

Abstract
Here, we use a mouse model (DBA/2J) to readdress the location of insult(s) to retinal ganglion cells (RGCs) in glaucoma. We localize an early sign of axon damage to an astrocyte-rich region of the optic nerve just posterior to the retina, analogous to the lamina cribrosa. In this region, a network of astrocytes associates intimately with RGC axons. Using BAX-deficient DBA/2J mice, which retain all of their RGCs, we provide experimental evidence for an insult within or very close to the lamina in the optic nerve. We show that proximal axon segments attached to their cell bodies survive to the proximity of the lamina. In contrast, axon segments in the lamina and behind the eye degenerate. Finally, the Wlds allele, which is known to protect against insults to axons, strongly protects against DBA/2J glaucoma and preserves RGC activity as measured by pattern electroretinography. These experiments provide strong evidence for a local insult to axons in the optic nerve.
Introduction
Vision loss in glaucoma is caused by the dysfunction and death of retinal ganglion cells (RGCs). The molecular pathways that damage RGCs in glaucoma are not understood, and it is unclear how or where the RGCs are insulted. It is possible that different parts of the RGC are directly insulted. A body of data supports an initial insult to the axons of RGCs in the lamina cribrosa region of the optic nerve, where they exit the eye. The lamina cribrosa is comprised of perforated sheets or plates of extracellular matrix (ECM, largely collagen) that provide support for the axons as they pass through the posterior wall of the eye (Anderson, 1969).
Important studies in humans and primates established that early damage to the RGC involves degenerative changes in the axon segment in the lamina cribrosa (e.g., Anderson and Hendrickson, 1974; Anderson and Hendrickson, 1977; Quigley and Anderson, 1976, 1977; Quigley and Addicks, 1980; Quigley et al., 1979, 1980, 1981, 1983). Bundles of axons are proposed to be vulnerable as they pass through the pores of the lamina. It is hypothesized that elevated IOP causes distortion or “bowing” of the ECM plates, which in turn damages axon bundles by mechanical stress (Quigley et al., 1983). Mechanical distortion of the ECM plates also has been suggested to contribute to glaucoma by affecting blood vessels (Quigley and Addicks, 1981; Maumenee, 1983; Fechtner and Weinreb, 1994). Local damage at the lamina has been suggested to account for abnormal axonal transport in glaucoma and its animal models (Anderson and Hendrickson, 1974). Importantly, the ECM plates of the lamina are covered by astrocytes that provide the axons with neurotrophic and other forms of support. Thus, an alternative hypothesis suggests that IOP elevation in some way alters these glial cells so that they damage the RGC axons (Hernandez, 2000). Loss of glial support functions may also be important for the neuronal compromise (Lappe-Siefke et al., 2003).
As a result of recessive mutations in two genes (Gpnmb and Tyrp1), DBA/2J mice develop elevated IOP and glaucoma with age (Chang et al., 1999; Anderson et al., 2002). Due to similarities to the human disease, this glaucoma is now widely used as a surrogate model for human glaucoma (Danias et al., 2003; Libby et al., 2005a; Schlamp et al., 2006; Steele et al., 2006). Using DBA/2J mice, we and others have provided evidence consistent with a direct insult to RGC axons in the optic nerve. First, in BAX-deficient DBA/2J mice, axon degeneration occurs but the RGC somata survive, indicating that axon degeneration is not a consequence of RGC death (Libby et al., 2005b). Second, RGC death is not uniform across the retina in DBA/2 glaucoma (Danias et al., 2003; Jakobs et al., 2005; Filippopoulos et al., 2006; Schlamp et al., 2006). Instead, RGCs degenerate in specific fan-shaped sectors of the retina (Jakobs et al., 2005; Schlamp et al., 2006) that are consistent with the injury of specific axon bundles in the optic nerve. The fan-shaped regions of cell loss are thought to be equivalent to the arcuate scotomas of human glaucoma. Arcuate scotomas resemble fans with a sideways bend and are also suggested to result from an axon injury (Shields, 1997). The difference in orientation of the fans is believed to mirror the different paths that the axons follow in the two species.
Although consistent with an insult to the axon, current data from humans, primates, and rodents fall short of experimentally demonstrating that the axon is insulted in the lamina in glaucoma. In the general case, it is well established that the first site of neuronal damage may be remote from the site of insult (for review see Conforti et al., 2007). For example, in transected motor axons, neuromuscular junctions that are many centimeters from the lesion degenerate first. The axons immediately adjacent to the lesion remain intact for two to three times longer than the distant terminals (Beirowski et al., 2005). Thus, locating the first degenerative changes to the optic nerve is not the same as demonstrating that the optic nerve is a site of insult in glaucoma.
Here, we use DBA/2J mice to reevaluate if the mouse optic nerve has a lamina and to determine if an insult to axons in the lamina is crucial in glaucoma. We developed substrains of DBA/2J that have specific mutations or a fluorescent marker that allowed us to assess the importance of axon injury. Additionally, we used a genetically matched DBA/2J control strain that does not develop glaucoma (Howell et al., 2007). Using these resources, we show that DBA/2J mice have an astrocytic or glial lamina, but it lacks the ECM plates of the human lamina cribrosa. We next provide evidence for a direct insult to RGC axons in the optic nerve, and likely within the lamina. Finally, we show that the Wallerian degeneration slow allele (Wlds) strongly protects against glaucoma in the DBA/2J strain. Together, our data provide strong experimental evidence that an important and early event in glaucoma is damage to the axons of retinal ganglion cells as they exit the eye through the lamina.
Results
A robust network of astrocytes enmeshes RGC axons as they exit the mouse eye
It is reported that mice do not have a lamina cribrosa (Tansley, 1956; Fujita et al., 2000; Morcos and Chan-Ling, 2000; May and Lutjen-Drecoll, 2002), yet the DBA/2J strain develops glaucoma with hallmarks of human glaucoma. Given the suggested importance of the lamina in glaucoma, we reevaluated the status of the lamina in aged DBA/2J mice (11–12 mo). It has been suggested that the ECM plates of the lamina become more robust with age (Hernandez et al., 1989). We initially used a substrain of DBA/2J that does not develop glaucoma (D2-Gpnmb +, Howell et al., 2007), which allowed initial studies of the lamina without any confounding effects of disease.
The optic nerve of the mouse has a clearly defined region with a robust network of glial fibrillary acidic protein (GFAP)–positive astrocytes (Figs. 1 and 2). The astrocytes of this region form an enmeshing network of glial cells through which the RGC axons pass, and they are intimately associated with axons (Fig. 2). This network begins as the nerve passes into the scleral canal and it extends posteriorly, ending before myelination of the majority of axons (diagrammatically shown in Fig. 3). There is no evidence of collagenous plates in this region, with abundant collagen only observed in blood vessel walls (Fig. 1 and 2). To reflect the equivalent location compared with the human lamina cribrosa, the similar arrangement of glial cells, but the absence of ECM plates, we call this region the glial lamina.
Figure 1.

Aged mice have a robust glial lamina that lacks ECM plates. (A–F) Optic nerves of 11–12-mo-old DBA/2J mice that do not develop glaucoma (D2-Gpnmb +) were serially sectioned in longitudinal or cross-sectional planes. A robust astrocyte meshwork surrounds the axons where they exit the mouse eye. The astrocyte meshwork (glial lamina, delineated by brackets) is clearly evident as abundant nuclei oriented at right angles to the long axis of the optic nerve (A and B) and a cellular network (C and D, arrowheads) with extensive processes that stain positive for GFAP (E and F). In contrast to the lamina cribrosa of the human optic nerve (see Quigley and Addicks [1981]), the mouse astrocytes do not cover a network of robust collagenous plates (G and H). Although collagens are clearly visible in blood vessel walls (arrowheads), there is no meshwork of collagen staining that mirrors the astrocyte meshwork. All presented cross sections are from within the lamina region. V, vessels. Sections were stained with hematoxylin and eosin (H&E) (A and B, cells and axons pink; nuclei purple-blue); Bodian's stain (C and D, axons pale blue; nuclei dark purple-blue; glial cell bodies and processes remain clear); anti-GFAP with hematoxylin counter stain (E and F, astrocyte bodies and processes brown; nuclei purple-blue); or Masson's trichrome (G and H, collagens stain blue). Bars, 50 μm.
Figure 2.

Intimate association of axons and astrocytes in the mouse glial lamina. (A–F) All electron micrographs are in the longitudinal plane and from the intensely GFAP-positive region delineated by the bracket. (B) In this region, bundles of axons (AX) are enclosed between columns of astrocytes (pial columns, PC) that run in a rostral-caudal direction. The glial meshwork of the lamina (GL) is oriented at right angles to the pial columns. N, astrocyte nucleus. (C–E) The axons and glia are closely packed and glial processes (E, arrows) have an intimate association with individual axons. (F) At higher magnification, microfibrils (MF) identify astrocyte processes that are easily differentiated from axons. No collagenous plate-like structures were associated with the astrocytes. (G and H) Confocal microscopic analysis of a cross section through the same region also indicates a very intimate association between glia and axons. Anti-neurofilament (axons), green; anti-GFAP (astrocytes), red; DAPI (nuclei), blue. V, vessels. (A) Bar, 50 μm. (B–F) Bars, 2 μm. (G and H) Bars, 20 μm.
Figure 3.

