

Abstract
An exactly solvable model based on the topology of a protein native state is applied to identify bottlenecks and key sites for the folding of human immunodeficiency virus type 1 (HIV-1) protease. The predicted sites are found to correlate well with clinical data on resistance to Food and Drug Administration-approved drugs. It has been observed that the effects of drug therapy are to induce multiple mutations on the protease. The sites where such mutations occur correlate well with those involved in folding bottlenecks identified through the deterministic procedure proposed in this study. The high statistical significance of the observed correlations suggests that the approach may be promisingly used in conjunction with traditional techniques to identify candidate locations for drug attacks.
Keywords: Protein-folding modeling, prediction of key folding sites, HIV-1 protease, drug resistance
One of the open fundamental questions in molecular biology is how to predict the folded state of a protein from the knowledge of its sequence. Despite a large increase in available computing power in the past years, it has been impossible to answer this question by means of computer simulations of various degrees of complexity and detail. However, an increasing amount of experimental (Fersht 1995; Plaxco et al. 1998; Riddle et al. 1998; Chiti et al. 1999; Martinez and Serrano 1999) and theoretical results (Alm and Baker 1999; Micheletti et al. 1999; Clementi et al. 2000; Hoang and Cieplak 2000; Maritan et al. 2000) supports the view that the folding of natural proteins into their native state is largely influenced by the native-state topology (for a brief review see Baker 2000). Accordingly, the folding process is regarded as a well-defined sequence of obligatory steps to be taken to reach the native state. Even if protein sequences have evolved to fold efficiently, the kinetics en-route to the native state might be hindered by the realization of particularly difficult (rate-limiting) steps, such as the formation of nonlocal amino-acid interactions (contacts) that usually requires the overcoming of large entropy barriers. Some nonlocal native contacts are rather crucial for the folding process, because their formation helps in establishing further native interactions and leads to a rapid progress along the folding pathway until another barrier is met. Their formation is associated to bottlenecks for the entire folding process. Strikingly, the amino acids involved in such crucial contacts are those for which the largest changes in the folding kinetics are observed in site-directed mutagenesis experiments (Fersht 1995), as first proven for CI2 and Barnase (Micheletti et al. 1999). This suggests that protein sequences have been optimized carefully so as to exploit the conformational entropy reduction accompanying the folding process (Wolynes et al. 1995) through the selection of the key amino acids. The number and importance of bottlenecks depends significantly on several factors. Among the most important are the contact order of the protein (Alm and Baker 1999) and whether it folds in two or more stages (Jackson 1998).
In previous studies (Cecconi et al. 2001; Settanni et al. 2001), we have shown how the most delicate folding stages can be identified within a molecular dynamics approach, by monitoring the formation probability of native and nonnative contacts from the unfolded to the native state. This can either be done as a function of time at a fixed temperature around the folding temperature or working at thermal equilibrium for a succession of decreasing temperatures (annealing). In principle, the two approaches need not be equivalent but, for the quantities we have investigated, they give consistent results. Then, concerning the identification of crucial contacts, one can safely concentrate on studying thermodynamic equilibrium at various temperatures. The main limitation of molecular dynamics (MD) and Monte-Carlo (MC) simulations, especially for long protein chains, is that they are extremely time demanding and plagued with statistical errors that can affect the predictions based on the study of the relative sensitivity of contact formation. Therefore it would be highly desirable to develop a suitable theoretical model, amenable to a deterministic (and computationally fast) treatment, thus resulting in a deeper understanding of the problem. Ideally, such a model should encompass all the "necessary ingredients" that usually are included in computer simulations: peptide-chain constraints, effective interactions between residues, favorable monomeric positions, and so forth. In the following, we describe a recently developed theoretical scheme (Micheletti et al. 2001a), that, while being very simplified and approximate compared to other schemes based on MD or MC simulations, can be treated analytically, leading to expressions that can be evaluated exactly. The calculated quantities rival those obtained through more sophisticated but computationally demanding MC and MD techniques. The purpose of this paper is to show how the model can be employed to yield helpful observables to identify the folding bottlenecks. In particular, we apply the method to the human immunodeficiency virus type 1 protease (HIV-1 PR), an enzyme that is crucially involved in the HIV infection (Condra et al. 1995). In general, the accurate knowledge of bottlenecks has important pharmaceutical ramifications because their knowledge may be exploited in a rational drug design. Because of the large amount of available clinical data, HIV-1 PR is a natural choice for a stringent test of our automated predictive scheme.
