

Abstract
A rare and unusual class of tripyrrolic compounds, violet-colored tripyrrin-1,14-diones, can be prepared easily and in moderately high yields from base (piperidine)-catalyzed condensation of 3-pyrrolin-2-ones with 2,5-diformylpyrroles. Dipyrrinones adopt the all syn-Z conformation leading to helical, lock-washer like structures, which form dimers that are held together by intermolecular hydrogen bonds in nonpolar solvents and in the crystal. Strong bathochromic spectral shifts of the tripyrrindione ∼480 nm long wavelength UV-visible absorption band are seen with added base: DBU, 615 nm; TFA, 573 nm; and Zn (OAc)2, 586 nm.
Keywords: Tripyrroles, hydrogen bonds, X-ray crystallography
1. Introduction
Tripyrrolic tripyrrindiones (Fig. 1A) were first reported in the synthetic work of Hans Fischer and Erich Adler in 19311 directed toward the syntheses of the tetrapyrrole, mesobilirubin-XIIIα, and its synthetic diyrrinone precursor, neoxanthobilirubinic acid. In the course of that study, and somewhat incidental to it, a brief study of tripyrranes was illustrated by report of two reactions: 4-carboethoxy-5-chloromethyl-3-methyl-3-pyrrolin-2-one with opsopyrrole and with opsopyrrole carboxylic acid to give unreported yields of brown-violet high melting tripyrrins (Fig. 1B). Although written as hydroxypyrrole tautomers of undesignated stereochemistry throughout, in Fischer’s convention, these two tripyrroles apparently constituted the first documented examples of tripyrrindiones. Nearly ten years later, Fischer and Reinecke,2 in connection with synthetic and analytical studies of glaucobilin, prepared the methyl ester of the carboxylic acid of Fig. 1B and concluded that the violet pigment of the Gmelin reaction of verdins was not due to a dihydroxytripyrrin (tripyrrindione). No further work on tripyrrindiones came from the Fischer lab, and they escaped further investigation until 1966,3a when a Fischer student, Henning von Dobeneck, prepared the hexamethyltripyrrindione of Fig. 1C3 in two steps by condensation of 5-bromo-3,4-dimethyl-2-formyl-1H-pyrrole with 2,3,7,8-tetramethyldipyrrinone to give a tripyrrole, followed by treatment with potassium acetate in acetic acid. The E-stereochemistry was designated.
Figure 1.

(A) Tripyrrindione carbon framework and numbering system. (B) Fischer and Adler’s brown-violet-colored synthetic tripyrrindiones (ref. 1) in the Fischer representations as hydroxypyrroles with no C=C stereochemistry denoted. (C) Hexamethyl tripyrrindione of von Dobeneck (ref. 2) as formulated in 1966 (left) and revised in 1971 (right) with the E stereochemistry as drawn by von Dobeneck. (D) The reddish urinary pigment, uroerythrin as represented in ref. 3. (E) Nakajima’s tripyrrolic pigments from urine, redrawn as represented (ref. 4). (F) The synthetic targets of the current work.
The first naturally-occurring tripyrrindione, as it was subsequently characterized,4 was isolated repeatedly, over many years, from urine and urate sediments, and named uroerythrin.5 The pigment was characterized as its dimethyl ester-lactam monomethyl ether, and its structure was proposed as shown in Fig. 1D.4 Apparently only the endo-vinyl isomer was isolated, and the E-stereochemistry was indicated throughout. Whether the predictably less stable exo-vinyl isomer was present is unclear. Subsequently, Nakajima et al.6 isolated two urinary pigments to which they assigned tripyrrindione structures (Fig. 1E), one of which is identical to that of Fig. 1D, the other is the “missing” exo-vinyl isomer, and for both the stereochemistry was not indicated. Apparently no further tripyrrindiones have been synthesized or isolated since 1994, leaving a total of 5-6 representatives in this class of compounds.
We were attracted to further investigate simple tripyrrindiones synthesized by a short synthetically logical route. We focused on the synthesis of hexaethyl of 1 (1, Figure 1F) by a more direct method than that employed in the synthesis of hexamethyl analog of Fig. 1C. We also synthesized tetramethyl analog 2 (Fig. 1E) for comparison and showed that 1 would prove to have better solubility in organic solvents, a lower melting point than the mp 315°C of the hexamethyl analog (Fig. 1C), and the potential for growing a crystal suitable for X-ray crystallographic analysis. The conformation of 1 in solution was examined by NMR methods and molecular mechanics calculations, and its molecularity was determined from vapor pressure osmometry (VPO) measurements. Throughout are described improved procedures for the monopyrrole starting materials.
2. Results and Discussion
2.1 Synthesis
Our approach to the syntheses of 1 and 2 was direct: 3,4-diethyl-2,5-diformyl-1H-pyrrole (3)7 would be condensed with either 3,4-diethyl-3-pyrrolin-2-one (4)8 or its dimethyl analog (5)9, as outlined in Scheme 1. Pyrrole dialdehyde 37,10 was prepared from the known ethyl 3,4-diethyl-1H-pyrrole-2-carboxylate (6)11 in 71% yield by reaction of carboxylic acid 7 with triethyl orthoformate in the presence of trifluoroacetic acid.7 Accordingly, we report herein improved procedures for the syntheses of 3-7: preparing 6 and 7 and converting 7 to 3, 88,12 to 4, and 3,4-dimethyl-1H-pyrrole to 5.9 Pyrrole dialdehyde 3 was condensed13 with excess pyrrolinone, catalyzed by piperidine in ethanol solvent (sealed tube) at 100°C. The reaction proceeds through the monocondensation product, 8 or 9, which reacts further to afford either 1 or 2. The progress of the reaction is monitored by following the disappearance of 8 or 9, which typically takes about 30 hours. Alternatively, and more conveniently on a larger scale, the use of the sealed tube can be dispensed with by carrying out the reaction in refluxing p-dioxane: with 3 + 4 a 57% yield of 1 + 10% of 10 was obtained after 6 days. When a 2:1 ratio of pyrrolinone 4:dialdehyde 3 was used in the sealed tube reaction, with a short reaction time, an 87% yield of dipyrrinone aldehyde 1014 was obtained.
Scheme 1.

Because of its better solubility and the ease in which 1 gave crystals suitable for X-ray studies, most of what follows in sections 2.3-2.6 was conducted with 1.