Optic nerve regions and damage levels. (A) Schematic showing the regions we defined as nerve fiber layer, pre-lamina, and lamina (not to scale). Axons (pink) course into the optic nerve head, pass out of the eye through the glial lamina (crosshatches, located within the scleral canal) and become myelinated behind the eye (horizontal lines). Ret, retina; V, vessels; Sc, sclera. (B) The severity of glaucomatous nerve damage was determined in a retro-orbital portion of each optic nerve (Materials and methods). Each assigned damage level is clearly different by axon counting. Axon counts were as follows: D2-Gpnmb + with no glaucoma (white bar) 52074 ± 451 (n = 10), DBA/2J mice (black bars), with no or early glaucoma 51862 ± 934 (n = 10), moderate glaucoma 33283 ± 2199 (n = 15), and severe glaucoma 5454 ± 1211 (n = 24). The axon counts for nerves with no or early damage were not statistically different to the no glaucoma D2-Gpnmb + controls. They are classified as no or early glaucoma because no glaucomatous nerve damage is detected behind the eye but at the assessed ages some of the eyes must be at early stages of glaucoma. The differences comparing the no or early, moderate, and severe damage levels were significant (P < 0.001). (C–J) Representative images of D2-Gpnmb + with no glaucoma (C and G) and DBA/2J with no or early glaucoma (D and H), moderate glaucoma (E and I), and severe glaucoma (F and J). Very mild, age-related damage, similar to that which occurs in other nonglaucomatous mouse strains, occurs in the D2-Gpnmb + strain (C, arrow shows a single damaged axon that stains darkly with PPD, Materials and methods). This age-related damage is also seen in DBA/2J optic nerves that were determined to have no or early glaucoma (D, arrow). Nerve fiber layer thinning (arrowheads) is evident in H&E-stained sections of moderately affected nerves and severe nerves have obvious nerve fiber layer loss and optic nerve excavation (asterisk). These changes are hallmarks of glaucoma. C–F are semi-thin sections stained with PPD. G–J are plastic sections stained with H&E. V, vessels. Bars, 100 μm.
The glial lamina is an important site of axon damage
Because the glial lamina of DBA/2J mice completely lacks ECM plates, we used a combination of electron microscopy and immunohistochemistry to identify the location of initial axon damage in this glaucoma model. We analyzed axons as they gather in the nerve fiber layer, run through the optic nerve head (prelamina region), and exit the eye through the lamina in mice that were 11–12 mo old. At these ages, the degree of glaucomatous damage varies between eyes (Libby et al., 2005a). We selected DBA/2J eyes that did not have detectable glaucoma. Some of these eyes must be at early stages of disease and are classified as having no or early glaucoma (Fig. 3 and Materials and methods).
In DBA/2J mice with no or early glaucoma, damaged axon segments (swollen regions with disorganization of axonal contents and accumulation of organelles and neurofilaments) were readily identified in the glial lamina (Fig. 4). The damaged axon regions are similar to dystrophic neurites reported in other conditions (Fig. 4, F–H) (Vickers, 1997). In the studied eyes, dystrophic neurites were not detected in the nerve fiber layer and were rare in the prelamina region. Consistent with previous reports, neurofilaments accumulated in damaged axon segments (Fig. 4, I and J). The damaged axon segments represented early glaucoma damage and not simply age-related changes because they were much less abundant in the D2-Gpnmb + eyes (Fig. 4, K and L). In some eyes, dystrophic neurites were only observed in the lamina, and in all eyes they were most abundant in the lamina (Fig. 4, K and L).
Figure 4.

Early damage to RGC axons occurs at the lamina. RGC axons were analyzed for glaucomatous damage at different locations as they extend across the retina and exit the eye. Damage was assessed in the nerve fiber layer (NFL), the prelamina, and glial lamina regions of the optic nerve. All mice were 11 mo old and had no or early glaucoma based on analysis of the retro-orbital optic nerve. (A–C) D2-Gpnmb + no glaucoma controls, the axons were healthy and had normal morphology at all locations based on electron microscopy. Only rare damaged axons were detected. This rare age-related damage occurs in aged mice of various strains that do not develop glaucoma. (D–F) The axons of age- and sex-matched DBA/2J mice with early glaucoma did not differ from controls at any intraocular location, as they extended from the cell body to the lamina. At the glial lamina, however, dystrophic neurites (F, arrows) were readily detected. (G and H) The dystrophic neurites (arrows) consisted of swollen and damaged axon segments containing an accumulation of organelles including swollen mitochondria (arrowhead). (I and J) Due to the accumulation and disorganization of axonal contents, dystrophic neurites were also detected using a combination of both nonphosphorylated (Smi32) and phosphorylated (Smi34) neurofilament antibodies (green). They were visible as abnormally large and bright staining, damaged axon regions (arrows) in the lamina. Although dystrophic neurites labeled with each antibody (not depicted), we used the combination to maximize the intensity of staining. GFAP-positive astrocytes are shown in red. (K and L) Counting dystrophic neurites indicates that the first site of morphological damage to the intraocular RGC axon occurs in the lamina. The average number of dystrophic neurites (DNs) per EM field in the lamina was significantly higher in DBA/2J mice with no or early glaucoma compared with the no glaucoma D2-Gpnmb + controls (K, left panel, P < 0.001, t test, n = 8 eyes per genotype). This clearly demonstrates that dystrophic neurites are part of the glaucoma process and not just age-related changes. Comparing the indicated regions, the number of dystrophic neurites was highest in the lamina (P < 0.001). (L) Neurofilament and GFAP staining were used to assess serial, optic nerve cross sections extending from the nerve fiber layer at the retinal surface and into the lamina. Counting neurofilament-stained dystrophic neurites confirmed the EM findings. The values for individual DBA/2J eye show that at this age some eyes were at an early stage of glaucoma with significantly more dystrophic neurites than control mice (P < 0.001 comparing the lamina region of DBA/2J to D2-Gpnmb +; n = 9 per genotype). (A–H) Bars, 2 μm. (I–J) Bars, 20 μm.
A CFP marker shows that most early damage is highly localized in the mouse lamina
To further define the location of initial axon damage, and to allow sensitive evaluation of entire intraocular axons from their cell bodies to the glial lamina, we developed a substrain of DBA/2J mice that expresses CFP in their RGCs (D2.Thy1-CFP, Materials and methods). Because it is well established that axonal components accumulate in regions of axonal damage, we reasoned that axonal CFP would behave similarly and allow ready visualization of damaged axon segments. A similar approach has been used by others (Beirowski et al., 2005).
The integrity of the axons was assessed from RGC somata to the lamina by confocal microscopy (Fig. 5). In eyes that had no or early glaucoma (Fig. 3), the somata were readily detected and had no obvious abnormalities. In the same eyes, however, discrete and focal swellings were present on individual axons in the glial lamina (Fig. 5). These swellings were characterized by very intense CFP signal. Some of these axons had severe swellings that were similar in diameter to dystrophic neurites (>2 μm, much larger than the majority of unmyelinated axons in this region of a healthy nerve). However, the great majority of damaged axons were less severely altered with mild swellings only slightly larger than normal axons (Fig. 5 B). In the eyes with early glaucoma, axon damage was localized to the glial lamina and was rarely seen in the prelamina or nerve fiber layer (Fig. 5). This damage was not present in young DBA/2J mice and increased in severity as glaucoma progressed. These results confirm that the first morphological damage to the ocular portion of the RGC axon occurs at the glial lamina.
Figure 5.

Early focal axon damage occurs in the glial lamina. To allow sensitive detection of axon damage along the entire axon from the soma to the lamina we analyzed D2.Thy1-CFP eyes (Materials and methods). For consistency with previous images, CFP-positive axons are pseudo-colored green. (A) Schematic demonstrating the location in the optic nerve head of the presented prelamina and lamina images (single focal plane) that are from the same eye for each damage level. In the prelamina region, vessels are apparent as a dark region in the center of the nerve. In the lamina, the vessels extend from the center to the edge of the nerve. (B) In an 11-mo-old eye with no or early glaucoma (Fig. 3), axon damage was detected as highly focal swelling of individual axons in the lamina. As previously reported for axonal contents, CFP accumulated in the swellings making them brightly fluorescent (arrowheads). The swellings were not present in other regions of the intraocular axon. (C–K) Representative examples of the prelamina and lamina regions in eyes with different degrees of glaucoma are shown. (C, F, and I) No glaucomatous damage was detected at any level in preglaucomatous young eyes (n = 4). (D, G, and J) Obvious axonal swellings were evident specifically in the lamina of eyes that were at early stages of glaucoma. These eyes were initially classified as having no or early glaucoma based on analysis of optic nerve from behind the eye (Fig. 3), but 5/8 were found to have early damage in the lamina. (E, H, and K) At moderate stages of glaucoma, the axonal damage had spread to portions of the axon in both the prelamina region and nerve fiber layer, and some axons had a highly abnormal morphology. The compressed lamina images represent a compressed Z stack of the entire lamina region that could be imaged in the mounted specimens (it was not possible to image the most posterior lamina). All images were collected using identical conditions. Bars, 20 μm.
Mild axon damage is restricted to the lamina but severe damage extends to the brain, early in glaucoma
Subsequently, we used D2.Thy1-CFP mice with early glaucoma to analyze RGC axons extending behind the eye from the glial lamina to the superior colliculus (see Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200706181/DC1). In these eyes, mild swellings were most numerous in the lamina and essentially absent at all other locations. This finding is also consistent with an early insult to axons within the glial lamina. In contrast to the distribution of mild damage, severely damaged axon segments with swellings of similar size to the dystrophic neurites were present in the lamina, the retrobulbar optic nerve, and all regions studied behind the eye to the superior colliculus (Fig. S1). Counts of these large swellings demonstrated that their number is the same in both the lamina and the retrobulbar optic nerve (unpublished data). The severe swelling was not uniform along each affected axon, but occurred focally (Fig. S1). This pattern is characteristic of axon degeneration in other diseases and after induced injury (Beirowski et al., 2005; Coleman, 2005). Wallerian degeneration is a process that results in the rapid degeneration of the entire axon segment distal to a lesion (Waller, 1850; Beirowski et al., 2005; Coleman, 2005). Overall, our data suggest that axons are insulted to different degrees within the lamina of an eye during early glaucoma. Some axons suffer a severe insult that appears to result in rapid Wallerian-like degeneration, while many more axons suffer a lesser insult that results in milder damage that is not sufficient to induce Wallerian-like degeneration of the distal axon (at least initially) (see Whitmore et al., 2005).
Regional loss/survival of lamina axons matches retinal loss/survival of RGCs
We, and others, have shown that RGC loss in the retinas of DBA/2 mice occurs regionally (Danias et al., 2003; Jakobs et al., 2005; Schlamp et al., 2006). Consistent with this, axon loss was not uniform in the lamina but occurred in discrete regions that varied from nerve to nerve (Fig. 6, A–F).
