

Abstract
Paclitaxel (Taxol ®) and docetaxel (Taxotere ®) are very important anti-tumor drugs in clinical use for cancer. However, their clinical utility is limited due to systemic toxicity, low solubility and inactivity against drug resistant tumors. To improve chemotherapeutic levels of these drugs, it would be highly desirable to design strategies which bypass the above limitations. In this respect various prodrug and drug targeting strategies have been envisioned either to improve oral bioavailability or tumor specific delivery of taxoids. Abnormal properties of cancer cells with respect to normal cells have guided in designing of these protocols. This review article records the designed biochemical strategies and their biological efficacies as potential taxoid chemotherapeutics.
Keywords: Taxoid, prodrug, drug conjugate, drug targeting, receptor, antibody, TPT, ADEPT, colloidal gold nanoparticles, cancer cells
1. Introduction
The diterpenoid natural product paclitaxel (PTX, Taxol®, 1) and its semi-synthetic analog docetaxel (DTX, Taxotere®, 2) are the leading anticancer drugs in clinical use today.1, 2 Taxoids (PTX and DTX) bind to tubulin polymer in a stoichiometric ratio and promote tubulin polymerization. This phenomenon completely disrupts tubulin polymerization dynamics, leading to cell cycle arrest at the G2/M phase and thence to apoptosis.3, 4 These two taxoid drugs were approved by the FDA for the treatment of several carcinomas including breast, advanced ovarian, non small cell lung (NSCLC), head and neck, colon, and AIDS-related Kaposi’s sarcoma.5 The combined annual sales of these two taxoids were well over $1 billion in the year 2001.6 While these taxoids perform well in the clinic for various types of cancer, several combination therapies of these drugs together with other anti-cancer agents that operate by different mechanisms of action to these taxoids (e.g. cisplatin and doxorubicin) have been explored to unravel synergistic effects that could lead to more effective therapy for tumor malignancies.7 These two taxoid drugs have also become the benchmark chemotherapeutic agents to evaluate the in vitro and in vivo efficacy of several newly discovered anticancer natural products such as the epothilones, 8 discodermolide, 9 eluetherobin,10 laulimalide,11 peloruside,12 and others that have a similar mechanism of action to that of PTX.
Despite the successful performance exhibited by these taxoids for several cancers, their utility in the clinic is hampered by severe limitations such as: poor aqueous solubility, non-selective toxicity to tumor cells, and inactivity against drug resistant (MDR) cell lines.13 Due to the poor water solubility of PTX (1), it had to be formulated and administered with Cremophor EL® (polyethoxylated caster oil and ethanol, 50:50) as a surfactant. Unfortunately, this formulation caused severe adverse allergic reactions due to histamine release14 and hypersensitivity reactions.15 Although DTX (2) was more soluble in water than PTX,16 it still had to be formulated with 15% dry ethanolic solution containing 80% polysorbate for clinical use. In an effort to eliminate the solubility issues associated with these taxoids, significantly more water soluble taxoids have been developed, some of which have entered into the preclinical development.17,18
The second limitation exhibited by these taxoids is their systemic toxicity, due to their non-selective cytotoxicity action on tumor cells over normal cells. This property resulted in severe adverse side effects, including bone marrow suppression, febrile nuetropenia, neurotoxicities, mucositis, ulceration to mouth, throat, as well as a variety of other cardiac abnormalities.19 This undesirable toxicity has restricted the administration and dose levels, which often lead to incomplete tumor eradication.
The ideal way to eliminate the side effects caused by these taxoids, would be to target them to the tumor site, in the right amount and also at the right time by means of a “magic bullet”. The success of this strategy would have significant implications for future chemotherapy, such as the elimination of systemic toxicity and an increased bioavailability of drug to the tumor, thus leading to a lower dose of drug being required. To achieve this goal it is essential to recognize the intrinsic physiological and morphological differences between normal and tumor cells. Fortunately some broad differences in cancer cells have been observed, such as a higher metabolic rate when compared to normal cells. This then leads to a hypoxic environment in tumor cells, which in turn induces anaerobic metabolism that ultimately leads to a lower pH in the cancer cells. Cancer cells also consume more primary metabolites, due to rapid cell proliferation. For this to occur, cancer cells overexpress several receptors that attract several primary metabolites (such as peptides, polysaccharides, fatty acids, etc.). Cancer cells also possess leaky vasculature that allows small molecules to enter tumor cells much more rapidly than normal cells.20 Tumors of breast and ovarian cancers also overexpress steroid hormone receptors which have specific interactions with the female hormone estrone.
Based on the above broad differences, several taxoid prodrug and targeted delivery protocols have been developed, and some of them have yielded better anti-cancer properties compared to the parent compound. Some of them have already entered into the preclinical development. In this article, we examine the prodrug strategies that have been explored to improve the oral bioavailability, and targeting methods which developed for tumor specific delivery of the taxoids (PTX and DTX). For the purpose of this review, the protocols are divided into two classes namely; prodrug protocols and drug targeting protocols.
Prodrugs in general consist of a taxoid with a hydrolysable and or a self immolative linker unit that would release the active drug at the tumor cells under the influence of the tumor environment such as pH or bio-reductive agents present in the cancer cells or a structurally modified inactive drug that rearranges to an active drug upon reaching the cancer cell (Fig. 1, path A). On the other hand, drug delivery protocol makes use of tumor markers affinity to certain ligands. The drug delivery would be realized by a drug conjugate which is made up of a cytotoxic drug (e.g. taxoid) connected directly or through a linker to a tumor recognizing agent (Fig 1, path B). The tumor recognizing agent could be a hormone, peptide, polysaccharide, enzyme, vitamin, gold nanoparticle, fatty acid, and antibody etc. that have a certain affinity to tumor cells. Ideally, the drug conjugate should have the following properties: selective binding affinity to cancer cells and essentially non-toxic to normal cells, stability throughout the blood stream, and upon reaching to cancer cell, it should be cleaved readily to release the active drug.
Fig. 1.
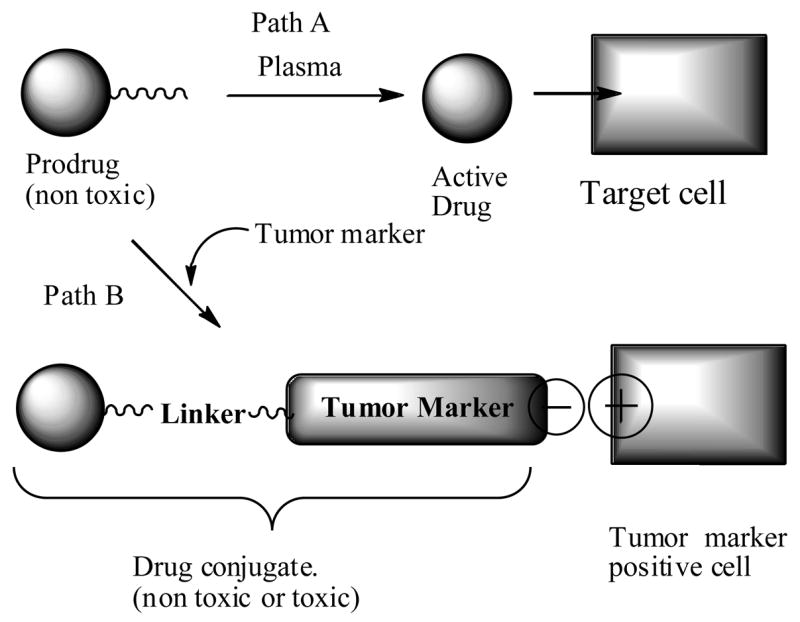
Schematic representation of prodrug and drug conjugate delivery to tumor cells.
2. Taxoid prodrug protocols
As outlined in the introduction, an ideal prodrug should have better aqueous solubility, prolong the circulation time, release the active drug only at the tumor site and also does not generate any byproducts which might show unpleasant properties. In this respect several prodrug protocols have been envisioned, based on the premise that when the prodrug passing by the tumor site, the physiological conditions of the cancer cells would cause release of the active drug.
A noteworthy point is that a majority of the taxoid prodrugs or drug conjugates are generated by linking to the C2′-OH position of PTX or DTX.21 The SAR studies22 and pharmacophore modeling analysis23 have determined that a free C2′-OH is essential for the bioactivity of taxoids, and either esterification or derivatization of this C2′-OH could lead to inactive compounds. This is a requisite property of a prodrug. Additionally C2′-OH derivatives (esters and carbamates etc.) are found to be much more accessible to various enzymes in the body and are able to undergo hydrolysis to release active drug. In few cases derivatives by alterations at C10-OAc (OH) and C7-OH of the taxoids have also been reported, however, these derivatives may or may not be able to release active taxoids, because they were found to be relatively more stable compared to C2′-esters of taxoids.24
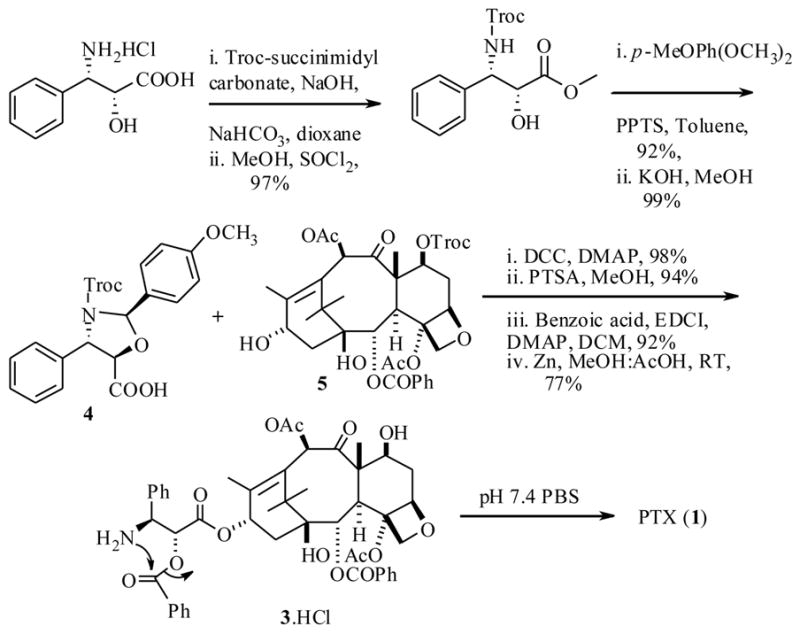
We begin with the recent report by Kiso et al.25 who synthesized a water soluble PTX derivative called isotaxel (3) as shown in scheme 1. The synthesis was accomplished in a convergent manner utilizing the troc-protected derivatives 4 and 5 respectively, which were prepared from a phenylisoserine and baccatin III starting materials, using DCC as a coupling reagent in the crucial step. This prodrug 3 has no additional water soluble functionalities and would not generate any byproduct during its conversion to parent drug PTX, but would merely undergo a rearrangement to produce the active drug PTX. Remarkably the isotaxel (3) showed 1800 fold greater aqueous solubility, due to the ionized amino group. This prodrug was also found to be stable in the pH range 2–5. Very importantly this prodrug released PTX at physiological pH 7.4 with a half life (t1/2) 15 min. This time was almost within the suggested systemic distribution period of the drug. However no in vivo efficacy of this isotaxel has been reported.
