

Abstract
Semaphorins are a large family of evolutionarily conserved morphogenetic molecules originally identified for their repelling role in axonal guidance. Intriguingly, semaphorins have recently been implicated in cancer progression (Neufeld, G., T. Lange, A. Varshavsky, and O. Kessler. 2007. Adv. Exp. Med. Biol. 600:118–131). In particular, semaphorin 3B (SEMA3B) is considered a putative tumor suppressor, and yet we found that it is expressed at high levels in many invasive and metastatic human cancers. By investigating experimental tumor models, we confirmed that SEMA3B expression inhibited tumor growth, whereas metastatic dissemination was surprisingly increased. We found that SEMA3B induced the production of interleukin (IL) 8 by tumor cells by activating the p38–mitogen-activated protein kinase pathway in a neuropilin 1–dependent manner. Silencing the expression of endogenous SEMA3B in tumor cells impaired IL-8 transcription. The release of IL-8, in turn, induced the recruitment of tumor-associated macrophages and metastatic dissemination to the lung, which could be rescued by blocking IL-8 with neutralizing antibodies. In conclusion, we report that SEMA3B exerts unexpected functions in cancer progression by fostering a prometastatic environment through elevated IL-8 secretion and recruitment of macrophages coupled to the suppression of tumor growth.
Semaphorins are a highly conserved family of molecular signals originally identified as guidance cues for axon navigation in neural development (1, 2). Moreover, several semaphorins have been implicated in angiogenesis, immune function, and cancer (for reviews see references 3, 4).
The functional role of semaphorins in tumor progression is still controversial. For example, semaphorin 3B (SEMA3B) and the homologous gene SEMA3F have been indicated as putative tumor suppressors because they are both located in the chromosomal region 3p21.3, a locus frequently deleted or hypermethylated in lung cancers (5, 6). SEMA3B and SEMA3F can furthermore inhibit tumor proliferation in vitro and in vivo (7–9) Moreover, SEMA3F has been shown to inhibit angiogenesis and metastatic dissemination (10, 11).
We have previously demonstrated that semaphorin function is mediated by a family of plasma membrane receptors, the plexins (12). However, most secreted semaphorins (including SEMA3B) do not bind directly to plexins but instead interact with plexin-associated coreceptors, denoted neuropilin (NP) 1 and 2. Moreover, NPs directly bind vascular endothelial growth factor (VEGF) family members and associate in complex with VEGF receptors (13), playing a crucial role in angiogenesis beyond axon guidance (for review see reference 14). SEMA3B interacts with both NP1 and NP2, whereas little is known about the requirement of specific plexins in the receptor complex. In general, different plexins of the A subfamily seem to be functionally redundant in mediating semaphorin signaling in association with the NPs.
Interestingly, SEMA3B has been shown to exert dual functions in the guidance of cortical axons, acting either as an attractant or a repellant on diverse neuronal populations (15). This finding is consistent with that reported for other family members and suggests that semaphorins may have antagonistic functions, depending on the target cells. This is particularly important in cancer biology, because tumors are known to be multifaceted tissues comprising several regulatory cells, in addition to tumor cells. Intriguingly, although previous experiments have analyzed SEMA3B activity on tumor cell proliferations, its potential effects on metastatic dissemination have not been addressed. We report that although expression of SEMA3B in tumor cells may delay tumor growth, it also promotes metastatic dissemination. Our data indicate that this activity of SEMA3B is mediated by NP1-dependent activation of p38–mitogen-activated protein kinase (MAPK), leading to the induction of p21 growth-inhibitory protein, as well as of the prometastatic cytokine IL-8. Moreover, we found that IL-8 activity is required to mediate the recruitment of tumor-associated macrophages (TAMs) and metastatic dissemination in SEMA3B-expressing tumors.
RESULTS
SEMA3B is commonly expressed in human cancer cells
Previous studies (5–9) have indicated SEMA3B as a putative tumor suppressor whose expression may be lost in cancer cells. Surprisingly, in a screen of >25 human tumor-derived cell lines, we found significantly high levels of SEMA3B mRNA in cells derived from different kinds of tumors, including some of metastatic origin such as colon carcinoma (LoVo), mammary carcinoma (SKBR-3), and melanomas (Sk-Mel-2 and MALME-3M; Fig. 1 A). We confirmed the expression of SEMA3B receptors NP1 and 2, as well as multiple plexins, in these cells (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1; and not depicted); however, we did not observe any significant (direct or inverse) correlation between SEMA3B expression and that of potential receptors in tumor cells. To investigate further a putative correlation between SEMA3B expression and tumor progression, we analyzed three distinct datasets derived from microarray gene expression profiling in breast cancers (16), lung cancers (17), and malignant melanomas (18). Surprisingly, we could not observe any direct correlation between low SEMA3B expression and poor patient survival; instead, high levels of SEMA3B seemed to associate with metastatic progression in a subset of lung carcinomas (P = 0.0062; Fig. S2 C). Importantly, malignant melanomas were found to express strikingly high levels of SEMA3B compared with benign nevi (P = 2.5 × 10−11; Fig. 1 B). This is consistent with our finding that established melanoma cell lines express high SEMA3B levels (e.g., Sk-Mel-2, MALME-3M, and UACC-257 in Fig. S1 A). Moreover, Hoek et al. reported a sixfold induction of SEMA3B gene expression in primary malignant melanoma cells compared with normal melanocytes (19).
Figure 1.

SEMA3B is significantly expressed in human cancers. (A) SEMA3B expression levels were measured by real-time PCR in 26 different human tumor cell lines, including colorectal carcinomas (HT-29, LoVo, SV80, HCT 116, and Difi), breast adenocarcinomas (SK-BR3, MDA-MB231, and T47D), osteosarcoma cells (U2OS, KHOS-NP, Saos-2, and MG-63), prostate adenocarcinomas (PC-3 and DU145), and melanomas (Sk-Mel-2, UACC-257, MALME-3M, and MDA-MB435; reference 20), as well as gastric carcinoma (GTL-16), hepatocellular carcinoma (HepG2), cervical adenocarcinoma (HeLa), chronic myelogenous leukemia (K-562), pancreatic cancer (SUIT-2), histiocytic lymphoma (U-937), neuroblastoma (IMR5), and nonsmall-cell lung carcinoma (A549). A Taqman probe for human β-actin was used as the internal control for each sample. SEMA3B expression levels in cancer cells were then normalized to the mean (REF = 1; dashed line) of values obtained in three different nontumor immortalized cell lines (mammary epithelial cells [MCF-10A], mesothelial cells [Met-5A], and retinal pigmented epithelium [RPE]). Data are reported as the mean ± SD of triplicates. The y axis is broken to include representation of high value data points. (B) SEMA3B expression in 45 individual samples of malignant melanoma and 18 samples of benign nevi. Reported values indicate normalized SEMA3B detection signals derived from a microarray dataset previously described by Talantov et al. (reference 18). The dashed line indicates the threshold of detection call. None of the nonmalignant samples expressed SEMA3B above the detection confidence threshold, whereas semaphorin expression is high in most melanoma samples (P = 2.5 × 10−11).
These findings seemed to challenge the idea that SEMA3B functions as a tumor suppressor and prompted us to experimentally test in vivo the functional role of SEMA3B in cancer progression and metastasis. For our experiments, we thus selected MDA-MB435 human tumor cells (of melanoma origin) (20), which are characterized by very low expression of SEMA3B (Fig. 1 A), a fast growth rate, and low metastatic potential, and A549 nonsmall-cell lung carcinoma cells that express intermediate levels of SEMA3B and form slowly growing metastatic tumors in nude mice.
SEMA3B inhibits tumor growth but induces metastasis in vivo
We transduced MDA-MB435 and A549 cells with lentiviral vectors either encoding SEMA3B or a noncoding empty vector (EV). SEMA3B-transduced cells did not undergo any clear morphological change compared with controls (unpublished data). Consistent with what has been previously shown in other tumor cell lines (7–9), we observed growth inhibition in MDA-MB435 and A549 cells upon SEMA3B expression (Fig. 2 A). Moreover, in accordance with previous data on MDA-MB435 cells (9), we found that NP1 receptor expressed in tumor cells was likely implicated to mediate this autocrine effect, because NP1-blocking antibodies could rescue SEMA3B-induced growth inhibition, whereas RNA interference (RNAi)–mediated suppression of NP2 could not (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1). Notably, we found no evidence of increased cell apoptosis in vitro upon SEMA3B expression, nor that SEMA3B expression significantly affected tumor cell motility or cell scattering in vitro (unpublished data).
Figure 2.

SEMA3B-expressing tumors display reduced growth but increased metastatic dissemination. (A) MDA-MB435 (MDA) and A549 cells transduced to express SEMA3B (3B) or a noncoding EV were grown in culture for 4 d in 1% FBS. Cell growth was evaluated daily in separate dishes by staining cells with crystal violet and measuring absorbance. Data shown represent the mean and SD of triplicates.(B and C) SEMA3B-transduced and EV control tumor cells were injected subcutaneously into nude mice. Graphs display tumor volume (measured externally during the experiment; B) and tumor weight (measured after excision at the end of the experiment; C), respectively. Data shown are the mean ± SEM from nine mice per each experimental group. ***, P < 0.0005. (D) The expression of SEMA3B (Myc tagged) was detected in tumors by staining tissue sections with anti-Myc antibodies (green), whereas nuclear counterstaining was done with DAPI (blue). Bar, 100 μm. SEMA3B was similarly detected in tumor lysates by Western blotting (bottom). (E) At the end of the experiment, the lungs of mice carrying tumor xenografts (as described above) were contrasted by airway perfusion with India ink, and superficial metastases were counted under a stereoscopic microscope. Numbers of metastases are given as the mean ± SD from nine mice for each experimental group (left). We further determined the metastatic index of SEMA3B-expressing and control tumors (right) by calculating the ratio between the number of metastatic foci and tumor weight. **, P < 0.01; ***, P < 0.001. (F) A549 cells expressing SEMA3B under control of a doxycycline-inducible promoter were injected subcutaneously in nude mice. Tumor cells expressing the transactivator rTTA alone were used as controls. After 46 d, the mice were killed, and we measured tumor burden, counted lung metastasis, and calculated the metastatic index as described. Data shown are the mean ± SEM. Western blot analysis on protein lysates from tumor samples (top) revealed a minimum level of recombinant SEMA3B expression in the absence of the doxycycline, which was strongly induced by treatment with the drug. *, P < 0.05; **, P < 0.01.