Figure 6.

Axon degeneration is regionalized in the lamina and corresponds to fan-shaped RGC loss in the retina. (A–F) Compressed Z stack images through the lamina of DBA/2J nerves with increasing levels of glaucomatous damage (neurofilaments, green; GFAP positive glia, red). Axon loss/survival is clearly regionalized to different areas of the optic nerve and not randomly distributed. (G and H) The retina and corresponding lamina region of an eye with moderate glaucoma. Discrete fan-shaped areas of RGC loss are apparent in the retina (dotted “V,” G) and appear to correspond to two regionalized areas of axon loss in the lamina (dotted boxes, H). This is consistent with localized damage to axons within the lamina leading to fan-shaped patterns of RGC loss in the retina but it was not possible to follow the region of axon loss unambiguously through the whole optic nerve head. Bars, 50 μm.
If local insults to axons determine the RGCs that degenerate, the regional loss/survival of axons within the lamina should correspond to regions of RGC loss/survival in the retina. Thus, we compared the pattern of loss/survival of RGCs and their associated axons in the retina with the pattern in the lamina. In eyes with early or moderate glaucoma, discrete regions of axon loss in the glial lamina appeared to correspond to distinct regions of RGC loss in the retina (Fig. 6, G and H). Additionally, in D2.Thy1-CFP eyes with moderate or severe glaucoma, we were able to trace axons from RGCs surviving within fan-shaped regions of the retina to the optic nerve head and into the lamina where they formed discrete bundles (Fig. 7). Thus, axons surviving in discrete regions of the lamina correspond to the fan-shaped sectors of RGCs surviving in the retina. These results are consistent with a sequence of events in which localized insults within the glial lamina damage bundles of axons and lead to fan-shaped patterns of RGC loss in the retina. Even though these results suggest that axon and cell death are linked to one another, by themselves they cannot distinguish which causes which.
Figure 7.

Axons from surviving RGCs track to discrete regions in the glial lamina, and many other axons that survive in the eye are lost in the lamina. Proving a direct relationship between the pattern of RGC survival/loss in defined regions of the optic nerve and retina, D2.Thy1-CFP–labeled axons were found to run from RGCs surviving in individual fan-shaped regions of the retina to individual local regions in the glial lamina. Mounted tissue was optically sectioned with a confocal microscope, starting at the retinal surface and capturing images at 0.5-μm intervals from the nerve fiber layer through to the glial lamina. (A) Compressed Z stack of a retina and its optic nerve. The pseudo-colored (green, yellow, blue and red) axons on the right side travel from fan-shaped regions in the retina to discrete regions in the glial lamina. The surviving axons on the left (pseudocolored in white) travel from the retina but end just in front of the lamina, clearly showing that some axons that survive in the eye are lost in the lamina. (B) Schematic indicating positions of optical sections shown in other panels. (C and F) Compressed Z stack with outline of the optic nerve at the glial lamina denoted with a dotted ring (C, pseudo-colored; F, raw grayscale). (D and G) Single layer image within the nerve fiber layer. (E and H) Single layer image within the glial lamina with the nerve outlined by the dotted ring. The dotted line indicates the blood vessels entering the optic nerve at this level. Bars, 100 μm.
Axons survive to the proximity of the lamina in D2.Bax−/− mice with severe glaucoma
Demonstrating that early axon damage occurs in the glial lamina, and that the pattern of axon loss/survival in the optic nerve corresponds to the loss/survival of RGCs in the retina, does not directly test the proposition that axons are insulted in the lamina. The first site of degeneration is not a reliable indicator of where the neuron was initially insulted (Conforti et al., 2007). Additionally, it is possible that the regional patterns of RGC death in glaucoma occur as a consequence of some other factor(s) that is not understood.
To experimentally test if an axon insult occurs in the glial lamina in DBA/2J mice, we designed a study based on the well-established pattern of axon degeneration and axon survival that occurs in response to direct and focal axon injury in the peripheral nervous system. In the peripheral nervous system, direct and focal axon injury results in degeneration of the entire length of the distal portion of the axon that is separated from the cell body by the lesion. The distal axon rapidly degenerates by Wallerian degeneration (Waller, 1850; Stoll et al., 2002). In contrast, the proximal portion of the axon that is attached to the cell body can survive up to the proximity of the axon insult, as long as the cell body survives. Similarly, one report using optic nerve transection suggested that proximal intraretinal axons survive until the RGC somata degenerate (Anderson, 1973). Because all RGC somata survive indefinitely in BAX-deficient DBA/2J mice (Libby et al., 2005b), we reasoned that the proximal axon may survive up to the proximity of any focal axon insult, even in eyes with severe glaucoma. Accordingly, the extent of proximal axon survival may act as a marker to locate the site of axon insult.
First, we tested if proximal RGC axons survive up to the site of insult in D2.Bax −/− mutant mice. The intraorbital optic nerves of 3-mo-old D2.Bax −/− (BAX-deficient) and D2.Bax +/+ (BAX-sufficient) mice were crushed as far behind the eye as possible. These mice were too young to have glaucoma. In D2.Bax +/+ eyes, the RGC somata and their axons degenerated along their entire length. In contrast, in D2.Bax −/− eyes, the proximal axon survived behind the eye and up to the proximity of insult for 30–60 d after crush was performed (see Fig. S2). Thus, in these nonglaucomatous eyes, the extent of axon survival was a reliable indicator of the site of insult to the axons of the optic nerve.
Second, we determined the extent of proximal axon survival in glaucoma. We used D2.Bax −/− mice selected to have >95% retro-orbital axon loss. To match the timing of proximal axon analysis to the crush experiment, we included mice that developed this severe loss of retro-orbital axons in the previous 30–60 d (Materials and methods). In these mice, proximal axons survived from the RGC bodies to the proximity of the anterior edge of the glial lamina inside the eye. In contrast, the vast majority of axon segments in the lamina had degenerated and those distal to the lamina had always degenerated (Fig. 8; Fig. S3). This study provides experimental evidence for a major axon insult occurring within or very close to the lamina in the optic nerve during DBA/2J glaucoma. Together with the demonstration that the first identifiable damage to many axons is in the lamina, this experiment strongly suggests than an insult occurs within the lamina early in glaucoma.
Figure 8.

Axons survive up to the lamina in BAX-deficient mice with severe glaucoma. (A–I) Representative images from an EM analysis of RGC axons running from the nerve fiber layer (NFL) to lamina are shown. (A–C) Control 12-mo-old D2-Gpnmb +. Tightly packed axons (AX) are present in the nerve fiber layer, which is covered by the internal limiting membrane and footplates of Muller cells (*, A). The axons continue in an orderly arrangement through the prelamina region and into the lamina. (D–F) Age- and sex-matched DBA/2J mouse with severe glaucomatous axon loss behind the eye. In the nerve fiber layer (D), only a few damaged and swollen axons remain. Muller cells are hypertrophic and fill much of the region. Their cytoplasm, adjacent to the internal limiting membrane, is thickened (*). In the prelamina region (E), axons are largely absent and replaced by thickened glial processes. Activated astrocytes are abundant (astrocyte nucleus, N). At the lamina (F), large glial processes have replaced essentially all axons. (G–I) Age- and sex-matched D2.Bax −/− (BAX-deficient) mouse with severe axon loss behind the eye. In the nerve fiber layer (G), the axons survive but appear swollen. The Muller cell cytoplasm (*) is not hypertrophic. In the prelamina region (H), the majority of axons are intact but large swellings (arrows) are found. In contrast, in the lamina (I) essentially all axons are lost and the glia are very hypertrophic. The damage is essentially the same as that in (F). (J–L) Neurofilament labeling confirms axon survival to the proximity of the lamina in D2.Bax −/− mice. Neurofilament-labeled axons (green) in longitudinal sections of the optic nerve are shown. In a young DBA/2J mouse (J), the axons clearly gather in the nerve fiber layer at the inner edge of the optic nerve and continue to pass through to the lamina. In a 10-mo-old DBA/2J mouse with severe axon loss behind the eye (K), axons are completely missing from the nerve fiber layer and entire optic nerve. In contrast, in a 10-mo-old BAX-deficient DBA/2J mouse (L), with severe axon loss behind the eye, the axons continue to run along the retina and survive up to the proximity of the lamina (within known retraction distances following local insult; Fig. S3; Kerschensteiner et al., 2005). As was seen by EM, there is an abrupt loss of axons at the lamina. Despite axon survival up to thelamina, the optic nerve head has still remodeled (become excavated) and so the morphology is not directly comparable to the control. Importantly, this pattern of axon survival up to the lamina provides strong experimental evidence for a direct insult to the axon at this location in this inherited glaucoma (see Fig. S3 for higher magnification images of J and L). (A, D, and G) Bar, 2 μm. All other EM bars, 500 nm. (J–L) Bar, 75 μm.
Wlds allele protects axons in DBA/2J glaucoma
To further assess the role of axonal injury in glaucoma, we developed a strain of DBA/2J mice with the Wlds allele (D2.Wlds, Materials and methods). The Wlds gene dominantly protects axons from degeneration induced by axonal trauma (Lunn et al., 1989; Perry et al., 1990, 1991; Ribchester et al., 1995; Mack et al., 2001). Because Bax heterozygosity (Bax +/−) protects the RGC somata in DBA/2J glaucoma, we also determined if inheriting both the Wlds and Bax − mutations would be beneficial (Fig. 9).
Figure 9.