Scheme 1.
Recently Scheeren et al.26 reported a few attractive PTX prodrugs with malic acid attached at the C2′ as well as the C7 positions. The synthesis of prodrug 6 as well as its sodium salt 7 and also 8 was achieved starting from a protected malic acid 9 and PTX (1) respectively, using the similar diimide mediated coupling, as shown in Scheme 2. Gratifyingly, all the prodrugs 6–8 exhibited 20–60 fold improved water solubility when compared to PTX and are stable during 48 h incubation in PBS buffer at pH 7.4 at 37 °C. No detectable amount of PTX was liberated from these prodrugs in the above time period. Interestingly, compound 6 and it sodium salt 7 generated PTX when they were incubated in human plasma, however the dimalic acid prodrug 8 did not produce any PTX under similar conditions. The latter result is consistent with the fact that C7-esters of PTX are much more stable than C2′ esters, because the C7 position of the PTX is in a hindered position and thus would not be as exposed to external biological agents. Nevertheless the sodium salt 7 showed a half life (t1/2) of 4 h in human plasma, suggesting that it would increase the circulation time of PTX in the body.
Scheme 2.

The prodrugs 6–8 were investigated for in vitro cytotoxicity against the MCF-7 and EVSA-T breast cancer cell lines (Table 1). Prodrugs 6 and 7 showed similar activity to PTX, whereas the dimalic acid PTX prodrug 8 was much less active than PTX. The bioactivity results are consistent with the above stability results encountered with this C7 ester prodrug 8, so its reduced activity can be attributed to its inability to release PTX in cell culture medium. The prodrug 7 was further evaluated for in vivo bioactivity against murine leukemia P388 tumor model, and it was found to cause dose-dependent increase of survival time. Optimum results were obtained with the compound injected at 25 mg/kg, and the results were significantly better than those with PTX. In this test no mouse survived after 30 days period with PTX, whereas there were 2/6 survivors after 45 days with compound 7.
Table 1.
Aqueous solubility, stability in plasma and in vitro cytotoxicities of prodrugs 6–8.
| Water Solubility | T 1/2 (h) | IC50 (ng/mL) | |||
|---|---|---|---|---|---|
| Compound | mg/ml | pH 7 | Plasma | MCF-7 | EVSA-T |
| PTX | 0.01 | <3 | <3 | ||
| 6 | 0.2 | >24 | 20 | <3 | <3 |
| 7 | 0.6 | No PTX | 4 | <3 | <3 |
| 8 | 0.5 | No PTX | No PTX | 69 | 59 |
To maximize drug bioavailability, and minimize the systemic toxicity poised by the cytotoxic drug, numerous drug delivery systems (DDS), such as polymeric micelles,27 colloidal nanoparticles,28 dendrimers,29 aerosol,30 and liposomes,31 have been sought with an effort to enhance the circulation time of the drug in plasma.32 These agents would protect the drug from plasma-induced transformations and also transport an adequate amount of drug to the appropriate site.31 Particularly relevant to the present topic are liposomes. These colloidal particles are made of biodegradable natural material, found to be non-toxic to the body, and composed of a lipid bilayer surrounding the aqueous pocket making a capsule-like carrier. Liposomes could carry both hydrophobic as well as hydrophilic molecules to the appropriate site. However, these drug delivery systems are recognized by mononuclear phagocytic system (MPS) and are thus trapped by the reticuloendothelial system (RES). In this process various plasma proteins and enzymes were found to be involved and to enhance the clearance of liposomes from the site, while releasing the encapsulated molecule at the site. To circumvent this rapid clearance of liposomes by the RES, various modified liposomal alternatives, especially with polyethylene glycol (PEG), glucuronic acid derivatives, palmitoyl-D-glucuronide (PGLcUA), were developed. All these liposomal delivery systems did indeed improve the circulation time and prevented RES-mediated trapping and clearance. 33
It is worth pointing out here that the size and charge of the liposomes could be controlled during their formulation, depending upon the requirements poised by the potential drug. Previously various cytotoxic drugs were administered for lung cancer treatment by liposomes as a delivery vehicle.34 This method has several advantages over other methods of drug administration such as increasing the drug concentration in the lungs, and has lower dosage requirements with overall reduced systemic toxicity.35 However, by this method the active drug would be released at once at the tumor site, which often causes the drug clearance by efflux pumps. In this respect, a slow releasing prodrug would have greater impact than a single bolus delivery.
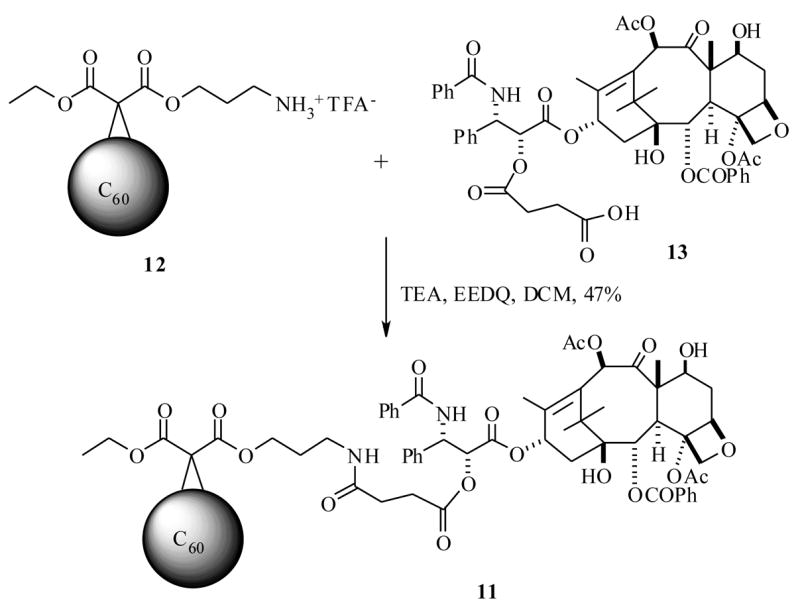
Recently, Wilson et al.36 envisioned that a C60 fullerene modified liposome could plausibly deliver the PTX in slow release fashion at the lungs, since fullerenes are found to be biologically stable and have the potential to form a slow drug releasing system. Thus the PTX prodrug 11, with glutaroyl and amino ether linkages between a C60 fullerene and PTX, was synthesized by an EEDQ-mediated coupling strategy between 12 and 13, which were in turn prepared from a C60 fullerene and PTX respectively, as shown in Scheme 3. The prodrug 11 was investigated to determine its efficacies. First the biodistribution was measured comparing with parent drug PTX. These results indicated that 11 increases PTX aggregation in aqueous (10% DMSO) solution. The prodrug 11 was determined to be stable at physiological pH, however when it was incubated with bovine plasma at 37 °C, it released PTX with a half life of 80 min. If 11 exhibits a similar half life in the lungs, it could enhance PTX exposure to lungs at least four fold as compared with the previously measured half life of PTX in lungs (20 min) when the drug was delivered by aerosol.35 Compound 11 was investigated for liposome formulation, and its antitumor activity against A549 (human epithelial lung carcinoma) cells was determined as a liposome suspension. The results show that it forms a stable liposome suspension with dilauroylphosphatidylcholine (DLPC), and this suspension did not let PTX precipitate. The cytotoxicity data of 11-DLPC, PTX-DLPC, and intermediates 12-, 13-DLPC were measured, and the results indicated that the 11-DLPC has almost equal potency to a PTX-DLPC formulation, with IC50 values of 410 and 253 nM respectively. In this study the intermediates 12 with DLPC and DLPC alone were found to be inactive, clearly indicating the C60 fullerene role in PTX delivery to the lung cancer cells. However, additional in vivo biological results are necessary to fully determine the efficacy of the prodrug 11.
Scheme 3.
As stated previously, many solid tumors have enhanced metabolic rates that cause hypoxia (oxygen deficiency), a phenomenon that could activate several biological enzymes to metabolize bioreductive prodrugs into toxic drugs. This metabolism occurs preferentially at absence of oxygen. Hypoxia also leads to anaerobic metabolism resulting in the formation of excess lactic acid and consequently lowering the intracellular pH.37 This is a distinguishing property of cancer cells, and presents a tool for designing prodrugs that could conceivably be cleaved, by low pH environment to release the active drugs upon reaching the cancer cells. In this respect several attempts have been made to design taxoid prodrugs that could use the hypoxic environment of tumor cells to generate the bioactive taxoid drug.
For example, Scheeren et al.38 have synthesized several PTX analogs, a typical structure is represented as 15, which is a mixed carbonate derivative formed between C2′-OH of the PTX and p-nitrocinnamyl alcohol. The synthesis of 15 was achieved from the reactive mixed carbonate 14 and PTX. Conceptually, the nitro group is expected to be reduced to amino derivative 16 by bio-reductive agents present in the hypoxic conditions in the cell (Scheme 4). Once the amino group is formed, it would under go self-emmolation (electronic reorganization) to release the active PTX. In this study several analogs similar to 15 were synthesized (varying aryl groups in place of p-nitrocinnamyl). Among them only 15 showed improved properties such as decreased cytotoxicity towards various cell lines with respect to PTX. All other derivatives (not shown) synthesized in this study showed almost equal cytotoxicity to PTX, because they were less stable and have released the PTX by esterases present in the assay cell culture. Only 15 showed less cytotoxicity against various cancer cell lines when compared to the parent drug PTX (Table 2), and it was stable for 24 h in the pH 7 buffered solution at 37 °C. Reduction of 15 by chemical methods such as Zn/AcOH/MeOH, resulted in the release of PTX, suggests it could be a potential prodrug, however, no detailed in vivo efficacies of this prodrug have been reported to date.
Scheme 4.
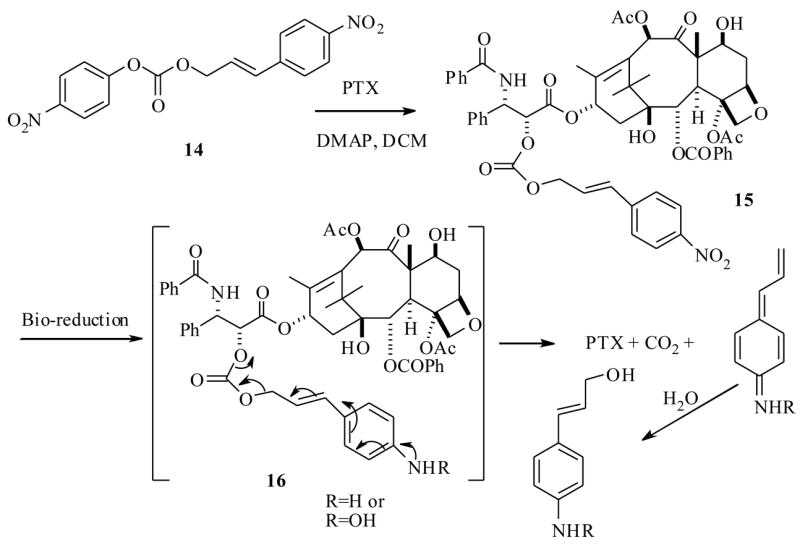
Table 2.