The tumorigenic potential of SEMA3B-expressing cells was analyzed in vivo by subcutaneous transplantation in nude mice. Tumor volume was periodically measured, and tumor weight was determined upon dissection at the end of the experiment. Tumor xenografts expressing SEMA3B displayed a significant reduction in volume and tumor burden (Fig. 2, B and C), yet they were surprisingly associated with a remarkable increase of spontaneous metastatic dissemination to the lungs (Fig. 2 E). The metastatic potential was quantified by scoring macrometastasis in the lungs (Fig. 2 E, left). Moreover, we found that the ratio between the number of metastatic foci and the size of respective primary tumors was increased >10 times in SEMA3B-expressing tumors versus controls (Fig. 2 E, right), indicating a strong increase in the metastatic potential of tumor cells. SEMA3B expression was confirmed in primary tumors by the immunostaining of cryostat sections (Fig. 2 D, top) and by Western blot analysis of protein lysates from tumor samples (Fig. 2 D, bottom). SEMA3B expression was also detected in metastatic cells by immunostaining (unpublished data), ruling out that they represented a selected subpopulation of cells that have lost SEMA3B expression.
To confirm the specificity of these effects, we expressed SEMA3B under the control of an rTTA- and doxycycline-inducible promoter in A549 cells. This system allowed us to obtain drug-regulated expression of SEMA3B in tumor xenografts in vivo. Notably, low levels of SEMA3B were detected in tumors in the absence of the drug, whereas they greatly increased upon treatment with doxycycline (Fig. 2 F, top). Semaphorin levels in xenografts correlated directly with the metastatic potential and inversely with the tumor burden (Fig. 2 F, bottom).
Tumor cell proliferation and angiogenesis in SEMA3B-expressing tumors
We hypothesized that the inhibition of tumor growth by SEMA3B could be accounted for by a primary effect on cell proliferation, consistent with our data in vitro. To reliably answer this question, we analyzed tumors of comparable size taken 15 d after transplant. Consistent with in vitro observations, SEMA3B-expressing tumor cells showed a >60% reduction in proliferation rate when transplanted in vivo (as assessed by anti–human Ki-67 staining; Fig. 3 A), whereas apoptosis was only moderately increased (detected by Tdt-mediated dUTP-biotin nick-end labeling [TUNEL]; Fig. 3 B); these results were further confirmed by analyzing tumors at a later stage (30 d; not depicted). Double-staining experiments with markers of endothelia and TAMs did not detect a significant number of mouse stromal cells undergoing apoptosis (unpublished data), consistent with those that were essentially represented by tumor cells. Because it has been shown previously that other semaphorins can inhibit VEGF-induced angiogenesis (for review see reference 21), we asked whether SEMA3B could similarly mediate inhibition of tumor angiogenesis. We could not find any significant difference in vessel area, as measured by lectin-perfused vessels, between SEMA3B xenografts and controls in tumors of comparable size (15 d after transplant); notably, the number of vessels was slightly increased in SEMA3B-expressing xenografts compared with controls, suggesting that vessel structure was altered (Fig. 3, C and D). However reduced angiogenesis was unlikely to be primarily responsible for delayed tumor growth. We observed a 25% reduction of angiogenesis in tumors expressing SEMA3B at later stages (day 30), when they had become remarkably smaller than controls (unpublished data). Moreover, in SEMA3B-expressing tumors, we observed notable defects of pericyte recruitment to blood vessels compared with control xenografts (Fig. 3 E).
Figure 3.

Tumor cell proliferation and angiogenesis in SEMA3B-expressing tumors. (A) Control or SEMA3B-expressing tumors (15 d after transplant) were immunostained to reveal KI-67 proliferation markers (red), whereas nuclei were visualized by DAPI (blue). Representative fields are shown. Bar, 20 μm. The bar graph shows the percentage of Ki-67–positive cells (mean ± SD) in six tumors per each experimental group. For all quantifications shown in this figure, at least three sections and 10 fields were counted for each tumor. **, P < 0.01. (B) Tumor sections were analyzed to reveal apoptotic cells using TUNEL green kit. Micrographs show representative fields. Bars, 20 μm. The bar graph indicates the percentage of TUNEL-positive cells (mean ± SD) in six tumors per each experimental group. (C) Micrographs show sections from tumors stained with SA-lectin to reveal endothelial cells (red), whereas nuclei were marked with DAPI (blue). Bar, 60 μm. (D) Quantification of the results in C. The graphs indicate the percent area occupied by vessels (left) or the vessel number (right), calculated in six fields per section. Values represent the mean ± SD from six tumors per each experimental group. *, P < 0.05. (E) Sections from tumors excised at day 21 after transplant were immunostained with anti-CD31 (red) and with an antibody revealing the chondroitin proteoglycan NG2 that is expressed on pericytes (green). Micrographs show representative fields at low and high magnifications (left and right, respectively). Bars: (left) 60 μm; (right) 20 μm.
Macrophage recruitment in SEMA3B tumors
Several important factors regulating tumor angiogenesis and metastatic dissemination are released by TAMs (22–24). Interestingly, by staining our cryostat sections with the selective antibody F4/80 for macrophages, we observed a twofold induction of macrophage recruitment into the tumor periphery (Fig. 4 A, top row) in SEMA3B xenografts, as well as an increased infiltration in the center of the tumor (Fig. 4 A, bottom row). In contrast, tumor-infiltrating granulocytes did not increase upon SEMA3B expression in vivo (based on Gr1 immunostaining; unpublished data).
Figure 4.

SEMA3B expression in tumor cells induces macrophage recruitment. (A) Sections from tumors excised at day 15 after transplant were immunostained with anti-F4/80 antibody to selectively detect macrophages (red), whereas nuclei were revealed with DAPI (blue). Macrophages were quantified in the periphery and in the center of the tumors. Bars, 60 μm. The graphs represent the mean ± SD from eight tumors for each experimental group (at least three sections and 10 fields occupied by macrophages were counted from each tumor). ***, P < 0.001; **, P < 0.01. (B) The migration of monocytes purified from human peripheral blood was analyzed in a Transwell migration assay. Increasing concentrations of CM (1x, 3x, or 10x, as indicated) derived from tumor cells transduced with SEMA3B (3B) or EV were included in the lower chamber (or alternatively in the upper chamber, as specified). Migrated monocytes were stained with Giemsa and counted in low magnification micrographs of the filters, as reported previously (reference 27). Values in the graph represent the mean ± SD of three independent experiments. 100 ng/ml MCP-1 in the lower chamber provided an internal positive control. n.t., nontreated cells.
These findings indicated that SEMA3B expression in tumor cells induces their ability to attract monocytes/macrophages. To further investigate this effect, we performed migration assays in vitro with monocytes purified from human peripheral blood. As shown in Fig. 4 B, the conditioned medium (CM) of MDA-3B cells strongly attracted monocytes in a dose-dependent manner compared with the medium collected from mock-transduced control cells. The effect was comparable to that induced by the well-known monocyte chemotactic protein 1 (MCP-1; Fig. 4 B). Intriguingly, monocyte chemotaxis was not affected by immunodepleting SEMA3B content in the CM (unpublished data), suggesting that the semaphorin had induced the release of further regulatory factors by tumor cells.
SEMA3B induces IL-8 expression in tumor cells
By performing microarray analysis of gene expression on mRNA derived from MDA-MB435 cells transduced with SEMA3B or EV, we found that the most significantly regulated gene was IL-8 (P < 0.001; Fig. 5 A). Interestingly, IL-8 is known to be a chemoattractant for leucocytes and endothelial and smooth muscle cells, which are further implicated in human cancer biology (25–27).
Figure 5.

SEMA3B induces IL-8 expression in tumor cells, which accounts for increased monocyte attraction. (A) mRNA from transduced MDA-MB435 cells was used to probe a microarray representative of the human genome. Results are shown as a dot plot of individual genes, comparing the expression levels found in EV– and SEMA3B-transduced cells. A selected panel of 622 genes, including cytokines and factors mediating inflammatory response (identified by keywords as described in Materials and methods), is highlighted in red. IL-8 is the only gene in the panel significantly regulated more than twofold. ***, P < 0.001) (B) Real-time quantitative PCR mRNA expression analysis of SEMA3B- and EV-transduced tumor cells using Taqman probes for IL-8 and for the housekeeping gene β-actin (internal reference). Reported values indicate fold induction of IL-8 expression in SEMA3B-expressing cells relative to the respective EV control for each cell line, and values correspond to the mean ± SD of three individual experiments. *, P < 0.05. (C) The CM from MDA-MB435 EV control and SEMA3B-expressing cells was collected and concentrated 300 times. 10 μl of this preparation was analyzed by Western blotting with anti–IL-8 antibodies. The indicated amounts of purified IL-8 were loaded for internal reference. The concentration of IL-8 in the original CM of MDA-3B cells was thus estimated to be in the range of 5–7 ng/ml. (D) Western blot analysis of A549 cells expressing SEMA3B under a drug-inducible promoter; control cells (EV) expressed the doxycycline-dependent transactivator rTTA alone. Upon doxycycline treatment, SEMA3B expression in tumor cells (revealed by anti-Myc tag antibodies) and IL-8 levels in the CM (revealed with MAB208) were increased; purified IL8 was loaded as an internal reference. (E) The CM of SEMA3B tumor cells and EV controls were assayed in monocyte migration experiments, similar to those shown in Fig. 4 B. Where indicated, IL-8 neutralizing antibodies (MAB208) or an unrelated control antibody (i.e., anti-CD19; C Ab), or 100 ng/ml of purified IL-8 were added in the lower well. The number of migrated monocytes was scored as in Fig. 4 B, and the graph shows the mean of three independent experiments. IL-8 is responsible for the increased monocyte-attracting activity released by SEMA3B-expressing tumor cells. **, P < 0.01.
We thus confirmed microarray data by real-time quantitative PCR in tumor cell lines transduced with SEMA3B (i.e., A549 lung carcinoma, HeLa cervical carcinoma, HT29 colon carcinoma, and GTL16 gastric carcinoma; Fig. 5 B). In addition, the CM of MDA-3B and control cells was collected and analyzed by Western blotting, revealing that IL-8 secretion was strongly induced upon SEMA3B expression (Fig. 5 C). Moreover, IL-8 protein levels were specifically up-regulated by treatment with doxycycline in A549 carcinoma cells expressing SEMA3B under the control of a drug-inducible promoter (Fig. 5 D).