Wlds profoundly protects from glaucomatous damage. (A) The IOP distributions of mice of each genotype overlap extensively. The boxes show the upper and lower quartiles and the bars show the extremes including outliers. The centerline of each diamond is the mean and the upper and lower points of each diamond represent the 95% confidence intervals of the mean. Mice with a single copy of the Wlds fusion gene are hemizygous (Hemi). At both 9 and 12 mo, there were no significant differences in IOP (ANOVA) between the neuroprotected, Wlds mice (see below), and mice of any other genotype. At 10.5 mo, the wild-type (WT) mice had an unusually high skewing of their IOP compared with typical values at this age (see Anderson et al., 2005; Libby et al., 2005a), and their IOP was significantly higher than mice of all other genotypes (P < 0.04 for all comparisons). This skewing did not account for the decreased axon loss in Wlds mice. This is clear because (1) there was extensive overlap between all genotypes, and (2) the IOP distribution of Wlds mice was extremely similar (P = 0.7) to Bax +/− heterozygotes mice (Het), whose axons were not protected (see below). (B) Axon protection in Wlds mice. Distributions of optic nerve damage in mice of the indicated genotypes are shown at two important time points (Materials and methods). The Wlds allele significantly rescues axons from glaucomatous degeneration at 12 mo of age (P < 0.0001, chi square comparing Wlds Hemi to their WT littermates). Importantly, Wlds increased the number of eyes with no detectable glaucoma. Bax +/− heterozygosity conferred no protective effect when compared with wild-type mice (right). Though not statistically significant, there tended to be a greater degree of protection in D2.WldsBax +/− double mutants (right), compared with Wlds single mutants (left). The number of eyes analyzed for each genotype at 10.5 mo was typically ≥40 but 20 for D2.Bax +/− heterozygotes. At 12 mo, typically ≥63 eyes were studied for each genotype but 21 for D2.Bax +/− heterozygotes. (C–F) Wlds preserves RGC axons and optic nerve morphology as seen in semi-thin sections. Most wild-type (C) and D2.Bax +/− nerves (E) had severe glaucoma with very substantial axon loss (see Fig. 3) and extensive glial scarring. In contrast, the majority of D2.Wlds (D) and D2.WldsBax +/− nerves (F) had no or early glaucoma, with <5% of axons damaged (Fig. 3) and no evidence of glial hypertrophy or scarring. (G) Axon counts for a randomly selected sample of DBA/2J and D2.Wlds nerves with no or early glaucoma (n = 6 for each genotype). The averages are not statistically different (P = 0.235), and none of the D2.Wlds nerves had decreased axon counts. Because Wlds more than doubled the number of eyes with no or early glaucoma, half of these counted eyes were rescued. (H–J) Retinal flatmounts showing that in Wlds protected eyes RGC somata survive (H, preglaucoma; I, severe glaucoma; J, no or early glaucoma). (K) This was confirmed by counting RGC layer cells in eyes with no or early glaucoma. Again, half of the D2-Wlds eyes were rescued and none had cell numbers below the range of wild-type values (n = 10 each genotype, P > 0.1). As expected, in both wild-type and D2.Wlds mice with severe optic nerve damage the majority of RGCs were lost. The retinal images shown are from a matched region of the superior, peripheral retina. (L) Despite the profound rescue, a mild degree of somal shrinkage (∼10%) was seen in D2.Wlds eyes with no detectable glaucoma. The shrinkage appears to occur generally across cells of different sizes (see Fig. S4). (M) Wlds strongly preserved PERG amplitude, a measure of RGC activity, in randomly selected mice (number of eyes = 16, 14, 18 for D2-Gpnmb+, DBA/2J, and D2.Wlds, respectively). Bars, 100 μm.
Clinical examinations from 6–12 mo of age (Materials and methods) showed that the onset, rate of progression, and severity of the glaucoma-inducing iris disease did not differ between mice with any of the tested genotypes. IOP was assessed at key ages relevant to the DBA/2J glaucoma (Libby et al., 2005a), and the IOP distribution of mice with each genotype overlapped extensively (Fig. 9).
Assessment of optic nerve damage severity at two important time points (Libby et al., 2005a) showed that the Wlds mutation has a strong protective effect on the survival of optic nerve axons (Fig. 9 B, left). At 12 mo of age, 68% of wild-type eyes (59/86) had severe glaucoma. In comparison, only 33% of D2.Wlds eyes (27/81) had severe glaucoma. Also at 12 mo, the Wlds mutation more than doubled the number of mice with no detectable glaucoma (from 22% wild type [19/86] to 49% Wlds [40/81]). Axon counts confirmed the protection (Fig. 9).
In addition to optic nerve damage severity, we assessed RGC soma number. Wlds is well established to directly protect axons but not somata (Glass et al., 1993; Deckwerth and Johnson, 1994; Deshmukh et al., 1996; Deckwerth et al., 1998; Ikegami and Koike, 2003; Adalbert et al., 2005). RGC somata survived in D2.Wlds eyes whose axons were spared (Fig. 9). Although the somata were spared, they were not completely protected as on average somal diameter had shrunk by ∼10% (Fig. 9). RGC somata were largely absent in D2.Wlds eyes with severe axon loss, indicating that Wlds cannot protect the soma from death if the axon degenerates.
To allow more sensitive evaluation of the protective effects of Wlds against RGC dysfunction in glaucoma, we assessed the response of RGCs using pattern electroretinography (PERG) (Porciatti et al., 2007). The PERG response is well documented to require RGC activity (see Porciatti, 2007). To determine the effects of glaucoma on PERG, we evaluated 10–11-mo-old DBA/2J mice with each stage of glaucoma. To control for possible effects of the pigment-dispersing iris disease on PERG, we also evaluated a congenic strain of B6 mice that has the same Tyrp1 and Gpnmb mutations as DBA/2J mice and develops the same degree of iris disease, but is resistant to IOP elevation and does not develop glaucoma (Anderson et al., 2006). PERG was not altered by the iris disease (Fig. S5 A) but was severely impaired by the glaucoma (Fig. S5 B). In fact, PERG proved very sensitive to the glaucoma and was impaired early in the disease before detectable axon loss. Wlds strongly protected RGC activity and preserved PERG to nonglaucomatous control values in randomly selected 10–12-mo-old mice (Fig. 9).
Discussion
The lamina cribrosa of the optic nerve is suggested to have a critical importance in glaucoma by contributing to the injury of RGC axons. In conflict with this, mice are reported to lack a lamina cribrosa but develop glaucoma with striking similarities to the human disease. In this paper, we reevaluate the optic nerve anatomy of aged mice, and we provide strong experimental evidence for a direct insult to RGC axons within the glial lamina in a mouse model of glaucoma.
First, we show that aged DBA/2J mice have a robust network of astrocytes in the optic nerve where it exits the eye. These findings are consistent with previous studies (Morcos and Chan-Ling, 2000; Ding et al., 2002; May and Lutjen-Drecoll, 2002; Petros et al., 2006). A network of astrocytes also exists in the human lamina cribrosa, and the astrocytes cover plates of ECM (Anderson, 1969; Hernandez et al., 1987). In mice, however, ECM plates are absent, leading to the concept that the mouse optic nerve does not have a lamina cribrosa (Fujita et al., 2000; Morcos and Chan-Ling, 2000; May and Lutjen-Drecoll, 2002). Instead, based on our presented data and to reflect both the similarities and differences to the human lamina cribrosa, we suggest that mice have a glial lamina. This implies conservation of at least some functions, as both the location of the glial lamina and the spatial arrangement of astrocytes are similar in the two species.
Various studies are consistent with a direct insult to the RGC axon in the optic nerve in glaucoma, but current data fall short of experimentally demonstrating that the axon is insulted in the lamina in glaucoma (Introduction). Here, we demonstrate that the first visible damage to the intraocular portion of the RGC axon occurs in the glial lamina of DBA/2J mice, where both dystrophic neurites and milder axonal swellings are detected. Unlike a recent study, we did not observe that the damage was more likely to be localized around the entrance of the major blood vessels (May and Mittag, 2006), which enter the nerve at the anterior edge of the glial lamina.
Because the RGC somata of BAX-deficient DBA/2J mice survive direct axon injury (Libby et al., 2005b), we could use their eyes to test the proposition that an insult occurs within the lamina in glaucoma. In the peripheral nervous system, direct and focal axon injury results in degeneration of the entire length of the distal portion of the axon that is separated from the cell body by the lesion. The distal axon rapidly degenerates by Wallerian degeneration (Waller, 1850; Stoll et al., 2002). In contrast, the proximal portion of the axon that is attached to the cell body can survive up to the region of axon insult, as long as the cell body survives. If this were true for RGC axons in BAX-deficient DBA/2J mice, then the point to which the proximal axons survive would be in the proximity of a glaucoma insult.
Using BAX-deficient mice, we show that the proximal axon segment extending from the cell body survives to the proximity of the glial lamina in DBA2J glaucoma, but the more distal axon completely degenerates. This argues that RGC axons are insulted within or very close to the glial lamina. Alternatively, when considering this experiment alone, it is possible that a primary insult is to the cell body, resulting in progressive dying-back of the axon, from the brain toward the cell body. In this case, the proximal axons would survive into the nerve head if they are differentially supported by unknown factors at this location. Arguing against this alternative explanation, proximal RGC axons survive behind the eye and up to the region of insult in BAX-deficient mice following optic nerve crush. Thus, at least after crush, the axons survive to the region of insult and do not simply degenerate toward the eye until they reach a supportive environment. Together with our demonstration that the first identifiable damage to many axons is in the lamina and that there is a fan-shaped pattern of RGC death, which is only easily explained by damage to axon bundles in the optic nerve, these data provide strong experimental evidence for an early insult within the lamina.
Importantly, in DBA/2J mice that are wild type for Bax, many axons survive at the retinal surface and into the optic nerve but suddenly terminate at the lamina (Fig. 7). Because axon degeneration is asynchronous, this argues that the RGC cell bodies and their proximal axons survive for a significant time after degeneration of the distal axon separates them from the brain.
In glaucoma, it is suggested that focal axon injury in the lamina results in rapid Wallerian degeneration of the distal axon (Quigley et al., 1983; Whitmore et al., 2005). Although our studies suggest that this is true for some axons, the manner in which individual axons are insulted and then degenerate may differ even within individual eyes (depending on the severity of the axon insult, see Whitmore et al., 2005). More severe insults can result in almost complete compartmentalization of the proximal and distal axon segments and subsequent rapid Wallerian degeneration of the distal axon (Conforti et al., 2007; Whitmore et al., 2005). Milder insults may allow greater functional connectivity between the soma, proximal axon, and the distal axon segments and degeneration via the slower process of axonal dying-back. Our data suggest that the majority of damaged axons suffer milder insults early in DBA/2J glaucoma. Most damaged axons exhibited only mild swellings in the lamina (Fig. 5, Fig. S1, and associated text in Results). Consistent with milder insults to many axons in DBA/2J glaucoma, a recent study demonstrated axonal dying-back later in the disease (Schlamp et al., 2006).
