

Abstract
The maintenance of memory T cells is central to the establishment of immunological memory, although molecular details of the process are poorly understood. In the absence of the polycomb group (PcG) gene Bmi1, the number of memory CD4+ T helper (Th)1/Th2 cells was reduced significantly. Enhanced cell death of Bmi1−/− memory Th2 cells was observed both in vivo and in vitro. Among various proapoptotic genes that are regulated by Bmi1, the expression of proapoptotic BH3-only protein Noxa was increased in Bmi1−/− effector Th1/Th2 cells. The generation of memory Th2 cells was restored by the deletion of Noxa, but not by Ink4a and Arf. Direct binding of Bmi1 to the Noxa gene locus was accompanied by histone H3-K27 methylation. The recruitment of other PcG gene products and Dnmt1 to the Noxa gene was highly dependent on the expression of Bmi1. In addition, Bmi1 was required for DNA CpG methylation of the Noxa gene. Moreover, memory Th2-dependent airway inflammation was attenuated substantially in the absence of Bmi1. Thus, Bmi1 controls memory CD4+ Th1/Th2 cell survival and function through the direct repression of the Noxa gene.
The quality of adaptive immune responses depends on the size of the antigen-specific memory T cell pool, which is regulated through specific homeostatic mechanisms controlling both cell survival and proliferation. Upon antigen recognition, naive CD4 T cells undergo a rapid clonal expansion, followed by the differentiation into functionally distinct Th cell subsets, such as effector Th1 and Th2 cells (1–3). These effector T cells undergo a dramatic contraction in numbers after antigen clearance, with 90–95% succumbing to apoptosis within weeks (4–6). However, some of the effector T cells are maintained as memory T cells for long periods in vivo (7, 8).
In contrast to CD8 memory T cells, CD4 memory T cells may not require the signals through common cytokine receptor γ chain (9, 10). However, critical regulatory roles of IL-7 in the generation and survival of memory CD4 T cells were reported recently (11, 12). In addition, the homeostasis of memory CD4 T cells is dependent on the signals through the TCR as well as the IL-7 receptor (13). Bcl-2 and Mcl-1 are reported to be the downstream targets of the IL-7 receptor and promote T cell survival (14–16).
Several properties that distinguish memory T cells from naive T cells have been described, such as increased longevity and enhanced capacity for recall response to cognate antigen (17). Memory T cells have several features associated with stem cells (18), and the similarity of the gene expression pattern between memory T/B cells and long-term hematopoietic stem cells was reported (19). Similar to hematopoietic stem cells, memory T cells appear to possess the ability to proliferate in response to homeostatic signals. The homeostatic signals may drive self-renewal, whereas antigenic signals drive effector cell differentiation and function.
The polycomb group (PcG) gene Bmi1 has recently been implicated in the maintenance of hematopoietic (20, 21), neural (22), and cancer stem cells (23). PcG gene products form multimeric complexes and maintain the early determined gene expression patterns of key developmental regulators, such as homeobox genes both in invertebrates and vertebrates (24, 25). Bmi1, Mel-18, M33, Pc2, Rae-28/Mph1, Mph2, and Ring1A/B are constituents of a multimeric protein complex similar to the polycomb repressive complex (PRC)1 identified in Drosophila. Another PcG complex, which contains Eed, Suz12, Ezh1, and Ezh2, is PRC2. PRC2 possesses an intrinsic histone H3-K27 methyltransferase activity (26–29), which implicates a likely mechanism for PcG-mediated gene silencing. Lack of individual components of PRC1 results in apoptosis and the loss of proliferative responses of immature lymphoid cells (30, 31). In mature lymphocytes, PcG gene products play several roles in differentiation and cell fate. Mel-18 controls Th2 cell differentiation through the regulation of GATA3 expression (32), and Bmi1 controls the stability of GTAT3 protein in developing Th2 cells (33). EzH2 is involved in B cell development and controls IgH V(D)J rearrangement (34).
We herein investigated the role of Bmi1 in the generation and maintenance of memory CD4+ Th1/Th2 cells. In the absence of Bmi1, the generation of both Th1 and Th2 memory cells was impaired as a consequence of increased Noxa expression. Because our previous study indicated that the expression of Bmi1 is higher in Th2 cells than Th1 cells, we sought to investigate the molecular mechanisms underlying the Bmi1/Noxa-mediated regulation of memory T cell generation by focusing primarily on memory Th2 cells. Our results indicate that Bmi1 controls memory CD4 T cell survival and function through the direct repression of the Noxa gene.
RESULTS
Generation of memory Th1/Th2 cells was impaired in the absence of Bmi1
In Bmi1−/− mice, although a reduction of cell numbers in the lymphoid organs was seen, substantial numbers of CD4 and CD8 T cells developed with normal expression of developmental cell surface antigens (not depicted). After in vitro antigenic stimulation, Bmi1−/− CD4 T cells were shown to proliferate well (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1) and differentiate normally into IL-4–producing effector Th2 cells under Th2 conditions or IFN-γ–producing effector Th1 cells under Th1 conditions (33). Under IL-4–limiting conditions, Th2 cell differentiation was impaired significantly in the absence of Bmi1, and the Bmi1-mediated regulation of the stability of GATA3 protein was demonstrated (33).
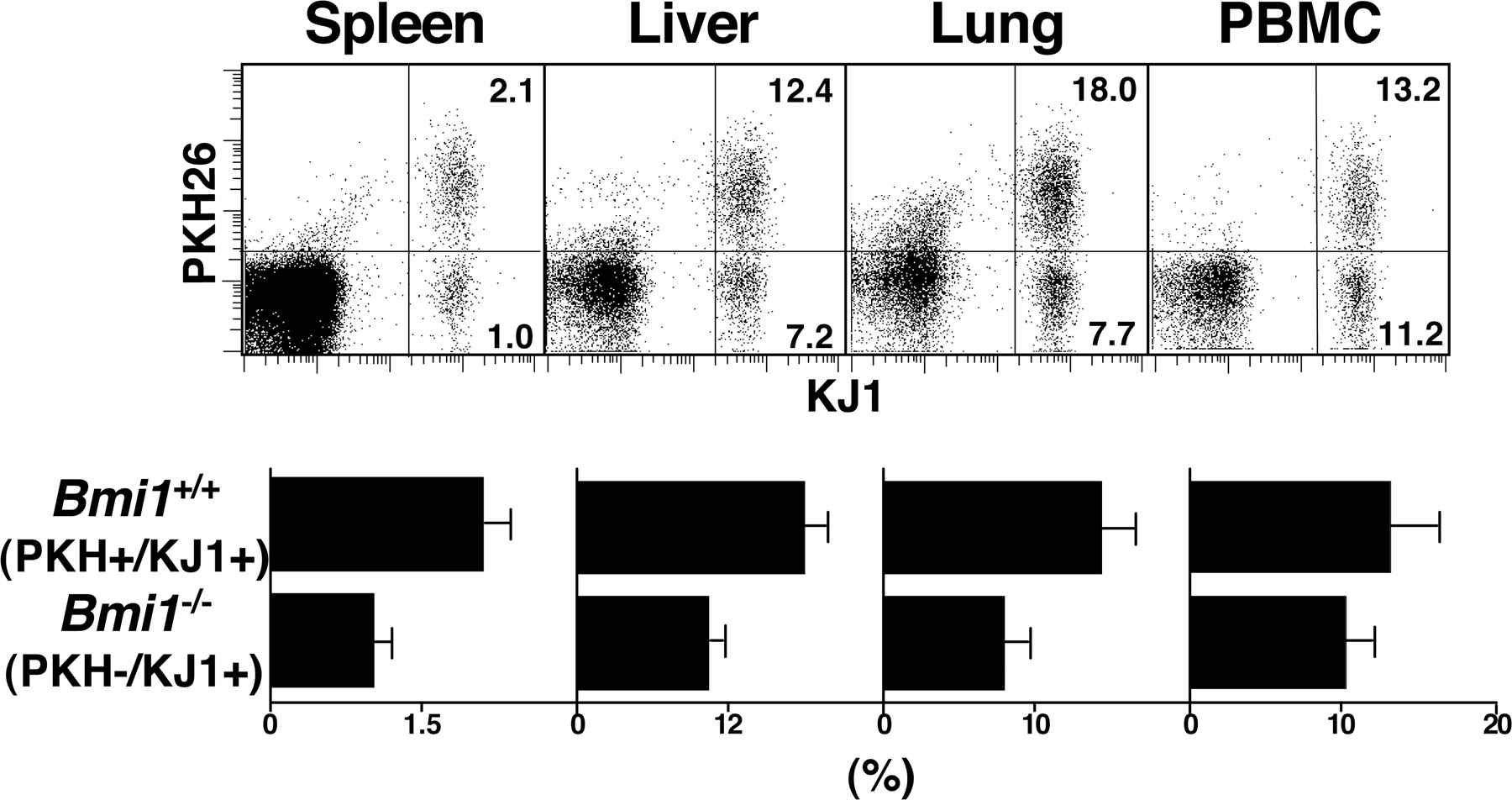
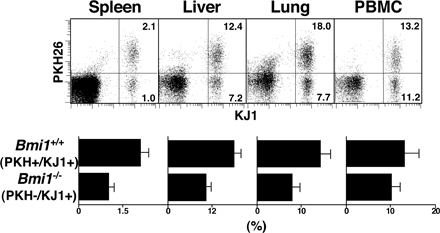
To investigate the role of Bmi1 in the generation and maintenance of memory Th1/Th2 cells, we used a “memory Th1/Th2 mouse” system (35, 36) in which OVA-specific αβTCR transgenic (Tg) CD4 T cells from Bmi1+/+, Bmi1+/−, or Bmi1−/− mice were stimulated with antigenic OVA peptide and wild-type antigen-presenting cells under Th2 or Th1 culture conditions in vitro for 6 d, and these effector Th cells were then adoptively transferred into syngeneic BALB/c nu/nu mice (memory Th1/Th2 mice). 5 wk after cell transfer, the numbers of donor-derived KJ1+ memory Th1/Th2 cells were assessed in various organs. As shown in Fig. 1 A, a Bmi1 gene dose-dependent affect on the numbers of memory Th2 cells was observed in all tissues tested (spleen, liver, lung, and PBMCs). It is worth noting that even heterozygous Bmi1+/− groups showed marked effects. Although the expression levels of IL-7Rα and IL-2Rβ were slightly lower on Bmi1−/− memory Th2 cells as compared with wild-type cells, other surface marker antigens were expressed normally (Fig. S1 B). Next, KJ1+ effector Th1 cells were transferred into BALB/c nu/nu mice. The memory Th1 cell generation from Bmi1−/− effector Th1 cells was also impaired in all tissues tested (Fig. 1 B). The reduction in the Bmi+/− group was not obvious. A similar loss of memory Th cell generation was observed in both Th2 (Fig. 1 C) and Th1 cells (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1) when Bmi1−/− effector cells were transferred into nonlymphopenic normal BALB/c mice. These results indicate that the generation of memory Th1/Th2 cells was impaired in the absence of Bmi1.