2.2 Structures and NMR Spectroscopy
The constitutional structures of 1 and 2 follow from the method of synthesis13 and the structures of the starting materials.7-12 The symmetry of 1 and 2 are recognizable by the (reduced) number of carbon signals (Table 1), and good correlation between the observed chemical shifts and those of the dipyrrinone analogs.8,15 Similar good correlations may be found in the 1H-NMR spectra of Table 1, which also reveals shifted pyrrole and lactam signals in CDCl3 and (CD3)2SO solvents. The deshielded signals relative to those found in monomeric dipyrrinones (lactam NH: 7.8-7.0 ppm; pyrrole NH: 8.1-7.8 ppm)16 point to hydrogen bonding interactions, with the solvent in (CD3)2SO and from intermolecular hydrogen bonding in CDCl3. The data from (CD3)2SO differ somewhat from those of typical diyrrinones in that the lactam NH chemical shift of 1 is ∼0.3-0.4 ppm more deshielded, while the pyrrole NH chemical shift is quite comparable to values found in typical dipyrrinones.15 The NH chemical shifts of 1 in (CD3)2SO are consistent with hydrogen bonding to solvent.16,17 The chemical shifts of 1 and 2 in CDCl3 are more like those found in bilirubin and semirubins (with the pyrrole and lactam NH signals at ∼10.7 and 9-9.8 ppm, respectively)12b,14c,17 than they are like dipyrrinone dimers (pyrrole and lactam NHs at ∼10.5 and ∼11 ppm, respectively) or rubin ester dimers (pyrrole and lactam NHs at ∼10.3 and ∼10.6 ppm respectively).17,18 The data for 1 in CDCl3 thus clearly point to intermolecular hydrogen bonding, but one where the lactam NH···O=C hydrogen bond is slightly weaker than in an intermolecularly hydrogen bonded dipyrrinone dimer, and/or a dimer in which the lactam NH is located in the shielding core of an aromatic ring or π-bond.
Table 1.
1H- and 13C-NMR chemical shiftsa of tripyrrindiones 1 and 2.
|
13C-NMR |
1H-NMR |
||||||
|---|---|---|---|---|---|---|---|
| Carbon | 1 |
2 |
1 |
2 |
|||
| CDCl3 | (CD3)2SO | CDCl3 | CDCl3 | (CD3)2SO | CDCl3 | ||
| 1/14 | C=O | 173.8 | 172.6 | 174.4 | ? | ? | ? |
| 2/13 | =C- | 132.6 | 132.2 | 132.6 | ? | ? | ? |
| 21 | CH2CH3 | 17.7 | 17.7 | 8.3 | 2.18 (q)b | 2.27 (q)b | 1.72 (s) |
| 22 | CH3 | 13.8 | 14.6 | ? | 0.95 (t)b | 1.12 (q)b | ? |
| 3/12 | =C- | 146.8 | 146.9 | 141.4 | ? | ? | ? |
| 31 | CH2/CH3 | 17.7 | 17.6 | 9.9 | 2.46 (q)b | 2.48 (q)b | 2.07 (s) |
| 32 | CH3 | 15.6 | 16.4 | ? | 1.17 (t)b | 1.02 (t)b | ? |
| 4/11 | =C- | 131.1 | 131.0 | 130.3 | ? | ? | ? |
| 5/10 | =CH- | 98.8 | 97.6 | 98.8 | 6.04 (s) | 5.93 (s) | 6.03 (s) |
| 6/9 | =C- | 130.1 | 129.7 | 126.1 | ? | ? | ? |
| 7/8 | =C- | 128.7 | 128.2 | 128.4 | ? | ? | ? |
| 71 | CH2 | 16.8 | 17.2 | 17.6 | 2.56 (q)b | 2.50 (q)b | 2.53 (q)b |
| 72 | CH3 | 16.5 | 17.1 | 16.5 | 1.19 (t)b | 1.04 (t)b | 1.17 (t)b |
| 15/17 | NH | ? | ? | ? | 9.73 (brs) | 10.10 (brs) | 9.5 (brs) |
| 16 | NH | ? | ? | ? | 10.46 (brs) | 10.73 (brs) | 10.6 (brs) |
Chemical shifts in δ, ppm for 10-3 M solutions at 22°C.
J = 7.5 Hz.
Nuclear Overhauser Effect (NOE) studies of 1 in CDCl3 and (CD3)2SO revealed a close proximity of the C(5) hydrogen to the flanking lactam ethyl (CH2 and CH3) hydrogens at C(31) and C(32) [by symmetry: the C(10) hydrogen to the C(121) and C(122) ethyl hydrogens, thereby indicating the Z-configuration at the C(4) and C(10) exocyclic carbon-carbon double bonds]. Strong NOEs were also found between the lactam and pyrrole NHs, and between the C(5)/C(10) hydrogens and the flanking pyrrole ethyl groups, confirming a syn-Z conformation, as shown in Fig. 2.
Figure 2.
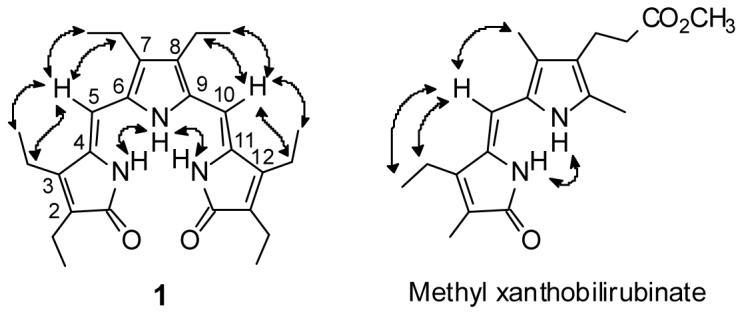
Nuclear Overhauser Effects (NOEs), indicated by curved arrows, seen in 1 in both CDCl3 and (CD3)2SO solvents. Similar NOEs are found in typical dipyrrinones, such as methyl xanthobilirubinate.
2.3 Molecularity in Solution
The suspected dimerization of 1, as revealed by 1H-NMR NH chemical shifts was further explored by vapor pressure osmometric measurement of 1 in CHCl3 solution. Calibrated vs a measured molecular weight of 214 ±11 g/mol of benzil (FW = 210.2 g/mol) in CHCl3 a MW for 1 (FW = 421.6 g/mol) was determined to be 831 ±30 g/mol in ∼10-3 M solution ? or very nearly twice the molecular weight. These data, together with the 1H-NMR data, are consistent with complete dimer formation held together by intermolecular hydrogen bonding.