Although our data strongly support the glial lamina as a key site of insult to RGC axons in DBA/2J glaucoma, they do not rule out additional sites of insult or early damage. It is possible that multiple parts of the RGC are insulted in glaucoma. Both microglia and oligodendrocytes may mediate damage at different locations (Tezel and Wax, 2004; May and Mittag, 2006; Nakazawa et al., 2006). Changes in RGC synapses in the retina and brain have been observed early in glaucoma, and dendritic changes were the earliest observed abnormalities in a primate model of glaucoma (Morgan et al., 1998; Weber et al., 2000; Whitmore et al., 2005; Gupta et al., 2007; Stevens et al., 2007). Nevertheless, the specific patterns of RGC death in glaucoma are only readily explained by a crucial axon insult in the lamina of the optic nerve.
In glaucoma, the nature of the axon insult in the lamina is not clearly defined. The ECM plates of the lamina cribrosa are suggested to mechanically damage axons in glaucoma, and individual variability in the structure of the plates is suggested to modulate glaucoma severity (Quigley et al., 1983). Our experimental data strongly support an axon insult in the glial lamina of DBA/2J mice, but the lamina of these mice lacks ECM plates at ages when glaucoma develops. This shows that ECM plates are not necessary to insult the axon in the lamina. Although it remains possible that the ECM plates may modulate damage in human glaucoma (as the nerve is much larger), these findings strongly focus attention on other components of the lamina such as the astrocytes. Astrocytes provide key support functions for neurons that may be lost in glaucoma (Newman, 2004; Jakobs et al., 2005; Perea and Araque, 2006). Alternatively, activated astrocytes may directly damage axons in the lamina (Johnson et al., 2000; Morgan, 2000).
Because an important insult damages the RGC axon in glaucoma, neuroprotective measures directed at the axon may be beneficial. The Wlds mutation can substantially slow axon degeneration. Axons in Wlds mice can conduct action potentials and release neurotransmitters up to 3 wk after transection (Crawford et al., 1995; Ribchester et al., 1995; Mack et al., 2001). Wlds is protective even in diseases that do not involve mechanical transection (Perry et al., 1991; Ferri et al., 2003; Samsam et al., 2003; Mi et al., 2005). Wlds can also protect synapses but unlike the axon protection, which persists in old mice, its beneficial effects on synapses substantially diminish with age (Gillingwater et al., 2002).
Here, we show that the Wlds mutation strongly protects against DBA/2J glaucoma with significant protection of both axons and somata at 12 mo of age. The Wlds mutation is known to protect against direct axon injury at similar ages, but no experiments have uncovered directly protective effects on the soma or synapse at this age (Glass et al., 1993; Deckwerth and Johnson, 1994; Deshmukh et al., 1996; Deckwerth et al., 1998; Gillingwater et al., 2002; Ikegami and Koike, 2003; Adalbert et al., 2005). Despite the profound protective effect of Wld s, it did not completely prevent shrinkage of RGC somata. The mild shrinkage may reflect a degree of trophic deprivation as a consequence of a persistent, partial decrease in axonal transport (Deshmukh et al., 1996; Deckwerth et al., 1998). In previous studies, Wlds has not completely prevented damage but rather delayed degeneration (Coleman, 2005). Further studies are needed to evaluate the duration of its protection in DBA/2J glaucoma, but our preliminary studies suggest a continued protection to 15 mo. Not all Wlds eyes were protected from glaucoma, suggesting that the degree of insult may vary in individual eyes, and/or that the level of WLDS protein may be close to a threshold for protection. There is some evidence that increasing the expression of Wlds confers a greater degree of protection (Mack et al., 2001). Future experiments will evaluate if mice homozygous for Wlds have an even greater degree of protection compared with the hemizygotes evaluated here.
In addition to protecting the axon and soma, the Wlds mutation prevented severe decline of PERG (at least until 12 mo). We found that PERG declines early in DBA/2J glaucoma, and before axon loss. The greatly diminished PERG may not reflect axon dysfunction because PERG is retained at high levels even in eyes with RGCs that have severely damaged and disconnected axons following axotomy (in mice that overexpress BCL2 (Cenni et al., 1996; Porciatti, 2007). Although we cannot discount a role of the proximal axon segment in maintaining PERG (the proximal axon is expected to survive in BCL2-overexpressing mice), it is clear that normally functioning, connected axons are not required for PERG. Thus, the severe decrease of PERG may reflect direct insult(s) to synapses, dendrites, or somata (Porciatti, 2007). Further studies are needed to determine the nature of the RGC compromise(s) resulting in decline of PERG and whether or not it precedes the axon insult in the glial lamina. It is possible that this early damage is a prerequisite for axon damage in the lamina. Alternatively, axon damage may be a completely independent process.
Materials and methods
Mouse husbandry
All experiments were conducted in accordance with the Association for Research in Vision and Ophthalmology's statement on the use of animals in ophthalmic research. Mice were maintained as previously described (Libby et al., 2005a).
Strain production and breeding
The original Tg(Thy1-CFP)23Jrs allele (Feng et al., 2000) was backcrossed to DBA/2J mice for 20 generations to create the DBA/2J.Tg(Thy1-CFP)23Jrs/Sj substrain (herein called D2.Thy1-CFP). The original Wlds allele (Lunn et al., 1989; Mack et al., 2001) was backcrossed into DBA/2J to create strain DBA/2J.BOla-Wld s/Sj (D2.Wlds). Initial assessment of Wlds and wild-type (nonWlds) littermates was at N5 with the majority of mice analyzed at higher generations (up to N10). To generate a sub-line of mice segregating for both the Wlds and Bax − alleles (Libby et al., 2005b), a D2.Bax +/− heterozygote (N16) was crossed to a D2.Wlds mouse and double mutants were selected for further backcrossing. Initial assessment of D2.WldsBax − double mutants and their single mutant or wild-type littermates was at N6 with many mice being analyzed at higher generations (up to N12). We produced the D2-Gpnmb + strain as recently described (Howell et al., 2007). For PERG, we used the control strain C57BL/6J-Tyrp1b.GpnmbR150X that develops the same iris disease as DBA/2J mice, but are resistant to developing high IOP and glaucoma (Anderson et al., 2006).
Analysis of glaucomatous damage
Eyes and intracranial portions of optic nerves were processed as previously reported (Smith et al., 2002). Nerves were stained with paraphenylenediamine (PPD), which differentially stains single damaged axons allowing sensitive detection of axon injury. Nerves were determined to have one of three damage levels that are readily distinguishable by axon counting (Fig. 3) (Anderson et al., 2005; Libby et al., 2005a,b). For the Wlds and Bax experiment, two masked investigators assigned the same damage level to 91% of the nerves (360 out of 399). The 39 nerves that were assigned different damage levels only differed by one level. A third masked investigator assessed these 39 nerves and always agreed with one of the original damage levels. The most commonly assigned level was used. Representative images were taken on an Olympus BX50 microscope.
Dissections and histochemical stains
Eyes with retrobulbar nerves attached were dissected from the orbit and fixed with 4% PFA in 0.1 M phosphate buffer as previously described (Smith et al., 2002). A square block of tissue (1 mm × 1 mm, consisting of optic nerve, flanking retina, choriod, and sclera) was dissected from the fixed posterior segment using a Supersharp scalpel blade. Tissue was embedded in paraffin and serially sectioned either longitudinally or in cross section. For most eyes, every fifth section was stained, and for a subset of eyes, every section was stained to allow a complete analysis of the anatomy and diagrammatic representation of the nerve as in Fig. 3. Histochemical stains included hematoxylin and eosin (H&E), Bodian, and Masson's trichrome (Luna, 1968). At least 10 nerves were analyzed with each of these stains for both DBA/2J and D2-Gpnmb + mice (11–12 mo old). Sections were imaged on an Olympus BX50 microscope.
Immunohistochemistry
Antibodies used were anti-neurofilaments (Smi-32 and Smi-34, Covance, 1:500) and anti-GFAP (Sigma-Aldrich, 1:500). Secondary antibodies were either Alexafluor 488 or 594 (Invitrogen). Nuclei were counterstained with DAPI (Sigma-Aldrich). Sections were analyzed on a Leica SP5 confocal microscope. Specific location within the optic nerve head was determined by staining every fifth section with Smi-32, Smi-34, and GFAP antibodies to identify landmarks, and by the position of the major blood vessels (shown diagrammatically in Fig. 3). Dystrophic neurites both stained strongly for Smi-32/Smi-34 (used in combination to maximize staining) and were greater in diameter (≥2 μm) than the largest nonmyelinated axons in the lamina of a normal nerve. Dystrophic neurite counts were performed using a 40× lens, with 3× digital zoom. For each region of interest (nerve fiber layer, prelamina, lamina, or retrolamina) from each eye, dystrophic neurites were manually counted in a 5-μm-thick section covering the entire area of the optic nerve.
Electron microscopy
Optic nerves were harvested as described above. The tissue and sections were processed as previously described (Smith et al., 2002). The entire nerve was serially sectioned longitudinally as follows. Several thick sections (1 μm) were cut followed by ∼60 thin sections (60 nm). This initial section set (thick and thin sections) was labeled “level 1.” Another set of thick and thin sections was then cut and labeled level 2. Sectioning progressed in this way across the entire nerve. Thick sections were used to identify the location of the most central portion of the optic nerve that included the full width of the scleral canal and the central retinal artery. In most cases, there were from two to four thick/thin section sets (central level sets) that included this area. The number of thin sections per grid varied, but was usually at least 12. The entire grid was scanned and selectively photographed. For any eye, at least two central levels and two different grids from each level were reviewed when determining anatomy. Therefore, for a specific nerve, 24–48 sections were scanned and photographed. Nerves were examined for 10 DBA/2J mice with no or early glaucoma (determined as described above), 10 D2-Gpnmb + mice, 6 DBA/2J mice with severe glaucoma, and 6 D2-Bax −/− mice with severe glaucoma. All mice were ∼11–12 mo old. All electron microscopy was performed on a JEOL-JEM1230 microscope.