Monocyte/macrophage recruitment induced by SEMA3B depends on IL-8
Consistent with previous reports (28–30), we observed a significant dose-dependent increase of monocyte migration in vitro in response to purified IL-8 (Fig. S4 A, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1). This monocyte-attracting activity of purified IL-8 was comparable to that induced by the CM of SEMA3B-expressing cells (Fig. S4 A); moreover, it was efficiently blocked by monoclonal anti–IL-8 neutralizing antibodies (MAB208; Fig. S4 B). Therefore, to test whether monocyte recruitment induced by SEMA3B-CM could actually depend on increased IL-8 secretion by tumor cells, we repeated migration assays, as in Fig. 4 B, in the presence of anti–IL-8 neutralizing antibodies. Fig. 5 E shows that this treatment abolished the functional difference between the chemotactic activities of SEMA3B-CM and control EV-CM, whereas an irrelevant antibody (α-CD19) was ineffective. These data clearly indicated that the cytokine IL-8, released by tumor cells in response to an autocrine loop of SEMA3B, was crucially implicated to increase monocyte recruitment.
Consistent with our evidence that SEMA3B regulates IL-8 production, we asked whether IL-8 levels might be affected upon knocking down endogenous SEMA3B in cancer cells. We thus stably transduced various tumor cells expressing medium to high levels of SEMA3B (i.e., A549, HeLa, LoVo, and Sk-Mel-2 cells) with constructs expressing short hairpin RNAs (shRNAs) targeting SEMA3B (shSEMA3B) or control unrelated sequences (shCTR). Importantly, we found that the knock down of SEMA3B expression led to reduced levels of IL-8 mRNA (Fig. 6 A). Furthermore, three different shRNA sequences for SEMA3B were tested with consistent results, ruling out off-target effects (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1).
Figure 6.

SEMA3B knock down in tumor cells affects IL-8 production and monocyte recruitment. (A) RNA was collected from LoVo, A549, HeLa, and Sk-Mel-2 cells transduced with shRNA against SEMA3B (to suppress gene expression; shSEMA3B) and unrelated sequences (shCTR). Real-time PCR was performed using Taqman probes for SEMA3B (results shown at the bottom) and IL-8 (in the graph). Expression levels were normalized to controls for each cell line and are represented as fold changes. Data shown are the mean ± SEM. (B) Monocyte migration to CM derived from Sk-Mel-2 melanoma cells transduced with shRNAs. IL-8 neutralizing antibodies (MAB208) or unrelated control antibodies (anti-CD19) were included in the lower chamber. Knock down of SEMA3B expression in Sk-Mel-2 cells abolished IL-8–dependent monocyte-attracting activity in the CM. The number of migrated monocytes represents the mean ± SEM of two independent experiments. **, P < 0.01.
To investigate the role of endogenous SEMA3B in monocyte recruitment by tumor cells, we performed migration assays with the CM of Sk-Mel-2 melanoma cells that basally secrete high levels of IL-8 (unpublished data). The medium collected from control cells strongly attracted monocytes compared with that of SEMA3B-deficient cells (shSEMA3B; Fig. 6 B). Importantly, this functional difference was entirely accounted for by the loss of IL-8 expression in shSEMA3B cells, because including IL-8 neutralizing antibodies abrogated the difference between monocyte migration induced by control CM and that of SEMA3B-deficient melanoma cells (Fig. 6 B).
IL-8 mediates macrophage infiltration and metastasis in SEMA3B-expressing tumors
Elevated IL-8 expression in tumors is a poor prognostic indicator correlating with invasive/metastatic progression (31–33); thus, we hypothesized that this cytokine could account for the metastatic progression observed in SEMA3B-expressing tumors. To this end, we exploited neutralizing antibodies (MAB208) to inhibit IL-8 function in vivo, as successfully done in previous studies (34, 35). MDA-EV and MDA-3B tumor xenografts were treated with MAB208 or with an irrelevant control antibody from the same day of transplant and twice a week for a period of 2 wk by direct injection at the tumor site. Tumor volume was not significantly altered by MAB208 treatment (Fig. 7 A). In contrast, the increased recruitment of macrophages into tumors and the metastatic dissemination induced by SEMA3B were totally prevented by MAB208 (Fig. 7, C and B, respectively), whereas tumors treated with control antibody were not affected. Moreover, tumor angiogenesis in SEMA3B-expressing tumors was strongly reduced upon MAB208 treatment and associated with sites of residual macrophage infiltration (Fig. 7, D and E). In contrast, there was no significant reduction in control tumors. These findings suggested a putative antiangiogenic activity of SEMA3B, masked in vivo by increased IL-8 release. Notably, other class 3 semaphorins have been shown to repel endothelial cells and to inhibit angiogenesis both in vitro and in vivo (10, 36). In fact, we could demonstrate a repelling activity of SEMA3B on human umbilical vein endothelial cells (HUVECs) in co-culture experiments and migration assays in vitro, even if this inhibitory effect was partly mitigated by the autocrine secretion of IL-8 induced by the semaphorin (Fig. S6, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1). Consistent with the reduced vascularization in SEMA3B-expressing tumors treated with MAB208, we also found a fourfold induction of apoptosis (Fig. 7 F), whereas tumors in all other conditions were not significantly affected. Importantly, we ruled out any direct detrimental effect of the antibody on the viability or proliferation of tumor cells by further examining the growth rate in the absence or presence of MAB208 in vitro (unpublished data).
Figure 7.

Neutralizing IL-8 in SEMA3B-expressing tumors blocks macrophage recruitment and metastasis. (A) Nude mice were injected subcutaneously with either MDA-EV or MDA-3B tumor cells together with 100 μg of neutralizing IL-8 antibody (MAB208) or 100 μg of control antibody (anti-CD19). Antibodies were administered locally into tumors again at days 2, 5, 8, and 11 after tumor transplant. Data shown are the mean tumor volume ± SD from 10 tumors per each experimental group at the day of excision (day 15). (B) To reveal metastatic dissemination from the tumors, the lungs of mice described in A were analyzed by real-time RT-PCR using species-specific Taqman probes for mouse HPRT and human HPRT transcripts. Plotted values indicate the mean ratio between human and mouse RNA levels in the lungs of five mice per each experimental group ± SD. *, P < 0.05. (C) Tissue sections from SEMA3B and EV tumors, treated with control antibody or MAB208 (from the same experiment as in A), were stained with anti-F4/80 to detect monocytes/macrophages (red), whereas nuclei were revealed with DAPI (blue). Bar, 60 μm. Each data point represents the mean percentage of the macrophage-occupied area ± SD from eight tumors per each experimental group (10 fields were counted from at least three different sections for each tumor). **, P < 0.01; ***, P < 0.001. (D) Blood vessels and macrophages in sections of MDA-EV or MDA-3B tumors treated as described were revealed with anti–VE-cadherin (red) and anti-F4/80 (green); nuclei were marked with DAPI. Bar, 60 μm. (E) Blood vessels in sections of MDA-EV or MDA-3B tumors treated as described were revealed with anti-CD31 antibody (red), whereas nuclei were marked by DAPI (blue). Bar, 60 μm. The graph shows the mean vessel area ± SD from eight tumors for each experimental group (at least three sections and 10 fields were quantified per tumor). ***, P < 0.0005. (F) Sections from MDA-EV or MDA-3B tumors treated as described were subjected to TUNEL staining to reveal apoptotic cells. Bar, 60 μm. The graph shows the mean percentage of TUNEL-positive cells (±SEM) from five tumors for each experimental group in at least three sections and 10 fields. *, P < 0.05.
SEMA3B-dependent IL-8 induction is mediated by NP1 and p38–MAPK activation
In an effort to identify the molecular mechanisms driving SEMA3B-dependent IL-8 induction in tumor cells, we investigated NF-κB and p38–MAPK, two known regulators of IL-8 expression at transcriptional and posttranscriptional levels (25). The nuclear translocation of NF-κB was not significantly increased in MDA-3B cells (unpublished data). However, SEMA3B-expressing cells displayed a striking increase in p38 phosphorylation upon serum starvation compared with controls (Fig. 8 A). Moreover, by treating MDA-MB435 or A549 cells with CM containing SEMA3B, we rapidly induced the phosphorylation of p38 and its major substrate, activating transcription factor 2 (ATF-2), whereas MAPK phosphorylation was not induced (Fig. 8, B and C). To investigate the requirement of p38–MAPK signaling for SEMA3B-dependent IL-8 induction, we cultured transduced cells in the presence of selective p38 inhibitors. As shown in Fig. 8 D, blocking p38 signaling abrogated IL-8 expression in SEMA3B-expressing cells.
Figure 8.

p38–MAPK signaling elicited by SEMA3B mediates IL-8 induction. (A) Immunoblotting analysis to reveal phospho-p38 protein levels in MDA-MB435 tumor cells expressing SEMA3B (or EV controls), grown in the presence or absence of 10% FBS for 48 h. Results are displayed as fold induction of the phosphorylated/total p38 ratio in SEMA3B cells versus controls, as measured by densitometric analysis. (B) Serum-starved MDA-MB435 cells were treated for 30 min with CM collected from SEMA3B or EV control cells. The levels of phospho-p38 and phospho–ATF-2 were analyzed in protein lysates by immunoblotting. The same filter was furthermore decorated to reveal vinculin, providing loading controls. Plotted values indicate fold increase of the phosphorylated p38/vinculin or ATF2/vinculin ratios in SEMA3B-treated cells versus controls. (C) Immunoblotting analysis of A549 cells treated as in B with SEMA3B or EV control CM. Filters were probed to reveal phospho-p38, phospho-p42/44–MAPK, and vinculin levels (as loading control). The graph shows fold increase of the phosphorylated p38/vinculin or phospho-p42/44–MAPK/vinculin ratios in SEMA3B-treated cells versus controls. (D) MAB208 antibody was used to detect IL-8 expression by immunofluorescence (top) or by Western blotting (bottom) in SEMA3B-expressing and control cells grown in the absence of serum for 48 h, in the presence or absence of 10 μM of the selective p38–MAPK inhibitor SB202190. Micrographs were acquired with a bioimager (Pathway; BD Biosciences) and quantified by Autovision 1.5 (10 images per each data point). Results are displayed as mean ± SD. ***, P < 0.001. (E) CDKI p21 expression was detected by Western blotting in A549 cells, transduced with SEMA3B or EV control, upon serum starvation (48 h in 1% FCS). Cells treated with 20 nM staurosporin (STS) for 24 h provided a positive control for p21 activation. β-Actin levels provided a reference for protein loading. Plotted values indicate fold induction of the p21/β-actin ratio. (F) A549 cells were serum starved for 24 h and then treated for 30 min with CM from SEMA3B or EV control cells in the presence of 20 ng/ml anti-NP1 blocking antibody or the unrelated antibody anti–VSV-G. Cells treated with 20 μm cisplatin for 2 h provided a positive control for p38 activation. The levels of phospho-p38 and vinculin (as loading control) were analyzed in protein lysates by immunoblotting. (G) Consistent with a previous report, many tumor cells could not survive RNAi-mediated knock down of NP1 (reference 65); however, we managed to stably express NP1-targeted shRNAs (shNP1) in HeLa cells previously transduced with SEMA3B or EV (Fig. 5 B). Real-time PCR analysis indicated that NP1 expression levels were knocked down to 20% compared with cells transduced with unrelated sequences (shCTR; not depicted). Notably, SEMA3B autocrine stimulation was unable to up-regulate IL-8 levels in NP1-deficient cells, indicating the requirement for this receptor in the signaling pathway. The graph shows fold increase of expression relative to control cells. Data shown are the mean ± SEM.