Figure 1.

Impaired generation of memory Th1/Th2 cells from Bmi1−/− effector Th2 cells. (A and B) Bmi1+/+, Bmi1+/−, or Bmi1−/− effector Th2 (A) or Th1 (B) cells with DO11.10 Tg background were intravenously transferred into BALB/c nu/nu mice (n = 5). 5 wk later, the number of KJ1+ memory Th2 cells was determined by flow cytometry. Typical staining patterns of KJ1/CD4 and percentages of KJ1+ cells are shown. Four independent experiments were performed with similar results. (C) Bmi1+/+, Bmi1+/−, or Bmi1−/− effector Th2 cells with DO11. 10 Tg background were intravenously transferred into BALB/c mice (n = 5). (D) Impaired semi-acute survival of Bmi1+/− Th2 cells. Thy1.1 Bmi1+/+ (Ly5.2 background) and Thy1.2 Bmi1+/− (Ly5.2 background) effector Th2 cells were mixed (1:1) and transferred (3 × 107 cells/mouse) into syngeneic C57BL/6 mice with a Ly5.1 background. The ratio of Thy1.2+/Thy1.1+ cells in Ly5.2+ cells is shown (n = 3). (E) Homeostatic proliferation of the splenic memory Th2 cells was assessed by the BrdU incorporation in vivo. Representative BrdU staining profiles of KJ1+ memory Th2 cells prepared 5 wk after cell transfer are shown. The percentages of BrdU+ cells with standard deviation are shown (n = 5). Three independent experiments were performed with similar results. (F) Apoptotic cell death of freshly prepared memory Th2 cells from the spleen was measured by a TUNEL assay. As a positive control, cells were treated with DNase I. Representative TUNEL profiles are shown with percentages of TUNEL+ cells. Two independent experiments were performed with similar results.
A kinetics study showed that the decrease in Bmi1−/− Th2 cells was observed from 2 d after cell transfer (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1). Consequently, we assessed the semi-acute survival of Bmi1+/− effector Th2 cells by a competition analysis using Bmi1+/+ (Thy1.1) and Bmi1+/− (Thy1.2) C57BL/6 Ly5.2 effector Th2 cells and Ly5.1 host animals with a C57BL/6 background. Typical staining patterns of mixed cells for cell transfer and of transferred donor-derived cells (Ly5.2) from the indicated tissues are shown in Fig. S4. Interestingly, a significant decrease in the ratio of Bmi1+/−/Bmi1+/+ (Thy1.2/Thy1.1) cells was already observed 2 d after cell transfer, and this decline continues through days 7–21 (Fig. 1 D). The kinetics of the preferential reduction of Bmi1+/− cells was similar in all tissues tested. The defect in semi-acute survival in Bmi1−/− Th2 cells was also observed under lymphopenic conditions (Fig. S5).
The homeostatic proliferation as measured by BrdU incorporation in vivo was decreased slightly in Bmi1−/− memory Th2 cells as compared with wild type (14.3 ± 0.4% vs. 9.1 ± 1.5%) (Fig. 1 E). Next, we assessed the extent of ongoing apoptotic cell death in vivo by a TUNEL assay. In Bmi1−/− memory Th2 cells, ongoing apoptosis was enhanced dramatically as compared with wild type (8.2 vs. 57.1%) (Fig. 1 F). In addition, increased annexin V+PI+ cells were detected in Bmi1−/− memory Th2 cells after restimulation in vitro, indicating that these memory cells are highly prone to cell death (Fig. S1 C).
Deletion of p16ink4a and p19arf genes failed to restore the decreased memory Th2 cell generation in the absence of Bmi1
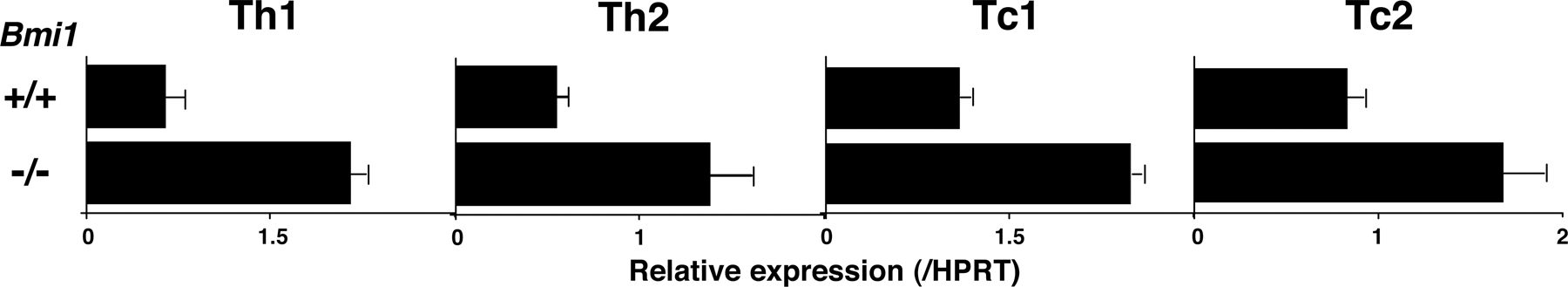
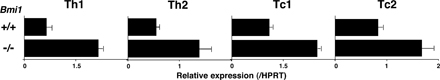
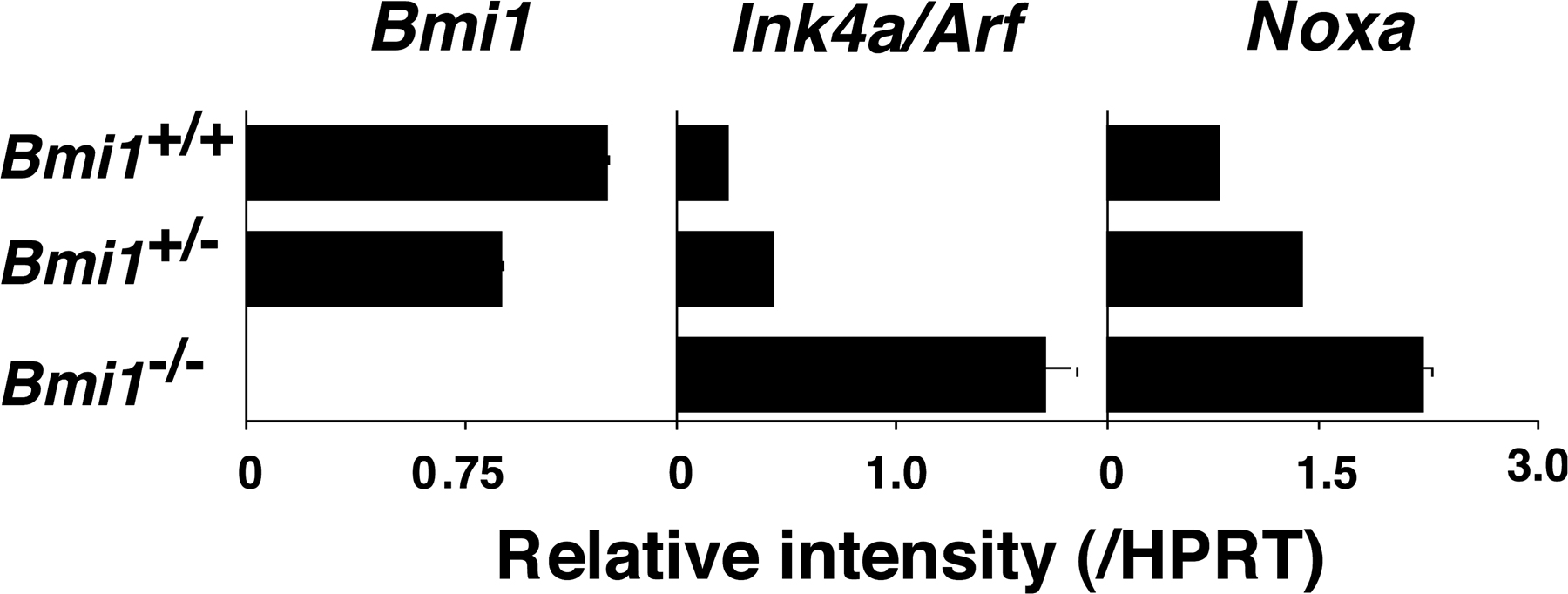
Bmi1 has been reported to promote cell proliferation, cell survival, and stem cell self-renewal by repressing the Ink4a/Arf locus (37). This locus codes for two proteins, p16ink4a and p19arf (Ink4a and Arf), through the use of alternative reading frames. Ink4a is a cyclin D–dependent kinase inhibitor that promotes cell cycle arrest after Rb activation. Arf induces p53 activation and p53-mediated cell death (38). In the Bmi1−/− effector Th2 cells, we confirmed the increased expression of the Ink4a/Arf gene after anti-TCR stimulation (Fig. 2 A). An increased mRNA expression of both Ink4a and Arf was confirmed (not depicted). In addition, various proapoptotic genes, which are known to be targets for p53, such as Bax, Puma, Noxa, Bim, Bad, Fas, and Fas ligand, were increased in Bmi1−/− effector Th2 cells (Fig. 2 A). In Bmi1−/− memory Th2 cells, increased expression of mRNA was observed in the Ink4a/Arf, Bax, Puma, Noxa, Bad, and Fas genes, whereas expression of antiapoptotic genes Bcl2, BclxL, and Mcl1 was unchanged (Fig. 2 B). To examine whether the increased expression of Ink4a/Arf in Bmi1−/− Th2 cells plays a role in the Bmi1-mediated maintenance of memory Th2 cells, Bmi1−/−mice were crossed with Ink4a−/−/Arf−/− mice, and effector Th2 cells generated from Bmi1−/−Ink4a−/−Arf−/− with Ly5.2 background were transferred into Ly5.1 mice. 5 wk later, the number of Ly5.2+ memory Th2 cells was assessed. Depletion of Ink4a and Arf genes itself had no effect on the generation of memory T cells (Fig. 2 C, panels 1 and 3) and failed to restore memory Th2 cell generation in Bmi1−/− background (Fig. 2 C, panels 2 and 4). Furthermore, the deletion of the p53 gene failed to rescue the Bmi1+/− memory Th2 cell numbers (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1). These results suggest that the Ink4/Arf and p53-dependent pathways are not key pathways responsible for the reduced memory Th2 cell generation in Bmi1−/− memory mice. To identify any possible proapoptotic genes whose expression is regulated by Bmi1 but not regulated by the Ink4/Arf and p53-dependent pathways, the expression levels of these proapoptotic genes were assessed in the Bmi1−/−/Ink4a−/−/Arf−/− Th2 cells. Among these proapoptotic genes, the level of Noxa mRNA increased in Bmi1−/− Th2 cells and remained high in Bmi1−/−/Ink4a−/−/Arf−/− background, whereas other targets such as Bax, Puma, Bim, Bad, Fas, and Fas ligand were at the normal expression levels (Fig. 2 D). Increased expression of Noxa mRNA was observed also in effector Th1, Tc1, and Tc2 cells in the absence of Bmi1 (Fig. S7). These results indicate that the expression of Noxa is suppressed by the expression of Bmi1, but it is independent from the control of the Ink4/Arf and p53 pathways. The results thus far obtained suggest that Bmi1 controls the generation of both Th1 and Th2 memory cells accompanied with an increased expression of Noxa. Our previous study indicated that the expression of Bmi1 is higher in Th2 cells than Th1 cells, and Bmi1 controls the generation of Th2 cells more profoundly as compared with Th1 cells (33). Therefore, to better understand the molecular mechanisms underlying the Bmi1/Noxa-mediated regulation of memory CD4 T cell generation, we focused much of our studies on memory Th2 cells.