4,000× magnification photographs were used to count dystrophic neurites. This magnification included ∼90% of the tissue contained within a single opening of a 200-mesh grid. Counts were performed for eight DBA/2J and eight D2-Gpnmb + mice. For each mouse, twenty 4,000× fields were counted for each of the nerve fiber layer, prelamina, and lamina regions (Fig. 3).
Detecting damage in CFP mice
D2.Thy1-CFP eyes were dissected free and fixed in 4% PFA. The optic nerve was trimmed to leave ∼100 μm attached behind the eye. The anterior segment, lens, and majority of sclera, choroid, and RPE were removed before the retina was mounted flat on a slide. A Leica SP5 confocal microscope was used to visualize CFP fluorescence. Location within the optic nerve head was determined using the position of the central retinal vessels as landmarks (Fig. 6).
To assess RGC axons from eye to brain, each eye was carefully removed with the proximal portion of the optic nerve attached and fixed in 4% PFA and mounted flat as described above. The intra-orbital optic nerve that extended from the cut site to the bony orbit (termed retro-lamina nerve) was dissected out and fixed in 4% PFA. The brain was dissected from the skull with the remaining portion of the optic nerve (retro-orbital to superior colliculus) attached. The retro-orbital portion of the optic nerve (up to the chiasm) was removed for assessment of glaucomatous damage. The brain, including optic nerve and optic tract from the chiasm to the superior colliculus, was fixed in 4% PFA and cryosectioned. For each mouse, five 20-μm sections from each region were analyzed for damage. The nerve fiber layer, lamina, retro-lamina, and chiasm were analyzed in cross section, and the beginning of the optic tract and the superior colliculus in longitudinal section. Eyes and nerves from three D2-CFP mice (9.5 mo) were assessed in this way (five eyes with “no or early” glaucoma, one with “moderate” glaucoma).
Agarose embedding and immunhistochemistry
The eyes were fixed in situ with 4% PFA and dissected, leaving the optic nerve and ring of sclera around the optic nerve head attached. The optic nerve was then cut directly behind the sclera with a scalpel blade. The whole tissue was incubated for 10 d with the primary antibodies (SMI-32 and anti-GFAP), and for 4 d with fluorescent secondary antibodies. Nuclei were counterstained with TOPRO3 (Invitrogen). The tissue was mounted in a slab of 4% agarose between two coverslips so both the retina and the optic nerve cross section could be imaged on a Leica SP5 confocal microscope.
Clinical Examination and IOP Measurement
Slit-lamp examination (Anderson et al., 2002) and IOP measurements (John et al., 1997; Savinova et al., 2001) were as described previously. At least 40 mice of each genotype were assessed at 9, 10.5, and 12 mo of age.
Controlled optic nerve crush
To determine if RGC axons survive up to the site of insult in D2.Bax −/− mutants, controlled crush was performed as previously described (Li et al., 1999; Libby et al., 2005b). Eyes were harvested 30 and 60 d after the procedure, ensuring that the window of cell death had already passed.
Assessing proximal axon survival in glaucoma
In our colony, ∼70% of mice with severe glaucoma at 10 mo of age developed it over the previous month (∼30 d, see Libby et al., 2005a). Thus, most mice with severe glaucoma at 10 mo of age lost the majority of their axons within the previous 30 d. Similarly, the majority of 11-mo-old mice with severe glaucoma lost their axons within the previous 60 d. Thus in terms of axon degeneration, our analysis of proximal axon survival in 10–11-mo-old mice (four 10-mo-old immunohistochenistry, six 11-mo-old EM) matches the timing of our similar analysis following optic nerve crush, where all mice lost their distal axons over the previous 30–60 d. After demonstrating that proximal axons ended very close to the lamina in 10- and 11-mo-old mice with severe glaucoma, we assessed some eyes at 13 mo of age (four eyes, plastic sections stained with H&E) to determine if there was greater die-back of the proximal axon. Finally, we analyzed three eyes at 19 mo of age. All given numbers refer to Bax −/− mice. Similar numbers of age-matched DBA/2J controls were analyzed.
Measurements of soma sizes
Nissl-stained retinas of DBA/2J or D2.Wlds mice were flat-mounted and eight representative images (two for each quadrant of the retina) were taken at 40× and imported into SigmaScan (Jandel Scientific). A square of 120 × 120 μm was placed onto the center of each image. Every cell body (RGCs and amacrine cells, exluding endothelial cells) either inside the square, or intersecting its upper or right border was measured, for a total of ∼600 somas. Soma sizes (in μm2) from the two groups (5 DBA/2J and 10 D2.Wlds retinas) were compared using the Mann and Whitney u-test.
Pattern electroretinography
Pattern electroretinography was measured as previously reported (Porciatti et al., 2007). Mice from our colony were shipped in a “masked” fashion to the University of Miami. Full-field, flash ERG was used as a control on all mice (an outer-retina signal that is not affected by retinal ganglion cells), and it did not differ significantly between control and glaucomatous DBA/2J mice.
Online supplemental material
The online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200706181/DC1. Fig. S1 shows the assessment of optic nerve damage from the retina to the superior colliculus in D2-CFP mice. Fig. S2 shows that proximal axons survive up to the site of optic nerve insult in D2.Bax −/− mice. Fig. S3 shows that proximal axons survive up to the lamina in D2.Bax −/− mice. Fig. S4 shows that the somal shrinkage in D2.Wlds mice is not limited to a few classes of somal size but occurs more generally. Fig. S5 shows that PERG is severely impaired early in DBA/2J glaucoma.
Supplemental Material
Acknowledgments
We thank L. Wilson, A. Bell, and I.M. Cosma for technical assistance with the experiments, J. Torrance for figure preparation, and G. Cox, P. Fuerst, S. Nair, and X. Zhu for critical reading of the manuscript. We also thank The Jackson Laboratory Scientific Services including P. Finger, J. Miller, and B. Mortimer for technical assistance.
Scientific support services are subsidized by a core grant from the National Cancer Institute (CA34196). This work was supported in part by EY017169 (R.H. Masland), F32EY014515 (R.T. Libby), and National Institutes of Health RO3 EY016322 and P30-EY14801 (V. Porciatti). R.H. Masland is a Senior Investigator of Research to Prevent Blindness. S.W.M. John is an Investigator of The Howard Hughes Medical Institute.
G.R. Howell and R.T. Libby contributed equally to this work.
R.T. Libby's present address is University of Rochester Eye Institute, University of Rochester Medical Center, Rochester, NY.
Abbreviations used in this paper: ECM, extracellular matrix; GFAP, glial fibrillary acidic protein; H&E, hematoxylin and eosin; PERG, pattern electroretinography; PPD, paraphenylenediamine; RGC, retinal ganglion cell.
References
- Adalbert, R., T.H. Gillingwater, J.E. Haley, K. Bridge, B. Beirowski, L. Berek, D. Wagner, D. Grumme, D. Thomson, A. Celik, et al. 2005. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur. J. Neurosci. 21:271–277. [DOI] [PubMed] [Google Scholar]
- Anderson, D.R. 1969. Ultrastructure of human and monkey lamina cribrosa and optic nerve head. Arch. Ophthalmol. 82:800–814. [DOI] [PubMed] [Google Scholar]
- Anderson, D.R. 1973. Ascending and descending optic atrophy produced experimentally in squirrel monkeys. Am. J. Ophthalmol. 76:693–711. [DOI] [PubMed] [Google Scholar]
- Anderson, D.R., and A. Hendrickson. 1974. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Invest. Ophthalmol. 13:771–783. [PubMed] [Google Scholar]
- Anderson, D.R., and A.E. Hendrickson. 1977. Failure of increased intracranial pressure to affect rapid axonal transport at the optic nerve head. Invest. Ophthalmol. Vis. Sci. 16:423–426. [PubMed] [Google Scholar]