In vitro cytotoxicity of PTX and prodrug 15
| Compound | IC50 (ng/mL)* | ||||||
|---|---|---|---|---|---|---|---|
| H226 | IGROV | MCF-7 | EVSA-T | WIDR | M19 | A498 | |
| PTX | <3 | 10 | <3 | <3 | <3 | <3 | <3 |
| 15 | 187 | 620 | 186 | 221 | 308 | 384 | 187 |
MCF-7, EVSA are breast carcinoma, WIDR are colon, IGROV are ovarian carcinoma, M19 are melanoma, A498 renal carcinoma, H226 non small cell lung carcinoma, cell lines
Along similar lines Vrudhula et al.39 have synthesized several C2′-ester-disulfide PTX derivatives (17a–c) (Scheme 5), based on the similar notion that the disulfide bond could be reduced to free thiol in the hypoxic environment of the tumor site. Subsequently the free thiol intermediate would undergo a self-immolative cyclization to release the bioactive PTX. The synthesis of disulfide prodrugs was accomplished as shown in Scheme 5, by reaction of excess of thioglucose, glutathione and captopril separately with pyridyl disulfide-2′O-PTX ester (18) under basic conditions to furnish 17a–c respectively.
Scheme 5.
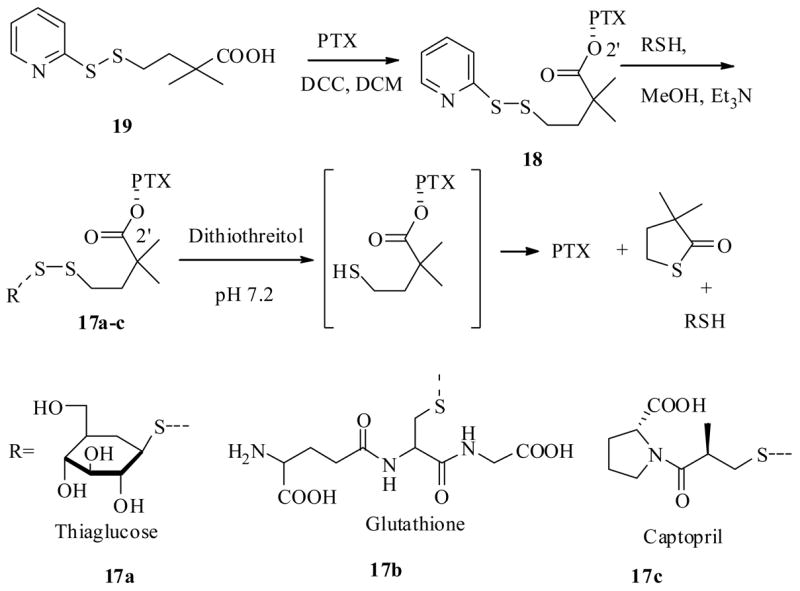
After having synthesized the prodrugs 17a–c and 18, they examined whether these prodrugs could release PTX in a reductive environment. Thus, treatment of prodrugs 17a–c and 18 with dithiothreitol (DTT), resulted in reduction followed by self-immolation to release PTX with half life periods from 4 min to 1 h. The cytotoxicity of the prodrugs was determined against the L2987 lung cancer cell line, and results indicated that prodrugs alone were significantly (30–650 fold) less cytotoxic than PTX. Interestingly, when tested in combination with reducing agent DTT, they all exhibited similar bioactivity to parent drug PTX except 17c, which is about ten times less than PTX, indicating they could serve as prodrugs.
All these prodrugs were also tested for their in vivo biological profile in athymic nude mice that had subcutaneous L2987 human lung adenocarcinoma xenografts. Except for prodrug 17c, containing a captopril unit on PTX, all other prodrugs proved to be not as effective as PTX at the dose range from 85–150 mg/kg. One possible reason could be that the C2′-ester hydrolysis of the prodrugs might have not occurred in mice at the tumor site. However, the prodrug 17c showed superior in vivo bioactivity when compared to PTX. First 17c was well tolerated at a 125 mg/kg dose with only 10–20% body weight loss, and this prodrug cured 60% of the tumors at the same dose. PTX had only a 30 mg/kg maximum tolerated dose, and this dose did not regress the tumor as was found with 17c. The possible reason for the better activity of 17c may be due to the captopril moiety, which was found to inhibit angiogenesis and slows down the tumor growth in rats.40
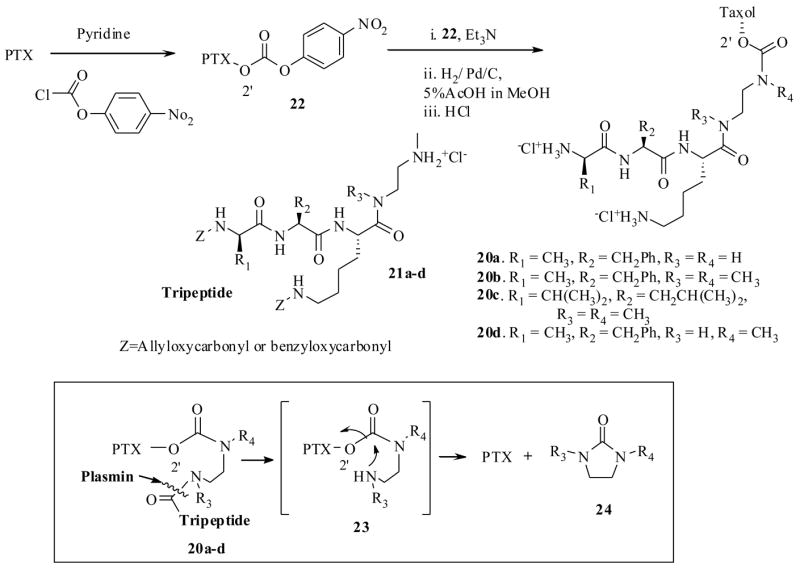
The serine protease, plasmin is overexpressed in tumor cells, and plays a significant role in the various stages of cancer.41 Normal cells do possess the enzyme plasmin, but inhibitors such as the α2-antiplasmin of normal cells inhibit its activity. So the presence of high concentrations of plasmin in tumor cells could well serve as a guide to design prodrugs which could be selectively cleaved by plasmin at tumor cells. This hypothesis was previously applied very effectively for the design of prodrugs of several other cytotoxic agents.42 Based on this principle, recently Scheeren et al.43 have synthesized the PTX-peptide prodrugs 20a–d, starting from the carbonate 22, which was derived from PTX and p-nitrophenyl chloroformate, and a tripeptide 21 (Scheme 6). The key concept here is that plasmin would attack and cleave the peptide bond of 20 to generate intermediate spacer 23, which would immediately undergo 1,6-elimination reaction to release the active PTX as depicted in Scheme 6. The driving force here (and in the previous case Scheme 5), is the formation of entropically favored cyclic 5-membered ring 24, with the expulsion of PTX. In a separate synthesis they also synthesized the prodrug 25 from the starting material 26 and PTX (Scheme 7).
Scheme 6.
Scheme 7.
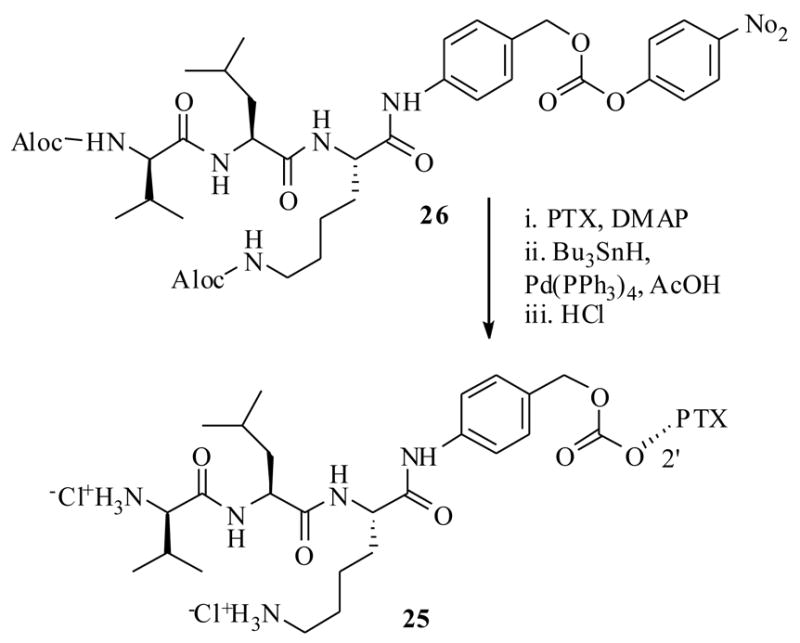
The prodrugs 20a–d were tested for their stability in Tris-buffer at pH 7.3 and were found to be stable for 3 days, except the prodrug 20a which was degraded to liberate baccatin III. To test the plasmin effect on the prodrugs 20a–d, they were incubated with human plasmin (in Tris-buffer at the same pH 7.3 for up to 3 days); only the prodrugs 20d and 25 released PTX, but not 20a–c under the same conditions. The determined half life in the above medium for carbamate prodrug 20d was only 3.5 min. However, it only produced peptide cleaved spacer 23, which has showed prolonged half life period 23 h to undergo complete 1,6-elimination to yield PTX. In contrast the carbonate prodrug 25 has shown 42 min half life with no detectable spacer unit indicating as soon as the plasmin hydrolysis had occurred it underwent instantaneous 1,6-elimination reaction.
All the prodrugs were subjected to in vitro cytotoxicity evaluations against various cell lines (Table 4), and were found to be several hundred orders of magnitude less potent than PTX. It is worth reiterating the point here that many of the C2′-O-esters and carbonates of PTX were found to be as active as PTX, since they undergo hydrolysis under cell culture medium to generate PTX,44 however, in this case the carbamate derivatives 20a–d and 25 possessed significant stability against the cell culture medium. The stability of the prodrugs 20a–d and 25 can be ascribed to blocking effects caused by the tri-peptide on the carbamate bond, which prevents the attack of several esterases present in the cell culture medium. Over all compound 25 could be potential prodrug candidate, if not on its own, by the ADEPT technology (discussed in Section 3.3) which is developed to localize enzymes at tumor site, since it possess less cytotoxicity against panel of cell lines and prolonged half life period in the cell cultures.
Table 4.