Figure 2.

Deletion of the p16ink4a and p19arf genes failed to rescue the impaired generation of memory Th2 cells in the absence of Bmi1. (A) mRNA expression of Ink4a/Arf and p53-related proapoptotic genes in Bmi1−/− effector Th2 cells was determined by quantitative RT-PCR. The relative intensity (/HPRT; mean of three samples) is shown with standard deviations. Three independent experiments were performed with similar results. (B) mRNA expression of Ink4a/Arf, p53-related proapoptotic genes, and antiapoptotic genes in Bmi1−/− memory Th2 cells was analyzed. The relative intensity (/HPRT; mean of three samples) is shown with standard deviations. Two independent experiments were performed with similar results. (C) Effects on p16ink4a and p19arf deficiency on the memory Th2 cell generation. The effector Th2 cells from the indicated mice (Ly5.2 background) were transferred into Ly5.1 host mice. 5 wk after cell transfer, the number of Ly5.2+ memory Th2 cells was determined. The mean values are shown with standard deviations (n = 5; right). The experiments were performed twice with similar results. (D) mRNA levels of proapoptotic genes in Bmi1−/−/Ink4a−/−/Arf−/− effector Th2 cells were determined by quantitative RT-PCR. The relative intensity (/HPRT; mean of three samples) is shown with standard deviations. The experiments were performed twice with similar results.
The expression of Noxa controls the generation of memory Th2 cells
Consequently, to test whether the changes in the level of Noxa affect the induction of cell death, effector Th2 cells were infected with a retrovirus containing the human NGFR and Noxa, and the number of annexin V+ cells was assessed after in vitro suspension culture for 24 h without cytokines. Noxa-introduced cells showed increased annexin V+ staining (70.0%) as compared with mock-infected (24.0%) or noninfected cell populations (Fig. 3 A). Next, KJ1+ effector Th2 cells infected with a retrovirus containing the GFP and Noxa genes were transferred into BALB/c nu/nu mice, and the numbers of Noxa-expressing (KJ1+GFP+) memory Th2 cells were assessed. Although the generation of KJ1+ memory Th2 cells was not affected, the number of KJ1+GFP+ memory Th2 cells and their GFP mean fluorescence intensity were clearly reduced in the Noxa-transduced group (Fig. 3 B). A gene dose-dependent increase in the expression of Noxa was detected in Bmi1+/− and Bmi1−/− memory Th2 cells (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1). Bmi1−/−Noxa−/− mice were not born despite extensive breeding attempts. Consequently, we used Bmi1+/−Noxa−/− mice and found that memory Th2 cell generation in the mice transferred with Bmi1+/− effector Th2 cells was restored by the deletion of the Noxa gene (Fig. 3 C). Furthermore, enhanced generation of memory Th2 cells was observed in the mice that received Noxa−/− effector Th2 cells (Fig. 3 D). Thus, we concluded that the reduction in the number of Bmi1−/− memory Th2 cells is at least in part due to the increased expression of Noxa in Bmi1−/− Th2 cells.
Figure 3.

Expression level of Noxa controls the generation of memory Th2 cells. (A) Enforced expression of Noxa-induced cell death in effector Th2 cells after cytokine depletion. Effector Th2 cells infected with a Noxa-IRES-hNGFR–containing retrovirus were cultured in vitro for 24 h without cytokines. hNGFR profiles (left) and annexin V staining profiles of the electronically gated hNGFR+ (gate #2) and hNGFR− (gate #1) populations are shown. Three independent experiments were performed with similar results. (B) KJ1+ effector Th2 cells infected with Noxa-IRES-EGFP–containing retrovirus were transferred into BALB/c nu/nu mice. 5 wk later, memory Th2 cell generation was determined by KJ1/EGFP expression. Expression of EGFP in pretransferred effector Th2 cells (top left) and a typical KJ1/GFP profile of freshly prepared memory Th2 cells (top right) are shown. In the bottom panels, the percentages of KJ1+ cells and GFP+ Noxa-overexpressing cells and the mean fluorescence intensity of the GFP+ cells are shown with standard deviations (n = 4). The experiments were performed twice with similar results. (C) The effector Th2 cells from Bmi1+/+/Noxa+/+, Bmi1+/−/Noxa+/+, and Bmi1+/−/Noxa−/− mice (Ly5.2) were transferred into Ly5.1 host mice, and the number of Ly5.2+ memory Th2 cells was determined. A typical staining pattern of CD4/Ly5.2 (top) and the percentages of Ly5.2+ cells among CD4 T cells are shown with standard deviations (n = 5; bottom). Three independent experiments were performed with similar results. (D) Deletion of the Noxa gene enhanced the generation of memory Th2 cells. In vitro–generated Noxa−/− effector Th2 cells (Ly5.2) were transferred into Ly5.1 host mice. 5 wk after cell transfer, the number of Ly5.2+ memory Th2 cells was determined. A representative CD4/Ly5.2 profile (left) and the mean values with standard deviations (n = 5; right) are shown. The experiments were performed twice with similar results.