It has been previously described that p38 regulates the expression of the cytokine-dependent kinase inhibitor (CDKI) p21-Waf (37). We therefore investigated p21 protein levels in SEMA3B-transduced cells and found that they were increased compared with controls (staurosporin was used as a positive control for p21 induction; Fig. 8 E). Because we had found that NP1 is required for SEMA3B-dependent growth inhibition (Fig. S3 A), we asked whether the same receptor was involved in SEMA3B-induced p38–MAPK activation. In fact, by incubating cells with anti-NP1 blocking antibodies, we demonstrated that the activation of this pathway requires NP1 (Fig. 8 F). Furthermore we exploited RNAi to knock down NP1 in tumor cells. Interestingly, SEMA3B overexpression in NP1-deficient cells could not up-regulate IL-8 levels (Fig. 8 G), indicating that the signaling pathway depends on this receptor. In contrast, RNAi-mediated knock down of NP2 did not affect SEMA3B-dependent IL-8 induction (unpublished data). Thus, the NP1 receptor and p38–MAPK signaling are required in the pathway mediating SEMA3B-dependent growth inhibition and IL-8 up-regulation.
DISCUSSION
Semaphorins, beyond their well-characterized function in neuronal guidance, have also been implicated in tumorigenesis. In particular, SEMA3B is considered a putative tumor suppressor gene. In fact, consistent with previous reports (7–9), we observed that SEMA3B is able to inhibit the proliferation of tumor cells in vitro and delay the growth of tumor xenografts in nude mice (Fig. 2, A–C). Based on these premises, high expression of SEMA3B in tumors may be thought to correlate with good patient prognosis and vice versa. However, by analyzing large datasets of human tumor samples, we could not find any statistically significant correlation between SEMA3B expression and patient survival. In contrast, our data indicated that subsets of tumors with elevated SEMA3B levels are associated with metastatic progression and poor prognosis. These unexpected findings prompted us to experimentally test in vivo the functional role of SEMA3B in cancer progression, especially because previous studies had not investigated its potential relevance in tumor invasion and metastasis. Interestingly, we found that an elevated expression of SEMA3B in MDA-MB435 and A549 human cancer cells induced spontaneous metastasis from subcutaneous tumors in mice. This effect was even more striking considering that SEMA3B-expressing primary tumors were remarkably smaller (Fig. 2 E). Moreover, unlike what was described for SEMA3E (38), tumor cells expressing SEMA3B did not show an increased ability to form metastatic lung colonies upon direct injection in the blood circulation (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1), indicating that SEMA3B acts by promoting cancer cell dissemination from the primary tumor, and not by increased cell survival or homing from the circulation.
Because SEMA3B expression in tumor cells did not seem to affect cell adhesion, cell migration, or cell scattering in vitro in a cell-autonomous manner (unpublished data), we postulated that the increased metastatic potential observed in vivo may implicate regulatory cells in the tumor microenvironment. In fact, we found that SEMA3B expression increased the recruitment of monocytes/macrophages into the tumor microenvironment (Fig. 4); notably these TAMs are well-known mediators of tumor invasion and metastasis (39, 24). Moreover, SEMA3B expression induced the secretion of IL-8 by tumor cells (Fig. 5 and Fig. 6), a cytokine also associated with tumor progression and metastasis (40). By means of neutralizing antibodies, we demonstrated that the activity of IL-8 was responsible for macrophage recruitment and for the prometastatic effect mediated by SEMA3B (Fig. 7). Finally, we identified the receptor and intracellular pathway, namely NP1 and p38–MAPK, required to mediate SEMA3B activities in cancer cells, leading to increased expression of IL-8 and the cell-cycle inhibitor p21-Waf (Fig. 8).
IL-8 is a pleiotropic cytokine produced by tumor cells and cells of the tumor microenvironment (40–44). IL-8 can also act as chemoattractant for neutrophils and monocytes (26–28, 35). Moreover, it has been shown to promote endothelial cell survival and angiogenesis in vivo, as well as tumor cell migration and survival (42, 43). Elevated IL-8 expression in tumors is a poor prognostic indicator, correlated with increased metastasis and to remarkable effects on the tumor microenvironment (e.g., leukocyte recruitment and tumor angiogenesis) (45, 46). Studies have shown that IL-8 levels are up-regulated through the activation of p38–MAPK and/or NF-κB independent pathways (25), as well as in response to RAS signaling (34). We now show for the first time that SEMA3B induces IL-8 expression in tumor and endothelial cells, thereby supporting a new and unexpected function of this semaphorin to promote tumor progression and metastasis. The specificity of SEMA3B-dependent regulation of IL-8 was confirmed in different ways: (a) SEMA3B overexpression in a variety of tumor cells—under constitutive or drug-regulated promoters—correlated with induced IL-8 levels (Fig. 5); (b) RNAi-mediated suppression of endogenous SEMA3B in tumor cells led to reduced levels of IL-8 (Fig. 6); and (c) one of the two SEMA3B receptors, NP1, was found to be required for SEMA3B-dependent activation of p38–MAPK and IL-8 induction (Fig. 8).
By means of IL-8-blocking antibodies, we have demonstrated that the functional difference between SEMA3B-expressing tumor cells and their respective controls, in terms of monocyte/macrophage-attracting activity and metastatic progression in vivo, was entirely dependent on the release of IL-8 (Fig. 5 E; Fig. 6 B; and Fig. 7, B and C). As expected, the CM of tumor cells basally contained additional monocyte chemoattractants, independently from SEMA3B regulation. This was consistent with our microarray expression analysis; moreover, we directly investigated by real-time PCR the expression levels of two major monocyte chemoattractants capable of mediating invasive/metastatic tumor progression, such as CSF-1 and MCP-1, in four different tumor cell lines transduced with SEMA3B. We found that these factors are basally expressed; however, unlike what was seen for IL-8, there was no consistent induction of MCP-1 or CSF-1 levels in cancer cells overexpressing SEMA3B (unpublished data). Moreover, by analyzing gene expression in a dataset derived from human melanoma samples, we found that SEMA3B expression is directly correlated with IL-8 levels and with the expression of the macrophage marker CD14 (Fig. S8, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1) (47). Intriguingly, we did not observe an increased recruitment of neutrophils in SEMA3B-expressing xenografts in vivo, although SEMA3B-CM increased the basal migration of neutrophils in vitro (by 1.5-fold) because of its content in IL-8 (unpublished data). This discrepancy may be explained by the concept that the recruitment of different leucocytes into growing tumors in vivo is further regulated by the activity of additional factors released in the microenvironment by cancer and stromal cells.
TAMs recruited from the peripheral blood play a crucial role in tumor angiogenesis and tumor progression (24), especially by releasing proangiogenic and proinvasive factors such as VEGF or epidermal growth factor (48–51). Notably, we have evidence in vitro that some of the factors released by TAMs, such as epidermal growth factor and VEGF, induce the migration of the tumor cells used in our studies (unpublished data). None of these mechanisms is likely to act alone to mediate invasion and metastasis in vivo. Thus, we propose that the prometastatic function of SEMA3B seen in vivo depends on the activity of IL-8 and on additional factors released by macrophages recruited in the tumor microenvironent.
Intriguingly, despite the observation that SEMA3B expression in cancer cells induced high levels of IL-8 and increased the recruitment of infiltrating macrophages, we found that the tumor-associated vessel area was not significantly affected. However, upon IL-8 neutralization, not only was SEMA3B-mediated macrophage recruitment inhibited, but vessel area was also markedly decreased compared with control tumors. These data suggested that IL-8 and the proangiogenic signals released by TAMs in SEMA3B tumors were in balance with some antiangiogenic activity. In fact, consistent with what was reported for other class 3 semaphorins (36, 10–11), we found that SEMA3B is capable of repelling HUVECs and inhibiting their migration in vitro, an activity that is probably balanced in vivo by the proangiogenic factors released by TAMs. Notably, although it was recently shown that another semaphorin, SEMA3F, inhibits VEGF expression in tumor cells (52), we demonstrated that SEMA3B does not affect VEGF levels in cancer cells in vitro or in vivo (Fig. S9, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1).
Blood vessels in SEMA3B-expressing tumors further revealed defects in pericyte recruitment, a mechanism that has been implicated in metastatic dissemination (53). In fact, although normal blood vessels get stabilized and remodeled by associated pericytes and other “mural” cells, pericyte recruitment is often affected in tumor vasculature; endothelial cell survival and morphogenesis thereby becomes dependent on the angiogenic factors released by tumor cells and by infiltrating TAMs (54). Notably, we found that most of the vessels in SEMA3B-expressing tumors were in close association with macrophages (Fig. 7 D). In conclusion, the metastatic potential of SEMA3B-expressing tumors seems to involve multiple coordinated mechanisms dependent on IL-8 secretion and on remarkable changes in the tumor microenvironment.
In our study, we also investigated receptors and signaling pathways implicated in SEMA3B-mediated functions in tumor cells. SEMA3B was reported to bind both NP1 and NP2. Our unpublished data show that SEMA3B cannot bind directly to any of the plexins, whereas several plexins are redundantly capable to associate with NP1 and mediate a functional response to SEMA3B in COS cells similar to that known for most secreted class 3 semaphorins (55). We thus focused on NPs as required receptors to mediate SEMA3B activity. Notably, NPs are widely expressed in cancer cells and in the tumor microenvironment (e.g., in endothelial cells). It was previously shown that SEMA3B and VEGF have antagonistic functions in regulating tumor growth, and this was explained by binding competition of the two factors for NP1 (9). We have obtained analogous results in A549 cells (unpublished data). However recent findings seem to refute the assumption that semaphorin and VEGF binding sites on NP1 are overlapping (56). In fact, none of these data ruled out the possibility that SEMA3B and VEGF may independently elicit two antagonistic signaling pathways. In this respect, we have demonstrated that NP1 is responsible for SEMA3B-dependent growth inhibition by blunting this effect with NP1 blocking antibodies (Fig. S3 A). In addition, we showed that SEMA3B triggers p38–MAPK activation and IL-8 induction in an NP1-dependent manner (Fig. 8, F and G). Notably, p38 signaling is associated with cell-cycle inhibition via the induction of the CDKI p21 (37), which was also up-regulated in response to SEMA3B in our experiments (Fig. 8 E). Thus, p38–MAPK activation and the consequent induction of the CDKI p21 could have a major role in growth inhibition induced by SEMA3B. By using RNAi, we could furthermore rule out the requirement of NP2 to mediate SEMA3B functions in tumor cells. For instance, upon RNAi-mediated knock down of NP2, the antiproliferatory effect of SEMA3B in A549 carcinoma cells was preserved or even increased (Fig. S3 B). These data suggest that NP1 and NP2 are alternative receptors for SEMA3B and trigger independent signaling pathways.