Theory
The model we adopt builds on the importance of the native-state topology in steering the folding process, that is, in bringing into contact pairs of amino acids that are found in interaction in the native state. A primary quantity of interest that we shall calculate is the probability that a given native contact is established at a definite stage of the folding process. Probably, the oldest attempt to calculate such quantity dates back to Flory, who tried to estimate the probability pij that two sites i and j in a long harmonic chain (the peptide) are in contact (Flory 1956). The approximation introduced by Flory was to neglect correlations between residues, which amounts to considering the chain embedded in a highly dimensional space. As a result, the pij's are a decreasing function of the sequence separation |i–j|. Clearly, this approximation is not apt to pinpoint the key folding sites, as it exploits the native topology at the simplest level; in fact, it takes into account only the contact order of native interactions. The Flory approach, however, can be refined by incorporating correlations between the formation of pairs, triplets, etc., of contacts (Chan and Dill 1990; Camacho and Thirumalai 1995; Debe and Goddard III 1999). Here, we use a recently introduced energy function that allows us to calculate the pij's within a self-consistent analytic scheme. The strategy is similar in spirit to that of Go and Scheraga (1976) where only the formation of native interactions is energetically rewarded and is common to all recent approaches, which exploits the native-state topology (Alm and Baker 1999; Micheletti et al. 1999; Clementi et al. 2000; Hoang and Cieplak 2000; Maritan et al. 2000).
We describe the proteins by the coordinates ri of the Cα atom of the i-th amino acids. The simplified energy functional for the chain of N residues is
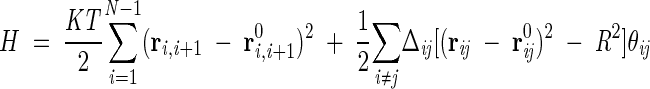 |
(1) |
where K is the strength of the peptide bonds, assumed to be harmonic, and T is the absolute temperature in units of the Boltzmann constant.
The relative position between amino-acid centroids is denoted by rij = ri−rj and the corresponding native positions are indicated with the superscript 0. Δ is the contact matrix, whose element Δij is 1 if residues i and j are in contact in the native state (i.e., their Cα separation is below the cutoff c = 6.5 Å) and 0 otherwise. The matrix Δij along with the set r0ij encodes the topology of the protein. The factor θij has the form
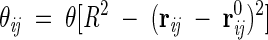 |
(2) |
where θ(✗) is the unitary step function and R is a distance cutoff defining the range of the interaction between nonconsecutive amino acids. In standard off-lattice approaches, the interaction V(d) between nonbonded amino acids at a distance d, is taken to be a square-well potential, or some type of Lennard-Jones interaction. Our choice in equation 1 is a sort of "harmonic well" which, while being physically sound and viable, is suitable for a self-consistent treatment, as explained below. The location of the outer rim of the well is controlled by R, which can be set to a few Angstroms (R = 3 Å in the present study) to penalize conformations where the separation of two residues differs significantly from the native one. In the native state, each θij is close to 1, while in the denaturated state, cases usually are negligible.
While the present form of the model does not accurately describe the effects of self-avoidance, this does not lead to a qualitatively wrong behavior in the highly denatured ensemble (large T ). The treatment of steric effects becomes progressively more accurate as temperature is lowered. In fact, the model guarantees that the native state is the true ground state, and therefore protein conformations found at low temperature inherit the native self-avoidance. The connectedness of the chain, as well as its entropy, are captured in a simple but nontrivial manner. The most significant advantage of the model is that it can be used to explore the equilibrium thermodynamics without being hampered by inaccurate or sluggish dynamics.
Two limit cases of the model described by equation 1 are worthy of notice. In the absence of any bias towards the target structure (i.e., when both Δij and the {r0}'s are removed) the model reduces to the standard Gaussian polymer model whose behavior is exactly known (Flory 1956; Kloczkowski and Jernigan 1999). Furthermore, the limit when T→0 (when all native contacts are established and the bonded-energy term fluctuations are negligible) the model reduces to the Gaussian network model that has been introduced and used to study the near-native vibrational properties of several proteins (Bahar et al. 1997, 1999; Keskin et al. 2000; Atilgan et al. 2001).
The thermodynamics of the model are fully determined by the partition function
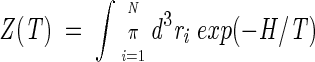 |
(3) |
In the integral of equation 3 and in the following, it is always meant that translational invariance is explicitly broken by fixing, for example, the center of mass of the system (see Appendix).
The integral (3) is still hard to treat analytically, because of the presence of nonquadratic interactions in the last term of Hamiltonian (1). We thus perform a further, but nontrivial, simplification by replacing H with the variational Hamiltonian H0
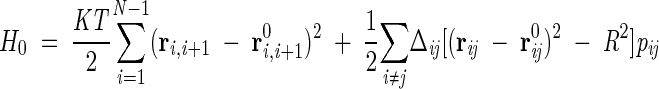 |
(4) |
where the factors θij are now substituted by parameters independent of the coordinates. Because of its quadratic form, the model described by equation 4 can be solved with the standard techniques for Gaussian integrals. Such parameters have to be optimally determined so as to ensure self-consistency:
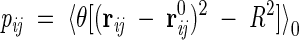 |
(5) |
The symbol 〈. . .〉0 indicates that the thermal averages are performed through the Hamiltonian H0. In such self-consistent approach, the problem is fully solved and we can compute the resulting partition function from which we extract all the thermal properties and averages. In particular, the logarithm of the partition function Z has the following explicit expression:
 |
(6) |
where the matrix M is defined as
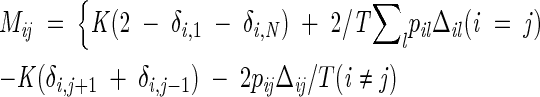 |
(7) |
and the prime in equation 6 denotes that the zero eigenvalue of M has to be omitted (see Appendix).