Bmi1 directly binds to the Noxa gene locus and regulates the histone modification
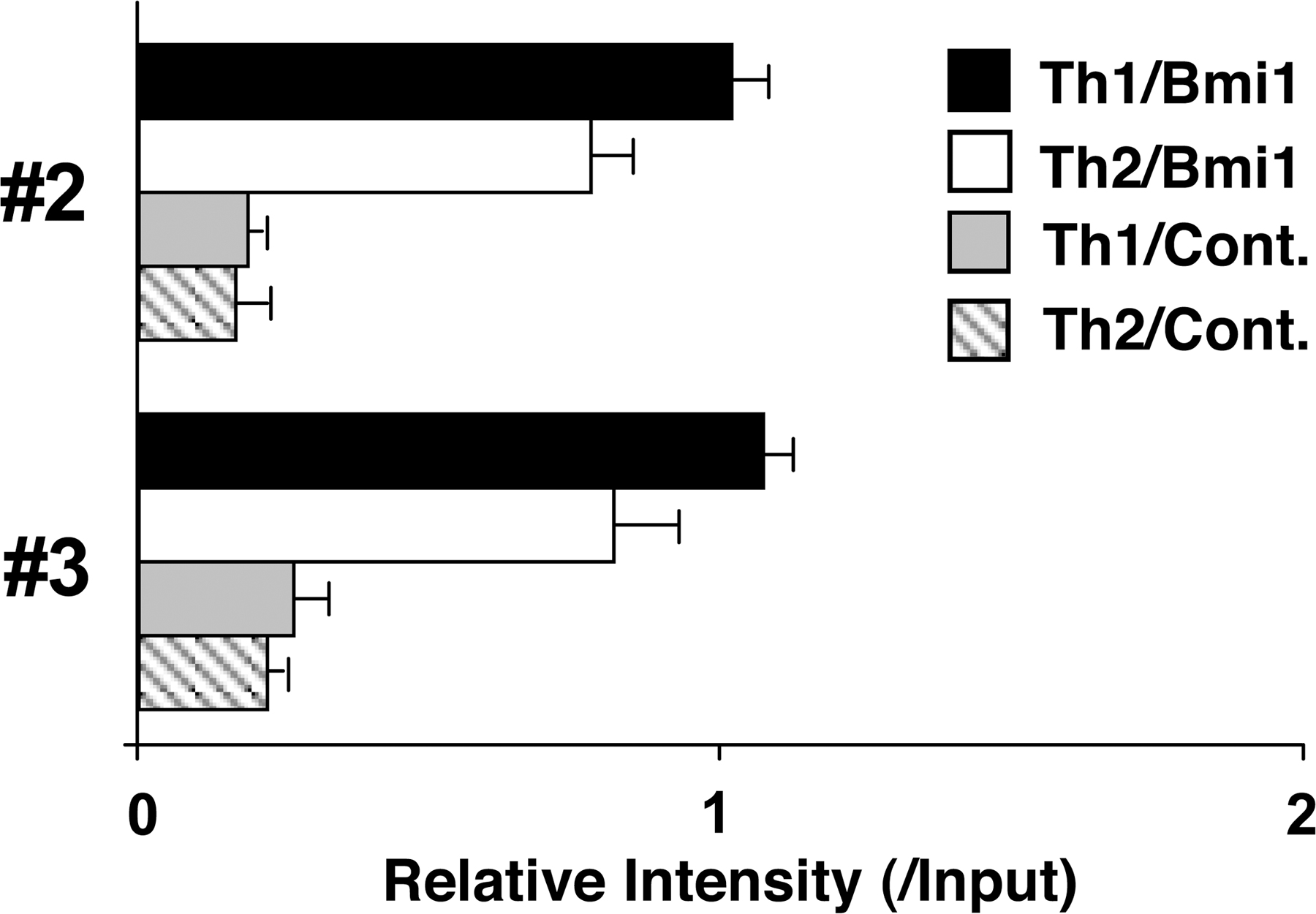
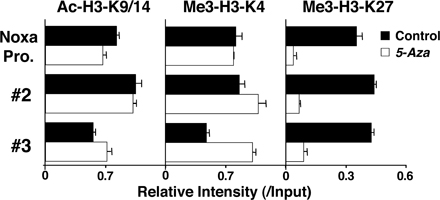
To assess the molecular mechanisms underlying the Bmi1-mediated repression of the Noxa gene, we performed chromatin immunoprecipitation (ChIP) assays with six primer pairs covering the Noxa gene (Fig. 4 A). The accumulation of Bmi1 was observed around the CpG island (Fig. 4 B, #2 and #3) of the Noxa locus. The binding of Bmi1 was confirmed by a ChIP assay with a quantitative PCR system (Fig. 4 C). Equivalent binding of Bmi1 was observed in Th1 cells (Fig. S9, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1). Histone H3-K9/14 acetylation and tri-methylation of histone H3-K4 were observed also around the CpG island (Fig. 4 D, #2 and #3). More interestingly, histone H3-K27 was highly tri-methylated in wild-type Th2 cells over broader regions in the Noxa locus, and the tri-methylation was apparently reduced in the Bmi1−/− Th2 cells (Fig. 4 D). The changes in histone modifications were also assessed using a quantitative PCR system. Modest increase in histone H3-K9/14 acetylation and a substantial decrease in H3-K27 tri-methylation at the Noxa gene locus were confirmed in the Bmi1−/− Th2 cells (Fig. 4 E) and Bmi1−/− Th1 cells (Fig. S10). Interestingly, the levels of histone H3-K27 tri-methylation at the Ink4a gene locus, another target gene of Bmi1, were not affected by the depletion of Bmi1 (Fig. 4 E). The levels of histone H3-K27 tri-methylation at the promoter regions of the Bim and Puma were not significantly detected in the presence or absence of Bmi1. The binding of other PcG gene products, such as Ring1b and Suz12, was detected around the same regions (Fig. 4 F, #2 and #3), and the binding was substantially decreased in the absence of Bmi1. Thus, a PcG gene product complex containing Bmi1 appears to bind to the Noxa gene directly and regulate histone modifications, such as tri-methylation of H3-K27 in Th1 and Th2 cells.
Figure 4.

Bmi1 binds to the Noxa gene and regulates tri-methylation of H3-K27. (A) Schematic representation of the Noxa gene locus. The location of primers (#1 to #6), a CpG island, and exons are indicated. (B) A ChIP assay of the Noxa gene locus was performed using anti-Bmi1 antibody (Bmi1) and control antibody (Cont.) in Bmi1+/+ and Bmi1−/− Th2 cells. Three independent experiments were performed with similar results. (C) A ChIP assay of the Noxa gene locus was performed as in B, and the levels of binding were assessed by a quantitative PCR analysis. (D) Histone modifications (Ac-H3-K9/K14; acetylation of histone H3-K9/K14 and Me3-H3-K4; tri-methylation of histone H3-K4 and Me3-H3-K27; tri-methylation of histone H3-K27) at the Noxa gene locus in Bmi1+/+ and Bmi1−/− effector Th2 cells. (E) A ChIP assay of the Noxa (#2 to #4), Ink4a (Ex.2, exon 2; Ex.3, exon 3), Bim (Bim pro., Bim promoter), and Puma (Puma pro., Puma promoter) locus was performed as in D, and the levels of binding were assessed by a quantitative PCR analysis. (F) The association of Ring1B and Suz12 at the Noxa gene locus in Bmi1+/+ and Bmi1−/− Th2 cells. The association of Bmi1, Ring1B, and Suz12 was determined by a ChIP assay using specific antibodies.
Bmi1 controls the CpG methylation at the Noxa gene locus and represses the mRNA expression of Noxa
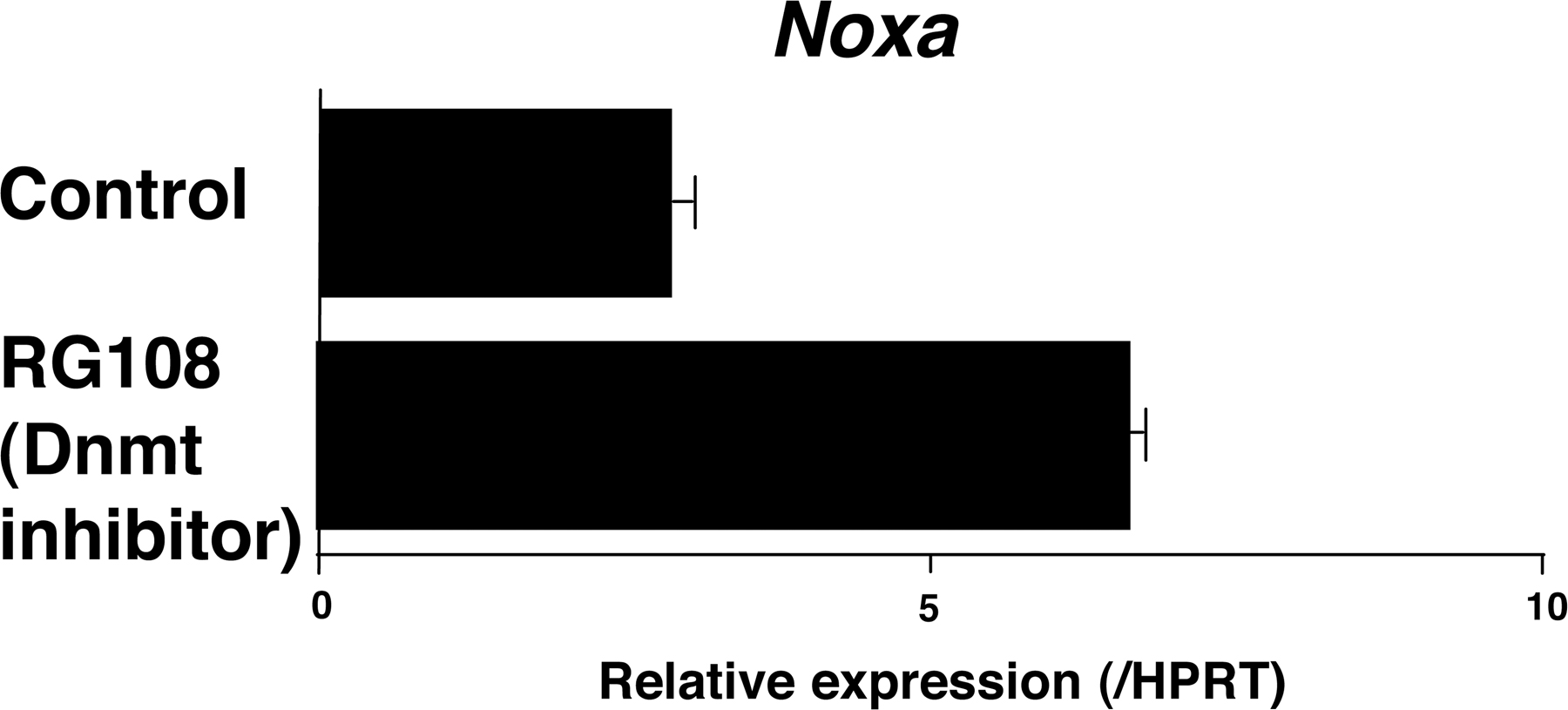
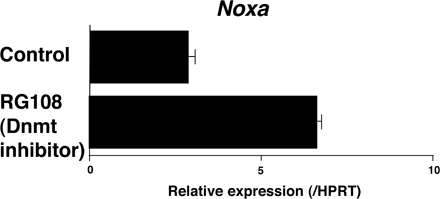
Next, to study the levels of CpG DNA methylation around the CpG island of the Noxa gene, a methylated DNA immunoprecipitation (MeDIP) assay was performed. As shown in Fig. 5 A, the 5′ region of the CpG island (promoter and #2) was methylated in wild-type effector Th2 cells, and the methylation levels were very low in the Bmi1−/− cells. A DNA methyltransferase Dnmt1 was found to bind at the CpG island (Fig. 5 B, #2 and #3), and the binding was also dependent on Bmi1 in Th2 cells (Fig. 5 B) and Th1 cells (Fig. S11, available at http://www.jem.org/cgi/content/full/jem.20072000/DC1). To assess a role of CpG methylation on Noxa expression, effector Th2 cells were treated with 5-Aza-2'-deoxycytidine (5-Aza), an inhibitor of CpG DNA methylation. As expected, the expression of Noxa mRNA was dramatically increased (Fig. 5 C) accompanied by the reduction of a tri-methylation level of histone H3-K27 (Fig. S12). Furthermore, the binding of Bmi1, Ring1b, and Suz12 was substantially reduced by the treatment with 5-Aza (Fig. 5 D). A similar increase in the expression of Noxa and the 5-Aza–dependent reduction in the binding of Bmi1 were observed in Th1 cells (Fig. S13). Finally, we established a knockdown system for Dnmt1 using a cultured T cell line, TG40, as described in Materials and methods. Up-regulation of Noxa mRNA (Fig. 5 E) and the reduction of Bmi1 binding at the Noxa gene locus (Fig. 5 F) were induced by the introduction of shRNA for Dnmt1. Enhanced Noxa mRNA expression was induced in primary Th2 cells by the treatment with a DNA methyltransferase inhibitor, RG108 (Fig. S14). These results indicate that a DNA methyltransferase Dnmt1 plays an important role in the recruitment of PcG gene products and the expression of the Noxa gene in Th2 cells.