In order to explore aspects of the dimerization of 1, we examined the NH chemical shifts at increasing dilution. Of course, no variation is found in (CD3)2SO over the concentration range 1.0 × 10-3 to 4.5 × 10-5. However, in CDCl3, over the range 8.2 × 10-4 to 4.1 × 10-5, only a small (∼0.1 ppm) upfield shift is observed (Table 2). The latter contrasts markedly with data for the dipyrrinone methyl xanthobilirubinate (Fig. 2) which also is dimeric in CDCl3 but shows an order of magnitude larger (∼1 ppm) upfield shift.16 Given that the association constant KA for dimerization of methyl xanthobilirubinate at room temperature was determined to be 25-30,000 M-1, it is clear that tripyrrindiones must have a much larger KA.
Table 2.
Concentration dependence of pyrrole and lactam NH chemical shifts of 1 in (CD3)2SO and in CDCl3 at 22°C compared with similar data for methyl xanthobilirubinate (XBR-Me).
| Pyrrole (P) and Lactam (L) Chemical Shifts (δ, ppm) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Conc. of 1 (mM) | (CD3)2SO |
Conc of 1 (mM) | CDCl3 |
Conc. of XBR-Me (mM) | CDCl3 |
|||
| PNH | LMH | PNH | LNH | PNH | LNH | |||
| 0.8907 | 10.084 | 10.215 | 0.8172 | 9.712 | 10.302 | 0.9766a | 10.136 | 10.933a |
| 0.4453 | 10.084 | 10.215 | 0.4086 | 9.686 | 10.283 | 0.4883 | 10.048 | 10.765 |
| 0.2227 | 10.084 | 10.215 | 0.2643 | 9.662 | 10.257 | 0.1808 | 9.700 | 10.371 |
| 0.0445 | 10.084 | 10.215 | 0.0409 | 9.584 | 10.186 | 0.09039 | 9.535 | 10.200 |
The data plateau at higher concentrations, 0.125 M; PNH: 10.401, LNH: 11.334.
In further support of dimeric association through hydrogen bonding in nonpolar solvents, we observed small deviations from Beer’s Law19 in CHCl3 and n-hexane but no deviations in (CH3)2SO.
2.4 X-ray Crystal Structure
Further support for the constitutional structure of 1 comes from X-ray crystallography, which confirms the syn-Z conformation as well as the hydrogen-bonded dimer (Fig. 3). In the dimer, each tripyrrindione adopts a helical conformation by twisitng about the C(5)-C(6) and C(9)-C(10) single bonds by ∼20°, and the pitch of the helix is ∼ 2.35 Å. In order to fit together in the dimer, the two tripyrrindiones adopt the same helicity: M binds to M, P to P; thus, each dimer is chiral. The unit cell includes four dimer pairs (8 tripyrrindiones; 4 M and 4 P). The bond angles and bond distances (Fig. 4) are very similar to those found in the X-ray crystal structure of 7,9-dimethyl-2,3,8-trimethyl-10H-dipyrrinone,20 which is found as a hydrogen bonded dimer in the crystal, with planar monomers.
Figure 3.
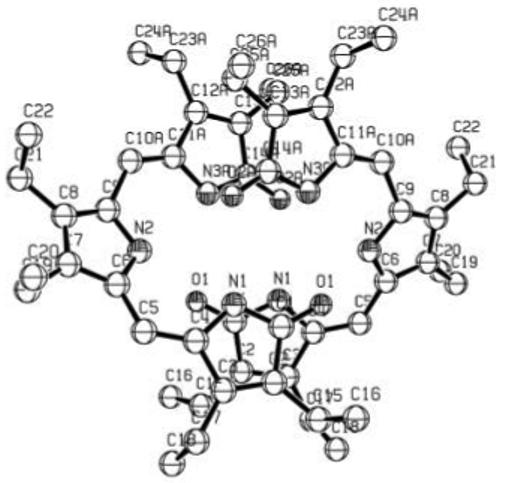
Structural drawing of intermolecularly hydrogen-bonded dimeric 1 in the crystal showing the atom numbering system; hydrogens are deleted for clarity of representation.
Figure 4.

Bond distances (Å) and angles (°) found in the X-ray crystallographic structure of tripyrrindione 1 hydrogen-bonded dimer, with numbering system used. Hydrogen bonding distances are indicated by dashed lines, and the following related main atom distances were found: O1-pyrrole N2 (3.19 Å), O1-lactam N3 (2.81 Å), 02-lactam N1 (2.84 Å). In a typical dipyrrinone dimer X-ray crystal structure, 7,9-dimethyl-2,3,8-triethyl-10H-dipyrrinone (ref. 20), exhibits lactam O to pyrrole N and lactam O to lactam N distances: 2.89 Å and 2.86 Å, corresponding to pyrrole NH···O=C lactam distances of 2.09Å and a lactam NH···O=C lactam distance of 2.09 Å and a lactam NH···O=C lactam distance of 1.97 Å. The bond lengths and bond angles are nearly identical.
2.5 Conformation from Molecular Dynamics Calculations
In support of the helical tripyrrindione structure found in the crystal, and consistent with the NMR spectroscopic analysis of pigment conformation, molecular dynamics calculations21 of 1 show a helix with comparable torsion angles about the C5-C6 and C9-C10 single bonds: C4-C5-C6-N2 ≅ N2-C9-C10-C11 ∼26°. The same sign of the angles indicates twisting in opposite directions so as to produce the helical structure. There is very little twisting about the exocyclic C=C bonds: N1-C4=C5-C6 ≅ C9-C10=C11-N3 ∼2°. However, an isoenergetic symmetric structure with C4-C5-C6-N2 and N2-C9-C10-C11 torsion angles ∼27° and only ∼-27° deviation from planarity in the N1-C4=C5-C6 and C9-C10=C11-N3 is also found. The calculations indicate conformational mobility in the lactam rings flipping up and/or down relative to the pyrrole ring. The helical structure is required for formation of the hydrogen-bonded dimer.
2.6 Optical spectroscopy
The UV-visible spectral data for 1 in solvents with a wide range of polarity are given in Table 3. Characteristic of orange-red colored solutions, an intense and broad long wavelength absorption band is found near 500 nm, with an equally intense but narrower band near 325 nm. The long wavelength band is characteristic of two overlapping electronic transitions, which becomes more apparent when one views the actual spectra (Fig. 5). The shape of the long wavelength band is reminiscent of exciton splitting seen in bilirubinoids and dipyrrinone dimers. And while one might view a dimeric tripyrrindione to be a molecular exciton, this is more likely to be seen in nonpolar solvents than in a solvent like (CH3)2SO, in which dimerization is not evident but which is where the band separation is most recognizable ? suggesting a non-excitonic origin. The data suggest overlapping transitions of an as yet unclear origin.
Table 3.