The quantities pij in equation 5 represent precisely the occurrence probability of a contact between residues i and j and indicate the frequency with which that native contact is established. At thermal equilibrium, their dependence on temperature reflects the status of compactness of the protein molecule. For instance, well below the folding temperature, Tf, each pij is expected to assume a value close to unity, as all native contacts are already formed. Instead, for temperatures much larger than Tf, all pij(T) tend to be very small, reflecting the low propensity of the protein to establish contacts. Thermodynamics quantities can be easily derived from the pij's. Another quantity necessary to characterize the folding transition is the specific heat, which exhibits one or more peaks in correspondence of significant structural rearrangements of the protein conformation. Because every energy change is mainly associated to the formation of native interactions, we address the question of which native contacts contribute mainly to the peak(s) of the specific heat. A clear answer to this question is found readily in the temperature behavior of frequencies pij. Indeed, each pij(T) exhibits a sigmoidal dependence of temperature, and the modulus of its temperature derivative develops a sharp maximum in correspondence to the point of inflection (crossover temperature). The importance of every native contact i–j turns out to be characterized by the crossover temperature and the maximum slope of its pij, which can be regarded as an indicator of its degree of cooperativity. In fact, the most important contacts are those with high crossover temperature and associated high cooperativity. This fact allows a complete identification and classification of the bottlenecks, because we are now able to identify those contacts that are thermodynamically relevant to peaks and shoulders of the specific heat.
Application to HIV-1 protease
The HIV encodes a protease, HIV-1 PR, whose inhibition is crucial to prevent the maturation of infectious HIV particles (Condra et al. 1995). The role of the protease in infection spreading is to act as a "molecular scissor", cleaving inactive viral polyproteins into smaller, functional proteins. In the presence of protease inhibitors, viral particles are unable to mature and are cleared rapidly. Extensive clinical trials have led to the development of the following five HIV-1 PR inhibitors that are approved by the Food and Drug Administration (FDA): Saquinavir mesylate (SAQ), Ritonavir (RIT), Indinavir sulfate (IND), Nelfinavir mesylate (NLF), and Amprenavir (APR) (Ala et al. 1998). Such drugs particularly are effective in short-term treatments, while resistance limits their long-term efficacy.
Indeed, mutants resistant to protease inhibitors can emerge in vivo after <1 year (Condra et al. 1995). Table 1 summarizes the list of HIV-1 PR known mutating sites causing drug resistance.
Table 1.
Mutations in the protease associated with FDA-approved drug resistance (Ala et al. 1998)
| Drug | Point mutations |
| RTN (Molla et al. 1996; Markowitz et al. 1995) | 20,33,35,36,46,54,63,71,82,84,90 |
| NLF (Patick et al. 1996) | 30,46,63,71,77,84 |
| IND (Condra et al. 1995; Tisdale et al. 1995) | 10,32,46,63,71,82,84 |
| SQV (Condra et al. 1995; Tisdale et al. 1995; Jacobsen et al. 1996) | 10,46,48,63,71,82,84,90 |
| APR (Reddy and Ross 1999) | 46,63,82,84 |
In an earlier work, the study of the near-native harmonic vibrations of the HIV-1 PR has shown that a number of sites that are paramount to the stability of the native enzyme are close to some of the residue of Table 1 (Bahar et al. 1999). The self-consistent scheme of equation 4 allows us to extend this result by modeling the partially folded ensemble at finite temperature.
In particular, we will be concerned with the characterization of such an ensemble near the folding transition temperature. The motivation to do so stems from a recent study (Cecconi et al. 2001) where we have shown that such mutating amino acids correspond, with high statistical significance, to sites involved in the folding kinetic bottlenecks. The rationale for this finding is that the most effective drugs can be eluded only by mutations occurring in correspondence of the key sites. Because of the sensitivity of the folded native conformation to these sites, only fine-tuned mutations are allowed in correspondence to these sites. Such mutations have to result in a native-like enzymatic activity and in the avoidance of the drug action. These constraints act as a severe selective pressure on the mutated proteases that the HIV virus is able to express. As a result, the mutations that ultimately will cause drug resistance are expected to occur in correspondence to the crucial sites. These residues are influenced heavily by the native topology and hence should display little dependence on the particular (effective) drug to be eluded.