We therefore focused on the intracellular mediators leading to IL-8 induction by SEMA3B in tumor cells. Previous data have shown that IL-8 expression is up-regulated through the activation of p38–MAPK (for review see reference 25). In this study, we show that p38–MAPK activity is significantly induced by SEMA3B; moreover, IL-8 induction was blocked by treatment with p38 inhibitors (Fig. 8). Interestingly, in addition to its role in cell-cycle arrest, a growing body of evidence implicates p38–MAPK in tumor progression and metastasis (for review see reference 57) This may be explained by a mechanism called “tumor dormancy,” by which cancer cells slow down proliferation to escape environmental stresses while maintaining a high invasive and metastatic potential. Consistent with this scenario, SEMA3B-induced p38 activation may feature an “escape” response of tumor cells, based on slow cell growth and gene induction promoting invasion and metastasis. Interestingly, there are other examples of putative tumor suppressors found to promote cancer progression, e.g., STAT1 (58) and the SMADs (59). It was also shown that hypoxic conditions put a brake on tumor growth, whereas they may in turn foster cancer cells that are more aggressive and metastatic (60). Moreover, it was recently reported that IL-8 induces angiogenesis and rescues tumor cells from undergoing apoptosis in colon cancer cells that lack HIF1-α expression (35). Notably, although hypoxic conditions have been reported to regulate the expression of IL-8 and its receptors (61, 62), we detected comparable levels of tissue hypoxia in SEMA3B-expressing tumors and in controls (by pimonidazol staining; unpublished data).
In conclusion, our results indicate that tumors expressing high SEMA3B levels grow slowly because of reduced proliferation of tumor cells, consistent with our finding that SEMA3B induces p38–MAPK activation. In addition, SEMA3B triggers an escape program from cancer suppression, mediated by p38-dependent IL-8 production by tumor cells, and by a stromal response that fosters cancer progression and metastatic dissemination (Fig. 9). These unexpected findings expand our knowledge of the functional activities mediated by SEMA3B in tumors and suggest a reconsideration of semaphorins as multifaceted regulators of cancer progression.
Figure 9.

Proposed mechanisms for autocrine SEMA3B signaling in tumor progression. Schematic diagram depicting our current model to explain multiple functional activities of SEMA3B in tumor cells. Autocrine SEMA3B signals, mediated by NP1 receptor (in association with plexins or other receptor subunits to be identified) leads to p38–MAPK activation. This results in growth inhibition (via CDKI p21 induction), as well as transcriptional activation of IL-8 expression (via ATF-2). In turn, the activity of IL-8 on stromal cells of the tumor microenvironment (including TAM) promotes invasion and metastasis.
MATERIALS AND METHODS
Cell lines.
Cell lines were obtained from American Type Culture Collection, except for the following: GTL-16 (provided by S. Giordano, IRCC, Torino, Italy), SUIT-2 (provided by M.F. Di Renzo, IRCC, Torino, Italy), and IMR5 (provided by V. Pistoia, Giannina Gaslini Institute, Genoa, Italy). Cell lines were grown in standard medium supplemented with l-glutamine and 10% FBS (Sigma-Aldrich).
Antibodies and reagents.
Myc-tagged proteins were detected by a biotin-conjugated anti-Myc antibody (9E10; Santa Cruz Biotechnology, Inc.). Anti-CD31 and anti-F4/80 were purchased from BD Biosciences. Other antibodies were as follows: anti–VE-cadherin (R&D Systems), anti–human Ki-67 antibody (Dako), anti-NG2 (Chemicon), and anti–IL-8 (catalog no. MAB208 [clone 6217]; R&D Systems). Antibodies to detect p38–MAPK, phospho-p38 (Thr180/Tyr182), and phospho–ATF-2 (Thr71) were from obtained from Cell Signaling Technology. Anti-p21 (F-5) was purchased from Santa Cruz Biotechnology, Inc., and anti–smooth muscle actin was purchased from Sigma-Aldrich. Anti-NP1 antibodies (63) were provided by A. Kolodkin (Johns Hopkins University, Baltimore, MD). The p38–MAPK inhibitor SB202190, staurosporin, cisplatin, and any other reagents, if not otherwise specified, were obtained from Sigma-Aldrich.
Gene expression in mammalian cells.
Mouse SEMA3B cDNA was subcloned, fused with an Myc epitope tag at the C terminus, into the lentiviral transfer plasmid pRRLsin.cPPT.hCMV.GFP.Wpre. Nonreplicating viral particles containing SEMA3B transfer plasmid (or an EV noncoding plasmid as control) were produced in 293T packaging cells and incubated with cultured human cells in the presence of 8 μg/ml polybrene, as previously described (64). This method ensured stable gene transfer with a very high efficiency (>95% transgene-positive cells, as determined by immunostaining), without need to select individual cell clones. In addition, to rule out the variability of biological responses, we tested at least three independent batches for each construct that was used to transduce cells. Regulated expression of SEMA3B was achieved by transducing tumor cells with a lentiviral expression construct under the control of an rTTA- and doxycycline-regulated promoter (the cloning plasmid was provided by E. Vigna, IRCC, Torino, Italy; reference 65). Semaphorin expression was induced by adding 0.1–1 mg/ml doxycycline in the cell-culture medium, and 1 mg/ml in the drinking water of mice. The CM harvested from transduced cells grown in the absence of fetal serum were concentrated and size fractionated to enrich for SEMA3B and other regulatory proteins using 20 U of Centricon Plus (Millipore).
Suppression of gene expression by RNAi.
SEMA3B expression was suppressed in tumor cells by lentiviral-mediated expression of shRNAs specifically targeting the SEMA3B transcript. To this end, we used three different shRNA-SEMA3B sequences with consistent results (Fig. S5): two were derived from the Sigma-Aldrich Mission library under accession no. NM_004636 (sequence 1, TRCN0000062898; and sequence 5, TRCN0000062899), whereas the other (sequence 3) was identified in our laboratory by computer-assisted analysis (5′-ACGTCCAAGTCTCCGAACA-3′). Results shown in the figures refer to sequence 1. NP1 expression was knocked down by lentiviral-mediated expression of gene-targeted shRNA sequences that were previously described (66), and lentiviral vectors expressing shRNA against NP2 were provided by S. Rizzolio (IRCC, Torino, Italy).
Xenograft tumor model.
All mice studies were conducted with 6–8-wk-old immunocompromised CD1−/− nude athymic female mice (Charles River Laboratories. 2 × 106 MDA-MB435 or 107 A549 cells were injected subcutaneously into the flank of anaesthetized animals. For the treatment with IL-8 neutralizing antibodies, four groups of five mice were injected on both sides with either MDA-EV or MDA-3B cells, together with 100 μg/mouse of either IL-8 neutralizing monoclonal antibody (MAB208) or the control antibody CD19 (Antigenix America). Tumor size was monitored every 2 d using calipers, and the length (l) and width (w) of the developing tumor was converted to volume (v) using the equation v = (w2 × l)/2. Mice were killed 15–30 d after transplant, and tumors were weighted after dissection. Superficial pulmonary metastases were contrasted by black India ink airway infusion before excision, and were counted on dissected lung lobes under a stereoscopic microscope, as previously described (67). Histological analysis was performed on paraffin-embedded sections stained with hematoxylin and eosin. In other experiments, lungs were snap frozen in liquid nitrogen for RNA analysis. All animal procedures were approved by the Ethical Commission of the University of Torino and by the Italian Ministry of Health.
Tissue analysis.
Samples were cut into 10-μm-thick sections and immunostained with the appropriate antibodies. Streptavidin and all secondary antibodies used were conjugated with Alexa Fluor 488 or Alexa Fluor 546 fluorochromes (Invitrogen). Cell nuclei were labeled with DAPI (1:2,000; Invitrogen). To detect apoptotic cells in tumor sections, we used an indirect TUNEL assay using the ApopTag Green In Situ Apoptosis Detection Kit (Chemicon). Slides were mounted with Fluoromount-G (SouthernBiotech). Independent fields were quantified for every section (as indicated specifically for each experiment in the corresponding figure legend), and different sections of every tumor (as indicated in the figure legends) were analyzed using a microscope (DM IRB; Leica) and quantified by ImageJ software (available at http://rsb.info.nih.gov/ij/). A t test was performed for all quantifications, and p-values were indicated for each experiment in the corresponding figure legend.
Growth curve assay.
Tumor cells were plated in multiple 96-well dishes (Costar) at an initial density of 103 cells per well. After 24 h of serum starvation, the cells were cultured in DMEM supplemented with l-glutamine and 1% FBS for 1, 2, 3, and 4 d. Cells were fixed with 11% glutheraldehyde and stained with 0.5% crystal violet in 20% methanol. The dye was eluted with 10% vol/vol glacial acetic acid, and the absorbance at 562 nm was determined by using a Multiskan reader (Titertek).
SDS-PAGE and Western immunoblotting.
Cellular proteins were solubilized in 1% Triton X-100–containing extraction buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 10% glycerol) containing 1 mM Na3VO4, 100 mM NaF, 1 mM PMSF, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 μg/ml pepstatin. The amount of proteins was quantified by bicinchoninic assay, and proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and blocked in phosphate-buffered saline, 0.1% Tween 20, 5% BSA. The membrane was incubated with appropriate dilutions of primary antibody, followed by the appropriate peroxidase-conjugated secondary antibody (Bio-Rad Laboratories). Final detection was done by enhanced chemiluminescence (GE Healthcare). Detected signals were digitally measured using QuantityOne (Bio-Rad Laboratories).
Monocyte migration assays.