Solvent dependence of the UV-visible spectral data for tripyrrindione 1.a
| λmax / nm (ε) /L?mol-1?cm-1 | |||||
|---|---|---|---|---|---|
| Hexane | Benzene | CHCl3 | CH3CN | CH3OH | (CH3)2SO |
| 498 (32,800) | 496 (23,600) | 488 (25,500) | 471 (27,300) | 479 (31,000) | 484 (31,700) |
| 331 (30,300) | 326 (32,800) | 325 (31,100) | 319 (32,300) | 321 (29,200) | 323 (30,900) |
Solutions are ∼1 × 10-5 M in 1 and contain 2% CHCl3.
Figure 5.

Solvent dependence of the UV-visible spectra of 1 at ∼1 × 10-5 M at 23°C. The curves are numbered corresponding to the solvents used: 1) methanol; 2) chloroform; 3) acetonitrile; 4) trifluoroacetic acid; 5) hexane; 6) dimethylsulfoxide; 7) benzene.
We observed pronounced UV-visible spectral changes in 1, especially with the ∼490 nm long wavelength band. Upon addition of TFA, the solution becomes more red as the long wavelength band bathochromically shifted and becomes less broad so that in pure TFA, λmax appears near 530 nm and ε rises to ∼53,000 (Fig. 6). The shorter wavelength band near 325 nm is barely changed. An even more striking effect is found upon addition of base. The organic base DBU was particularly effective (Fig. 6) in shifting the long wavelength band, which is well-separated into two bands in neat DBU. Solutions become blue and a pink fluorescence is observed. The data are summarized in Table 6 and might be interpreted in terms of a protonated lactam in TFA and a lactim enolate ion in DBU.
Figure 6.

Influence of acid (curve 3: trifluoroacetic acid, neat) and base (curve 2: DBU, 1,8-diazabicylclo[5.4.0]undec-7-ene, neat) on the UV-visible spectra of 1, compared with curve 1: CHCl3 for ∼ 1 × 10-5 M solutions at 23°C.
Fischer and coworkers reported a color shift in their tripyrrindiones (Fig. 1B) upon addition of Zn(OAc)2 to alcohol solutions of the pigment.1,2 Thus, upon addition of alcoholic Zn(OAc)2 to an alcoholic solution of their tripyrrindiones the color shifted from violet to blue, but showed no fluorescence. Upon addition of ammonium hydroxide it became yellow and intensely fluorescent. Our pigment 1 behaved in a similar way (Fig. 7). The bathochromic shift, blue coloration and absence of fluorescence upon addition of Zn(OAc)2 are qualitatively similar to those found from 1 in DBU. The shift to yellow and strong fluorescence upon addition of NH4OH to the solution is again similar. However, attempts to isolate the Zn salt or its product following addition of NH4OH have been unsuccessful. We assume that the zinc complexes with a lactim tautomer of 1, as it does with dipyrrylmethenes.
Figure 7.

Influence of added zinc acetate (curve 2) followed by added conc. NH4OH (curve 3) on the UV-visible spectrum of 1 in CH3OH (curve 1) at ∼1 × 10-5 M concentration and 23°C.
3. Concluding Comments
Tripyrrindione 1, synthesized smoothly from monopyrrole components, adopts an all-syn-Z lock-washer-like conformation in solution and in the crystal. In chloroform solution, 1 is dimeric, with strong evidence for intermolecular hydrogen bonding. In the crystal, 1 forms intermolecularly hydrogen-bonded pairs of M to M and P to P helical dimers.
4. Experimental Section
4.1. General Procedures
Nuclear magnetic resonance (NMR) spectra were obtained on a Varian Unity Plus 500 MHz spectrometer in CDCl3 solvent (unless otherwise specified) at 25°C. Chemical shifts were reported in δ ppm referenced to the residual CHCl3 1H signal at 7.26 ppm and 13C signal at 77.0 ppm. A combination of heteronuclear multiple bond correlation (HMBC) spectra and 1H{1H} NOE data were used to assign 1H and 13C NMR spectra. UV-visible spectra were recorded on a Perkin-Elmer Lambda-12 spectrophotometer. Melting points were taken on a Mel Temp capillary apparatus and are corrected. Combustion analyses were carried out by Desert Analytics, Tucson, Az. Analytical thin layer chromatography was carried out on J. T. Baker silica gel IB-F plantes (125 μ layers). Radial chromatography was carried out on Merck silica gel PF254 with gypsum preparative layer grade, using a Chromatotron (Harrison Research, Palo Alto, CA). Spectral data were obtained in spectral grade solvents (Aldrich or Fisher). Deuterated chloroform and dimethylsulfoxide were from Cambridge Isotope Laboratories. 3,4-Diethyl-1H-pyrrole (6)11 and 3,4-dimethyl-1H-pyrrole (7)22,23 were prepared as reported previously from ethyl 3,4-diethyl-1H-pyrrole-2-carboxylate11 and ethyl 3,4-dimethyl-1H-pyrrole-2-carboxylate,22,23 respectively.
4.1.1. Ethyl 3,4-Diethyl-1H-pyrrole-2-carboxylate (6)
The following is an improved procedure, which avoids the use of DBU base.11,12b 3-Acetoxy-4-nitrohexane11,12b (2.4 g) and ethyl isocyanoacetate11,12b,24 (1.4 g) were placed in a 100-mL round bottom flask together with a 1:1 mixture of THF and ethanol (30 mL). Anhydrous potassium carbonate (3.5 g) was added in portions while the mixture was stirred vigorously. The reaction mixture was then stirred at room temperature for 3 days. After ascertaining that the reaction was complete (TLC), the mixture was poured onto water (100 mL), acidified to pH 5 with 5% HCl, and extracted with diethyl ether (4 × 25 mL). The solvent was removed at reduced pressure (rotovap) after drying over Na2SO4. The resulting oil was passed through a short column of silica gel using 1% CH3OH in CH2Cl2 as eluent. The eluate was collected, and the solvent was evaporated to yield a brown oil (one spot on TLC, eluent CH2Cl2): 2 g, 81% [lit.11 oil]. It had 1H-NMR (CDCl3) δ: 1.16 (t, J = 6.9 Hz, 3H), 1.2 (t, J = 7.7 Hz, 3H), 1.36 (t, J = 7.7 Hz, 3H), 2.5 (q, J = 7.7 Hz, 2H), 2.8 (q, J = 7.7 Hz, 2H), 4.3 (q, J = 6.9 Hz, 2H), 6.7 (d, J = 2.9 Hz, 1H), 9.1 (bs, 1H) ppm; 13C-NMR (CDCl3) δ: 14.3 (q), 14.8 (q), 15.3 (q), 17.9 (t), 18 (t), 59.6 (t), 118.5 (s), 119 (d), 126 (s), 132 p (s) ppm.