In vitro cytotoxicity of PTX and prodrugs 20a–d and 25
| Compound | ID50 (ng/mL)* | ||||||
|---|---|---|---|---|---|---|---|
| H226 | IGROV | MCF7 | EVSA-T | WIDR | M19 | A498 | |
| PTX | <0.003 | 0.010 | <0.003 | <0.003 | <0.003 | <0.003 | <0.003 |
| 20a | 41 | 19 | 16 | 11 | 13 | 45 | 40 |
| 20b | 21 | 11 | 6.6 | 7.2 | 10 | 17 | 38 |
| 20c | 25 | 26 | 6.9 | 5.2 | 11 | 24 | 45 |
| 20d | 48 | 45 | >63 | >63 | 47 | 47 | >63 |
| 25 | 16 | 27 | 9.2 | 9.3 | 11 | 20 | >60 |
MCF-7, EVSA are breast carcinoma, WIDR are colon, IGROV are ovarian carcinoma, M19 are melanoma, A498 renal carcinoma, H226 non small cell lung carcinoma, cell lines
3. Taxoid drug targeting strategies
Prodrugs described in the previous section could only improve the oral bioavailability of drug, however, there is no guarantee that they may be selective for cancer cells. So, the aim of identifying a magic bullet, which is extremely stable during the circulation in the entire body and exclusively releases the active drug at tumor cell, is highly ambitious. Therefore it would be highly advantageous, if we could identify a tumor marker exclusively localized at the tumor (i.e. proteins/receptors/hormones etc.) and attach a drug molecule to it. On this premise, several drug targeting protocols have been envisioned and few attempts in this direction have already produced promising results. The following sections would describe some of the efforts and achievements.
3.1 Estradiol as a taxoid targeting agent
Nuclear proteins, such as Estrogen Receptors (ERs) are found to selectively attract and localize their ligands and/or ligand cognates in cells, where they are overexpressed. ERs are also found to play a pivotal role in modulating several biological effects of estrogens and anti-estrogens.45,46 They are overexpressed in cancer cells such as breast, ovarian and gonads that are the primary target organs for estrogen action.47 Thus ERs provide us with a plausible target for the selective delivery of cytotoxic agents, such as taxoids, through affinity of the ligands like estradiol (26). In fact this concept has been explored for the selective targeting of other cytotoxic drugs in the past48 and the results indicate that the selective toxicity to ER positive MCF-7 breast cancer cells has occurred, when compared to ER negative MDA-MB-231 cells.49
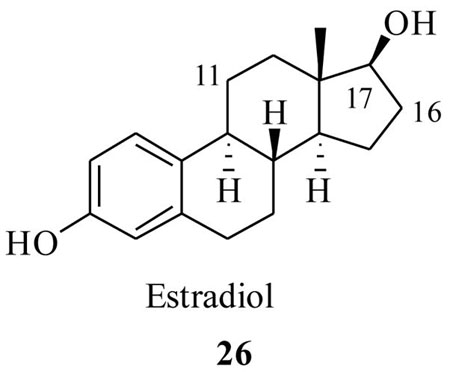
Thus, Kingston et al.50 have designed and synthesized few PTX-estradiol conjugates as shown in Scheme 8. It was anticipated that the estradiol would transport the PTX to the breast ER (α) positive breast cancer cells, since it has a high binding interaction with ERs. The synthesis was achieved starting from estrone 27, which was synthetically functionalized at two different positions by the reported methods, to furnish estradiol derivatives 28 and 30. These derivatives were then coupled to 2′-OTBS protected PTX by EDCI mediated couplings, followed by deprotection, resulting in the PTX-estradiol conjugates 31 and 32 respectively (Scheme 8). Estradiol conjugates from the C7 and C10 position of the PTX (33 and 34) were also prepared by a similar synthetic strategy. It is worth reiterating that, from the SAR studies of estradiol, the structural modifications at C11 and C16 positions do not cause any changes in binding affinities to ER positive cells.51 Also from the SAR of PTX, it is well known that the derivatization at C2′ and C7 could lead to less bioactive taxoids and modifications at the C10 position may or may not change the bioactivity of PTX.
Scheme 8.

The PTX-estradiol conjugates 31–34 were tested against both ER positive PC3 prostate and MCF-7 breast, ER negative A2780 ovarian and MDA-MD-231 breast cancer cell lines. From Table 5, it is clear that the parent compound PTX has similar cytotxicity towards ER positive and ER negative cell lines. It is also evident that all the drug conjugates showed significantly lower cytotoxicities against both breast cancer cells, and also ovarian caner cells. However, they showed nearly equal activity against prostate cancer cells, when compared to PTX. The conjugates also did not show any selective cytotoxicity to ER positive breast cancer cells (vs. ER negative), except the conjugate 34 which exhibited moderately 3-fold higher cytotoxicity to ER positive MCF-7 breast cancer cells vs. ER negative MDA-MB-231 cells.
There are various factors contribute to lower activities of the above drug conjugates. First, the conjugates should be able to be hydrolyzed to release the PTX in the cell culture, as it happened with many other cell lines in the case of C2′ esters of PTX. Similarly, the fact that C2′ ester conjugates 31 and 32 exhibited slightly higher activities when compared to 33 and 34 (which are C7 and C10 esters of PTX respectively, found to be much more stable than C2′O-PTX esters), confirms the previous assertion that they might have undergone hydrolysis to release the PTX, which actually contributes to the cytotoxicity. The estradiol might have lost its binding affinity to ER positive receptors, since it was structurally modified and conjugated to PTX. Nevertheless, additional biological evaluations are necessary to fully understand the latter point.
The success of this strategy would depend on the conjugate ability to bind to ER positive cells, internalization, thence to release the active drug. However, the conjugates (except 34, which showed 3-fold selective toxicity to ER (α) positive cells) were unable kill ER positive breast cancer cells indicates that they bind poorly to ER positive cells. If the latter point is true, their binding ability could be improved by coupling the taxoid drug at 17α position of estradiol rather than C11 and C16 positions, since other cytotoxic agents coupled from this position retained the binding abilities comparable to estradiol.51 Likewise, increasing the length of the linker between taxoid and estradiol would also help conjugates bind well to the ER positive cells.
3.2 Antibody as a taxoid drug targeting agent (TPT and or PMT technologies)
Antigens are overexpressed at the surface of various tumor cells and have a high binding affinity to certain antibodies. Conceptually, this is an attractive feature aiding in the design of drug conjugates by attaching the cytotoxic drug to an antibody and selectively targeting the drug to tumor cells.52 In fact, this concept has led the development of the various recombinant antibodies that would selectively localize tumor antigens. The field has been rapidly expanding and is now in advanced stages since the development of monoclonal antibodies (mAbs).53 mAbs are highly specific antibodies, created by discrete colonies of hybridized cells, and show high binding specificity for tumor-specific antigens that could transport the cytotoxic drugs very well to the tumor site.52 When the mAb-drug conjugate (also called as immunoconjugate) reaches the tumor site it will be internalized by an antigen-mediated process called endocytosis.54,55 The latter process would trigger several biochemical processes in the cells to release the active drug. This strategy was previously coined as either tumor-activated prodrug therapy (TPT) or prodrug monotherapy (PMT).
The success of the TPT strategy depends heavily on the internalization of the immunoconjugate, release of the active drug, the nature of cytotoxic agent, and finally on tumor specific affinity of mAb. It was previously discovered that only a fraction of mAbs are localized to the tumor site from the total injection dose, and to achieve satisfactory therapeutic levels from immunoconjugate therapy, the drug conjugate must have a higher potency than the clinically used free drug (estimates indicate that the drug must have an IC50 = < 1×10−10 M).52 In the past these mAbs were effectively used to target other cytotoxic drugs such as doxorubicin, calicheamicin, and maytansinoids to the tumor cells and laid the foundation to exploit various other new cytotoxic drugs such as taxoids for TPT delivery. 56,57 Several efforts for taxoid drug targeting by the above concept have been reported thus far.
As one example, the tyrosine kinase receptors P140 TrKA and p75 are found to be overexpressed in many cancer cells such as neuroblastoma, non small cell lung, B-cell lymphoma, and melanoma.58 They are also expressed in normal cells such as neurons, however in very low density. In this respect, separate investigations by Saragovi et al.59 and Shooter et al.60 developed anti-TrkA (mAb 5C3) and anti-p75 (mAb MC192) antibodies as tumor marking agents.
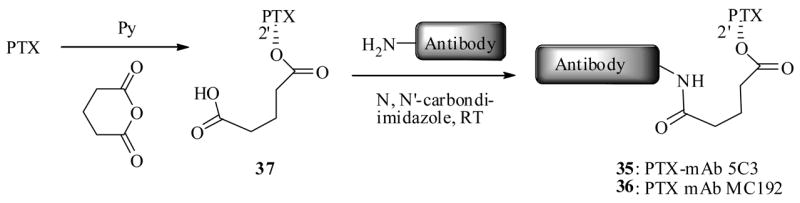
Recently Saragovi et al.61 synthesized two PTX-immunoconjugates, 35 and 36, by a carbodiimidazole mediated coupling of C2′-glutaric acid PTX-ester 37 separately with two antibodies, mAb53 and mAbMC192. The precursor 37 was generated from a simple reaction between PTX and glutaric anhydride as shown in Scheme 9. In this report, they have systematically investigated the various properties of the immunoconjugate PTX-MC192 (36) as delineated below.
Scheme 9.
First, the binding affinity of PTX-MC192 immunoconjugate (36) against two cancer cell lines 4-3.6 and B104, that express p75 receptors, was investigated in FACS (fluorescent activated cells scan) assays. Conjugate 36 was found to retain the overall binding ability of the antibody itself. The in vitro cytotoxicity of 36 was determined and results indicated that it was better than free PTX against B104 cells at equimolar doses. Selective bioactivity of the 36 was evaluated against the cancer cells that do not express p75 receptors. Results indicated that 36 was inactive, where as parent PTX showed dose dependent cytotoxicity against the same cancer cells. Together the data confirms the selectivity of 36 towards cells that express p75 receptors. Its specificity was established by competition experiments. Thus 10nM concentrations of 36 (equivalent to free 10nM PTX) showed effective B104 cell death, where as addition of the 40nM antibody MC192 itself, which competes for p75, blocked the effect of 36. And at the same time addition of a non- specific antibody, which does not compete for p75, did not show any effect on the conjugate bioactivity. Similar experiments on free PTX, with 40nM of MC192, did not have any effect on the PTX cytotoxcity confirming the 36 specific binding ability to the cells that express p75 receptors. In this study they investigated the parent antibody (MC192 alone) pharmacological role as adjuvant in enhancing the cytotoxicity of the immunoconjugate 36, compared to free PTX. Results showed that mAb-MC192 alone neither enhance or decrease the cytotoxicity of the free PTX at various concentrations and also the fixed concentration of PTX with varying concentration of the antibody. Lastly, the 36 was subjected to in vivo evaluations against neuroblastoma xenograft in nude mice. Studies indicate that the conjugate was effective in reducing the tumor growth, compared with control, where as free PTX alone or in combination with MC192 was not able to reduce the tumor growth. Additionally, the immunoconjugate 36 has prolonged the survival of the mice on average by 30% compared with free PTX. Like wise, in vivo experiments corroborate their in vitro findings of 36, making it a potential candidate for the treatment of tumors expressing p75 receptors.