PBMCs were isolated from heparinized blood from healthy volunteers by density gradient centrifugation using Lymphoprep (Nycomed). Monocytes were positively purified with a magnetically labeled monoclonal antibody to CD14 (Miltenyi Biotec). Purity of the CD14+ fraction was always >94%; contaminating granulocytes were between 2–3%. To test migration, a Transwell system (8-μm pores; Costar) was used as previously described (68–70). In brief, 2 × 105 monocytes from healthy donors were placed in the upper chamber of the insert (in the presence of 1% FCS) and were allowed to migrate through a semipermeable membrane toward CM in the lower chamber. Unless otherwise specified in the figures, we used 10× concentrated CM (containing approximately 60 ng/ml IL-8; see legend to Fig. 5 C for quantification). When indicated in the figures, 0.5–1 μg/ml anti-IL8 antibody (MAB208), 1 μg/ml anti-CD19 control antibody, or the specified cytokines were included in the lower chamber. After 90 min of migration, cells adherent to the upper side of the membrane were mechanically removed. To score for monocyte migration, Transwell inserts were fixed in methanol, and the cells migrated to the lower side of the membrane were stained with Giemsa. By microscopic analysis, we confirmed that >90% of these cells were clearly mononuclear. Low-power microscopic images of the Transwell membranes were then analyzed using QuantityOne to score the number of migrated cells. Because granulocytes did not adhere to the membrane, migrated neutrophils were identified and counted by flow cytometry in the medium from the lower chamber.
RNA isolation and real-time PCR.
Total RNA from tumor cell lines or tissues was isolated by using the RNeasy Protect Mini Kit (QIAGEN) according to the manufacturer's instructions. cDNA preparation was performed according to standard procedures using M-MLV RT (Promega) and oligo-dT (Sigma-Aldrich) primers. The expression of SEMA3B, IL-8, NPs, and plexins were analyzed using Taqman gene-specific probes from Applied Biosystems (Table S1, available at http://www.jem.org/cgi/content/full/jem.20072509/DC1). Moreover, we used SYBR green real-time PCR primers to measure the expression of h-MCP1 (forward, 5′-TCTGTGCCTGCTGCTCATAG-3′; reverse, 5′-GCTTCTTTGGGACACTTGCT-3′; 168 bp), H-CSF-1 (forward, 5′-GCAAGAACTGCAACAACAGC-3′; reverse, 5′-CAGAGTCCTCCCAGGTCAAG-3′; 177 bp), and VEGF-A (forward, 5′-CCTTGCTGCTCTACCTCCACC-3′; reverse, 5′-TCCTCCTTCTGCCATGGG-3′). Real-time PCR analysis was performed (7900HT Fast Real-time PCR System; Applied Biosystems).
RNA extraction and processing for microarray analysis.
Total RNA from tumor cell lines was isolated by using RNeasy Protect Mini Kit according to the manufacturer's instructions. RT and biotinylated cRNA synthesis were performed using the Illumina TotalPrep RNA Amplification Kit (Ambion), according to the manufacturer's protocol. Hybridization of the cRNAs was performed for 18 h on Human-6 Expression BeadChips (48K v1.0; Illumina). Hybridized arrays were stained and scanned in a Beadstation 500 (Illumina) according to the manufacturer's protocols.
Microarray data analysis.
MIAME-compliant microarray data are publicly available at the National Center for Biotechnology Information Gene Accession Omnibus under accession no. GSE10431. BeadStudio software (Illumina) was used to analyze raw data grouped by experimental condition. After rank-invariant normalization, genes were filtered for detection (>0.95 in at least one of the two experimental groups) and assessed for statistically significant differential expression using the Illumina custom test (iterative robust least squares fit). To select a panel of genes related to inflammation and leukocyte chemotaxis, a series of keywords were used to search the Gene Ontology annotation of the analyzed probes. The keywords were as follows: interleukin, chemo*, cytokine, extracell*, solub*, chemota*, adhesi*, motility, inflammat*, and immun*.
Online supplemental material.
Fig. S1 shows NP1 and NP2 expression in human cancer cells. Fig. S2 shows SEMA3B expression in human tumors. Fig. S3 demonstrates that NP1, but not NP2, is required for SEMA3B-mediated growth inhibition. Fig. S4 shows that IL-8 stimulates the migration of monocytes. Fig. S5 demonstrates that RNAi-mediated knock down of endogenous SEMA3B suppresses IL-8 production in tumor cells. Fig. S6 shows SEMA3B repelling activity on endothelial cells. Fig. S7 demonstrates that SEMA3B expression does not increase the formation of lung colonies upon tail-vein injection of transduced tumor cells. Fig. S8 shows that SEMA3B expression in human melanoma samples correlates with the expression of CD14 macrophage marker and elevated IL-8 levels. Fig. S9 demonstrates that VEGF expression in tumor cells is not affected by SEMA3B. Supplemental materials and methods describes MTT assays, HUVEC co-culture experiments, and endothelial cell migration. Table S1 shows Taqman probes used for real-time quantitative PCR. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20072509/DC1.
Supplementary Material
Acknowledgments
We are grateful to A. Kolodkin for providing anti-NP1 antibodies, and to E. Vigna and S. Rizzolio for the lentiviral tet-inducible expression vector and lentiviral vectors expressing shRNA to NP2, respectively. We would like to thank S. Artigiani, P. Fazzari, J. Penachioni, D. Cantarella, B. Martinoglio, E. Tenaglia, A. Bertotti, C. Isella, A. Elia, A. Camperi, L. Palmas, and R. Albano, as well as L. Naldini and M. De Palma, for help and support.
The financial support of the Italian Association for Cancer Research (to L. Tamagnone, P.M. Comoglio, and E. Medico) and of Regione Piemonte (to A. Casazza and L. Tamagnone) is acknowledged. C. Rolny is a Fellow of the European Union–Marie Curie Association.
The authors have no conflicting financial interests.
Abbreviations used: ATF-2, activating transcription factor 2; CDKI, cytokine-dependent kinase inhibitor; CM, conditioned medium; EV, empty vector; HUVEC, human umbilical vein endothelial cell; MAPK, mitogen-activated protein kinase; MCP-1, monocyte chemotactic protein 1; NP, neuropilin; RNAi, RNA interference; SEMA3B, semaphorin 3B; shRNA, short hairpin RNA; TAM, tumor-associated macrophage; TUNEL, Tdt-mediated dUTP-biotin nick-end labeling; VEGF, vascular endothelial growth factor.
L. Capparuccia and A. Casazza contributed equally to this work.
C. Rolny's present address is Dept. of Genetics and Pathology, Unit of Vascular Biology, Rudbeck Laboratory, Uppsala University, S-751 85 Uppsala, Sweden.
References
- 1.Luo, Y., D. Raible, and J.A. Raper. 1993. Collapsin: a protein in brain that induces the collapse and paralysis of neuronal growth cones. Cell. 75:217–227. [DOI] [PubMed] [Google Scholar]
- 2.Kolodkin, A.L., D.J. Matthes, and C.S. Goodman. 1993. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell. 75:1389–1399. [DOI] [PubMed] [Google Scholar]
- 3.Tamagnone, L., and P.M. Comoglio. 2004. To move or not to move? Semaphorin signalling in cell migration. EMBO Rep. 5:356–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kruger, R.P., J. Aurandt, and K.L. Guan. 2005. Semaphorins command cells to move. Nat. Rev. Mol. Cell Biol. 6:789–800. [DOI] [PubMed] [Google Scholar]
- 5.Sekido, Y., S. Bader, F. Latif, J.Y. Chen, F.M. Duh, M.H. Wei, J.P. Albanesi, C.C. Lee, M.I. Lerman, and J.D. Minna. 1996. Human semaphorins A(V) and IV reside in the 3p21.3 small cell lung cancer deletion region and demonstrate distinct expression patterns. Proc. Natl. Acad. Sci. USA. 93:4120–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuroki, T., F. Trapasso, S. Yendamuri, A. Matsuyama, H. Alder, N.N. Williams, L.R. Kaiser, and C.M. Croce. 2003. Allelic loss on chromosome 3p21.3 and promoter hypermethylation of semaphorin 3B in non-small cell lung cancer. Cancer Res. 63:3352–3355. [PubMed] [Google Scholar]
