Wright, P.E. 1989. Assignment of the proton NMR spectrum of reduced and oxidized thioredoxin: Sequence-specific assignments, secondary structure, and global fold. Biochemistry 28 7074–7087. [DOI] [PubMed] [Google Scholar]
- Eliezer, D., Yao, J., Dyson, H. J., and Wright, P.E. 1998. Structural and dynamic characterization of partially folded states of apomyoglobin and implications for protein folding. Nat. Struct. Biol. 5 148–155. [DOI] [PubMed] [Google Scholar]
- Englander, S.W. 1998. Native-state HX. Trends Biochem. Sci. 23 379–381. [DOI] [PubMed] [Google Scholar]
- Englander, S.W., Sosnick, T.R., Englander, J.J., and Mayne, L. 1996. Mechanisms and uses of hydrogen exchange. Curr. Opin. Struct. Biol. 6 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander, S.W., Mayne, L., Bai, Y., and Sosnick, T.R. 1997. Hydrogen exchange: The modern legacy of Linderstrom-Lang. Protein Sci. 6 1101–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge, V., Hoshino, M., Kuwata, K., Arai, M., Kuwajima, K., Batt, C.A., and Goto, Y. 2000. Is folding of β-lactoglobulin non-hierarchic? Intermediate with native-like β-sheet and non-native α-helix. J. Mol. Biol. 296 1039–1051. [DOI] [PubMed] [Google Scholar]
- Freund, S.M., Wong, K.B., and Fersht, A.R. 1996. Initiation sites of protein folding by NMR analysis. Proc. Natl. Acad. Sci. 93 10600–10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garel, J.R. and Baldwin, R.L. 1973. Both the fast and slow refolding reactions of ribonuclease A yield native enzyme. Proc. Natl. Acad. Sci. 70 3347–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgescu, R.E., Li, J., Goldberg, M.E., Tasayco, M.L., and Chaffotte, A.F. 1998. Proline isomerization-independent accumulation of an early intermediate and heterogeneity of the folding pathways of a mixed α/β protein, Escherichia coli thioredoxin. Biochemistry 37 10286–10297. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. R. and Shortle, D. 1997. Characterization of long-range structure in the denatured state of staphylococcal nuclease. II. Distance restraints from paramagnetic relaxation and calculation of an ensemble of structures. J. Mol. Biol. 268 170–184. [DOI] [PubMed] [Google Scholar]
- Hoang, L., Bedard, S., Krishna, M.M., Lin, Y., and Englander, S.W. 2002. Cytochrome c folding pathway: Kinetic native-state hydrogen exchange. Proc. Natl. Acad. Sci. A. 99 12173–12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodsdon, M.E. and Frieden, C. 2001. Intestinal fatty acid binding protein: The folding mechanism as determined by NMR studies. Biochemistry 40 732–742. [DOI] [PubMed] [Google Scholar]
- Holmgren, A. 1979. Thioredoxin catalyzes the reduction of insulin disulphides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254 9627–9632. [PubMed] [Google Scholar]
- Holmgren, A., Soderberg, B.O., Eklund, H., and Branden, C.I. 1975. Three-dimensional structure of Escherichia coli thioredoxin-S2 to 2.8 Å resolution. Proc. Natl. Acad. Sci. 72 2305–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino, M., Katou, H., Hagihara, Y., Hasegawa, K., Naiki, H., and Goto, Y. 2002. Mapping the core of the β(2)-microglobulin amyloid fibril by H/D exchange. Nat. Struct. Biol. 9 332–336. [DOI] [PubMed] [Google Scholar]
- Hvidt, A. and Nielsen, S.O. 1966. Hydrogen exchange in proteins. Adv. Protein Chem. 21 287–386. [DOI] [PubMed] [Google Scholar]
- Itzhaki L.S., Neira, J.L., and Fersht, A.R. 1997. Hydrogen exchange in chymotrypsin inhibitor 2 probed by denaturants and temperature. J. Mol. Biol. 270 89–98. [DOI] [PubMed] [Google Scholar]
- Jackson, S.E. and Fersht, A.R. 1991. Folding of chymotrypsin inhibitor 2. 1. Evidence for a two-state transition. Biochemistry 30 10428–10435. [DOI] [PubMed] [Google Scholar]
- Jeng, M.F. and Dyson, H.J. 1995. Comparison of the hydrogen-exchange behavior of reduced and oxidized Escherichia coli thioredoxin. Biochemistry 34 611–619. [DOI] [PubMed] [Google Scholar]
- Juneja, J. and Udgaonkar, J.B. 2002. Characterization of the unfolding of ribonuclease a by pulsed hydrogen exchange study: Evidence for competing pathways for unfolding. Biochemistry, 41 2641–2654. [DOI] [PubMed] [Google Scholar]