- Anderson, M.G., R.S. Smith, N.L. Hawes, A. Zabaleta, B. Chang, J.L. Wiggs, and S.W. John. 2002. Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat. Genet. 30:81–85. [DOI] [PubMed] [Google Scholar]
- Anderson, M.G., R.T. Libby, D.B. Gould, R.S. Smith, and S.W. John. 2005. High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc. Natl. Acad. Sci. USA. 102:4566–4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, M.G., R.T. Libby, M. Mao, I.M. Cosma, L.A. Wilson, R.S. Smith, and S.W. John. 2006. Genetic context determines susceptibility to intraocular pressure elevation in a mouse pigmentary glaucoma. BMC Biol. 4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beirowski, B., R. Adalbert, D. Wagner, D.S. Grumme, K. Addicks, R.R. Ribchester, and M.P. Coleman. 2005. The progressive nature of Wallerian degeneration in wild-type and slow Wallerian degeneration (WldS) nerves. BMC Neurosci. 6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenni, M.C., L. Bonfanti, J.C. Martinou, G.M. Ratto, E. Strettoi, and L. Maffei. 1996. Long-term survival of retinal ganglion cells following optic nerve section in adult bcl-2 transgenic mice. Eur. J. Neurosci. 8:1735–1745. [DOI] [PubMed] [Google Scholar]
- Chang, B., R.S. Smith, N.L. Hawes, M.G. Anderson, A. Zabaleta, O. Savinova, T.H. Roderick, J.R. Heckenlively, M.T. Davisson, and S.W. John. 1999. Interacting loci cause severe iris atrophy and glaucoma in DBA/2J mice. Nat. Genet. 21:405–409. [DOI] [PubMed] [Google Scholar]
- Coleman, M. 2005. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 6:889–898. [DOI] [PubMed] [Google Scholar]
- Conforti, L., R. Adalbert, and M.P. Coleman. 2007. Neuronal death: where does the end begin? Trends Neurosci. 30:159–166. [DOI] [PubMed] [Google Scholar]
- Crawford, T.O., S.T. Hsieh, B.L. Schryer, and J.D. Glass. 1995. Prolonged axonal survival in transected nerves of C57BL/Ola mice is independent of age. J. Neurocytol. 24:333–340. [DOI] [PubMed] [Google Scholar]
- Danias, J., K.C. Lee, M.F. Zamora, B. Chen, F. Shen, T. Filippopoulos, Y. Su, D. Goldblum, S.M. Podos, and T. Mittag. 2003. Quantitative analysis of retinal ganglion cell (RGC) loss in aging DBA/2NNia glaucomatous mice: comparison with RGC loss in aging C57/BL6 mice. Invest. Ophthalmol. Vis. Sci. 44:5151–5162. [DOI] [PubMed] [Google Scholar]
- Deckwerth, T.L., and E.M. Johnson Jr. 1994. Neurites can remain viable after destruction of the neuronal soma by programmed cell death (apoptosis). Dev. Biol. 165:63–72. [DOI] [PubMed] [Google Scholar]
- Deckwerth, T.L., R.M. Easton, C.M. Knudson, S.J. Korsmeyer, and E.M. Johnson Jr. 1998. Placement of the BCL2 family member BAX in the death pathway of sympathetic neurons activated by trophic factor deprivation. Exp. Neurol. 152:150–162. [DOI] [PubMed] [Google Scholar]
- Deshmukh, M., J. Vasilakos, T.L. Deckwerth, P.A. Lampe, B.D. Shivers, and E.M. Johnson Jr. 1996. Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE family proteases. J. Cell Biol. 135:1341–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, L., K. Yamada, C. Takayama, and Y. Inoue. 2002. Development of astrocytes in the lamina cribrosa sclerae of the mouse optic nerve, with special reference to myelin formation. Okajimas Folia Anat. Jpn. 79:143–157. [DOI] [PubMed] [Google Scholar]
- Fechtner, R.D., and R.N. Weinreb. 1994. Mechanisms of optic nerve damage in primary open angle glaucoma. Surv. Ophthalmol. 39:23–42. [DOI] [PubMed] [Google Scholar]
- Feng, G., R.H. Mellor, M. Bernstein, C. Keller-Peck, Q.T. Nguyen, M. Wallace, J.M. Nerbonne, J.W. Lichtman, and J.R. Sanes. 2000. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 28:41–51. [DOI] [PubMed] [Google Scholar]
- Ferri, A., J.R. Sanes, M.P. Coleman, J.M. Cunningham, and A.C. Kato. 2003. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr. Biol. 13:669–673. [DOI] [PubMed] [Google Scholar]
- Filippopoulos, T., J. Danias, B. Chen, S.M. Podos, and T.W. Mittag. 2006. Topographic and morphologic analyses of retinal ganglion cell loss in old DBA/2NNia mice. Invest. Ophthalmol. Vis. Sci. 47:1968–1974. [DOI] [PubMed] [Google Scholar]
- Fujita, Y., T. Imagawa, and M. Uehara. 2000. Comparative study of the lamina cribrosa and the pial septa in the vertebrate optic nerve and their relationship to the myelinated axons. Tissue Cell. 32:293–301. [DOI] [PubMed] [Google Scholar]
- Gillingwater, T.H., D. Thomson, T.G. Mack, E.M. Soffin, R.J. Mattison, M.P. Coleman, and R.R. Ribchester. 2002. Age-dependent synapse withdrawal at axotomised neuromuscular junctions in Wld(s) mutant and Ube4b/Nmnat transgenic mice. J. Physiol. 543:739–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass, J.D., T.M. Brushart, E.B. George, and J.W. Griffin. 1993. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J. Neurocytol. 22:311–321. [DOI] [PubMed] [Google Scholar]
- Gupta, N., T. Ly, Q. Zhang, P.L. Kaufman, R.N. Weinreb, and Y.H. Yucel. 2007. Chronic ocular hypertension induces dendrite pathology in the lateral geniculate nucleus of the brain. Exp. Eye Res. 84:176–184. [DOI] [PubMed] [Google Scholar]
- Hernandez, M.R. 2000. The optic nerve head in glaucoma: role of astrocytes in tissue remodeling. Prog. Retin. Eye Res. 19:297–321. [DOI] [PubMed] [Google Scholar]
- Hernandez, M.R., X.X. Luo, F. Igoe, and A.H. Neufeld. 1987. Extracellular matrix of the human lamina cribrosa. Am. J. Ophthalmol. 104:567–576. [DOI] [PubMed] [Google Scholar]
- Hernandez, M.R., X.X. Luo, W. Andrzejewska, and A.H. Neufeld. 1989. Age-related changes in the extracellular matrix of the human optic nerve head. Am. J. Ophthalmol. 107:476–484. [DOI] [PubMed] [Google Scholar]
- Howell, G.R., R.T. Libby, J.K. Marchant, L.A. Wilson, I.M. Cosma, R.S. Smith, M.G. Anderson, and S.W. John. 2007. Absence of glaucoma in DBA/2J mice homozygous for wild-type versions of Gpnmb and Tyrp1. BMC Genet. 8:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegami, K., and T. Koike. 2003. Non-apoptotic neurite degeneration in apoptotic neuronal death: pivotal role of mitochondrial function in neurites. Neuroscience. 122:617–626. [DOI] [PubMed] [Google Scholar]
- Jakobs, T.C., R.T. Libby, Y. Ben, S.W. John, and R.H. Masland. 2005. Retinal ganglion cell degeneration is topological but not cell type specific in DBA/2J mice. J. Cell Biol. 171:313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, S.W., J.R. Hagaman, T.E. MacTaggart, L. Peng, and O. Smithes. 1997. Intraocular pressure in inbred mouse strains. Invest. Ophthalmol. Vis. Sci. 38:249–253. [PubMed] [Google Scholar]
- Johnson, E.C., L.M. Deppmeier, S.K. Wentzien, I. Hsu, and J.C. Morrison. 2000. Chronology of optic nerve head and retinal responses to elevated intraocular pressure. Invest. Ophthalmol. Vis. Sci. 41:431–442. [PubMed] [Google Scholar]
- Kerschensteiner, M., M.E. Schwab, J.W. Lichtman, and T. Misgeld. 2005. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 11:572–577. [DOI] [PubMed] [Google Scholar]
- Lappe-Siefke, C., S. Goebbels, M. Gravel, E. Nicksch, J. Lee, P.E. Braun, I.R. Griffiths, and K.A. Nave. 2003. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat. Genet. 33:366–374. [DOI] [PubMed] [Google Scholar]
- Li, Y., C.L. Schlamp, and R.W. Nickells. 1999. Experimental induction of retinal ganglion cell death in adult mice. Invest. Ophthalmol. Vis. Sci. 40:1004–1008. [PubMed] [Google Scholar]
- Libby, R.T., M.G. Anderson, I.H. Pang, Z.H. Robinson, O.V. Savinova, I.M. Cosma, A. Snow, L.A. Wilson, R.S. Smith, A.F. Clark, and S.W. John. 2005. a. Inherited glaucoma in DBA/2J mice: pertinent disease features for studying the neurodegeneration. Vis. Neurosci. 22:637–648. [DOI] [PubMed] [Google Scholar]
- Libby, R.T., Y. Li, O.V. Savinova, J. Barter, R.S. Smith, R.W. Nickells, and S.W. John. 2005. b. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS. Genet. 1:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna, L. 1968. Manual of Histological Staining Methods of the AFIP. McGraw-Hill, New York. 199 pp.