Figure 5.

Bmi1 is required for the DNA CpG methylation of the Noxa gene. (A) Decreased DNA CpG methylation at the CpG island of the Noxa gene locus in Bmi1−/− Th2 cells. DNA CpG methylation level was determined by MeDIP assay. MeDIP assay was performed using an anti–5-methyl cytidine antibody (5-meC) and a control antibody (Cont.). Mean values with standard deviation are shown (n = 3). (B) The association of Dnmt1, a DNA methyltransferase, at the CpG island of the Noxa gene locus. The association of Dnmt1 was determined by a ChIP assay using a specific antibody. (C) Increased mRNA expression of Noxa after the treatment with 5-Aza, an inhibitor of DNA methyltransferase. Th2 cells were treated with 5-Aza for 2 d, and Noxa mRNA expression was determined by a quantitative RT-PCR system. Three independent experiments were performed with similar results. (D) Dissociation of the PcG complex from the Noxa gene locus after the treatment with 5-Aza. The association of Bmi1, Ring1B, and Suz12 was determined by a ChIP assay using specific antibodies. Three independent experiments were performed with similar results. (E) mRNA expression of Noxa and Dnmt1 in a Dnmt1 knockdown (KD) T cell line. Dnmt1-KD and control-KD stable TG40 cell lines were generated using a lentivirus gene transduction system as described in Materials and methods. The levels of mRNA were determined by a quantitative RT-PCR. The relative intensity (/HPRT; mean of three samples) is shown with standard deviations. (F) Decreased binding of Bmi1 at the Noxa gene in Dnmt1-KD TG40 cells. A ChIP assay with an anti-Bmi1 antibody was performed, and the levels of Bmi1 binding were assessed by a quantitative PCR analysis.
The expression of Bmi1 is required for memory Th2 cell–dependent immune responses and inflammation in vivo
Finally, to examine functional defects in Bmi1−/− memory Th2 mice, we used a memory Th2 cell–dependent allergic airway inflammation model (36). Memory Th2 mice were generated and simply challenged by inhalation with OVA four times. The OVA-specific IgE and IgG1 (Th2-dependent isotypes) antibody production were decreased in the Bmi1−/− memory Th2 mice, whereas only a marginal decrease in the levels of Th1-dependent IgG2a was seen (Fig. 6 A). Next, we examined the levels of airway inflammation after OVA inhalation. The extent of inflammatory leukocyte infiltration in the peri-bronchiolar region (Fig. 6 B) and the infiltrated eosinophils, lymphocytes, and macrophages in the bronchioalveolar lavage (BAL) fluid (Fig. 6 C) was reduced significantly in the Bmi1−/− memory Th2 mice as compared with wild-type mice. The expression of Th2 cytokines (IL-4, IL-5, and IL-13) and Eotaxin 2 in the lung of OVA-inhaled Bmi1−/− memory Th2 mice was also reduced (Fig. 6 D). The periodic-acid-Schiff staining and the measurement of mRNA expression of Gob5, Muc5a/c, and Muc5b in the lung indicated a decrease in mucus hyperproduction in Bmi1−/− memory Th2 mice (Fig. 7, A and B). Furthermore, the airway hyperresponsiveness measured using a whole-body plethysmograph was not significantly induced in the Bmi1−/− memory Th2 mice (Fig. 7 C). In addition, by a direct invasive assay for lung resistance (RL), increase in the RL and decrease in the dynamic compliance were observed in the Bmi1−/− memory Th2 mice (Fig. 7 D). Collectively, these results indicate that the memory Th2 cell–dependent allergic responses were thus compromised in the Bmi1−/− memory Th2 mice. We also assessed the eosinophilic infiltration in DO11.10 Tg Bmi1+/+ and Bmi1+/− mice without cell transfer, and as expected, the level of eosinophilic infiltration was significantly milder in the Bmi1+/− mice (not depicted).
Figure 6.

Defects in the memory Th2 cell–dependent immune responses and inflammation in Bmi1−/− memory Th2 mice. OVA-specific Bmi1+/+ and Bmi1−/− effector Th2 cells with DO11. 10 Tg background were intravenously transferred into BALB/c nu/nu mice. 5 wk later, the mice were challenged four times by inhalation with OVA on days 0, 2, 8, and 10. (A) The serum concentrations of OVA-specific Igs with the indicated isotype after OVA inhalation (on day 11) were determined by ELISA. The mean values with standard deviations (five animals per group) are shown. *, P < 0.01; **, P < 0.05. The control represents BALB/c nu/nu mice without Th2 cell transfer. The experiments were performed twice with similar results. (B) On day 11, the lungs were fixed and stained with hematoxylin an eosin (HE). The number of infiltrated leukocytes in the peribronchiolar region (mean cell numbers/mm2 with standard deviations; n = 5) is also shown (bottom). The experiments were performed twice with similar results. Bars, 100 μm. (C) On day 12, BAL fluid was collected and May-Grunwald-Giemsa staining was performed. The absolute cell numbers of eosinophils (Eos.), neutrophils (Neu.), lymphocytes (Lym.), and macrophages (Mac.) in the BAL fluid are shown with standard deviations (n = 5). The results were obtained using the values of cell counting, the percentage of the cells, the total cell number per milliliter, and the volume of the BAL fluid recovered. *, P < 0.01. The experiments were performed twice with similar results. (D) The mRNA expression of IL-4, IL-5, IL-13, and Eotaxin 2 was determined by quantitative RT-PCR. The relative intensity (/HPRT; mean of three samples) is shown with standard deviations. The experiments were performed twice with similar results.
Figure 7.

Decreased mucus hyperproduction and airway hyperresponsiveness in Bmi1−/− memory Th2 mice. Bmi1+/+ and Bmi1−/− memory Th2 mice were challenged by inhalation with OVA as in Fig. 5. (A) 1 d after the last OVA inhalation (day 11), the lungs were fixed and stained with periodic-acid-Schiff (PAS). A representative staining pattern is shown. The control represents BALB/c nu/nu mice without Th2 cell transfer. Bars, 100 μm. (B) On day 12, total RNA was prepared from the lung, and the expression of Gob5, Muc5a/c, and Muc 5b (molecular makers for Goblet cell hyperplasia and mucus production) was determined by a quantitative PCR analysis. The relative intensity (/HPRT; mean of three samples) is shown with standard deviations. (C) OVA-induced airway hyperresponsiveness in Bmi1−/− memory Th2 mice. On day 11, the airway hyperresponsiveness in response to increasing doses of methacholine was measured in a whole-body plethysmograph. The mean values (n = 5) are shown with standard deviations. PBS, PBS-inhaled control; OVA, OVA-inhaled. *, P < 0.01. The experiments were performed twice with similar results. (D) On day 11, changes in the RL (left) and the dynamic compliance (Cdyn; right) were assessed. Mean values (six mice per group) are shown with standard deviations. *, P < 0.01; **, P < 0.05.
DISCUSSION
Here, we demonstrate that Bmi1 plays a crucial role in the generation and maintenance of memory CD4 T cells. Such a Bmi1-mediated regulation was seen in both memory Th1 and Th2 cells. In the absence of Bmi1, the generation of both Th1 and Th2 memory cells was impaired (Fig. 1 and Fig. S2) by the increased Noxa expression (Fig. 2 and Fig. S7). Bmi1 binds to the Noxa gene locus in both Th1 and Th2 cells (Fig. 4 and Fig. S9), and directly represses its transcription to promote memory Th2 cell survival. The involvement of H3-K27 tri-methylation and DNA CpG methylation in the repression of Noxa was revealed. The Th2-dependent allergic airway inflammatory responses were compromised in Bmi1−/− memory Th2 mice, suggesting a physiological role of Bmi1 in the establishment of Th2 cell–mediated memory responses.
Noxa is a member of a BH3-only protein family that initiates programmed cell death in various cells, including lymphocytes (39–41). Noxa is known to regulate selectively the pro-survival activity of Mcl1 and A1/Bfl-1 (42), and an important role of Mcl1 in the survival of lymphocytes was reported (43). The mRNA expression of antiapoptotic genes Mcl1, Bclx, and BclxL was not changed in Bmi1−/− memory (Fig. 2 B) and induced normally in effector Th2 cells by the IL-7 treatment (unpublished data). Enforced expression of Noxa in effector Th2 cells resulted in the decreased generation of memory Th2 cells (Fig. 3 B), suggesting that the overproduction of Noxa prevents memory Th2 cell generation even in the presence of normal abundance of antiapoptotic proteins. Thus, it is likely that Bmi1 attenuates Noxa-mediated inhibition of Mcl1 pro-survival activity and promotes memory T cell generation.