- ———. 2003. NMR characterization of disordered states of proteins on folding pathways. Curr. Sci. 84 157–172. [Google Scholar]
- Katti, S.K., LeMaster, D.M., and Eklund, H. 1990. Crystal structure of thioredoxin from Escherichia coli at 1.68 Å resolution. J. Mol. Biol. 212 167–184. [DOI] [PubMed] [Google Scholar]
- Kelley, R.F. and Richards, F.M. 1987. Replacement of proline-76 with alanine eliminates the slowest kinetic phase in thioredoxin folding. Biochemistry 26 6765–6774. [DOI] [PubMed] [Google Scholar]
- Kelley, R.F. and Stellwagen, E. 1984. Conformational transitions of thioredoxin in guanidinium chloride. Biochemistry 23 5095–5120. [DOI] [PubMed] [Google Scholar]
- Kelley, R.F., Wilson, J., Bryant, C., and Stellwagen, E. 1986. Effects of guanidinium chloride on the refolding kinetics of denatured thioredoxin. Biochemistry 25 728–732. [DOI] [PubMed] [Google Scholar]
- Kelley, R.F., Shalongo, W., Jagannadham, M.V., and Stellwagen, E. 1987. Equilibrium and kinetic measurements of the conformational transition of reduced thioredoxin. Biochemistry 26 1406–1411. [DOI] [PubMed] [Google Scholar]
- Klein-Seetharaman, J., Oikawa, M., Grimshaw, S.B., Wirmer, J., Duchardt, E., Ueda, T., Imoto, T., Smith, L.J., Dobson, C.M., and Schwalbe, H. 2002. Long-range interactions within a nonnative protein. Science 295 1719–1722. [DOI] [PubMed] [Google Scholar]
- Klimov, D.K. and Thirumalai, D. 1998. Lattice models for proteins reveal multiple folding nuclei for nucleation-collapse mechanism. J. Mol. Biol. 282 471–492. [DOI] [PubMed] [Google Scholar]
- Liu, K., Kelly, J.W., and Wemmer, D.E. 2002. Native state hydrogen exchange study of suppressor and pathogenic variants of transthyretin. J. Mol. Biol. 320 821–832. [DOI] [PubMed] [Google Scholar]
- Llinas, M., Gillespie, B., Dahlquist, F.W., and Marqusee, S. 1999. The energetics of T4 lysozyme reveal a hierarchy of conformations. Nat. Struct. Biol. 6 1072–1078. [DOI] [PubMed] [Google Scholar]
- Loh, S.N., Prehoda, K.E., Wang, J., and Markley, J.L. 1993. Hydrogen exchange in unligated and ligated staphylococcal nucleate. Biochemistry 32 11022–11028. [DOI] [PubMed] [Google Scholar]
- Matouschek, A., Kellis Jr., J.T., Serrano, L., Bycroft, M., and Fersht, A.R. 1990. Transient folding intermediates characterized by protein engineering. Nature 346 440–445. [DOI] [PubMed] [Google Scholar]
- Mayo, S.L. and Baldwin, R.L. 1993. Guanidinium chloride induction of partial unfolding in amide proton exchange in ribonuclease A. Science 262 873–876. [DOI] [PubMed] [Google Scholar]
- Mok, Y.-K., Kay, C.M., Kay, L.E., and Forman-Kay, J.D. 1999. NOE data demonstrating a compact unfolded state for an SH3 domain under non-denaturing conditions. J. Mol. Biol. 289 619–638. [DOI] [PubMed] [Google Scholar]
- Nall, B. 1985. Proline isomerization and protein folding. Mol. Cell. Biophys. 3 123–143. [Google Scholar]
- Neira, J.L., Sevilla, P., Menendez, M., Bruix, M., and Rico, M. 1999. Hydrogen exchange in ribonuclease A and ribonuclease S: Evidence for residual structure in the unfolded state under native conditions. J. Mol. Biol. 285 627–643. [DOI] [PubMed] [Google Scholar]
- Neri, D., Billeter, M., Wider, G., and Wüthrich, K. 1992. NMR determination of residual structure in a urea-denatured protein, the 434-repressor. Science 257 1559–1563. [DOI] [PubMed] [Google Scholar]
- Nolting, B., Golbik, R., Neira, J.L., Soler-Gonzalez, A.S., Schreiber, G., and Fersht, A.R. 1997. The folding pathway of a protein at high resolution from microseconds to seconds. Proc. Natl. Acad. Sci. 94 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveberg, M. and Fersht, A.R. 1996. Thermodynamics of transient conformations in the folding pathway of barnase: Reorganization of the folding intermediate at low pH. Biochemistry 35 2738–2749. [DOI] [PubMed] [Google Scholar]
- Rodriguez, H.M., Robertson, A.D., and Gregoret, L.M. 2002. Native state EX2 and EX1 hydrogen exchange of Escherichia coli CspA, a small β-sheet protein. Biochemistry 41 2140–2148. [DOI] [PubMed] [Google Scholar]
- Rumbley, J., Hoang, L., Mayne, L., and Englander, S.W. 2001. An amino acid code for protein folding. Proc. Natl. Acad. Sci. 98 105–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saab-Rincon, G., Gualfetti, P.J., and Matthews, C.R. 1996. Mutagenic and thermodynamic analyses of residual structure in the α subunit of tryptophan synthase. Biochemistry 35 1988–1994. [DOI] [PubMed] [Google Scholar]