It is worth mentioning here that investigators prepared immunoconjugates 35 and 36 by very simple synthesis and established all its biochemical properties, so that it could well serve as a guide for newly discovered antibody mediated drug delivery protocols to establish and identify an ideal candidate for TPT chemotherapy.
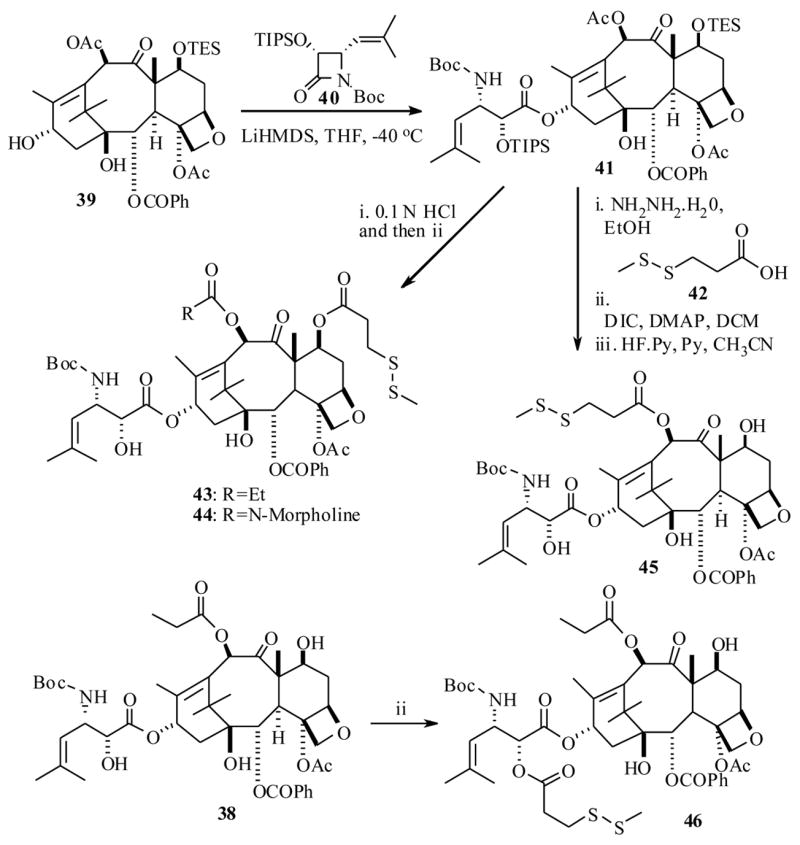
Along similar lines Ojima et al.62 also reported a successful protocol, through use of mAbs as taxoid drug delivery agents. For the execution of their strategy, they have used highly active taxoid 38, is a second generation DTX (2) analog, which exhibited remarkably 2–3 orders of magnitude higher potencies than DTX against various cell lines in vitro.63 In order to be able to couple the taxoid 38 to antibodies, they had to be functionalized synthetically with a 3-methyldisulfide (MDS)-propanoyl linker unit. Initially, they resorted to developing a SAR study on 38 with a MDS linker unit by attaching it at the C2′, C10 and C7 positions respectively to identify potential sites for antibody incorporation. The synthesis was accomplished as shown in Scheme 10, by a straightforward synthesis starting from a baccatin III derivative 39 and a β-lactam derivative 40, which has resulted in the taxoid derivatives 43–46.
Scheme 10.
The in vitro cytotoxicity of the taxoid analogs 43–46 against epidermoid A431, and non small cell lung A549 cancer cell lines, indicated that the C10-MDS-propanoyl taxoid 45 showed higher bioactivity than other taxoids, so it was selected for coupling to an antibody. The disulfide bond of the taxoid 45 was readily cleaved by treating with dithiothreitol (DTT) to furnish taxoid 47 with a free thiol unit. And at the same time the antibodies were also functionalised with N-succinimidyl-4-(2-pyridylthio)-petanoate to provide the intermediates, which were then treated with 47 in a buffered solution at pH 6.5 to yield immunoconjugates 47a–c (Scheme 11).
Scheme 11.

The epodermoid growth factor receptor (EGFR) is found to be overexpressed in several cancers such as head and neck, lung, breast, and human squamous cancers.64 Several immunoglobulin class G antibodies such as KS61 (IgG2a), KS77 (IgG1), and KS78 (IgG2a) were identified which could be localized at EGFRs on the tumor cell. In this regard, Ojima et al. selected the three antibodies and individually coupled then to a taxoid 47 to yield immunoconjugates 47-KS77, 47-KS61, 47-KS78 respectively as shown in Scheme 11. For the comparison purpose an immunoconjugate of 47 with antibody mN901, which does not bind to EGFR positive cancer cells, was also prepared.
The in vitro cytotoxicty evaluations of the immunoconjugate 47-KS78 were conducted against the EGFR expressing human epidermoid carcinoma cell line A431, and results indicated that the immunoconjugate 47-KS78 exhibited an IC50 1.5 nM. For the same cell line the immunococnjugate mN901-47 did not show any cytotoxicty. The addition of a large excess of anti-EGFR antibody (e.g. KS61) to the 47-KS78 eliminated the cytotoxicty caused by the immunoconjugate 47-KS78, clearly demonstrate the antibody mediated binding had occurred.
The in vivo efficacies of the immunoconjugates 47-KS61 and 47-KS77 were evaluated against human xenografts in severe combined nude mice (SUID) inoculated with A431 cancer cells. The immunoconjugates inhibited tumor growth in all the treated animals for the duration of experiment. In contrast, the mAb free taxoid 45 showed no therapeutic effect. Additionally there was no systemic toxicity to the mouse was noticed at the administered dose (10 mg/kg) of immunocojugate 47-KS77.
One point to be noted here that the conjugate described above is a C-10 derived taxoid (rather than usual C2′-derived taxoid) so additional experiments needs be fully investigated, about the exact nature of the conjugate internalization and release of the active taxoid drug after endocytosis. Nonetheless, the principal concept of antibody mediated drug targeting (TPT) seems very promising. As demonstrated by the above two reports discussed in this section, the results raise the hope for better taxoid chemotherapies for various cancers.
3.3. Taxoid targeting by ADEPT technology
The challenge for highly effective targeted delivery protocols is to suppress the non tumor cytotoxicity and improve the cytotoxic drug concentrations at the tumor site (vide infra). An alternative way to address this issue in a broader sense is an antibody directed enzyme prodrug therapy (ADEPT) technology. Envisioned two decades ago,65 this technology continues to gain momentum.66 By ADEPT technology an enzyme would be targeted first to the tumor site by means of an enzyme-antibody conjugate. Once this conjugate is localized at tumor site and cleared out from the systemic, then a less cytotoxic prodrug would be administered. When the prodrug reaches the tumor site the enzyme thus concentrated mainly at the tumor site would act on the prodrug to release the active drug. The ADEPT technology offers several advantages over other prodrug and targeted delivery protocols (TPT) described above such as, it would allow us to design and develop more water soluble prodrugs with better pharmacokinetic properties, however, it also would suffer from a limitation such as the number of enzymes would be limited, since very few enzyme antibody conjugates would be localized at the tumor cell. Unless the enzyme turnover number is very high, sufficient quantity of the active drug may not be made available by the enzyme present at the tumor site.
Conceptually, it is also possible to prepare a ‘super conjugate’ with all three (i.e. prodrug-antibody-enzyme) units as a cargo, and test the tumor specific delivery by the antibody interaction with the corresponding antigens present in the tumor cells. This idea is based on perspective that the enzyme would be in an inactive form in antibody-enzyme conjugate, which then can be coupled to a prodrug with a spacer unit. The active enzyme would be released only after endocytosis at tumor cell. In this case the cargo would carry sufficient number enzymes per molecule of prodrug, however, the synthesis of such a conjugate would be complicated one and thus this protocol has not been explored so far.
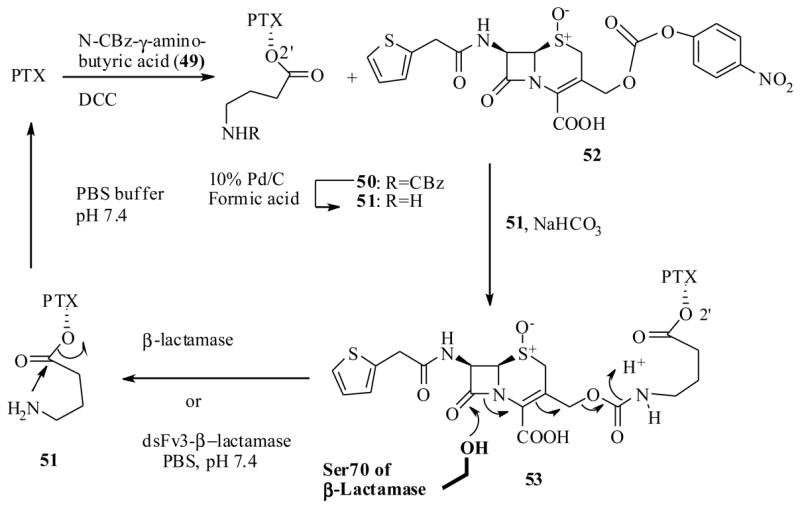
There are several enzymes such as β-lactamase, β-glucuronidase, carboxypeptidase-G2, and carboxypeptidase-A, cytosine deaminase, penicillin amidase, alkaline phaphatase and cysteine proteases (cathepsin B, -L) that could be bound to antibodies. These enzymes have been used in the past to specifically target tumor cells and explored in ADEPT for other cytotoxic drugs.67 Rodrigues et al. have prepared a fusion protein, dsFv3-β-lactamase, from the humanized antibody (humAb4D5-8), and β-lactamase (REM-1) to target the other cytotoxic drugs in the past.68 In that study they observed that β-lactamase shows high catalytic activity, large substrate specificity and would act on the β-lactam ring moiety of the prodrug to release the bioactive drug.68 They have now explored the PTX for the tumor specific delivery through this technology.69
The synthesis of PTX-prodrug conjugate 53 was achieved as shown in Scheme 12, by a coupling reaction between β-lactam derivative, cephalosporin 52, and PTX-2′-O-aminobutyryl ester 51, which in turn was prepared from PTX and N-CBz -γ-aminobutyric acid (49).
Scheme 12.