It is therefore our purpose to apply the scheme introduced in the previous section and identify the key residues within our topology-based scheme. The method, being completely analytic, is free from statistical uncertainty, common to all MC and MD simulation methods, or from difficulty (as a result of spatial restraints) to reach the target native state below the folding temperature.
Results and Discussion
The structural model at the basis of our analysis is the free enzyme (Condra et al. 1995). It is a homodimer with C2 symmetry, each subunit being composed of 99 residues (Fig. 1 ▶). Previous studies (Cecconi et al. 2001) have shown that geometrically important residue positions can be obtained by considering a single monomer. Indeed, the specific heat of the whole homodimer on decreasing the temperature shows a peak in correspondence of the folding of each subunit, and then at lower temperature, another peak signals the aggregation of the two subunits. Thus, in the following, we will be concerned only with a single monomer. The specific heat is obtained through numeric differentiation of the average internal energy, which has the following explicit analytic expression in terms of the pij(T)'s and the quantities introduced before:
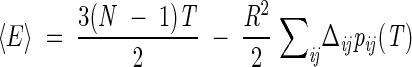 |
(8) |
The study of Go and Scheraga (1976) showed that systems described by energy-scoring functions that reward the formation of native contacts display cooperative (all-or-none) folding transitions with an associated peak(s) in the specific heat. Consistently with these expectations, the specific heat calculated by differentiating equation 8 with respect to T shows a single peak (Fig. 2 ▶), thus providing an unambiguous criterion for identifying the folding transition temperature TF. The width of the specific heat peak at the folding transition in Figure 2 ▶ is larger than the typical one found in experimental (Jackson 1998) and theoretical studies (Kaya and Chan 2000, 2001). The cooperativity of the transition can be enhanced by intervening on the actual value of K in equation 1; in fact, a decrease of K leads to sharper transitions. An alternative criterion for fixing the value of K is provided by its influence on the average amount of native structure that is formed at the native state. Because we are particularly interested in monitoring the progressive establishment of native contacts, we adopted this second possibility to set the value of K. In fact, by choosing K = 1/15 in equation 1, we ensure that, at TF, the average fractional occupation of native contacts, q:
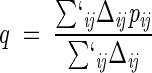 |
(9) |
is about 50% (see Fig. 2 ▶), as established in several experiments and numerical studies. The primed summation symbol indicates that the sum is not carried out over consecutive pairs. The degree of native similarity, q is a useful overall indicator to monitor the progress toward the native state in a folding process (Camacho and Thirumalai 1993; Sali et al. 1994). While the ultimate quantities of interest are the pij's, it is useful to consider an intermediate level of description and focus on the whole network of contacts that a given site takes part in. A natural order parameter is provided by the "average environment formation" (Lazaridis and Karplus 1997; Galzitskaya and Finkelstein 1999) which, for a generic site, i is defined as
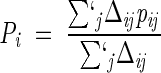 |
(10) |
Pi is a measure of the fraction of established native contacts the i-th residue precipitates to (clearly, Pi is defined only when the denominator of equation 10 is nonzero). The environment profiles for three different temperatures are shown in Figure 3 ▶. The irregular behavior of the profiles results from a complex interplay of the burial of the sites and the locality of their contacts. The hierarchical formation of secondary structures at high temperature is clearly visible. It is instructive to correlate the location of the sites known to cause resistance to drug treatments (see Table 1) with the features of the profiles. In particular, several mutating sites responsible for drug resistance (see Table 1) can be found in correspondence of the peaks of the environments (see, in particular, sites 20, 63, 71, 77, and 84). The most precise way to identify the key residues is, however, through the analysis of the fractional occupation of native contacts and not through the environments, as they only carry averaged information. Typical pij curves as a function of temperatures are shown in Figure 4 ▶. As anticipated in the Theory section, all pij's have monotonic sigmoidal shapes that mainly reflect the sequence separation, |i–j| and the native burial of each of the residues. In general, each contact is established at a different crossover temperature and with different intensity (Cecconi et al. 2001). The data relative to the frequencies of native-contact formation is conveniently summarized in the color-coded contact maps of Figure 5 ▶. A bright red color is used to highlight those contacts with the largest crossover temperatures above TF, see Figure 5A ▶, or highest intensity in Figure 5B ▶. Both of these intuitive notions can be used to identify the key folding contacts. The inspection of Figure 5 ▶ reveals that several kinetic bottlenecks (red regions) are located three to four contacts downstream the three β -turns in HIV-1 PR. In addition, the formation of contacts around residues 84 and 30, despite being so far away along the sequence, appears to be a crucial folding stage because it allows the