- 7.Tse, C., R.H. Xiang, T. Bracht, and S.L. Naylor. 2002. Human Semaphorin 3B (SEMA3B) located at chromosome 3p21.3 suppresses tumor formation in an adenocarcinoma cell line. Cancer Res. 62:542–546. [PubMed] [Google Scholar]
- 8.Tomizawa, Y., Y. Sekido, M. Kondo, B. Gao, J. Yokota, J. Roche, H. Drabkin, M.I. Lerman, A.F. Gazdar, and J.D. Minna. 2001. Inhibition of lung cancer cell growth and induction of apoptosis after reexpression of 3p21.3 candidate tumor suppressor gene SEMA3B. Proc. Natl. Acad. Sci. USA. 98:13954–13959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro-Rivera, E., S. Ran, P. Thorpe, and J.D. Minna. 2004. Semaphorin 3B (SEMA3B) induces apoptosis in lung and breast cancer, whereas VEGF165 antagonizes this effect. Proc. Natl. Acad. Sci. USA. 101:11432–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bielenberg, D.R., Y. Hida, A. Shimizu, A. Kaipainen, M. Kreuter, C.C. Kim, and M. Klagsbrun. 2004. Semaphorin 3F, a chemorepulsant for endothelial cells, induces a poorly vascularized, encapsulated, nonmetastatic tumor phenotype. J. Clin. Invest. 114:1260–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kessler, O., N. Shraga-Heled, T. Lange, N. Gutmann-Raviv, E. Sabo, L. Baruch, M. Machluf, and G. Neufeld. 2004. Semaphorin-3F is an inhibitor of tumor angiogenesis. Cancer Res. 64:1008–1015. [DOI] [PubMed] [Google Scholar]
- 12.Tamagnone, L., S. Artigiani, H. Chen, Z. He, G.I. Ming, H. Song, A. Chedotal, M.L. Winberg, C.S. Goodman, M. Poo, et al. 1999. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 99:71–80. [DOI] [PubMed] [Google Scholar]
- 13.Soker, S., H.Q. Miao, M. Nomi, S. Takashima, and M. Klagsbrun. 2002. VEGF165 mediates formation of complexes containing VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J. Cell. Biochem. 85:357–368. [DOI] [PubMed] [Google Scholar]
- 14.Guttmann-Raviv, N., O. Kessler, N. Shraga-Heled, T. Lange, Y. Herzog, and G. Neufeld. 2006. The neuropilins and their role in tumorigenesis and tumor progression. Cancer Lett. 231:1–11. [DOI] [PubMed] [Google Scholar]
- 15.Falk, J., A. Bechara, R. Fiore, H. Nawabi, H. Zhou, C. Hoyo-Becerra, M. Bozon, G. Rougon, M. Grumet, A.W. Puschel, et al. 2005. Dual functional activity of semaphorin 3B is required for positioning the anterior commissure. Neuron. 48:63–75. [DOI] [PubMed] [Google Scholar]
- 16.van 't Veer, L.J., H. Dai, M.J. van de Vijver, Y.D. He, A.A. Hart, M. Mao, H.L. Peterse, K. van der Kooy, M.J. Marton, A.T. Witteveen, et al. 2002. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 415:530–536. [DOI] [PubMed] [Google Scholar]
- 17.Bhattacharjee, A., W.G. Richards, J. Staunton, C. Li, S. Monti, P. Vasa, C. Ladd, J. Beheshti, R. Bueno, M. Gillette, et al. 2001. Classification of human lung carcinomas by mRNA expression profiling reveals distinct adenocarcinoma subclasses. Proc. Natl. Acad. Sci. USA. 98:13790–13795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Talantov, D., A. Mazumder, J.X. Yu, T. Briggs, Y. Jiang, J. Backus, D. Atkins, and Y. Wang. 2005. Novel genes associated with malignant melanoma but not benign melanocytic lesions. Clin. Cancer Res. 11:7234–7242. [DOI] [PubMed] [Google Scholar]
- 19.Hoek, K.S., N.C. Schlegel, P. Brafford, A. Sucker, S. Ugurel, R. Kumar, B.L. Weber, K.L. Nathanson, D.J. Phillips, M. Herlyn, et al. 2006. Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment Cell Res. 19:290–302. [DOI] [PubMed] [Google Scholar]
- 20.Rae, J.M., C.J. Creighton, J.M. Meck, B.R. Haddad, and M.D. Johnson. 2007. MDA-MB-435 cells are derived from M14 melanoma cells–a loss for breast cancer, but a boon for melanoma research. Breast Cancer Res. Treat. 104:13–19. [DOI] [PubMed] [Google Scholar]
- 21.Neufeld, G., T. Lange, A. Varshavsky, and O. Kessler. 2007. Semaphorin signaling in vascular and tumor biology. Adv. Exp. Med. Biol. 600:118–131. [DOI] [PubMed] [Google Scholar]
- 22.Porta, C., B. Subhra Kumar, P. Larghi, L. Rubino, A. Mancino, and A. Sica. 2007. Tumor promotion by tumor-associated macrophages. Adv. Exp. Med. Biol. 604:67–86. [DOI] [PubMed] [Google Scholar]
- 23.Condeelis, J., and J.W. Pollard. 2006. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 124:263–266. [DOI] [PubMed] [Google Scholar]
- 24.Lin, E.Y., A.V. Nguyen, R.G. Russell, and J.W. Pollard. 2001. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 193:727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann, E., O. Dittrich-Breiholz, H. Holtmann, and M. Kracht. 2002. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 72:847–855. [PubMed] [Google Scholar]
- 26.Mukaida, N. 2000. Interleukin-8: an expanding universe beyond neutrophil chemotaxis and activation. Int. J. Hematol. 72:391–398. [PubMed] [Google Scholar]
- 27.Xie, K. 2001. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 12:375–391. [DOI] [PubMed] [Google Scholar]
- 28.Gerszten, R.E., E.A. Garcia-Zepeda, Y.C. Lim, M. Yoshida, H.A. Ding, M.A. Gimbrone Jr., A.D. Luster, F.W. Luscinskas, and A. Rosenzweig. 1999. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 398:718–723. [DOI] [PubMed] [Google Scholar]
- 29.Smythies, L.E., A. Maheshwari, R. Clements, D. Eckhoff, L. Novak, H.L. Vu, L.M. Mosteller-Barnum, M. Sellers, and P.D. Smith. 2006. Mucosal IL-8 and TGF-beta recruit blood monocytes: evidence for cross-talk between the lamina propria stroma and myeloid cells. J. Leukoc. Biol. 80:492–499. [DOI] [PubMed] [Google Scholar]
- 30.Liu-Bryan, R., S. Pay, I.U. Schraufstatter, and D.M. Rose. 2005. The CXCR1 tail mediates beta1 integrin-dependent cell migration via MAP kinase signaling. Biochem. Biophys. Res. Commun. 332:117–125. [DOI] [PubMed] [Google Scholar]
- 31.Kubo, F., S. Ueno, K. Hiwatashi, M. Sakoda, K. Kawaida, K. Nuruki, and T. Aikou. 2005. Interleukin 8 in human hepatocellular carcinoma correlates with cancer cell invasion of vessels but not with tumor angiogenesis. Ann. Surg. Oncol. 12:800–807. [DOI] [PubMed] [Google Scholar]
- 32.Chen, J.J., P.L. Yao, A. Yuan, T.M. Hong, C.T. Shun, M.L. Kuo, Y.C. Lee, and P.C. Yang. 2003. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin. Cancer Res. 9:729–737. [PubMed] [Google Scholar]
- 33.Arenberg, D.A., S.L. Kunkel, P.J. Polverini, M. Glass, M.D. Burdick, and R.M. Strieter. 1996. Inhibition of interleukin-8 reduces tumorigenesis of human non-small cell lung cancer in SCID mice. J. Clin. Invest. 97:2792–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sparmann, A., and D. Bar-Sagi. 2004. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 6:447–458. [DOI] [PubMed] [Google Scholar]
- 35.Mizukami, Y., W.S. Jo, E.M. Duerr, M. Gala, J. Li, X. Zhang, M.A. Zimmer, O. Iliopoulos, L.R. Zukerberg, Y. Kohgo, et al. 2005. Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat. Med. 11:992–997. [DOI] [PubMed] [Google Scholar]
- 36.Serini, G., D. Valdembri, S. Zanivan, G. Morterra, C. Burkhardt, F. Caccavari, L. Zammataro, L. Primo, L. Tamagnone, M. Logan, et al. 2003. Class 3 semaphorins control vascular morphogenesis by inhibiting integrin function. Nature. 424:391–397. [DOI] [PubMed] [Google Scholar]
- 37.Mikule, K., B. Delaval, P. Kaldis, A. Jurcyzk, P. Hergert, and S. Doxsey. 2007. Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat. Cell Biol. 9:160–170. [DOI] [PubMed] [Google Scholar]
- 38.Christensen, C., N. Ambartsumian, G. Gilestro, B. Thomsen, P. Comoglio, L. Tamagnone, P. Guldberg, and E. Lukanidin. 2005. Proteolytic processing converts the repelling signal Sema3E into an inducer of invasive growth and lung metastasis. Cancer Res. 65:6167–6177. [DOI] [PubMed] [Google Scholar]
- 39.Wyckoff, J.B., Y. Wang, E.Y. Lin, J.F. Li, S. Goswami, E.R. Stanley, J.E. Segall, J.W. Pollard, and J. Condeelis. 2007. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 67:2649–2656. [DOI] [PubMed] [Google Scholar]