Once the PTX-prodrug conjugate 53 was synthesized they investigated the free β-lactamase and a tumor targeting fusion protein dsFv3-β-lactamase to mediate the hydrolysis of 53 to release the PTX linker 51. The results show that 53 was rapidly hydrolyzed by both the above enzymes to release 51, which showed longer half life period 16h to release PTX as it was observed with the spacer unit 23 (cf. scheme 6). It is interesting see the same spacer unit 51 when treated with β-lactam derivative 52 under mild basic conditions (with NaHCO3) did not release any PTX. This can be attributed to the changes in the chemical reaction to a biological environment.
The conjugate 53 was subjected to in vitro cytotoxicity and tubulin polymerization activity in the presence and absence of β-lactamase. The results show that conjugate on its own had no effect on the tubulin polymerization, however, when it was activated overnight with the β-lactamase enzyme, it showed similar tubulin polymerization activities. A brief period of exposure of the 53 to β-lactamase did not show any tubulin activity, and this result is consistent with the slow expulsion mechanism of PTX from linker 51.
The cytotoxicity against breast cancer cell line, SK-BR-3, revealed that conjugate 53 is 10 fold less potent than PTX. In sharp contrast, similar activities to PTX were observed in the presence of targeting fusion protein dsFv-3-β-lactamase. Thus proof of concept for the PTX drug targeting by ADEPT seems to be working, though the in vivo results are to be published on this conjugate 53.
Recently Scheeren et al. 70 also investigated the PTX drug targeting by ADEPT technology using a human enzyme β-glucuronidase, which is a lisosomal enzyme and found to have no activity in the blood pH. In this study they synthesized the prodrug conjugates 60–62 as shown in Scheme 13, by DIPC mediated coupling of the PTX to the intermediate carbohydrate linked carboxylic acids 58 and 59, which in turn were prepared from the protected sugar unit 55. The prodrug conjugates 60–62 have 6–8 spacer atoms between PTX and the carbohydrate moiety. Like before, the β-glucuronidase enzyme is expected to cleave glycosidic bond as shown in Scheme 13, to liberate the PTX linker 63 at the tumor site.
Scheme 13.

The biological results revealed that the prodrugs 60–62 were several hundred times more soluble in water when compared with PTX, and were found to be stable at pH 6.8 at 37 °C, except the prodrug conjugate 60, which was readily cleaved by the non specific hydrolysis. As a result only conjugates 61 and 62 were tested further in their study. The enzyme-catalyzed prodrug activation experiments were carried out with 100 μM prodrugs 61–62 versus 10 μM of human β-glucuronidase at 37 °C. The prodrugs, 61 and 62, released the PTX with a half life of 45 min and 2 h respectively. It is worth mentioning here, that in this experiment no 63 was detected by HPLC indicting that the spacer unit in 63 had spontaneously undergone cyclisation to release PTX. This result is in sharp contrast with results obtained by linker 53 in Scheme 12. This clearly emphasizes how important the linker design is in this tumor delivery protocol. The only difference between 63 is an extra gem-dimethyl group compared to 53.
The in vitro cytotoxicities of the prodrugs 61 and 62 against OVCAR-3 cells were tested and the IC50 values of the prodrugs 61 and 62 indicates that they were about 2-orders of magnitude less cytotoxic than parent PTX, but prodrug 60 had the same activity as PTX. Importantly the prodrugs upon activation with enzyme β-glucuronidase exhibited nearly equal cytotoxicity to the parent (Table 7)
Table 7.
In vitro cytotoxicity of PTX and prodrug conjugates 60–62
| Compound | OVCAR-3 cells IC50 (nM) | |
|---|---|---|
| Without β-glucuronidase | With β-glucuronidase | |
| PTX | 0.2 | NA |
| 60 | 0.23 | 0.23 |
| 61 | 27 | 1.1 |
| 62 | 20 | 0.6 |
They further determined the activities of prodrug conjugates 61–62 against OVCAR-3 cells, which were pretreated with a conjugate derived from murine anti-pancarcinoma monoclonal antibody 323/A3 and human β-glucuronidase. The results revealed that antibody-enzyme-conjugate specifically targeted to OVCAR-3 cells and showed enzyme activity on the prodrugs, which showed IC50 values only two fold higher than the prodrug. Nonetheless, it was found that the amount of enzyme bound to the above cells was too low to completely activate the prodrugs. This observation is quite consistent with speculated limitation by the ADEPT technology now witnessed by present investigation.
Monneret and Schmidt et al.71 have made extensive efforts in designing prodrugs of both PTX and DTX for tumor specific delivery by ADEPT technology. They have synthesized PTX conjugate 66 as shown in Scheme 14, starting from carbohydrate-p-nitro phenyl ether 63.71a They have also synthesized the PTX conjugate 74 as shown in Scheme 15, starting from 69 in a straightforward synthesis.71b Recently they also synthesized DTX conjugates 75 and 76 by similar synthetic reactions depicted in Scheme 15.71c It is worth clarifying a point here that conjugates 66 and 74-76 are two different classes, the former has only one linker unit, while the latter have two linker units between carbohydrate moiety and taxoid. They have also envisioned testing these prodrugs by β-glucuronidase mediated activation.
Scheme 14.

Scheme 15.

In vitro cytotoxicity assay results of the prodrug conjugates 66, 74–76, against LoVO (human colon cancer) cell line, indicated that they all have significantly lower cytotoxicities, when compared to their respective parents (PTX or DTX), and more importantly, upon activation with β-glucuronidase enzyme, the prodrugs showed equal cytotoxicity to PTX (Table 8). The liberated by product 68, however, has very low cytotoxicty compared to prodrug IC50 values suggesting that it does not have much impact on the values obtained by the prodrugs after β-glucoronidase mediated activation. In addition, the conjugates 66 and 74–76 showed inherent prodrug properties, for instance, they were found to be stable in a phasphate buffer at pH 7.2 at 37 °C for 24 h, and also found to be more aqueous soluble than PTX. Special mention should be made for the prodrug conjugate 66 showing 2000 fold more aqueous solubility than PTX, so it was selected to test further for it efficacy in ADEPT technology.
Table 8.
In vitro cytotoxicity of taxoid prodrug conjugates 66, 74–76
| Taxoid | IC50 Compound/IC50 of the parent | IC50 of compound + β-glucuronidase |
|---|---|---|
| PTX | 90nM | |
| 66 | 722 | 100nM |
| 74 | 153 | NA |
| 75 | 337 (DTX) | NA |
| 76 | 187 | NA |
| 68 | 3000 | NA |
To determine the kinetic properties of the prodrug 66 in vitro, they used E.coli β-glucuronidase enzyme that was found be analogous to human β-glucuronidase. 72 At the concentration of 100 μg/mL of β-glucuronidase per 250 μg/mL of prodrug 66, the ezyme catalysed reaction had occurred and prodrug 66 released the PTX with half life near 2h. In this experiment, no trace of intermediate (PTX-linker 67) was found, clearly representing that as soon as the enzyme activation had happened (cleavage of sugar unit), the resulting intermediate 67 underwent rapid self immolative cyclization to release PTX and a byproduct 68.
In the case of 74–76, the enzyme activation experiments were conducted at low concentrations due to their low solubility. At 30 μg/mL β-glucuronidase per 30 μg/mL of produrg, all of them showed only 10 min of half lives even at such low enzyme concentration. The difference in the half life periods of 74–76, compared to 66 can be explained by steric factors. In the case of 74–76, the carbohydrate unit and PTX were attached to para-position of aromatic ring which makes the glucuronate unit more accessible to enzyme compared to 66, in which both units were attached at ortho position, which would make unfavorable interactions with approaching enzyme. Also in the case of 74, the PTX-ethylene diamine linker unit 77 was detected (by HPLC). At enzyme concentrations 50 μg/mL and up 77 showed half life 13 min, in sharp contrast to 67 which was not detected.
It is worth noting a point here that the prodrugs 74–76 contain a double spacer unit which upon initial cleavage by enzyme would undergo 1,6-elimination to generate a transient N,N-dimethyl ethelene diamine spacer unit, which was found to undergo a cyclisation-elimination process much faster than a single N-substituted spacer unit present in 23d (cf. scheme 6).73 Though the preliminary results of these conjugates are encouraging, a detailed in vivo bioactivity data would help to understand the efficacy of these prodrugs.
3.4. Folic acid as taxoid drug targeting agent
Various cancer cells except prostate, pancreatic, bladder, and lymphoid cancer cells overexpress folic acid receptors (FRs).74 FRs also known as folate binding proteins (FBPs), have high binding affinity to folic acid (FA, a vitamin B) and play pivotal role in cellular uptake of folic acid.75 Folic acid was discovered to play prominent roles in the formation of new cells and tissues, especially at the prenatal and postnatal stages as well as during childhood. That is why gynecologists highly recommend FA to pregnant women. A hydrofolate form of folic acid is found to be abundant in the blood, and does not cause any side effects and immunogenicity to the human body.
The FRs were found to exist in various forms such as α-FBP, β-FBP, γ-FBP and δ-FBP. Among these α-FBP and β-FBP are membrane-associated folate receptors.76 The high binding affinity interactions between FRs and FA77 could conceptually enables us to design a cytotoxic drug-folic acid complex and selectively target it to the FR expressing tumor cells. Excellent reviews covering the topic of folates (FA) and their role in the targeted therapy have recently appeared in a series of reports.78 Once the folic acid-drug conjugate reaches the FR positive tumor cells, it will enter into the cells via a receptor mediated process called endocytosis.79 The explicit mechanism of how endocytosis of the drug-FA-conjugate occurs in the cell to release the bioactive drug has been represented by Leamon and Reddy.78a In brief, once the folate drug conjugate binds to the cell the plasma membrane surrounds the ‘folate drug conjugate-FR complex’ forming an early stage endosome, then the pH of the endosome drops suddenly to ~5 by the action of proton pumps, that are localized in the endosome membrane. Subsequently, the protonation of the carbonyl groups of FR by changes in pH, would enhance the conformational change of the receptor that would trigger the release of folate molecule.
In fact, dropping of cell pH upon FR mediated FA drug conjugate internalization is a highly useful property for designing prodrugs as well as linkers that could be selectively cleaved at lower pH 5. In the past the FA mediated drug targeting attempts have been explored for other cytotoxic agents.80 Few reports on taxoid drug targeting by FA have been appeared but Fuchs et al.81 have designed and synthesized several PTX-FA conjugates from the C2′ and C7 positions of PTX.
The synthesis of PTX-FA conjugates 85–88 was accomplished as shown in Scheme 16. Thus, the linker starting materials 80 and 81 were separately coupled to PTX by DIPC, to furnish the PTX linker intermediates 83 (after deprotection of 82), which then were coupled to a FA derivative 84 to yield FA-PTX- conjugates 85 and 86. Like wise the conjugates 87 and 88 were also prepared by similar syntheses.
Scheme 16.