collapse of the individual secondary structure motifs. It is striking that these results make an excellent parallel with those of Cecconi et al. 2001, where long and delicate MD simulations of the unfolding/refolding of HIV-1 PR were carried out using a much more sophisticated energy-scoring function. This provides a crossvalidation for the robustness of the results obtained both in the stochastic and the present, analytic, scheme. The emphasis is on the exactness of the present approach that allows us to determine easily the pij's with an arbitrary accuracy. The absence of stochastic noise allows us to compile Table 2, which shows the top contacts ranked according to crossover temperature and intensity. Sites that are known to cause drug resistance through mutations are highlighted in boldface. It is apparent that a high fraction of the top key folding contacts do, indeed, contain key mutating sites. To test the significance of such matches, we compare the number of marked mutating sites contained in each column of Table 2 with the number of those contained in a randomly compiled table. We expect a random list of t elements extracted among N, m of which are marked, to contain an average of tm/N marked elements with a square deviation of tm(N−m)(N−t)/[N2(N−1)]. For the case of HIV-1 PR, the total number of contacts (excluding consecutive residues) within a cutoff radius of 6.5 Å is N = 180 and the number of those that include at least one known mutating site is m = 60. By applying this analysis to the contacts of Table 2 (selected according to crossover temperature or cooperativity of formation) it shows that that the number of matches observed among the top sites typically exceeds that expected from a random choice by one standard deviation (the precise difference depends on how many top sites, t, are considered). An alternative and more stringent approach is to identify independent groups of highly correlated contacts, and then search for the key residues in each group. To a first approximation, the correlated sets of interacting pairs may be identified with the clusters in the contact map. This leads to define six main groups, the three β -sheets, the helix, and the two sets of long-range contacts, around contacts 14–60 and 23–84, respectively (see Fig. 5 ▶). The four contacts in each group with the highest intensity of formation above TF are summarized in Table 3. Out of the 24 contacts, 12 of them involve a key site, which is two standard deviations away from the number of matches expected on a random basis (7.9 ±2.1). Again, this testifies to both the reliability of the general scheme followed here and also to its robustness in the different possible implementations.
Fig. 1.

Structure of HIV-1 PR dimer (Condra et al. 1995). The highlighted locations indicate residues where mutations causing drug resistance are observed.
Fig. 2.

Specific heat, Cv, and fractional occupation of native contacts, q, of a monomer of HIV-1 PR. The temperature is scaled with the temperature Tf where the specific heat peak occurs.
Fig. 3.

Plot of Pi, the degree to which amino acid i is in a native-like conformation, versus i. In ascending order the curves are calculated at T/TF = 1.5, 1.0, and 0.5. The bar at the bottom shows the secondary structure associated with amino acid i.
Fig. 4.

Typical behavior of contact probabilities. pij versus T/TF for four native contacts involving pairs of sites with different sequence separation and degree of native burial.
Fig. 5.


Color-coded contact map of HIV-1 PR monomer. (A) Contacts with a large (small) crossover temperature are shown in red (blue). (B) Contacts with a large (small) cooperativity of formation above Tf are shown in red (blue).
Table 2.
The top contacts ranked according to the crossover temperature (first column) and cooperativity of formation above Tf(second column)
| Crossover temperature | Cooperativity |
| 25–86 | 14–66 |
| 28–86 | 14–64 |
| 58–76 | 10–23 |
| 58–77 | 14–65 |
| 57–77 | 13–66 |
| 13–66 | 12–66 |
| 30–86 | 87–91 |
| 32–84 | 13–65 |
| 32–76 | 23–84 |
| 29–86 | 10–22 |
| 31–84 | 56–77 |
| 23–84 | 57–77 |
| 14–66 | 23–83 |
| 25–85 | 22–84 |
| 14–65 | 57–78 |
| 45–56 | 86–89 |
| 89–91 | 34–78 |
| 13–65 | 58–77 |
| 87–89 | 30–88 |
| 84–86 | 32–75 |
| 56–58 | 32–76 |
| 25–84 | 31–76 |
| 86–88 | 42–58 |
| 64–71 | 90–94 |
| 57–76 | 87–90 |
Table 3.
The four contacts with the highest cooperativity of formation above Tffor each of the six clusters of the contact map
| Bottlenecks | Key contacts |
| β 1 | 10–23 |
| β 1 | 10–22 |
| β 1 | 14–20 |
| β 1 | 12–20 |
| β 2 | 42–58 |
| β 2 | 45–58 |
| β 2 | 43–58 |
| β 2 | 43–57 |
| β 3 | 56–77 |
| β 3 | 57–77 |
| β 3 | 58–77 |
| β 3 | 57–76 |
| Other1 | 14–66 |
| Other1 | 14–64 |
| Other1 | 14–65 |
| Other1 | 13–66 |
| Other2 | 23–84 |
| Other2 | 23–83 |
| Other2 | 22–84 |
| Other2 | 30–88 |
| Helix | 87–91 |
| Helix | 86–89 |
| Helix | 90–94 |
| Helix | 87–90 |
Interestingly, the results of Table 3 account better than those of Table 2 for the heterogeneous location of the key folding sites. The emerging conclusion is that a complete description of the crucial contacts can be obtained only by monitoring all the key stages of the folding process. In standard MC and MD simulations of protein unfolding/refolding, it is the simulated dynamics that reveal which, and how many, delicate stages exist. In the present approach, the folding process is characterized analytically, thus the complete set of folding bottlenecks follows from the study of distinct groups of interrelated contacts.