- Lunn, E.R., V.H. Perry, M.C. Brown, H. Rosen, and S. Gordon. 1989. Absence of Wallerian degeneration does not hinder regeneration in peripheral nerve. Eur. J. Neurosci. 1:27–33. [DOI] [PubMed] [Google Scholar]
- Mack, T.G., M. Reiner, B. Beirowski, W. Mi, M. Emanuelli, D. Wagner, D. Thomson, T. Gillingwater, F. Court, L. Conforti, et al. 2001. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 4:1199–1206. [DOI] [PubMed] [Google Scholar]
- Maumenee, A.E. 1983. Causes of optic nerve damage in glaucoma. Robert N. Shaffer lecture. Ophthalmology. 90:741–752. [DOI] [PubMed] [Google Scholar]
- May, C.A., and E. Lutjen-Drecoll. 2002. Morphology of the murine optic nerve. Invest. Ophthalmol. Vis. Sci. 43:2206–2212. [PubMed] [Google Scholar]
- May, C.A., and T. Mittag. 2006. Optic nerve degeneration in the DBA/2NNia mouse: is the lamina cribrosa important in the development of glaucomatous optic neuropathy? Acta Neuropathol. 111:158–167. [DOI] [PubMed] [Google Scholar]
- Mi, W., B. Beirowski, T.H. Gillingwater, R. Adalbert, D. Wagner, D. Grumme, H. Osaka, L. Conforti, S. Arnhold, K. Addicks, et al. 2005. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain. 128:405–416. [DOI] [PubMed] [Google Scholar]
- Morcos, Y., and T. Chan-Ling. 2000. Concentration of astrocytic filaments at the retinal optic nerve junction is coincident with the absence of intra-retinal myelination: comparative and developmental evidence. J. Neurocytol. 29:665–678. [DOI] [PubMed] [Google Scholar]
- Morgan, J.E. 2000. Optic nerve head structure in glaucoma: astrocytes as mediators of axonal damage. Eye. 14(Pt 3B):437–444. [DOI] [PubMed] [Google Scholar]
- Morgan, J.E., G. Jeffery, and A.J. Foss. 1998. Axon deviation in the human lamina cribrosa. Br. J. Ophthalmol. 82:680–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa, T., C. Nakazawa, A. Matsubara, K. Noda, T. Hisatomi, H. She, N. Michaud, A. Hafezi-Moghadam, J.W. Miller, and L.I. Benowitz. 2006. Tumor necrosis factor-α mediates oligodendrocyte death and delayed retinal ganglion cell loss in a mouse model of glaucoma. J. Neurosci. 26:12633–12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, E.A. 2004. Glial modulation of synaptic transmission in the retina. Glia. 47:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea, G., and A. Araque. 2006. Synaptic information processing by astrocytes. J. Physiol. (Paris). 99:92–97. [DOI] [PubMed] [Google Scholar]
- Perry, V.H., M.C. Brown, E.R. Lunn, P. Tree, and S. Gordon. 1990. Evidence that very slow Wallerian degeneration in C57BL/Ola mice is an intrinsic property of the peripheral nerve. Eur. J. Neurosci. 2:408–413. [DOI] [PubMed] [Google Scholar]
- Perry, V.H., M.C. Brown, and E.R. Lunn. 1991. Very slow retrograde and Wallerian degeneration in the CNS of C57BL/ola mice. Eur. J. Neurosci. 3:102–105. [DOI] [PubMed] [Google Scholar]
- Petros, T.J., S.E. Williams, and C.A. Mason. 2006. Temporal regulation of EphA4 in astroglia during murine retinal and optic nerve development. Mol. Cell. Neurosci. 32:49–66. [DOI] [PubMed] [Google Scholar]
- Porciatti, V. 2007. The mouse pattern electroretinogram. Doc. Ophthalmol. 115:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porciatti, V., M. Saleh, and M. Nagaraju. 2007. The pattern electroretinogram as a tool to monitor progressive retinal ganglion cell dysfunction in the DBA/2J mouse model of glaucoma. Invest. Ophthalmol. Vis. Sci. 48:745–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley, H.A., and E.M. Addicks. 1980. Chronic experimental glaucoma in primates. II. Effect of extended intraocular pressure elevation on optic nerve head and axonal transport. Invest. Ophthalmol. Vis. Sci. 19:137–152. [PubMed] [Google Scholar]
- Quigley, H.A., and E.M. Addicks. 1981. Regional differences in the structure of the lamina cribrosa and their relation to glaucomatous optic nerve damage. Arch. Ophthalmol. 99:137–143. [DOI] [PubMed] [Google Scholar]
- Quigley, H., and D.R. Anderson. 1976. The dynamics and location of axonal transport blockade by acute intraocular pressure elevation in primate optic nerve. Invest. Ophthalmol. 15:606–616. [PubMed] [Google Scholar]
- Quigley, H.A., and D.R. Anderson. 1977. Distribution of axonal transport blockade by acute intraocular pressure elevation in the primate optic nerve head. Invest. Ophthalmol. Vis. Sci. 16:640–644. [PubMed] [Google Scholar]
- Quigley, H.A., J. Guy, and D.R. Anderson. 1979. Blockade of rapid axonal transport. Effect of intraocular pressure elevation in primate optic nerve. Arch. Ophthalmol. 97:525–531. [DOI] [PubMed] [Google Scholar]
- Quigley, H.A., R.W. Flower, E.M. Addicks, and D.S. McLeod. 1980. The mechanism of optic nerve damage in experimental acute intraocular pressure elevation. Invest. Ophthalmol. Vis. Sci. 19:505–517. [PubMed] [Google Scholar]
- Quigley, H.A., E.M. Addicks, W.R. Green, and A.E. Maumenee. 1981. Optic nerve damage in human glaucoma. II. The site of injury and susceptibility to damage. Arch. Ophthalmol. 99:635–649. [DOI] [PubMed] [Google Scholar]
- Quigley, H.A., R.M. Hohman, E.M. Addicks, R.W. Massof, and W.R. Green. 1983. Morphologic changes in the lamina cribrosa correlated with neural loss in open-angle glaucoma. Am. J. Ophthalmol. 95:673–691. [DOI] [PubMed] [Google Scholar]
- Ribchester, R.R., J.W. Tsao, J.A. Barry, N. Asgari-Jirhandeh, V.H. Perry, and M.C. Brown. 1995. Persistence of neuromuscular junctions after axotomy in mice with slow Wallerian degeneration (C57BL/WldS). Eur. J. Neurosci. 7:1641–1650. [DOI] [PubMed] [Google Scholar]
- Samsam, M., W. Mi, C. Wessig, J. Zielasek, K.V. Toyka, M.P. Coleman, and R. Martini. 2003. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J. Neurosci. 23:2833–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinova, O.V., F. Sugiyama, J.E. Martin, S.I. Tomarev, B.J. Paigen, R.S. Smith, and S.W. John. 2001. Intraocular pressure in genetically distinct mice: an update and strain survey. BMC Genet. 2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlamp, C.L., Y. Li, J.A. Dietz, K.T. Janssen, and R.W. Nickells. 2006. Progressive ganglion cell loss and optic nerve degeneration in DBA/2J mice is variable and asymmetric. BMC Neurosci. 7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields, M.B. 1997. Shields' Textbook of Glaucoma. Williams and Wilkins, Baltimore, MD. 588 pp.
- Smith, R.S., A. Zabaleta, S.W. John, L.S. Bechtold, and L. Ikeda. 2002. General and specific histopathology. In Systematic Evaluation of the Mouse Eye. R.S. Smith, editor. CRC Press, New York. 265–297.
- Steele, M.R., D.M. Inman, D.J. Calkins, P.J. Horner, and M.L. Vetter. 2006. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest. Ophthalmol. Vis. Sci. 47:977–985. [DOI] [PubMed] [Google Scholar]
- Stevens, B., N.J. Allen, L.E. Vazquez, K.S. Christopherson, N. Nouri, G.R. Howell, D.K. Micheva, A.K. Mehelow, A.D. Huberman, B. Stafford, et al. 2007. The classical complement cascade mediates developmental CNS synapse elimination. Cell. In press. [DOI] [PubMed]
- Stoll, G., S. Jander, and R.R. Myers. 2002. Degeneration and regeneration of the peripheral nervous system: from Augustus Waller's observations to neuroinflammation. J. Peripher. Nerv. Syst. 7:13–27. [DOI] [PubMed] [Google Scholar]
- Tansley, K. 1956. Comparison of the lamina cribrosa in mammalian species with good and with indifferent vision. Br. J. Ophthalmol. 40:178–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel, G., and M.B. Wax. 2004. The immune system and glaucoma. Curr. Opin. Ophthalmol. 15:80–84. [DOI] [PubMed] [Google Scholar]
- Vickers, J.C. 1997. A cellular mechanism for the neuronal changes underlying Alzheimer's disease. Neuroscience. 78:629–639. [DOI] [PubMed] [Google Scholar]
- Waller, A. 1850. Experiments on the section of glossopharyngeal and hypoglossal nerves of the frog and observations of the alternatives produced thererby in the structure of their primitive fibres. Philos. Trans. R. Soc. Lnd. Biol. 140:423. [Google Scholar]
- Weber, A.J., H. Chen, W.C. Hubbard, and P.L. Kaufman. 2000. Experimental glaucoma and cell size, density, and number in the primate lateral geniculate nucleus. Invest. Ophthalmol. Vis. Sci. 41:1370–1379. [PubMed] [Google Scholar]
- Whitmore, A.V., R.T. Libby, and S.W. John. 2005. Glaucoma: thinking in new ways-a role for autonomous axonal self-destruction and other compartmentalised processes? Prog. Retin. Eye Res. 24:639–662. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
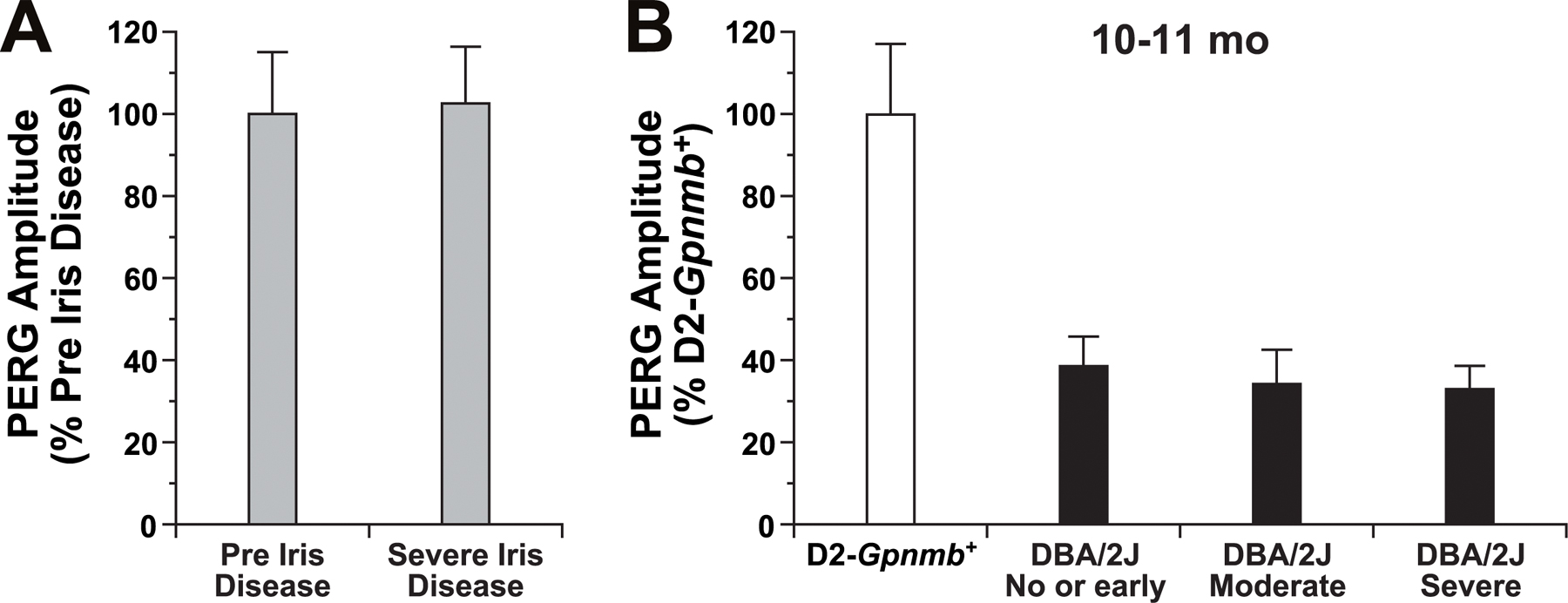{kind=link}
{kind=link}