The PTX-C2′O-FA conjugates 85 and 86 showed shorter half lives at pH 7 and pH 5 when compared to the corresponding conjugates 87 and 88 that were connected from the C7 position of the PTX (Table 9). This observations consistent with the previous point that the C2′O-esters of PTX are more labile than the C7 esters. Cytotoxicity results of the PTX-FA conjugates indicated that they were significantly more potent than PTX against 3 different cell lines in vitro (Table 9). The binding affinity of 88 was investigated against labeled 3H-folic acid, in comparison with free folate. Conjugate 88 retained most of the binding affinity of free folic acid against receptor positive murine M109 and human KB tumor cell lines. Efforts to discover any receptor-mediated specific cytotoxicity (in vitro) against receptor positive KB tumor cells (by comparing with parent PTX), revealed that the conjugate 88 was about 50 fold less cytotxic than PTX. At the same time concurrent addition of a 500 fold excess of free folic acid did not change the bioactivity of the conjugate, meaning that the folate moiety attached in the conjugate 88 is not responsible for cell internalization.
Table 9.
In vitro cytotoxicity of PTX-FA conjugates 85–88.
| (ED50 μg/mL) | T½ (h) at 37°C | ||||
|---|---|---|---|---|---|
| A549 lung | MCF-7 breast | HT-29 colon | pH 7 | pH 5 | |
| PTX | 2 × 10−2 | 5 × 10−2 | 3 × 10−2 | ||
| 85 | 1.6 ×10−3 | 3 × 10−3 | 1.2 × 10−3 | 1 | 17 |
| 86 | 1.2 × 10−3 | 2.4 × 10−3 | 1 × 10−3 | 9 | 38 |
| 87 | 1.7 × 10−3 | 3.3 × 10−3 | 2.9 × 10−3 | 40 | >300 |
| 88 | 1.6 × 10−3 | 2.6 × 10−3 | 1.1 × 10−3 | 109 | 197 |
At the same time 88 and PTX were treated with KB and M109 cells, and FA negative A549 cancer cells at constant drug concentrations. In this experiment conjugate 88 neither showed more potency than PTX, nor any folate receptor mediated selectivity in the above cell lines (its activity was found to be nearly equal against 3 different cell lines). Like wise, in vivo experiments on mice implanted with folate receptor positive M109 tumors did not show any useful level of increase in the life span of the mice, when compared to PTX.
The only interesting feature observed by the conjugate 88 is its lack of toxicity in tumor free mice. PTX treated mice showed 20% body weight loss, whereas mice treated with 88 showed no body weight loss. Overall, 88 is the best of the four PTX-FA conjugates synthesized in this study, but it failed to selectively kill the receptor expressing cells in vitro and did not show any superior activity in vivo against the FR positive tumor cells when compared to PTX. Thus, the concept of taxoid drug targeting by FA mediated drug delivery has not yielded conclusive evidence with the above conjugates. The results led to investigation of other types of linkers for making the PTX-FA conjugates, that might be cleaved at lower pH and also increase the folate mediated selective internalization, rather than non-selective entry into the cells. From Table 9, it is clear that the conjugates 85–88 were cleaved much faster at pH 7 rather than pH 5, and this result is in contrast to anticipated results knowing that tumor cell pH drops to 5 upon FA-mediated internalization.
Very recently, Majoros et al.82 reported a FA-mediated successful PTX drug delivery protocol to FR positive cell lines. In this study they have utilized a dendrimer PAMAM (polyamidoamine) as a linker unit between the PTX and FA as well as a flourescein isothiocyanate moiety (FITC) used as a florescent probe for cancer cell imaging.
The conjugate 89 was synthesized as shown in Scheme 17 starting from the advanced intermediates of PTX 90 and folic acid-dendrimer-complex 91. The average number of PTX molecules attached to conjugate 89 was determined to be 3. The PTX-FA-PAMAM-FITC-conjugate 89 was subjected to the cytotoxicity assay against FA receptor positive KB cells, at various concentrations (100, 50, 25 nM). A FA conjugate without PTX was also prepared in this study and used as a control in a parallel experiment. The results indicated that the conjugate 89 showed cytotoxicity against FR positive KB cells (human epidermoid carcinoma) at the highest concentration (100 nM). Flow cytometry analysis of cells revealed that the PTX-drug conjugate and drug free conjugate was taken up in to the FR positive cells, however, only the PTX-conjugate 89 killed the cancer cells, but not the drug free conjugate. Importantly, when subjected the same drug and drug free FA conjugates to the FR negative cells, they neither internalized in to the cells nor showed any cytotoxicity. The properties clearly emphasize that the FA-mediated targeted delivery has occurred.
Scheme 17.
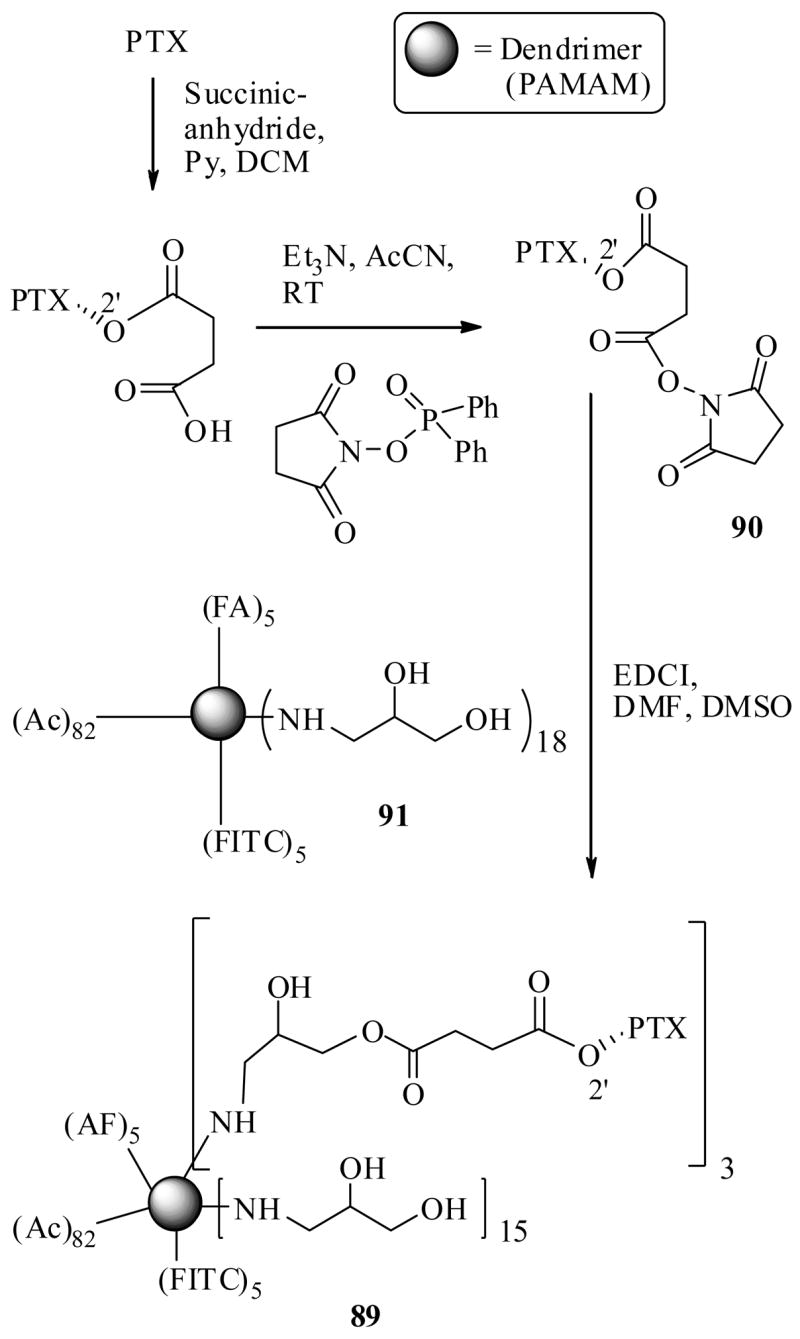
Furthermore XTT proliferation assay was performed to verify the efficacy of the drug conjugate 89 and drug free control. At the concentration of 200 nM, the PTX-FA-conjugate showed cytotoxic effects against both FR positive and FR negative cells, however, at lower concentrations (50 nM) the conjugate showed selective cytotoxicity against positive cells, leaving the FR negative KB cells unaffected. The control (drug free conjugate), however, had no cytotoxic effects at concentration 200 nM.
Several alluring properties of the dendrimer (multi-functional polymeric units) used in this study are note worthy. First, they not only are capable of transporting more than one drug molecule per molecule of the conjugate to the tumor site. Second, they have many additional synthetically available sites for attaching the fluorescent or radio-labeled probes to monitor the properties of a drug conjugate. Third, when they subjected to FR positive and FR negative cells without FA molecule (in the above study), they had virtually no association between the cells, thus explaining that they do not interfere with FA mediated drug conjugate binding efficiency. Over all, the taxoid drug targeting concept by folic acid seems promising, however, detailed in vivo efficacies are still to be reported.
3.5. Peptide dependent taxoid drug targeting
Numerous proteins and peptides (such as αvβ3, αvβ5, APN, VEGFR, MMPs) have been identified which are selectively localized in the tumor endothelial cells.83 These proteins are found to play a very important role in tumor angiogenesis and have specific affinity to various peptides.84 In particular, the GRP (gastrin releasing peptide), which belongs to bombesin (BBN) peptide family, is overexpressed in small cell lung cancer cells (SCLC) when compared to normal cells.85 In this regard, efforts have been directed to achieve tumor specific delivery of cytotoxic agents by means of a peptide conjugate.
Thus, Safavy et al.86 synthesized a PTX-PEG-GRP conjugate 92 as shown in Scheme 18 starting from peptide 93 and PTX-C2′-O-succinate. This conjugate 92 was expected to be localized by a specific receptor peptide binding at the tumor cell surface, then release the active drug upon hydrolytic cleavage of the peptide at tumor site. For the comparison study they also synthesized a peptide free PTX-PEG derivative 95 by a similar method used in the synthesis of 92.
Scheme 18.
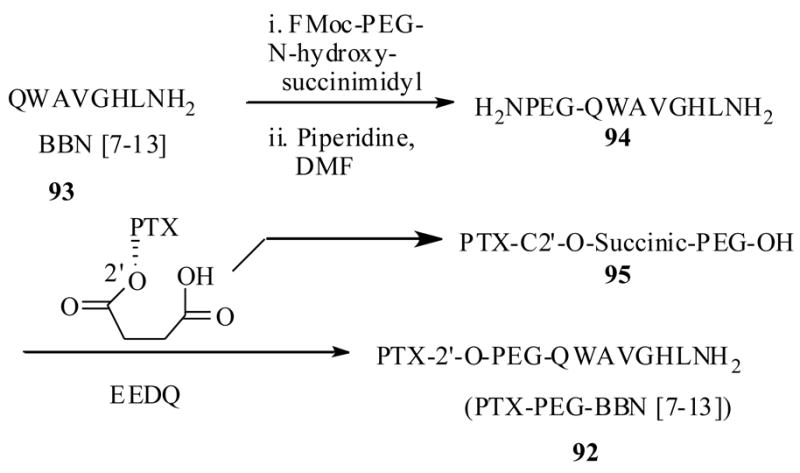
The conjugate 92 showed an interesting solubility profile. For example, consistent with previous reports, that resulted in significantly more soluble PTX derivatives when coupled PEGs, the conjugate 92 also showed better solubility than PTX, and it was readily soluble in aqueous media at 25mg/mL concentrations. Additionally it was soluble in organic solvents such methylene chloride, chloroform, and methanol where as the unconjugated peptide 93 (without PEG) was only soluble in dimethyl formamide and methanol, clearly showing the PEG role in solubulizing the PTX as well as peptide units.