Finally, we remark that the determination of the key contacts does not uniquely provide the key folding sites, as two sites are involved in each pairwise contact. This ambiguity can, in several cases, be resolved either by selecting those sites that take part in several crucial contacts, or by examining their distribution on the three-dimensional native structure for clues that may help breaking the ambiguity.
Conclusions
We have used an analytical technique to study and characterize the folding process of globular proteins. This deterministic method allows the automated identification of contacts involved in folding rate-limiting steps. As a result, the whole folding process is particularly sensitive to mutations occurring at sites involved in such crucial contacts. We test our scheme and its usefulness in pinpointing the crucial sites by applying it to HIV-1 protease. For this enzyme, extensive clinical trials have allowed the identification of several sites involved in drug-resistance mutations. Such sites have a meaningful overlap with the key folding sites predicted by our scheme with a modest computational effort compared to more sophisticated stochastic simulations techniques. This indicates that the available inhibiting drugs are quite effective because they can be eluded only by mutations of the (sensitive) key sites of the protease.
The proposed approach to identifying the crucial residues is quite general and ought to be useful in identifying the kinetic bottlenecks of other viral enzymes of pharmaceutical interest, thus aiding in the development of novel effective inhibitors. We expect to focus our future efforts on improving the present approach by taking into account the propensities of different amino acids to form contacting pairs. This limitation can be overcome by introducing physically viable (attractive) pairwise interactions (Maiorov and Crippen 1992; Sippl 1995; Seno et al. 1998; Miyazawa and Jernigan 1999; Micheletti et al. 2001b). In the present approach, this possibility was deliberately avoided to highlight the influence of the native-state topology alone on the kinetic bottlenecks, irrespective of the different chemical nature and strength of the effective amino-acid interactions. We expect that the inclusion of such effects, while not distorting the overall picture presented here, may change the relative strength of spatially close contacts. This may improve the agreement between Table 1 and Tables 2 and 3 by resolving those cases were a site adjacent to a mutating one is selected.
Acknowledgments
We are indebted to Paolo Carloni for several illuminating discussions and for having stimulated the present work. This work was supported by INFM, Murst Cofin2001.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked "advertisement" in accordance with 18 USC section 1734 solely to indicate this fact.
Appendix
In this appendix we discuss how the translation invariance of a quadratic energy-scoring function can be explicitly broken by fixing the center of mass of the system in the origin. The constrained partition function is written as
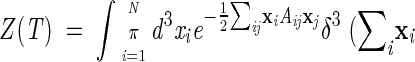 |
(11) |
where the matrix A incorporates the quadratic dependence of H0 in equation 4 from the space coordinates (and also includes the 1/T factor to yield the usual Boltzmann weight). The translation invariance of H0 implies that A satisfies the property: ΣjAij = 0, which amounts to say that the uniform vector, v1 ≡ N−1/2(1,1,1,1. . .,1) is an eigenvector of A with eigenvalue λ1 = 0. We assume that H0 is invariant only for the simultaneous translation of all the coordinates, {xi}. In this case, all other eigenvalues, {λi>1} are strictly positive and the corresponding eigenvectors vi>1 are all orthogonal to the zero mode v1.
By rewriting the Dirac-δ constraint as
 |
the partition function takes on the form 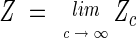 where
where
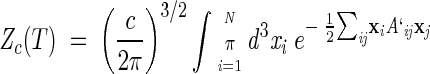 |
(12) |
where A′ij = Aij + c. It is straightforward to see that A` admits the same eigenvectors of A. Only the zero mode eigenvalue will change from zero to cN, while the others will be unmodified. Upon performing the Gaussian integrations in Zc we obtain
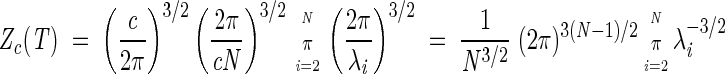 |
This shows that Zc is effectively independent of c and, therefore, the partition function Z simplifies to
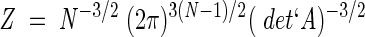 |
where the prime denotes that the determinant is calculated omitting the zero mode eigenvalue.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.3360102.