4.1.2. 3,4-Diethyl-3-pyrrolin-2-one (4)
The procedure described below provides 4 in twice the yield of converting 8 → 4 as previously reported.8 Pyrrole 6 (2.0 g, 10.3 mmol) was dissolved in ethanol (20 mL), then a solution of NaOH (1.2 g) in water (8 mL) was added, and the mixture was heated at reflux overnight. The volume was reduced under vacuum (rotovap), and the resulting solid was dissolved in a minimum amount of water and acidified with 5% HCl to pH 5 at 0°C. The precipitated carboxylic acid (7) was quickly filtered, washed with water, and air-dried overnight to yield 1.5 g (88%) of almost white, slightly pink solid with 1H-NMR (CDCl3) δ: 1.03 (t, J = 7.7 Hz, 3H), 1.1 (t, J = 7.7 Hz, 3H), 2.34 (q, J = 7.7 Hz, 2H), 2.63 (q, J = 7.7 Hz, 2H), 6.64 (d, J = 2.9 Hz, 1H), 11.06 (s, 1H), 11.96 (s, 1H) ppm; 13C-NMR (CDCl3) δ: 15.37 (q), 16.1 (q), 17.8 (t), 118.7 (s), 119.6 (d), 125.1 (s), 131 (s), 162.7 (s) ppm. The acid was taken directly to the next step.
Pyrrole acid 7 (4.0 g, 24 mmol) was added to water (100 mL) and then steam distilled. The distillate was extracted with ether (5 × 20 mL) and dried over Na2SO4. The ether solution was filtered and then evaporated at reduced pressure to yield 2.0 g (68%) of 812 as a yellow oil that is very sensitive to air and light; 1H-NMR (CDCl3) δ: 1.23 (t, J = 7.7 Hz, 6H), 2.5 (q, J = 7.7 Hz, 4H), 6.56 (d, J = 2.5 Hz, 2H), 8 (bs, 1H) ppm; 13C-NMR (CDCl3) δ: 14.4 (q), 18.25 (t), 114.2 (d), 124.4 (s) ppm. 3,4-Diethylpyrrole 8 was used directly in the next step.
3,4-Diethyl pyrrole 8 (0.5 g, 4.1 mmol) was heated at reflux with H2O2 (1 mL), dry pyridine (1 mL) and methanol (about 0.3 mL) for about 25 min; then more H2O2 (0.5 mL) was added, and the mixture was heated at reflux for a short period (until no more starting material was detected by TLC: eluent 1% MeOH:99% CH2Cl2). After the reaction was complete, the solvent volume was reduced under vacuum (rotovap), and the residue was dissolved in diethyl ether, washed with water and dilute (3%) HCl, followed by sat. NaCl. The ether layer was evaporated (rotovap) to yield 0.44 g (80%) of the pyrrolinone product 4 as yellow oil.8 It had 1H-NMR (CDCl3) δ: 1.1 (m, 6H), 2.3 (q, J = 7.7 Hz, 2H), 2.42 (m, 2H), 3.84 (s, 2H), 6.8 (bs, 1H) ppm. GCMS, m/z: 139, 124, 110, 96, 92, 82, 67, 55 amu.
4.1.3. 3,4-Diethyl-2,5-diformyl-1H-pyrrole (3)
The procedure reported below gives a higher yield of 3 than that reported earlier.7 Pyrrole acid 7 (0.5 g, 3.0 mmol) was dissolved in TFA (20 mL) at room temperature in the dark; then triethyl orthoformate (20 mL) was added dropwise at a rate such that the temperature remained close to room temperature. The mixture was then stirred for about 1 h (or until the reaction was complete); then the solvent volume was reduced (rotovap) and the remaining liquid was poured onto water and neutralized with sat. aq. NaHCO3. After ether extraction (5 × 10 mL), followed by evaporation of the solvent (rotovap), the solid, yellowish product (3) was found to be pure. The yield was 0.38 g (71%), mp 105-107°C [lit.7 mp 106°C]. The 1H- and 13C-NMR matched those previously reported.7
4.1.4 2,3,7,8,12,13-Hexaethyl-15H,17H-tripyrrin-1,14-dione (1)
Procedure A
3,4-Diethyl-3-pyrrolin-2-one (1.40 g, 10.0 mmol) was added to 3,4-diethyl-2,5-diformyl-1H-pyrrole (0.300 g, 1.68 mmol) dissolved in 20 mL of p-dioxane. Piperidine (30 drops) was then added slowly, and the reaction was heated at reflux under N2 for 7 days. After cooling to room temperature, the solvent was evaporated in vacuo (rotovap) and the residue was taken up in diethyl ether (100 mL). The aqueous layer contained the side product dipyrrinone (10) which was set aside for further purification. The organic layer was washed with 5% aq. HCl, saturated aq. NaHCO3, water and brine and dried over Na2SO4. The ether was evaporated, and the residue was separated using radial chromatography, 5% MeOH in CH2Cl2 as eluent to afford 339 mg (48%) of 1, mp 215-217°C; IR (film) ν: 3228, 2968, 2934, 2874, 1770, 1709, 1676 cm-1; NMR data in Table 1.
Anal. Calcd for C26H35N3O2 (421.6): C, 74.07; H, 8.37; N, 9.97.
Found: C, 73.84; H, 8.46; N. 9.85.
Procedure B
3,4-Diethyl-3-pyrrolin-2-one 4 (0.28 g, 1.93 mmol) was dissolved in 3 mL of a 1:1 mixture of absolute ethanol and CH2Cl2 in a glass pressure tube, to which piperidine (0.1 mL, 1 mmol) was added. The pressure tube reaction was immersed in a steam bath, and a solution of 3,4-diethyl-2,5-diformyl-1H-pyrrole 3 (0.088 g, 0.49 mmol) in CH2Cl2 (about 1 mL) was added dropwise into the reaction mixture. The progress of the reaction was checked periodically by TLC. When there were no starting materials (colorless) detected, and the yellow-green spot corresponding to dipyrrinone 10 was minimal in size (usually after about 24 h), the reaction was worked up as follows. The solvent was removed (rotovap), and the residue was dissolved in CH2Cl2, washed with 5% HCl, followed by 3 washings with sat. NaHCO3, then sat. NaCl. The resulting CH2Cl2 solution was dried over Na2SO4, filtered, and evaporated at reduced pressure. The residue was purified by radial chromatography using as eluent a mixture of CH3OH and CH2Cl2 with the concentration of CH3OH being increased gradually from 1% to 15%. The procedure yielded 115 mg (57%) of a dark red, almost black solid (1) that was pure by TLC and had mp 215-219°C; NMR in Table 1, and FAB-MS: calcd. for C26H35N3O2 (421.2729), found: 421.270 [M+].