- 40.Yuan, A., J.J. Chen, P.L. Yao, and P.C. Yang. 2005. The role of interleukin-8 in cancer cells and microenvironment interaction. Front. Biosci. 10:853–865. [DOI] [PubMed] [Google Scholar]
- 41.Salcedo, R., M. Martins-Green, B. Gertz, J.J. Oppenheim, and W.J. Murphy. 2002. Combined administration of antibodies to human interleukin 8 and epidermal growth factor receptor results in increased antimetastatic effects on human breast carcinoma xenografts. Clin. Cancer Res. 8:2655–2665. [PubMed] [Google Scholar]
- 42.Charalambous, C., L.B. Pen, Y.S. Su, J. Milan, T.C. Chen, and F.M. Hofman. 2005. Interleukin-8 differentially regulates migration of tumor-associated and normal human brain endothelial cells. Cancer Res. 65:10347–10354. [DOI] [PubMed] [Google Scholar]
- 43.Varney, M.L., A. Li, B.J. Dave, C.D. Bucana, S.L. Johansson, and R.K. Singh. 2003. Expression of CXCR1 and CXCR2 receptors in malignant melanoma with different metastatic potential and their role in interleukin-8 (CXCL-8)-mediated modulation of metastatic phenotype. Clin. Exp. Metastasis. 20:723–731. [DOI] [PubMed] [Google Scholar]
- 44.Ramjeesingh, R., R. Leung, and C.H. Siu. 2003. Interleukin-8 secreted by endothelial cells induces chemotaxis of melanoma cells through the chemokine receptor CXCR1. FASEB J. 17:1292–1294. [DOI] [PubMed] [Google Scholar]
- 45.Ivarsson, K., A. Ekerydh, I.M. Fyhr, P.O. Janson, and M. Brannstrom. 2000. Upregulation of interleukin-8 and polarized epithelial expression of interleukin-8 receptor A in ovarian carcinomas. Acta Obstet. Gynecol. Scand. 79:777–784. [PubMed] [Google Scholar]
- 46.Shi, Q., J.L. Abbruzzese, S. Huang, I.J. Fidler, Q. Xiong, and K. Xie. 1999. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin. Cancer Res. 5:3711–3721. [PubMed] [Google Scholar]
- 47.Taylor, P.R., L. Martinez-Pomares, M. Stacey, H.H. Lin, G.D. Brown, and S. Gordon. 2005. Macrophage receptors and immune recognition. Annu. Rev. Immunol. 23:901–944. [DOI] [PubMed] [Google Scholar]
- 48.Coussens, L.M., C.L. Tinkle, D. Hanahan, and Z. Werb. 2000. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell. 103:481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergers, G., R. Brekken, G. McMahon, T.H. Vu, T. Itoh, K. Tamaki, K. Tanzawa, P. Thorpe, S. Itohara, Z. Werb, et al. 2000. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2:737–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wyckoff, J., W. Wang, E.Y. Lin, Y. Wang, F. Pixley, E.R. Stanley, T. Graf, J.W. Pollard, J. Segall, and J. Condeelis. 2004. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64:7022–7029. [DOI] [PubMed] [Google Scholar]
- 51.De Palma, M., M.A. Venneri, R. Galli, L. Sergi Sergi, L.S. Politi, M. Sampaolesi, and L. Naldini. 2005. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 8:211–226. [DOI] [PubMed] [Google Scholar]
- 52.Potiron, V.A., G. Sharma, P. Nasarre, J.A. Clarhaut, H.G. Augustin, R.M. Gemmill, J. Roche, and H. Drabkin. 2007. Semaphorin SEMA3F affects multiple signaling pathways in lung cancer cells. Cancer Res. 67:8708–8715. [DOI] [PubMed] [Google Scholar]
- 53.Xian, X., J. Hakansson, A. Stahlberg, P. Lindblom, C. Betsholtz, H. Gerhardt, and H. Semb. 2006. Pericytes limit tumor cell metastasis. J. Clin. Invest. 116:642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benjamin, L.E., and E. Keshet. 1997. Conditional switching of vascular endothelial growth factor (VEGF) expression in tumors: induction of endothelial cell shedding and regression of hemangioblastoma-like vessels by VEGF withdrawal. Proc. Natl. Acad. Sci. USA. 94:8761–8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yaron, A., P.H. Huang, H.J. Cheng, and M. Tessier-Lavigne. 2005. Differential requirement for Plexin-A3 and -A4 in mediating responses of sensory and sympathetic neurons to distinct class 3 Semaphorins. Neuron. 45:513–523. [DOI] [PubMed] [Google Scholar]
- 56.Appleton, B.A., P. Wu, J. Maloney, J. Yin, W.C. Liang, S. Stawicki, K. Mortara, K.K. Bowman, J.M. Elliott, W. Desmarais, et al. 2007. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. EMBO J. 26:4902–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Engelberg, D. 2004. Stress-activated protein kinases-tumor suppressors or tumor initiators? Semin. Cancer Biol. 14:271–282. [DOI] [PubMed] [Google Scholar]
- 58.Kovacic, B., D. Stoiber, R. Moriggl, E. Weisz, R.G. Ott, R. Kreibich, D.E. Levy, H. Beug, M. Freissmuth, and V. Sexl. 2006. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell. 10:77–87. [DOI] [PubMed] [Google Scholar]
- 59.Kang, Y., W. He, S. Tulley, G.P. Gupta, I. Serganova, C.R. Chen, K. Manova-Todorova, R. Blasberg, W.L. Gerald, and J. Massague. 2005. Breast cancer bone metastasis mediated by the Smad tumor suppressor pathway. Proc. Natl. Acad. Sci. USA. 102:13909–13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pennacchietti, S., P. Michieli, M. Galluzzo, M. Mazzone, S. Giordano, and P.M. Comoglio. 2003. Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell. 3:347–361. [DOI] [PubMed] [Google Scholar]
- 61.Kim, K.S., V. Rajagopal, C. Gonsalves, C. Johnson, and V.K. Kalra. 2006. HIF-1 and NF-kappaB-mediated upregulation of CXCR1 and CXCR2 expression promotes cell survival in hypoxic prostate cancer cells. J. Immunol. 177:7211–7224. [DOI] [PubMed] [Google Scholar]
- 62.Maxwell, P.J., R. Gallagher, A. Seaton, C. Wilson, P. Scullin, J. Pettigrew, I.J. Stratford, K.J. Williams, P.G. Johnston, and D.J. Waugh. 2007. Oncogene. 26:7333–7345. [DOI] [PubMed] [Google Scholar]
- 63.Kolodkin, A.L., D.V. Levengood, E.G. Rowe, Y.T. Tai, R.J. Giger, and D.D. Ginty. 1997. Neuropilin is a semaphorin III receptor. Cell. 90:753–762. [DOI] [PubMed] [Google Scholar]
- 64.Follenzi, A., and L. Naldini. 2002. HIV-based vectors. Preparation and use. Methods Mol. Med. 69:259–274. [PubMed] [Google Scholar]
- 65.Vigna, E., S. Cavalieri, L. Ailles, M. Geuna, R. Loew, H. Bujard, and L. Naldini. 2002. Robust and efficient regulation of transgene expression in vivo by improved tetracycline-dependent lentiviral vectors. Mol. Ther. 5:252–261. [DOI] [PubMed] [Google Scholar]
- 66.Bachelder, R.E., E.A. Lipscomb, X. Lin, M.A. Wendt, N.H. Chadborn, B.J. Eickholt, and A.M. Mercurio. 2003. Competing autocrine pathways involving alternative neuropilin-1 ligands regulate chemotaxis of carcinoma cells. Cancer Res. 63:5230–5233. [PubMed] [Google Scholar]
- 67.Michieli, P., M. Mazzone, C. Basilico, S. Cavassa, A. Sottile, L. Naldini, and P.M. Comoglio. 2004. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 6:61–73. [DOI] [PubMed] [Google Scholar]
- 68.Cignetti, A., A. Vallario, I. Roato, P. Circosta, G. Strola, C. Scielzo, B. Allione, L. Garetto, F. Caligaris-Cappio, and P. Ghia. 2003. The characterization of chemokine production and chemokine receptor expression reveals possible functional cross-talks in AML blasts with monocytic differentiation. Exp. Hematol. 31:495–503. [DOI] [PubMed] [Google Scholar]
- 69.Matsushima, K., C.G. Larsen, G.C. DuBois, and J.J. Oppenheim. 1989. Purification and characterization of a novel monocyte chemotactic and activating factor produced by a human myelomonocytic cell line. J. Exp. Med. 169:1485–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Traves, S.L., S.J. Smith, P.J. Barnes, and L.E. Donnelly. 2004. Specific CXC but not CC chemokines cause elevated monocyte migration in COPD: a role for CXCR2. J. Leukoc. Biol. 76:441–450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
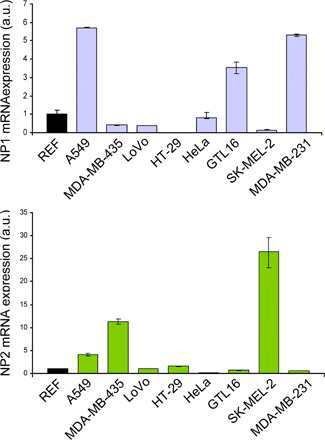{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
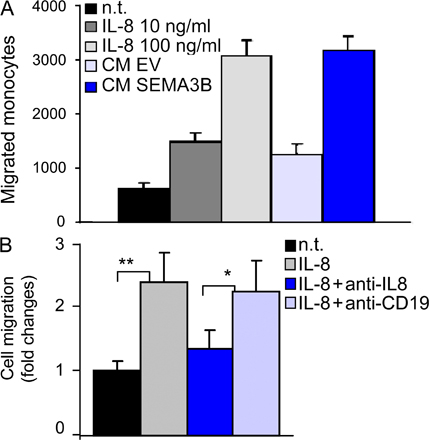{kind=link}
{kind=link}
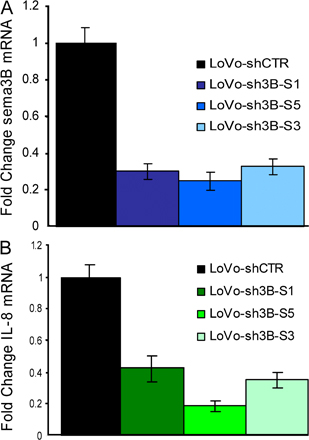{kind=link}
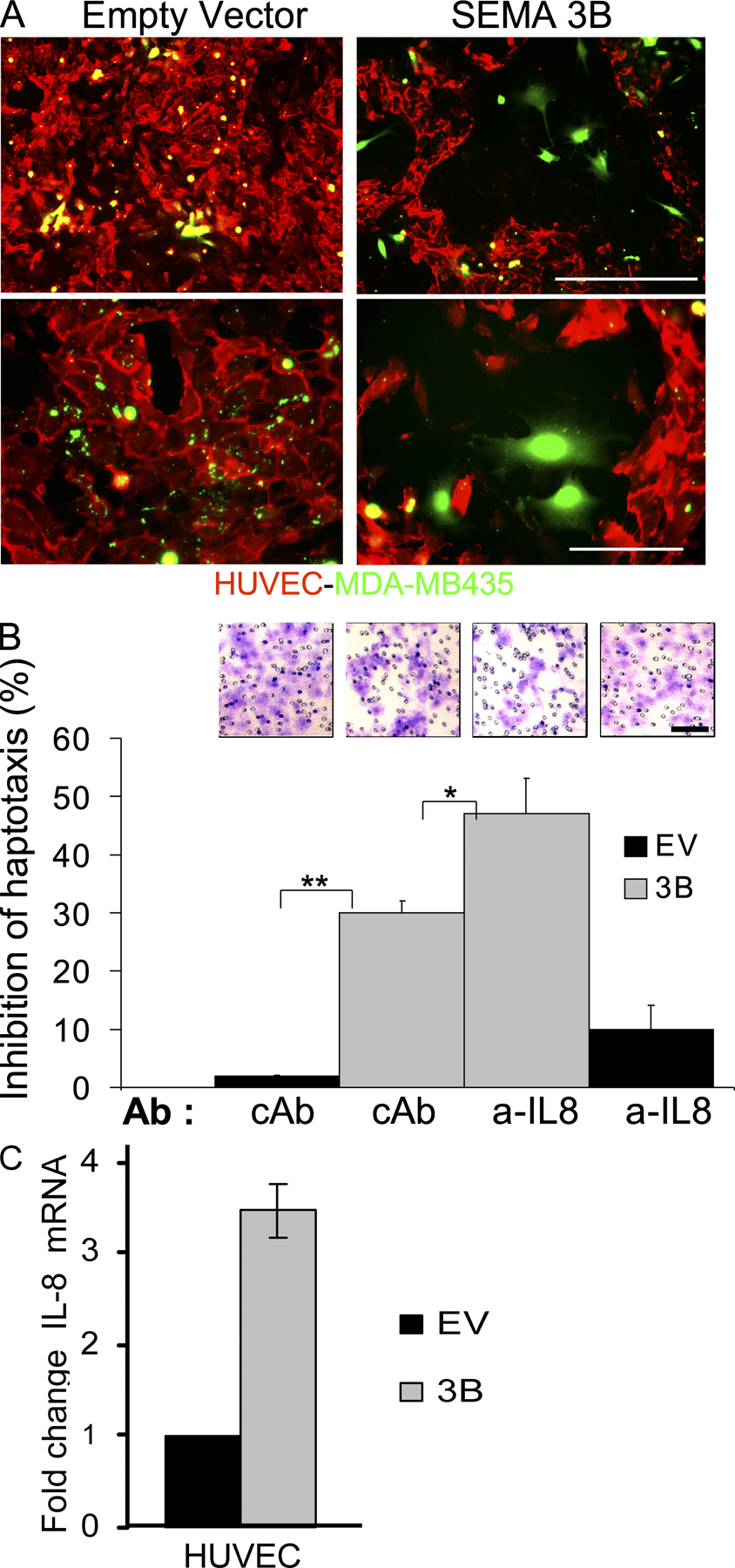{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
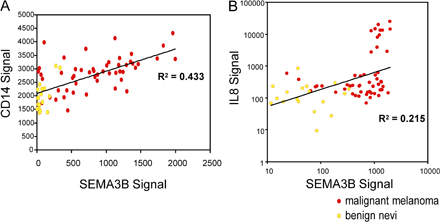{kind=link}
{kind=link}
{kind=link}