References
- Ala, P.J., Huston, E.E., Klabe, R.M., Jadhav, P.K., Lam, P.Y., and Chang, C.H. 1998. Counteracting HIV-1 protease drug resistance: Structural analysis of mutant proteases complexed with XV638 and SD146, cyclic urea amides with broad specificities. Biochemistry 37 15042–15049. [DOI] [PubMed] [Google Scholar]
- Alm, E. and Baker, D. 1999. Prediction of protein folding mechanisms from free energy landscapes derived from native structures. Proc. Natl. Acad. Sci. 96 11305–11310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atilgan, A.R., Durell, S.R., Jernigan, R.L., Demirel, M.C., Keskin, O., and Bahar, I. 2001. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 80 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar, I., Atilgan, A.R., and Erman, B. 1997. Direct evaluation of thermal fluctuations in proteins using a single parameter harmonic potential. Folding and Design 2 173–181. [DOI] [PubMed] [Google Scholar]
- Bahar, I., Erman, B., Jernigan, R.L., Atilgan, A.R., and Covell, D.G. 1999. Collective motions in HIV-1 reverse transcriptase: Examination of flexibility and enzyme function. J. Mol. Biol. 285 1023–1037. [DOI] [PubMed] [Google Scholar]
- Baker, D.A. 2000. Surprising simplicity to protein folding. Nature 405 39–42. [DOI] [PubMed] [Google Scholar]
- Camacho, C.J. and Thirumalai, D. 1993. Kinetics and thermodynamics of folding in model proteins. Proc. Natl. Acad. Sci. 90 6369–6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho, C.J. and Thirumalai, D. 1995. Theoretical predictions of folding pathways by using the proximity rule, with applications to bovine pancreatic trypsin inhibitor. Proc. Natl. Acad. Sci. 92 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi, F., Micheletti, C., Carloni, P., and Maritan, A. 2001. Molecular dynamics studies of HIV-1 protease: Drug resistance and folding pathways. Proteins: Structure Function and Genetics 43 365–372. [PubMed] [Google Scholar]
- Chan, H.S. and Dill, K.A. 1990. The effects of internal constraints on the configurations of chain molecules. J. Chem. Phys. 92 3118–3135. [Google Scholar]
- Chiti, F., Taddei, N., White, P.M., Bucciantini, M., Magherini, F., Stefani, M., and Dobson, C.M. 1999. Mutational analysis of acylphosphatase suggests the importance of topology and contact order in protein folding. Nat. Struct. Biol. 6 1005–1009. [DOI] [PubMed] [Google Scholar]
- Clementi, C., Nymeyer, H., and Onuchic, J.N. 2000. Topological and energetic factors: What determines the structural details of the transition state ensemble and `en-route' intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 298 937–953. [DOI] [PubMed] [Google Scholar]
- Condra, J.H., Schleif, W.A., Blahy, O.M., Gabryelski, L.J., Graham, D.J., Quintero, J.C., Rhodes, A., Robbins, H.L., Roth, E., Shivaprakash, M., et al. 1995. In-vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374 569–571. [DOI] [PubMed] [Google Scholar]
- Debe, D.A. and Goddard III, W.A. 1999. First principles prediction of protein folding rates. J. Mol. Biol. 294 619–625. [DOI] [PubMed] [Google Scholar]
- Fersht, A.R. 1995. Optimization of rates of protein folding—the nucleation condensation mechanism and its implications. Proc. Natl. Acad. Sci. 92 10869–10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flory, P.J. 1956. Theory of elastic mechanisms in fibrous proteins. J. Am. Chem. Soc. 78 5222–5235. [Google Scholar]
- Galzitskaya, O.V. and Finkelstein, A.V. 1999. A theoretical search for folding/unfolding nuclei in 3D protein structure. Proc. Natl. Acad. Sci. 96 11299–11304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go, N. and Scheraga, H.A. 1976. On the use of classical statistical mechanics in the treatment of polymer chain conformations. Macromolecules 9 535–542. [Google Scholar]
- Hoang, T.X. and Cieplak, M. 2000. Sequencing of folding events in go-type proteins. J. Chem. Phys. 113 8319–8328. [Google Scholar]
- Jackson, S.E. 1998. How do small single-domain proteins fold? Folding and Design 3 R81–R91. [DOI] [PubMed] [Google Scholar]
- Jacobsen, H., Hanggi, M., Ott, M., Duncan, I.B., Owen, S., Andreoni, M., Vella, S., and Mous, J. 1996. In vivo resistance to a human immunodeficiency virus type 1 Proteinase inhibitor: Mutations, kinetics, and frequencies. J. Infect. Dis. 173 1379– 1387. [DOI] [PubMed] [Google Scholar]
- Kaya, H. and Chan, H.S. 2000. Energetic components of cooperative protein folding. Phys. Rev. Lett. 85 4823–4826. [DOI] [PubMed] [Google Scholar]
- ——2001. Polymer principles of protein calorimetric two-state cooperativity. Proteins: Structure Function and Genetics 43 523. [Google Scholar]