Noxa was originally identified as a downstream target of p53 (44). Arf/Mdm2-mediated stabilization of p53 protein and the resulting mRNA expression of the p53 target genes were reported (45, 46). Therefore, it was likely that the enhancement of cell death observed in Bmi1−/− Th2 cells is p53 dependent. However, the deletion of the p53 gene failed to rescue the decreased generation of memory Th2 cells in the absence of Bmi1 (Fig. S6). Thus, an increased apoptotic cell death observed in Bmi1−/− memory Th2 cells appears to be p53 independent.
The Bmi1-mediated repression of the Ink4a/Arf gene observed in the hematopoietic and neural stem cells (45, 46) appears to operate in Th2 cells (Fig. 2, A and B). It is known that Arf induces p53 activation (38), resulting in the induction of p53-dependent genes, including Puma and Bim. An important role of Puma and Bim in the cell death of activated T cells was reported (47, 48), and the expression of these genes was up-regulated in Bmi1−/− effector and memory Th2 cells (Fig. 2, A and B). However, Bmi1−/−/Ink4a−/−/Arf−/− Th2 cells, in which the levels of Puma and Bim expression were not increased (Fig. 2 D), failed to generate normal numbers of memory Th2 cells (Fig. 2 C). Although the expression of Puma and Bim mRNA was up-regulated, the levels were considerably low compared with Noxa in Bmi1−/− effector Th2 cells (Fig. 2 A). This might explain the predominant effect of Noxa in the cell death of Bmi1−/− Th2 cells. However, the current experimental data do not allow us to make any conclusions to be drawn in regards to the relative contribution of each of the factors in the survival of CD4 T cells. Thus, although Puma and Bim may play a role in the survival of memory Th2 cells, Noxa appears to be a critical target for the Bmi1-mediated regulation of memory Th2 cell survival.
Semi-acute survival of transferred Bmi1−/− effector Th2 cells was substantially impaired (Fig. 1 D and Figs. S3–S5). On the other hand, high numbers of apoptotic cells were detected in the spleen even 5 wk after effector Bmi1−/− Th2 cell transfer (Fig. 1 F). The extent of the defect in homeostatic proliferation was modest (Fig. 1 E). The low number of Bmi1−/− memory CD4 T cells persisted for at least 2 mo (unpublished data). These results suggest that the long-term survival of memory Th2 cells is also dependent on the expression of Bmi1. Collectively, we conclude that Bmi1 controls both semi-acute survival of effector Th2 cells and long-term survival of memory Th2 cells.
The PRC2 was reported to possess an intrinsic histone H3-K27 methyltransferase activity (26–29). The PRC1, including Bmi1 and Ring1, was shown to possess an activity of histone H2A ubiquitination (49–51), whereas there has been no report indicating that the PRC1 possess an intrinsic histone H3-K27 methyltransferase activity. Accumulating evidence supports a sequential binding model (52), in which PRC2-mediated H3-K27 methylation serves as a binding site for the recruitment of PRC1 complex through the specific recognition of the H3-K27 methyl mark by the chromo domain of the polycomb protein, such as M33 (53, 54). In our study, however, the levels of tri-methylation of histone H3-K27 were substantially decreased in Bmi1−/− Th1 and Th2 cells (Fig. S10 and Fig. 4, D and E). In addition, the levels of binding of Suz12, a component of PRC2, at the Noxa gene locus were decreased in Bmi1−/− Th2 cells (Fig. 4 F). Thus, in Th2 cells, Bmi1 may play an important role for the recruitment of PRC2 and subsequent histone H3-K27 methylation at the Noxa gene locus. Alternatively, the Bmi1-containing PcG complex in Th2 cells may associate with a unique molecule possessing an intrinsic histone H3-K27 methyltransferase activity. In any event, tri-methylation of histone H3-K27 at the Noxa gene locus is strictly regulated by the expression of Bmi1.
The expression of Bmi1 is required for the DNA CpG methylation of the Noxa gene (Fig. 5 A). The binding of Dnmt1 at the CpG island was Bmi1 dependent (Fig. 5 B and Fig. S11). The treatment with a 5-Aza resulted in the dissociation of Bmi1, Ring1B, and Suz12 from the CpG island (Fig. 5 D) and the up-regulation of Noxa mRNA (Fig. 5 C and Fig. S13 A). Increased expression of Noxa mRNA and reduced binding of Bmi1 were confirmed in Dnmt1 knockdown T cells (Fig. 5, E and F). Recently, the PcG complex was reported to be associated with DNA methyltransferases, including Dnmt1 (55, 56). Thus, a DNA methyltransferase, such as Dnmt1, may play a critical role in the recruitment and the repressive function of the PcG complex at the Noxa gene.
In this study, we demonstrate that the generation and maintenance of memory Th1/Th2 cells (Fig. 1) and the memory Th2 cell–dependent airway inflammation are controlled by the expression of Bmi1 (Figs. 6 and 7). Our preliminary experiments demonstrate that at least Mel-18, Mph1/Rae28, and M33 appear to be involved in the regulation of memory Th2 cell generation (unpublished data), suggesting that the survival of memory Th2 cells is regulated by an epigenetic mechanism involving the Bmi1-containing PcG complex. We have recently reported that Bmi1 stabilizes GATA3 protein through the direct interaction with GATA3 (33). Mel-18 is involved in the induction of GATA3 expression in developing Th2 cells (32). Collectively, PcG gene products appear to control the development of effector and memory Th2 cells at multiple steps in a distinct manner and govern the Th2-type immune responses and inflammation.
MATERIALS AND METHODS
Mice.
Bmi1-deficient mice were provided by M. van Lohuizen (The Netherlands Cancer Institute, Amsterdam, Netherlands) (30). p16Ink4a/p19Arf double-deficient mice were provided by R.A. DePinho (Harvard Medical School, Boston, MA) (37). Noxa-deficient mice were provided by T. Taniguchi (The University of Tokyo, Tokyo, Japan) (44). The animals used in this study were backcrossed to BALB/c or C57BL/6 mice 10 times. Anti–OVA-specific TCR-αβ (DO11.10) Tg mice were provided by D. Loh (Washington University School of Medicine, St. Louis, MO) (57). All mice were used at 4–8 wk old. BALB/c and BALB/c nu/nu mice were purchased from Clea Inc. Ly5.1 mice were purchased from Sankyo Laboratory. All mice used in this study were maintained under specific pathogen-free conditions. All experiments using mice received approval from the Chiba University Administrative Panel for Animal Care. Animal care was conducted in accordance with the guidelines of Chiba University.
Reagents.
The reagents used in this study are as follows: FITC- or APC-conjugated anti-CD4 mAb (GK1.5), FITC-conjugated anti-Ly5.2 mAb (104), and PE-conjugated KJ1 (anti-clonotypic mAb for DO11.10 TCR) were purchased from BD Biosciences. Anti-FcRγII and III mAb (2.4G2) and unconjugated anti–IL-4 mAb (11B11), anti–IL-12 mAb (C17.8), and anti–IFN-γ (R4-6A2) were used as culture supernatants. Recombinant mouse IL-4 was from TOYOBO. The OVA peptide (residues 323–339; ISQAVHAAHAEINEAGR) was synthesized by BEX Corporation.
The generation of effector and memory Th1/Th2 cells.
Effector and memory Th1/Th2 cells were generated as described previously (35, 36). In brief, splenic KJ1+CD4+ T cells from DO11.10 OVA-specific TCR Tg mice were stimulated with 1 μM of an OVA peptide (Loh15) plus APC (irradiated splenocytes) under the Th1 or Th2 culture conditions for 6 d in vitro. Th1 condition: 25 U/ml IL-2, 10 U/ml IL-12, and anti–IL-4 mAb. Th2 condition: 25 U/ml IL-2, 10 U/ml IL-4, anti–IL-12 mAb, and anti–IFN-γ mAb. In some experiments, splenic CD4 T cells were stimulated with 3 μg/ml of immobilized anti–TCR-β mAb plus 1 μg/ml anti-CD28 mAb. These effector Th1/Th2 cells (3 × 107) were transferred intravenously into BALB/c nu/nu, BALB/c, or Ly5.1 C57BL/6 recipient mice. 5 wk after cell transfer, the generation of memory Th2 cells was assessed using donor cell–specific mAbs (KJ1 and anti-Ly5.2).
TUNEL assay.
TUNEL assay was performed with In Situ Cell Detection kit (Roche).
Measurement of BrdU incorporation in vivo.
The memory Th2 mice were treated twice with 1 mg BrdU on days 0 and 2. BrdU incorporation in splenic KJ1+ memory Th2 cells was assessed on day 4 using the BrdU Flow kit (BD Biosciences).
5-Aza treatment.
Developing Th2 cells were treated with 10 μM 5-Aza (Sigma-Aldrich) for 3 d, and then total RNA was prepared.
ELISA.
Serum OVA–specific Ig concentrations were determined by ELISA as described previously (36).
Quantitative RT-PCR.
Total RNA was isolated using the TRIzol reagent (Invitrogen). cDNA was synthesized using oligo (dT) primer and Superscript II RT (Invitrogen). Quantitative RT-PCR was performed as described previously using ABI PRISM 7000 Sequence Detection System (36). The primers for TaqMan probes for the detection were purchased from Applied Biosystems. The expression was normalized using the HPRT signal.
Retrovirus infection.
Retrovirus vector, pMXs-IRES-GFP, was provided by T. Kitamura (The University of Tokyo, Tokyo, Japan). The method for the generation of virus supernatant and the infection was described previously (32). Infected cells were collected 4 d after infection and transferred into recipient mice.
Lentivirus infection.