4.1.5. 9-Formyl-2,3,7,8-tetraethyl-10H-dipyrrinone (10)
The following procedures differ from that reported earlier,14 which added the 9-formyl group to the 9H-dipyrrinone.
Procedure A
The aqueous layer from Procedure A for the synthesis of 1 was allowed to evaporate and the residue was taken up in CH2Cl2, washed with 5% aq. HCl, sat. aq. NaHCO3, water and brine and dried over Na2SO4. Dipyrrinone 10 was isolated by radial chromatography (5% CH3OH in CH2Cl2 eluent) to yield 51 mg (10%), with mp 162-163°C. It had IR (film) ν: 3434, 2966, 2945, 1769, 1708, 1673, 1602 cm-1; 1H-NMR (CDCl3) δ: 1.13-1.28 (m, 12H), 2.45 (q, J = 7.6 Hz, 2H), 2.56 (m, 4H), 2.8 (q, J = 7.6 Hz, 2H), 5.97 (s, 1H), 9.7 (s, 1H), 10.4 (s, 1H), 10.75 (s, 1H) ppm; 13C-NMR (CDCl3) δ: 13.8 (q), 15.45 (q), 16.29 (q), 16.9 (t), 17.1 (t), 17.15 (t), 17.36 (q), 17.8 (t), 94.8 (d), 128.8 (s), 130 (s), 130.2 (s), 131.7 (s), 134.2 (s), 135 (s), 146.8 (s), 171.9 (s), 177.4 (d) ppm.
Procedure B
3,4-Diethyl-2,5-diformyl pyrrole 3 (0.086 g, 0.48 mmol) and 3,4-diethyl-3-pyrrolin-2-one 4 (0.135 g, 0.96 mmol) were dissolved in 3 mL of absolute ethanol in a resealable glass pressure tube (Ace Glass); then piperdine (0.1 mL, 1 mmol) was added. The tube was then sealed and put into a steam bath. The reaction progress was monitored from time to time by TLC, 3% CH3OH:97% CH2Cl2 eluent. When there was no trace of the starting material, and the only observed spot was a yellow-green one for 10 (usually within 1 h), the reaction was worked up as follows. Ethanol was removed by vacuum (rotovap), and the residue was dissolved in CH2Cl2, washed with 5% HCl, followed by 3 washings with sat. NaHCO3, then sat. NaCl. The resulting CH2Cl2 solution was dried over Na2SO4, filtered, and evaporated at reduced pressure (rotovap). The residue was passed through a short plug of silica gel, using CH2Cl2 as eluent, to afford 125 mg (85%) of a crystalline yellow-green solid, which was pure by TLC and had mp 161-169°C. FAB-MS: calcd. for C18H24N2O2 (300), found: 300 [M+].
4.1.6 Ethyl 3,4-Dimethyl-1H-pyrrole-2-carboxylate
The following is a different, higher yield synthesis than that reported earlier.22,23 2-Acetoxy-3-nitrobutane (19 g, 0.12 mol) and ethyl isocyanoacetate (13.3 g, 0.12 mol) were placed in a 1-L round bottom flask together with a 1:1 mixture of THF and ethanol (300 mL). Potassium carbonate (35 g) was added in portions while the mixture was stirred vigorously. The reaction was stirred at room temperature for 2 days, while the reaction progress was checked by TLC (eluent: 1% CH3OH:99% CH2Cl2). At completion the reaction mixture was poured into water, acidified to pH 5 with 5% HCl, and extracted with diethyl ether (4 × 75 mL). The solvent was removed under reduced pressure after drying over Na2SO4, and the solid residue was passed through a short column of silica gel (1% CH3OH eluent). The eluate was collected, and the solvent was evaporated (rotovap) to give the desired product, which was recrystallized from CH3OH-H2O to yield a white crystalline solid, 17.3 g (88%) that showed one spot on TLC (CH2Cl2 eluent). It had mp 91-92°C (lit.23 mp 90-91 °C). The 1H- and 13C-NMR data matched those previously reported.23
4.1.7 3,4-Dimethyl-1H-pyrrole (9)
Ethyl 3,4-dimethyl-1H-pyrrole-2-carboxylate from above (4.7 g, 28 mmol) was dissolved in ethanol (40 mL), then a solution of NaOH (3.5 g) in water (15 mL) was added, and the mixture was heated at reflux overnight. After 16 h the reaction was complete, the volume was reduced (rotovap), and the resulting solid was dissolved in a minimum amount of water and acidified at 0°C with 5% HCL to pH 5. The precipitated product was filtered quickly, washed with water and air dried overnight to yield 3.9 g (100%) of a white solid. It had 1H-NMR (CDCl3) δ: 1.9 (s, 3H), 2.15 (s, 3H), 6.65 (d, 1H), 11.0 (s, 1H), 12.0 (s, 1H) ppm; 13C-NMR (CDCl3) δ: 9.6 (q), 118.4 (s), 118.7 (s), 120.4 (d), 124.7 (s), 162.2 (s) ppm and was used directly in the next step.
The pyrrole acid (3.9 g, 28 mmol) was added to water, heated to boiling and ∼500 mL of water was co-distilled with decarboxylated pyrrole. The distillate was extracted with ether (5 × 75 mL) and dried over Na2SO4. The ether solution was filtered and evaporated (rotovap) to yield 1.7 g (65%) of a white solid that is very sensitive to air and light. It had mp 30-31°C (lit.23 mp 30-31°C)..
4.1.8. 3,4-Dimethyl-3-pyrrolin-2-one (5)
3,4-Dimethyl-1H-pyrrole (1.7 g, 18 mmol) was heated at reflux with H2O2 (4 mL), dry pyridine (4 mL) in CH3OH (10 mL) for about 15 min; then more H2O2 (4 mL) was added, and the mixture was heated at reflux for a short while (until no more starting material was detected by TLC, 1% CH3OH:99% CH2Cl2 as eluent). The solvent volume was reduced (rotovap), and the residue was dissolved in diethyl ether, washed with water and dilute (3%) HCl, followed by sat. aq. NaCl. The ether was evaporated (rotovap) to yield 1.7 g (95%) of the desired product (5) as white, low melting crystals, mp 93-94°C (lit.23 mp 93-95°C). It had 1H-NMR (CDCl3) δ: 1.8 (s, 3H), 2.0 (s, 3H), 3.8 (s, 2H), 6.25 (bs, 1H) ppm.