- Keskin, O., Bahar, I., and Jernigan, R.L. 2000. Proteins with similar architectures exhibit similar large-scale dynamic behavior. Biophys. J. 78 2093–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloczkowski, A. and Jernigan, R.L. 1999. Contacts between segments in the random-flight model of polymer chains. Comp. Theor. Pol. Sci. 9 285–294. [Google Scholar]
- Lazaridis, T. and Karplus, M. 1997. "New view" of protein folding reconciled with the old through multiple unfolding simulations. Science 278 1928–1931. [DOI] [PubMed] [Google Scholar]
- Maiorov, V.N. and Crippen, G.M. 1992. Contact potential that recognizes the correct folding of globular proteins. J. Mol. Biol. 227 876–888. [DOI] [PubMed] [Google Scholar]
- Maritan, A., Micheletti, C., and Banavar, J.R. 2000. Role of secondary motifs in fast folding polymers: A dynamical variational principle. Phys. Rev. Lett. 84 3009–3012. [DOI] [PubMed] [Google Scholar]
- Markowitz, M., Mo, H., Kempf, D.J., Norbeck, D.W., Bhat, T.N., Erickson, J.W., Ho, D.D. 1995. Selection and analysis of human immunodeficiency virus type 1 variants with increased resistance to ABT-538, a novel protease inhibitor. J. Virol. 69 701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez, J.C. and Serrano, L. 1999. The folding transition state between SH3 domains is conformationally restricted and evolutionarily conserved. Nat. Struct. Biol. 6 1010–1016. [DOI] [PubMed] [Google Scholar]
- Micheletti, C., Banavar, J.R., Maritan, A., and Seno, F. 1999. Protein structures and optimal folding from a geometrical variational principle. Phys. Rev. Lett. 82 3372–3375. [Google Scholar]
- Micheletti, C., Banavar, J.R., and Maritan, A. 2001a. Protein conformations in equilibrium. Phys. Rev. Lett. 87 DOI:088102–1. [DOI] [PubMed] [Google Scholar]
- Micheletti, C., Seno, F., Banavar, J.R., and Maritan, A. 2001b. Learning effective amino acid interactions through iterative stochastic techniques. Proteins: Structure Function and Genetics 42 422–431. [DOI] [PubMed] [Google Scholar]
- Miyazawa, S. and Jernigan, R.L. 1999. Residue-residue potentials with a favorable contact pair term an unfavorable high packing density term, for simulation and threading. J. Mol. Biol. 256 623–644. [DOI] [PubMed] [Google Scholar]
- Molla, A., Korneyeva, M., Gao, Q., Vasavanonda, S., Schipper, P.J., Mo, H.M., Markowitz, M., Chernyavskiy, T., Niu, P., Lyons, N., Hsu, A., Granneman, G.R., Ho, D.D., Boucher, C.A., Leonard, J.M., Norbeck, D.W., and Kempf, D.J. 1996. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 2 760–766. [DOI] [PubMed] [Google Scholar]
- Patick, A.K., Mo, H., Markowitz, M., Appelt, K., Wu, B., Musick, L., Kalish, V., Kaldor, S., Reich, S., Ho, D., Webber, S. 1996. Antiviral and resistance studies of AG1343, an orally bioavailable inhibitor of human immunodeficiency virus protease. Antimicrob. Agents Chemother. 40 292–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaxco, K.W., Simons, K.T., and Baker, D. 1998. Contact order and transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277 985–994. [DOI] [PubMed] [Google Scholar]
- Reddy, P. and Ross, J. 1999. Amprenavir—A protease inhibitor for the treatment of patients with HIV-1 infection. Formulary 34 567–675. [Google Scholar]
- Riddle, D.S., Grantcharova, V.P., Santiago, J.V., Alm, E., Ruczinski, I., and Baker, D. 1998. Experiment and theory highlight role of native state topology in SH3 folding. Nat. Struct. Biol. 6 1016–1024. [DOI] [PubMed] [Google Scholar]
- Sali, A., Shakhnovich, E., and Karplus, M. 1994. How does a protein fold. Nature 369 248–251. [DOI] [PubMed] [Google Scholar]
- Seno, F., Micheletti, C., Maritan, A., and Banavar, J.R. 1998. Variational approach to protein design and extraction of interaction potentials. Phys. Rev. Lett. 81 2172. [Google Scholar]
- Settanni, G., Cattaneo, C., and Maritan, A. 2001. Role of native state topology in the stabilization of intracellular antibodies. Biophys. J. 80 2935–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sippl, M.J. 1995. Knowledge based potentials for proteins. Curr. Opin. Struct. Biol. 5 229–235. [DOI] [PubMed] [Google Scholar]
- Tisdale, M., Myers, R.E., Maschera, B., Parry, N.R., Oliver, N.M., Blair, E.D. 1995. Cross-resistance analysis of human immunodeficiency virus type 1 variants individually selected for resistance to 5 different protease inhibitors. Antimicrob. Agents Chemother. 39 1704–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolynes, P.G., Onuchic, J.N., and Thirumalai, D. 1995. Navigating the folding routes. Science 267 1619–1620. [DOI] [PubMed] [Google Scholar]