Lentivirus vectors, pLKO.1 (SHC002) and pLKO.1 mouse Dnmt1 (TRCN39024), were purchased from Sigma-Aldrich. pCAG-HIVgp and pCMV-VSV-G-RSV-Rev vectors were provided by H. Miyoshi (RIKEN Bioresource Center, Ibaraki Japan). Recombinant lentiviruses were generated using a three-plasmid system as described previously (58). In brief, 293 T cells were transfected with self-inactivating lentiviral vector, pCAG-HIVgp vector, and pCMV-VSV-G-RSV-Rev vector. Virus containing culture supernatant was collected 48 h after transfection and used for infection. T cell line TG40 (2.5 × 105/well) was infected and selected using puromycin.
ChIP assay.
ChIP assay was performed as described previously (59). The antibodies using ChIP assay are as follows; anti-trimethyl histone H3-K4 (ab8580; Abcam), anti-trimethyl histone H3-K9 (Abcam), anti-trimethyl histone H3-K27 (Millipore), anti-Bmi1 (Santa Cruz Biotechnology, Inc.), anti-Dnmt1 (sc-20701; Santa Cruz Biotechnology, Inc.), anti-Suz12 (Abcam), and normal rabbit IgG (Santa Cruz Biotechnology, Inc.). An mAb specific for mouse Ring1b was provided by H. Koseki (Riken Research for Allergy and Immunology, Yokohama, Japan). The specific primers used in ChIP assay are shown in Table S1, which is available at http://www.jem.org/cgi/content/full/jem.20072000/DC1.
MeDIP.
MeDIP was performed using a METHYL kit (Diagenode). In brief, genomic DNA was purified from effector Th2 cells and sheared by sonication to reduce DNA lengths to between 200 and 1,000 bp. The sheared DNA was diluted and incubated with antiserum specific for the 5-methyl cytidine. Next, immune complexes were precipitated with protein A agarose. The precipitated DNA was purified using QIAquick PCR Purification kit (QIAGEN).
Assessment of memory Th2 cell function in vivo.
OVA-specific wild-type and Bmi1−/− effector Th2 cells (107 cells) were intravenously transferred into BALB/c nu/nu mice. 5 wk after cell transfer, the mice were challenged four times by inhalation with OVA on days 0, 2, 8, and 10. The serum Ig levels, lung histology, mRNA expression in the lung, and airway hyperresponsiveness were then assessed on day 11 as described previously (36, 60). BAL fluid was collected on day 12.
Statistical analysis.
Student's t test was used.
Online supplemental material.
Fig. S1 shows the phenotypic and functional characterization of Bmi1−/− Th2 cells. Fig. S2 shows the impaired generation of memory Th1 cells from Bmi1−/− effector Th1 cells. Fig. S3 depicts the kinetic analysis of the number of Th2 cells after adoptive transfer of effector Th2 cells, and Fig. S4 displays the competitive analysis for memory Th2 cell generation from Bmi1+/+ and Bmi1+/− effector Th2 cells under nonlymphopenic conditions. Fig. S5 shows a competitive analysis for memory Th2 cell generation from Bmi1+/+ and Bmi1−/− effector Th2 cells under lymphopenic conditions. Fig. S6 shows that deletion of the p53 gene failed to restore the generation of Bmi1+/− memory Th2 cells. In Fig. S7, mRNA expression of Noxa in Bmi1+/+ and Bmi1−/− effector Th1, Th2, Tc1, and Tc2 cells is shown. Fig. S8 displays mRNA expression of the Ink4a/Arf and Noxa in Bmi1+/+, Bmi1+/−, and Bmi1−/− effector Th2 cells. In Fig. S9, binding of Bmi1 at the Noxa gene locus in effector Th1 and Th2 cells is shown. Fig. S10 depicts histone modifications at the Noxa gene locus in Bmi1+/+and Bmi1+/− effector Th1 cells. Fig. S11 shows binding of Dnmt1 at the Noxa gene locus in Bmi1+/+and Bmi1+/− effector Th1 cells, and Fig. S12 shows histone modifications at the Noxa gene locus after 5-Aza treatment. Fig. S12 displays Noxa mRNA expression and the binding of Bmi1 at the Noxa gene locus in effector Th1 cells after 5-Aza treatment. In Fig. S14, mRNA expression of Noxa in Th2 cells treated with RG108, a DNA methyltransferase inhibitor is shown. Table S1 lists the primer pairs used for the ChIP assay. The online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20072000/DC1.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. Ralph T. Kubo for helpful comments and constructive criticism in the preparation of the manuscript. We thank Ms. Hikari Asou, Ms. Satoko Norikane, Ms. Kaoru Sugaya, and Mr. Toshihiro Ito for their excellent technical assistance.
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (Japan) (Grants-in-Aid: for Scientific Research on Priority Areas no. 17016010; Scientific Research [B] no. 17390139, Scientific Research [C] nos. 18590466, 19590491, and 19591609, Exploratory Research no. 19659121, Young Scientists [Start-up] no. 18890046, and Special Coordination Funds for Promoting Science and Technology), and the Ministry of Health, Labor and Welfare (Japan), Kanae Foundation, Uehara Memorial Foundation, Mochida Foundation, Yasuda Medical Foundation, Astellas Foundation, and Sagawa Foundation.
The authors have no conflicting financial interests.
Abbreviations used: 5-Aza, 5-Aza-2'-deoxycytidine; BAL, bronchioalveolar lavage; ChIP, chromatin immunoprecipitation; MeDIP, methylated DNA immunoprecipitation; PcG, polycomb group; PRC, polycomb repressive complex; RL, lung resistance; Tg, transgenic.
References
- 1.Mosmann, T.R., H. Cherwinski, M.W. Bond, M.A. Giedlin, and R.L. Coffman. 1986. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 136:2348–2357. [PubMed] [Google Scholar]
- 2.Seder, R.A., and W.E. Paul. 1994. Acquisition of lymphokine-producing phenotype by CD4+ T cells. Annu. Rev. Immunol. 12:635–673. [DOI] [PubMed] [Google Scholar]
- 3.Reiner, S.L., and R.M. Locksley. 1995. The regulation of immunity to Leishmania major. Annu. Rev. Immunol. 13:151–177. [DOI] [PubMed] [Google Scholar]
- 4.Kaech, S.M., E.J. Wherry, and R. Ahmed. 2002. Effector and memory T-cell differentiation: implications for vaccine development. Nat. Rev. Immunol. 2:251–262. [DOI] [PubMed] [Google Scholar]
- 5.Schluns, K.S., and L. Lefrancois. 2003. Cytokine control of memory T-cell development and survival. Nat. Rev. Immunol. 3:269–279. [DOI] [PubMed] [Google Scholar]
- 6.Lanzavecchia, A., and F. Sallusto. 2002. Progressive differentiation and selection of the fittest in the immune response. Nat. Rev. Immunol. 2:982–987. [DOI] [PubMed] [Google Scholar]
- 7.Dutton, R.W., L.M. Bradley, and S.L. Swain. 1998. T cell memory. Annu. Rev. Immunol. 16:201–223. [DOI] [PubMed] [Google Scholar]
- 8.Sprent, J., and C.D. Surh. 2002. T cell memory. Annu. Rev. Immunol. 20:551–579. [DOI] [PubMed] [Google Scholar]
- 9.Lantz, O., I. Grandjean, P. Matzinger, and J.P. Di Santo. 2000. γ chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat. Immunol. 1:54–58. [DOI] [PubMed] [Google Scholar]
- 10.Tan, J.T., B. Ernst, W.C. Kieper, E. LeRoy, J. Sprent, and C.D. Surh. 2002. Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J. Exp. Med. 195:1523–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondrack, R.M., J. Harbertson, J.T. Tan, M.E. McBreen, C.D. Surh, and L.M. Bradley. 2003. Interleukin 7 regulates the survival and generation of memory CD4 cells. J. Exp. Med. 198:1797–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li, J., G. Huston, and S.L. Swain. 2003. IL-7 promotes the transition of CD4 effectors to persistent memory cells. J. Exp. Med. 198:1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seddon, B., P. Tomlinson, and R. Zamoyska. 2003. Interleukin 7 and T cell receptor signals regulate homeostasis of CD4 memory cells. Nat. Immunol. 4:680–686. [DOI] [PubMed] [Google Scholar]
- 14.Kaech, S.M., J.T. Tan, E.J. Wherry, B.T. Konieczny, C.D. Surh, and R. Ahmed. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198. [DOI] [PubMed] [Google Scholar]
- 15.Maraskovsky, E., L.A. O'Reilly, M. Teepe, L.M. Corcoran, J.J. Peschon, and A. Strasser. 1997. Bcl-2 can rescue T lymphocyte development in interleukin-7 receptor-deficient mice but not in mutant rag-1−/− mice. Cell. 89:1011–1019. [DOI] [PubMed] [Google Scholar]
- 16.Opferman, J.T., H. Iwasaki, C.C. Ong, H. Suh, S. Mizuno, K. Akashi, and S.J. Korsmeyer. 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 307:1101–1104. [DOI] [PubMed] [Google Scholar]
- 17.Masopust, D., S.M. Kaech, E.J. Wherry, and R. Ahmed. 2004. The role of programming in memory T-cell development. Curr. Opin. Immunol. 16:217–225. [DOI] [PubMed] [Google Scholar]
- 18.Fearon, D.T., P. Manders, and S.D. Wagner. 2001. Arrested differentiation, the self-renewing memory lymphocyte, and vaccination. Science. 293:248–250. [DOI] [PubMed] [Google Scholar]
- 19.Luckey, C.J., D. Bhattacharya, A.W. Goldrath, I.L. Weissman, C. Benoist, and D. Mathis. 2006. Memory T and memory B cells share a transcriptional program of self-renewal with long-term hematopoietic stem cells. Proc. Natl. Acad. Sci. USA. 103:3304–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park, I.K., D. Qian, M. Kiel, M.W. Becker, M. Pihalja, I.L. Weissman, S.J. Morrison, and M.F. Clarke. 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 423:302–305. [DOI] [PubMed] [Google Scholar]