- Santoro, M.M. and Bolen, D.W. 1992. A test of the linear extrapolation of unfolding free energy changes over an extended denaturant concentration range. Biochemistry 31 4901–4907. [DOI] [PubMed] [Google Scholar]
- Sayle, R.A. and Milner-White, E.J. 1995. RASMOL: Biomolecular graphics for all. Trends Biochem. Sci. 20 374. [DOI] [PubMed] [Google Scholar]
- Scaley, M.L., Yi, Q., Gu, H., McCormack, A., Yates 3rd, J.R., and Baker, D. 1997. Kinetics of folding of the IgG binding domain of peptostreptococcal protein L. Biochemistry 36 3373–3382. [DOI] [PubMed] [Google Scholar]
- Schmid, F.X. 1982. Proline isomerization in unfolded ribonuclease A. The equilibrium between fast-folding and slow-folding species is independent of temperature. Eur. J. Biochem. 128 77–80. [PubMed] [Google Scholar]
- Schwalbe, H., Fiebig, K.M., Buck, M., Jones, J.A., Grimshaw, S.B., Spencer, A., Glaser, S.J., Smith, L.J., and Dobson, C.M. 1997. Structural and dynamical properties of a denatured protein. Heteronuclear 3D NMR experiments and theoretical simulations of lysozyme in 8 M urea. Biochemistry 36 8977–8991. [DOI] [PubMed] [Google Scholar]
- Smith, L.J., Fiebig, K., Schwalbe, H., and Dobson, C. M. 1996. The concept of a random coil. Residual structure in peptides and denatured proteins. Fold. Des. 1 95–106. [DOI] [PubMed] [Google Scholar]
- Srimathi, T.S., Kumar, T.K., Chi, Y.H., and Yu, C. 2002. Characterization of the structure and dynamics of a near-native equilibrium intermediate in the unfolding pathway of an all β-barrel protein. J. Biol. Chem. 277 47607–47516. [DOI] [PubMed] [Google Scholar]
- Stryer, L., Holmgren, A., and Reichard, P. 1967. Thioredoxin. A localized conformational change accompanying reduction of the protein to the sulfhydryl form. Biochemistry 41016–1020. [DOI] [PubMed] [Google Scholar]
- Takei, J., Chu, R.A., and Bai, Y. 2000. Absence of stable intermediates on the folding pathway of barnase. Proc. Natl. Acad. Sci. 97 10796–10801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasayco, M.L. and Chao, K. 1995. NMR study of the reconstitution of the β-sheet of thioredoxin by fragment complementation. Proteins 22 41–44. [DOI] [PubMed] [Google Scholar]
- Tasayco, M.L., Fuchs, J., Yang, X.M., Dyalram, D., and Georgescu, R.E. 2000. Interaction between two discontiguous chain segments from the β-sheet of Escherichia coli thioredoxin suggests an initiation site for folding. Biochemistry 3910613–10618. [DOI] [PubMed] [Google Scholar]
- Wang, Y. and Shortle, D. 1996. A dynamic bundle of four adjacent hydrophobic segments in the denatured state of staphylococcal nuclease. Protein Sci. 5 1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong, K.B., Freund, S.M., and Fersht, A.R. 1996. Cold denaturation of barstar: 1H, 15N and 13CNMR assignment and characterisation of residual structure. J. Mol. Biol. 259 805–818. [DOI] [PubMed] [Google Scholar]
- Woodward, C.K. and Hilton, B.D. 1980. Hydrogen isotope exchange kinetics of single protons in bovine pancreatic trypsin inhibitor. Biophys. J. 32 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward, C.K. and Rosenberg, A. 1971. Studies of hydrogen exchange in proteins. VI. Urea effects on ribonuclease exchange kinetics leading to a general model for hydrogen exchange from folded proteins. J. Biol. Chem. 246 4114–4121. [PubMed] [Google Scholar]
- Wrabl, J. and Shortle, D. 1999.A model of the changes in denatured state structure underlying m value effects in staphylococcal nuclease. Nat. Struct. Biol. 6 876–883. [DOI] [PubMed] [Google Scholar]
- Yao, J., Chung, J., Eliezer, D., Wright, P.E., and Dyson, H.J. 2001. NMR structural and dynamic characterization of the acid-unfolded state of apomyoglobin provides insights into the early events in protein folding. Biochemistry 40 3561–3571. [DOI] [PubMed] [Google Scholar]
- Yi, Q., Scaley, M.L., Simons, K.T., Gladwin, S.T., and Baker, D. 1997. Characterization of the free energy spectrum of peptostreptococcal protein L. Fold. Des. 2 271–280. [DOI] [PubMed] [Google Scholar]