4.1.9. 2,3,12,13-Tetramethyl-7,8-diethyl-15H,17H-tripyrrin-1,14-one (2)
3,4-Dimethyl-3-pyrroline-2-one (5) (0.21 g, 1.9 mmol) was dissolved in 3 mL of a 1:1 mixture of absolute ethanol and CH2Cl2 in an unsealed glass pressure tube, to which piperidine (0.1 mL, 1 mmol) was added, and the reaction tube was immersed into a steam bath. A solution of 2,5-diformyl-3,4-diethyl-1H-pyrrole (3) (0.085 g, 0.47 mmol) in CH2Cl2 (∼1 mL) was added dropwise into the reaction mixture, and the reaction progress was monitored by TLC (3% CH3OH:97% CH2Cl2 eluent). When no more starting materials (colorless) were no longer detected, and the yellow-green spot due to dipyrrinone was minimal in size (usually after about 30 h), the reaction was worked up as follows. The solvent was removed (rotovap), and the residue was dissolved in CH2Cl2, washed with 5% aq. HCl, followed by 3 washings with sat. aq. NaHCO3, then sat. aq. NaCl. The resulting CH2Cl2 solution was dried over Na2SO4, filtered and evaporated at reduced pressure. The resulting solid mixture was purified by radial chromatography using gradually increasing concentration of CH3OH in CH2Cl2 as eluent to afford 102 mg (59%) of a dark red, almost black solid, mp 282-284°C, that was pure by TLC. It had 1H-NMR (CDCl3) δ: 1.17 (t, J =7.5 Hz, 6H), 1.72 (s, 6H), 2.07 (s, 6H), 2.53 (q, J ∼7.5 Hz, 4H), 6.03 (s, 2H), 9.5 (bs, 1H), 10.6 (bs, 2H) ppm; 13C-NMR (CDCl3) δ: 8.3 (q), 9.87 (q), 16.48 (q), 17.6 (t), 98.8 (d), 126.1 (s), 128.4 (s), 130.3 (s), 132.6 (s), 141.4 (s), 174.35 ppm; FAB-MS: calcd. for C22H27N3O2 (365.2103), found: 365.210 [M+] amu.
4.2 X-ray Structure and Solution
Crystals of 1 were grown by slow evaporation of CH2Cl2. A crystal was placed into the tip of a 0.1 mm diameter glass capillary and mounted on a Bruker SMART Apex system for data collection at 100(2) K. A preliminary set of cell constants was calculated from reflections harvested from 3 sets of 20 frames for 1. These intial sets of frames were oriented such that orthogonal wedges of reciprocal space were surveyed (final orientation matrices determined from global least-squares refinement of 3524 reflections). The data collection was carried out using MoKα radiation (0.71073 Å graphite monochromator) with a frame time of 20 s and a detector distance of 4.94 cm. A randomly oriented region of reciprocal space was surveyed to the extent of 2 hemispheres and to a resolution of 0.66 Å. Four major sections of frames were collected with 0.5° steps in ω at 600 different ϕ settings and a detector position of 27° in 2θ. The intensity data were corrected for absorption and decay (SADABS).25 Final cell constants were calculated from the xyz centroids of strong reflections from the actual data collection after integration (SAINT 6.45, 2003).9 Crystal data and refinement information for 1 may be found in Table 5.
Table 5.
Crystal data and structural refinement for 1.
| Empirical Formula | C27H37Cl2N3O2 |
| Formula Weight | 506.50 |
| Temperature | 100 (2) K |
| Wavelength | 0.71073 Å |
| Crystal System | Monoclinic |
| Space Group | C2/c |
| Unit Cell Dimensions | a = 26.878(6) Å α = 90° |
| b = 13.650(3) Å β = 120.198° | |
| c = 17.082(5) Å γ = 90° | |
| Volume | 5417(2) |
| Z | 8 |
| Density | 1.242 Mg/m3 |
| Absorption Coefficient | 0.268 mm-1 |
| F(000) | 2160 |
| Crystal Size | 0.20 × 0.10 × 0.03 mm3 |
| Theta range for data collection | 1.73 to 22.50° |
| Index ranges | -28<h<28, -14<k<13, -18<l<18 |
| Reflections collected | 3524 |
| Independent reflections | 3524 [R(int) = 0.1275] |
| Completeness to theta = 22.50° | 99.2% |
| Absorption correction | SADABS |
| Max. and min. transmission | 0.9920 and 0.9484 |
| Refinement method | Full matrix least-squares on F2 |
| Data \ restraints \ parameters | 3524 \ 41 \ 350 |
| Goodness-of-fit on F2 | 1.061 |
| Final R indices [I>2 signal (I)] | R1 = 0.0658, wR2 = 0.1206 |
| R indices (all data) | R1 = 0.1889, wR2 = 0.1423 |
| Largest diff. peak and hole | 0.518 and -0.462 Å-3 |
The structure was solved and refined using SHELXTL.27 The monoclinic space group C2/c was determined based on systematic absences and intensity statistics. A direct-methods solution was calculated which provided most non-hydrogen atoms from the E-map. Full-matrix least squares/difference Fourier cycles were performed for structure refinement. All non-hydrogen atoms were refined with anisotropic displacement parameters unless stated otherwise. Hydrogen atom positions were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters (a C-H distance fixed at 0.96 Å and a thermal parameter 1.2 times the host carbon atom). Tables of atomic coordinates, bond lengths and angles, anisotropic displacement parameters, hydrogen coordinates and isotropic displacement parameters have been deposited at the Cambridge Crystallographic Data Centre, CCDC No. 651743.
Table 4.
Influence of acid, base and zinc on the UV-visible spectra of 1.a
| λmax / nm (ε) /L?mol-1?cm-1 | |||
|---|---|---|---|
| CF3CO2H | DBUb | CH3OH-Zn(OAc)2c | CH3OH-Zn(OAC)2 + NH4OHd |
| 537 (52,900) | 615 (41,300) | 586 (20,200) | 454 (13,000) |
| 324 (28,100) | 355 (24,000) | 376 (19,300) | 417 (14,000) |
| 312 (21,500) | 276 (25,400) | ||
Solutions are 1 × 10-5 M in 1.
Contains 385,000 equivs DBU.
Contains 1 equivalent of Zn(OAc)2.
With 1 mole equivalent of added conc. NH4OH.