- 21.Iwama, A., H. Oguro, M. Negishi, Y. Kato, Y. Morita, H. Tsukui, H. Ema, T. Kamijo, Y. Katoh-Fukui, H. Koseki, et al. 2004. Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity. 21:843–851. [DOI] [PubMed] [Google Scholar]
- 22.Molofsky, A.V., R. Pardal, T. Iwashita, I.K. Park, M.F. Clarke, and S.J. Morrison. 2003. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature. 425:962–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lessard, J., and G. Sauvageau. 2003. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 423:255–260. [DOI] [PubMed] [Google Scholar]
- 24.Satijn, D.P., and A.P. Otte. 1999. Polycomb group protein complexes: do different complexes regulate distinct target genes? Biochim. Biophys. Acta. 1447:1–16. [DOI] [PubMed] [Google Scholar]
- 25.van Lohuizen, M. 1999. The trithorax-group and polycomb-group chromatin modifiers: implications for disease. Curr. Opin. Genet. Dev. 9:355–361. [DOI] [PubMed] [Google Scholar]
- 26.Cao, R., L. Wang, H. Wang, L. Xia, H. Erdjument-Bromage, P. Tempst, R.S. Jones, and Y. Zhang. 2002. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 298:1039–1043. [DOI] [PubMed] [Google Scholar]
- 27.Kuzmichev, A., K. Nishioka, H. Erdjument-Bromage, P. Tempst, and D. Reinberg. 2002. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16:2893–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Czermin, B., R. Melfi, D. McCabe, V. Seitz, A. Imhof, and V. Pirrotta. 2002. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 111:185–196. [DOI] [PubMed] [Google Scholar]
- 29.Muller, J., C.M. Hart, N.J. Francis, M.L. Vargas, A. Sengupta, B. Wild, E.L. Miller, M.B. O'Connor, R.E. Kingston, and J.A. Simon. 2002. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 111:197–208. [DOI] [PubMed] [Google Scholar]
- 30.van der Lugt, N.M., J. Domen, K. Linders, M. van Roon, E. Robanus-Maandag, H. te Riele, M. van der Valk, J. Deschamps, M. Sofroniew, M. van Lohuizen, et al. 1994. Posterior transformation, neurological abnormalities, and severe hematopoietic defects in mice with a targeted deletion of the bmi-1 proto-oncogene. Genes Dev. 8:757–769. [DOI] [PubMed] [Google Scholar]
- 31.Akasaka, T., K. Tsuji, H. Kawahira, M. Kanno, K. Harigaya, L. Hu, Y. Ebihara, T. Nakahata, O. Tetsu, M. Taniguchi, and H. Koseki. 1997. The role of mel-18, a mammalian Polycomb group gene, during IL-7-dependent proliferation of lymphocyte precursors. Immunity. 7:135–146. [DOI] [PubMed] [Google Scholar]
- 32.Kimura, M., Y. Koseki, M. Yamashita, N. Watanabe, C. Shimizu, T. Katsumoto, T. Kitamura, M. Taniguchi, H. Koseki, and T. Nakayama. 2001. Regulation of Th2 cell differentiation by mel-18, a mammalian polycomb group gene. Immunity. 15:275–287. [DOI] [PubMed] [Google Scholar]
- 33.Hosokawa, H., M.Y. Kimura, R. Shinnakasu, A. Suzuki, T. Miki, H. Koseki, M. van Lohuizen, M. Yamashita, and T. Nakayama. 2006. Regulation of Th2 cell development by Polycomb group gene bmi-1 through the stabilization of GATA3. J. Immunol. 177:7656–7664. [DOI] [PubMed] [Google Scholar]
- 34.Su, I.H., A. Basavaraj, A.N. Krutchinsky, O. Hobert, A. Ullrich, B.T. Chait, and A. Tarakhovsky. 2003. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat. Immunol. 4:124–131. [DOI] [PubMed] [Google Scholar]
- 35.Yamashita, M., R. Shinnakasu, Y. Nigo, M. Kimura, A. Hasegawa, M. Taniguchi, and T. Nakayama. 2004. Interleukin (IL)-4-independent maintenance of histone modification of the IL-4 gene loci in memory Th2 cells. J. Biol. Chem. 279:39454–39464. [DOI] [PubMed] [Google Scholar]
- 36.Yamashita, M., K. Hirahara, R. Shinnakasu, H. Hosokawa, S. Norikane, M.Y. Kimura, A. Hasegawa, and T. Nakayama. 2006. Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity. 24:611–622. [DOI] [PubMed] [Google Scholar]
- 37.Jacobs, J.J., K. Kieboom, S. Marino, R.A. DePinho, and M. van Lohuizen. 1999. The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature. 397:164–168. [DOI] [PubMed] [Google Scholar]
- 38.Lowe, S.W., and C.J. Sherr. 2003. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr. Opin. Genet. Dev. 13:77–83. [DOI] [PubMed] [Google Scholar]
- 39.Strasser, A. 2005. The role of BH3-only proteins in the immune system. Nat. Rev. Immunol. 5:189–200. [DOI] [PubMed] [Google Scholar]
- 40.Alves, N.L., I.A. Derks, E. Berk, R. Spijker, R.A. van Lier, and E. Eldering. 2006. The Noxa/Mcl-1 axis regulates susceptibility to apoptosis under glucose limitation in dividing T cells. Immunity. 24:703–716. [DOI] [PubMed] [Google Scholar]
- 41.Marrack, P., and J. Kappler. 2004. Control of T cell viability. Annu. Rev. Immunol. 22:765–787. [DOI] [PubMed] [Google Scholar]
- 42.Willis, S.N., and J.M. Adams. 2005. Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17:617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Opferman, J.T., A. Letai, C. Beard, M.D. Sorcinelli, C.C. Ong, and S.J. Korsmeyer. 2003. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 426:671–676. [DOI] [PubMed] [Google Scholar]
- 44.Oda, E., R. Ohki, H. Murasawa, J. Nemoto, T. Shibue, T. Yamashita, T. Tokino, T. Taniguchi, and N. Tanaka. 2000. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 288:1053–1058. [DOI] [PubMed] [Google Scholar]
- 45.Bruggeman, S.W., M.E. Valk-Lingbeek, P.P. van der Stoop, J.J. Jacobs, K. Kieboom, E. Tanger, D. Hulsman, C. Leung, Y. Arsenijevic, S. Marino, and M. van Lohuizen. 2005. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 19:1438–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Molofsky, A.V., S. He, M. Bydon, S.J. Morrison, and R. Pardal. 2005. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 19:1432–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bauer, A., A. Villunger, V. Labi, S.F. Fischer, A. Strasser, H. Wagner, R.M. Schmid, and G. Hacker. 2006. The NF-κB regulator Bcl-3 and the BH3-only proteins Bim and Puma control the death of activated T cells. Proc. Natl. Acad. Sci. USA. 103:10979–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.You, H., M. Pellegrini, K. Tsuchihara, K. Yamamoto, G. Hacker, M. Erlacher, A. Villunger, and T.W. Mak. 2006. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 203:1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Napoles, M., J.E. Mermoud, R. Wakao, Y.A. Tang, M. Endoh, R. Appanah, T.B. Nesterova, J. Silva, A.P. Otte, M. Vidal, et al. 2004. Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell. 7:663–676. [DOI] [PubMed] [Google Scholar]
- 50.Wang, H., L. Wang, H. Erdjument-Bromage, M. Vidal, P. Tempst, R.S. Jones, and Y. Zhang. 2004. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 431:873–878. [DOI] [PubMed] [Google Scholar]
- 51.Cao, R., Y. Tsukada, and Y. Zhang. 2005. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 20:845–854. [DOI] [PubMed] [Google Scholar]
- 52.Wang, L., J.L. Brown, R. Cao, Y. Zhang, J.A. Kassis, and R.S. Jones. 2004. Hierarchical recruitment of polycomb group silencing complexes. Mol. Cell. 14:637–646. [DOI] [PubMed] [Google Scholar]
- 53.Fischle, W., Y. Wang, S.A. Jacobs, Y. Kim, C.D. Allis, and S. Khorasanizadeh. 2003. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17:1870–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Min, J., Y. Zhang, and R.M. Xu. 2003. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 17:1823–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vire, E., C. Brenner, R. Deplus, L. Blanchon, M. Fraga, C. Didelot, L. Morey, A. Van Eynde, D. Bernard, J.M. Vanderwinden, et al. 2006. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 439:871–874. [DOI] [PubMed] [Google Scholar]
- 56.Negishi, M., A. Saraya, S. Miyagi, K. Nagao, Y. Inagaki, M. Nishikawa, S. Tajima, H. Koseki, H. Tsuda, Y. Takasaki, et al. 2007. Bmi1 cooperates with Dnmt1-associated protein 1 in gene silencing. Biochem. Biophys. Res. Commun. 353:992–998. [DOI] [PubMed] [Google Scholar]
- 57.Murphy, K.M., A.B. Heimberger, and D.Y. Loh. 1990. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 250:1720–1723. [DOI] [PubMed] [Google Scholar]
- 58.Miyoshi, H., U. Blomer, M. Takahashi, F.H. Gage, and I.M. Verma. 1998. Development of a self-inactivating lentivirus vector. J. Virol. 72:8150–8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamashita, M., M. Ukai-Tadenuma, M. Kimura, M. Omori, M. Inami, M. Taniguchi, and T. Nakayama. 2002. Identification of a conserved GATA3 response element upstream proximal from the interleukin-13 gene locus. J. Biol. Chem. 277:42399–42408. [DOI] [PubMed] [Google Scholar]
- 60.Kamata, T., M. Yamashita, M. Kimura, K. Murata, M. Inami, C. Shimizu, K. Sugaya, C.R. Wang, M. Taniguchi, and T. Nakayama. 2003. src homology 2 domain-containing tyrosine phosphatase SHP-1 controls the development of allergic airway inflammation. J. Clin. Invest. 111:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}