Acknowledgments
We thank the U.S. National Institutes of Health (HD 17779) for generous support of this research. We also thank the National Science Foundation (CHE-0226402) for providing funding to purchase the X-ray diffractometer used in this work, Mr. Cameron Hilton for providing assistance in the X-ray analysis, and Prof. V.J. Catalano for achieving the resolution necessary to solve the apparent structure disorder in the crystal.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
4.0 References and notes
- 1.Fischer H, Adler E. Hoppe-Seyler’s Z. physiol. Chem. 1931;200:209–231. [Google Scholar]
- 2.Fischer H, Reinecke H. Hoppe-Seyler’s Z. physiol. Chem. 1940;265:9–21. [Google Scholar]
- 3(a).v. Dobeneck H, Schnierle F. Tetrahedron Lett. 1966:5327–5330. [Google Scholar]; (b) v. Dobeneck H, Messerschmitt T, Brunner E, Winderer U. Liebig’s Ann. Chem. 1971;751:40–49. [Google Scholar]
- 4.Berüter J, Colombo J-P, Schlunegger UP. Eur. J. Biochem. 1975;56:239–244. doi: 10.1111/j.1432-1033.1975.tb02226.x. [DOI] [PubMed] [Google Scholar]
- 5(a).Proust L. Ann. Chem. 1800;36:265. [Google Scholar]; (b) Simon A. Anthropochemie. (Berlin): 1842. p. 343. [Google Scholar]; (c) Pane N. J. Am. Med. Assoc. 1916;66:1433–1434. [Google Scholar]; (d) Borrien V. J. Pharm. et Chemie. 1917;16:45–51. [Google Scholar]; (e) Weiss M. Deutsch. Arch. Klin. Med. 1930;166:331–348. [Google Scholar]
- 6.Yamaguchi T, Shioji I, Sugimoto A, Komoda Y, Nakajima H. J. Biochem. 1994;116:298–303. doi: 10.1093/oxfordjournals.jbchem.a124523. [DOI] [PubMed] [Google Scholar]
- 7.Tradieux C, Bolze F, Gros CP, Guilard K. Synthesis. 1998:267–268. [Google Scholar]
- 8(a).Quistad GB. The Photooxidation of Alkylpyrroles and Bilirubin. 1972. UCLA Ph.D. thesis. [Google Scholar]; (b) Lightner DA, Quistad GB, Pak C-S. Synthesis. 1976:335–336. [Google Scholar]
- 9(a).Seidel W. Liebigs Ann. Chem. 1943;554:144–161. [Google Scholar]; (b) Plieninger H, Bauer H, Katritzky AR. Liebigs Ann. Chem. 1962;654:165–180. [Google Scholar]; (c) de Groot JA, van der Steen R, Fokkens R, Lugtenburg J. Recl. Trav. Chim. Pays-Bas. 1982;101:35–40. [Google Scholar]; (d) Byun YS, Lightner DA. J. Org. Chem. 1991;56:6027–6033. [Google Scholar]; (e) Xie M, Lightner DA. Tetrahedron. 1993;49:2185–2200. [Google Scholar]
- 10(a).Vogel E, Jux N, Rodriguez-Val E, Lex J, Schmickler H. Angew. Chem. Intl. Ed. Engl. 1990;291:1387–1390. [Google Scholar]; (b) Sessler JL, Mody TD, Lynch V. Inorg Chem. 1992;31:529–531. [Google Scholar]
- 11.Ono N, Kawamura H, Bougauchi M, Maruyama K. Tetrahedron. 1990;46:7483–7496. [Google Scholar]
- 12(a).Whitlock HW, Hanauer R. J. Org. Chem. 1968;33:2169–2171. [Google Scholar]; (b) Sessler JL, Mozaffari A, Johnson MR. Organic Syntheses. 1998;9:242–247. Coll. [Google Scholar]
- 13(a).Sun L, Tran N, Liang C, Tang F, Rice A, Schreck R, Waltz K, Shawver LK, McMahon G, Tang C. J. Med. Chem. 1999;42:5120–5130. doi: 10.1021/jm9904295. [DOI] [PubMed] [Google Scholar]; (b) Brower JO, Lightner DA, McDonagh AF. Tetrahedron. 2001;57:7813–7827. [Google Scholar]
- 14.Schoenleber RW, Kim Y, Rapoport H. J. Am. Chem. Soc. 1984;106:2645–2651. [Google Scholar]
- 15(a).Salzameda NT, Lightner DA. Monatsh. Chem. 2006;137:319–337. [Google Scholar]; (b) Salzameda NT, Huggins MT, Lightner DA. Tetrahedron. 2006;62:8610–8619. [Google Scholar]; (c) Huggins MT, Lightner DA. Monatsh. Chem. 2001;132:203–221. [Google Scholar]; (d) Huggins MT, Lightner DA. Tetrahedron. 2000;56:1797–1810. [Google Scholar]
- 16.Nogales DF, Ma J-S, Lightner DA. Tetrahedron. 1993;49:2361–2372. [Google Scholar]
- 17(a).Kaplan D, Navon G. Israel J. Chem. 1983;23:177–186. [Google Scholar]; (b) Kaplan D, Navon G. Biochem. J. 1982;201:605–613. doi: 10.1042/bj2010605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boiadjiev SE, Lightner DA. J. Heterocyclic Chem. 2000;37:863–870. [Google Scholar]
- 19(a).Huggins MT, Lightner DA. J. Org. Chem. 2000;65:6001–6008. doi: 10.1021/jo000393k. [DOI] [PubMed] [Google Scholar]; (b) Huggins MT, Lightner DA. Tetrahedron. 2001;57:2279–2287. [Google Scholar]
- 20.Cullen DL, Black PS, Meyer EF, Lightner DA, Quistad GB, Pak C-S. Tetrahedron. 1977;33:477–483. [Google Scholar]
- 21(a).Person RV, Peterson BR, Lightner DA. J. Am. Chem. Soc. 1994;116:42–59. [Google Scholar]; (b) Molecular mechanics and dynamics calculations employed to find the global energy minimum conformations of 1 were run on an SGI Octane workstation using vers. 7.0 of the Sybyl forcefield as described in refs 15 and 21a.
- 22.Kleinsplein GG. J. Am. Chem. Soc. 1995;77:1546. [Google Scholar]
- 23.Xie M, Lightner DA. Tetrahedron. 1993;49:2185–2200. [Google Scholar]
- 24.Chen Q, Huggins MT, Lightner DA, Norona W, McDonagh AF. J. Am. Chem. Soc. 1999;121:9253–9264. For an improved synthesis of ethyl isocyanoacetate. [Google Scholar]
- 25.Sheldrick GM. SADABS, vers. 2.1. Bruker Analytical X-ray Systems; Madison. WI: 2003. [Google Scholar]
- 26.SAINT, vers. 6.45. Bruker Analytical Systems; Madison, WI: 2003. [Google Scholar]
- 27.Sheldrick GM. SHELXTL, vers 6.14. Bruker Analytical X-ray Systems; Madison, WI: 1997. [Google Scholar]