To determine the tumor specific cell binding efficacy of 93, it was tested against two different cell lines. BNR-11 cell line, which was previously used to evaluate the binding affinity of the [125I]-Try3-BBN, and NCI-H1299 cell line, which has relatively lower number of BBN receptors.87 The results indicated that the conjugate 92 had the same impact on the inhibitory effect of [125I]-Try3-BBN to the above cell lines when compared to PTX free peptide 93 (Table 10), indicating neither PTX, nor the PEG units had any effect on the peptide binding. Then conjugate 92 was tested for its stability at physiological pH and in human plasma. In both of the above conditions, the conjugate released the PTX with half-lives 154 and 133 min. respectively. The in vitro cytotoxicity of 92 along with parent PTX and as well as peptide-free drug 95 was investigated against NCI-H1299 cells and results indicated that the conjugate 92 exhibited 50% better cytotoxicity at both 15 nM and 30 nM concentrations respectively, when compared to peptide-free PTX 95.
Table 10.
In vitro cytotoxicity and binding efficacy of 92 and peptide free PTX-PEG-OH (95).
| Compound | % binding of [125I]-Try3-BBN cell lines | IC50 (nM) | ||
|---|---|---|---|---|
| BNR 11 cell line | NCI-H1299 cell line | NCI-H1299 cell line | ||
| 12h | 96h | |||
|
|
||||
| PTX | - | - | 35 | 15 |
| No inhibitor | 70 | 19 | ND | ND |
| BBN [7-13] | 14 | 3.6 | ||
| PTX-PEG- | ND | ND | 72 | 44 |
| OH (95) | ||||
| PTX-PEG- | 5.4 | 4.5 | 14 | 6 |
| BBN (92). | ||||
The IC50 values were determined for 92 at various time points against the same cell line and showed in table 10. The results clearly indicates that the cytotoxicity values obtained for 92 are due to receptor mediated drug localization, since the peptide free taxoid 95 and parent PTX had a weaker IC50 values in the same experiment. Overall the results clearly demonstrate proof of concept that targeting a taxoid drug by a peptide. The in vivo efficacy of this drug conjugate has yet to be reported.
3.6 Gold nanoparticles as taxoid delivery agent
Nanoparticles and nanomaterials continue to a gain great deal of attention because of their significant biomedical applications.88 Particularly, gold nanoparticles initially discovered in nearly a century and half ago by Faraday,89 are now rediscovered for interesting physicochemical properties. In this regard numerous syntheses for the gold nanoparticles, with controlled size and shape in aqueous and non aqueous environments have been developed.90 At the same time the chemistry related to their surface modification has been investigated. Ligands containing either a thiol or amine groups strongly bind to the surface of the gold nanoparticles, presumably though a dative covalent bond formation between either thiol or amine functionality and surface of colloidal gold nanoparticles.
The biocompatibility of the gold nanoparticles was also investigated and studies report that they do not cause any cytotoxicity to the body, instead they exhibit useful properties such as: dose dependent reduction in cellular RNS (Reactive Nitrite Species) and ROS (Reactive Oxygen Species) levels when administered at 10 μM and above in RAW264.7 macrophages cells. They additionally cease the stress induced secretion of proinflammatory cytokine such as TNF-α, IL-1β inside the macrophages cells, suggesting that they could be potentially implicated in various future therapies. 91 In addition, gold nanoparticles posses attractive characteristics such as generally small size (as low as 32 nm diameter), large surface area to accommodate vast number of organic molecules, and more importantly, they have a propensity to evade phagocytic clearance by the reticuloendothelial system (RES) especially when coordinated with polymeric units such as thiolated polyethyleglycol (PEG-SH).90 It is also now known that the gold nanoparticles enter the tumor interstitium via its leaky vasculature (passive cellular uptake) and tend to reside at the interstitial space in tumor tissue, which is called as EPR (enhanced permeability and retention) effect. It was also reported that the gold nanoparticles could enter the tumor cells by endocytosis (active cellular uptake) especially when they are combined to another tumor marker such as TNF-α.92
In this respect Paciotti et al.92 have envisioned gold nanoparticles as a plausible alternative platform for the tumor targeted delivery of cytotoxic agents. TNF-α (tumor necrosis factor-α), a potent cytokine anticancer agent which causes severe systemic toxicity, was subjected to tumor specific delivery by means of gold nanoparticles (cAu) and results revealed that the cAu-TNFα vector was effective in reducing the tumor burden; however, it was rapidly cleared out by RES.92 Subsequent efforts from the same investigators, a vector of TNF and thiolated polyethylene glycol (PT) embedded on to gold nanoparticles resulted in the synthesis of a PT-cAu-TNF vehicle. They demonstrated that this vehicle escapes the RES-mediated rapid clearance and specifically increases the concentrations of TNF-α within the solid tumor.92 An attractive factor of gold nanoparticle mediated delivery, is the relative ease of tracking the vehicle once it is injected in to the body. In each physical state, the gold nanoparticles will have a typical color. For example in completely stabilized monodispersed state the particles will appear a red-burgudy color, while in a precipitated state they appear black.92
Encouraged by the above results, very recently Paciotti et al.93, 94 prepared a PTX-PT-cAu-TNF vehicle (96), which was composed of both TNF-α, thialated PTX prodrug 18 and peglated gold nanoparticles (PT-cAu). The synthesis of the vector 96 is achieved as shown in Scheme 19. When the PTX-PT-cAu-TNF vehicle (96) localized in to the solid tumor, it is expected to release the PTX by the intracellular glutathione mediated cleavage via the transition state 97 in a similar to fashion discussed in Scheme 5.
Scheme 19.

The PTX-PT-cAu-TNF vehicle (96) was extensively investigated for it its biological efficacy. First, the dithiothreitol (DTT) induced liberation of PTX from 96 and subsequently the bioactivity of liberated PTX was determined in vitro, against B16F10 melanoma cells. Second, for determining the extent of tumor localization of 96, 500 μL of it was injected to B16/F10 tumor burdened C57/BL6 mice. Tumor and blood samples were collected after animals were sacrificed and estimated. The results indicate that 96 had delivered at least 7 times more of TNF-α and 10 times more of PTX to tumors, when compared TNF-α and thiolated PTX prodrug 18 alone. Third, to find out the role played by TNF-α in the same tumors they prepared a PT-cAu-PTX vector (without TNF), and administered with equal doses of vectors (one with TNF and without TNF). These results indicated that the TNF-α does play a role in the tumor uptake of the vector (by active targeting).
Given the fact that, DTT is not available inside the tumor and also in the event that the intracellular glutathione may be available to release the PTX from 96 inside the tumor cell; they have envisioned to conduct the prodrug activation experiment with an external disulfide bond activator by an intra-tumor injection on in vivo tumor model. At this juncture, ‘cysteamine’ (2-Aminoethane thiol) is an approved therapeutic, was found to activate disulfide bonds, posed it could be used as an activator for the prodrug 96 in vivo.95 The experimental results indicate that 96 indeed was activated by cysteamine and has released the active drug, in agreement with in vitro DTT–mediated observations. Finally, the PTX-PT-cAu-TNF 96 caused tumor regression in a TNF-α sensitive tumor model.
Overall the results of this study indicate two following points: First nonoparticles mediated drug delivery takes advantage of EPR effect due to gold nanoparticles as well as active cellular uptake (targeting) due to TNF-α. Also lay the foundation to investigate multiple cytotoxic drug delivery by colloidal gold nanoparticles for a combination therapy, since gold nanoparticles possess intrinsically vast surface area and accommodate several organic molecules.
3.7 Hyaluronic acid as taxoid delivery agent
Extracellular and cell-surface-associated polysaccharide ‘hyaluronic acid’ (HA) was once believed as synovial fluid of joints, cartilages and connective tissue, now has emerged as a very important target because HA mediates a vast array of biological functions such as cell proliferation, migration, inflammation, wound healing and tumor development.96 HA (98) is a polymer composed of repeating disaccharide units of glucuronic acid and N-acetyl glucosamine [-β-(1,4)-GlcUA-β (1,3)-GlcNAc-]n is ubiquitously found in extracellular as well as intracellular matrices of many types of tumor cells. Often, high HA levels in cells are useful in a prognosis for malignant progression.97 Additionally, cancer cells overexpress surface receptors such as CD44, HAS2, Hyal-2 and RHAMM, all of which are implicated in tumor invasiveness, cell migration, and proliferation.98 The functions of the above receptors are found to be mediated through their specific interactions with HA (98). All the properties suggest that HA has potential as a targeting agent delivering cytotoxic agents to the tumor site. Since HA is produced by the body, no immunogenic responses are anticipated in using it as drug delivery agent. In this regard, several studies have used HA to directly target the cytotoxic drugs including taxoids and results seem encouraging. 99, 100
Prestwich et al., have synthesized a PTX-HA conjugate with an adipic acid dihydrazide (ADH) linker as shown in scheme 20, starting from a high molecular weight HA-polymer and PTX-2′-hemisuccinate-NHS ester.100 In the synthesis, a low molecular weight HA was first generated from degradation of a high molecular weight HA-polymer by using the enzyme HASe. Then it was covalently linked to ADH (99) using EDCI as coupling reagent to provide HA-ADH (100). Finally HA-ADH was coupled to PTX-hemisuccinate in buffer solution to yield the drug conjugate PTX-HA (101).
Scheme 20.

The conjugate, PTX-HA (101) has been investigated for its in vitro cytotoxicities against four very different, SK-OV-3 (ovarian), HBL-100 (breast), HCT-116 (colon) and NIH-3-T-3 (untransformed fibroblast) cell lines, of which the first 3 cell lines overexpress HA receptors CD44.101 The conjugate 101 showed activity towards only first 3 cell lines, but not towards NIH-3-T-3 cell line. This suggests HA-PTX conjugate has an affinity to HA-receptors. They have tested the activity of conjugate 101 in detail, relative to free PTX as well as premixed PTX and HA-ADH sample, against HCT-116 colon cancer cell line. The results show that conjugate 101 had better activity compared to latter. In a control experiment, HA-ADH did not show any activity at the concentrations 10-times more than conjugate 101 concentration. To further confirm the activity of conjugate 101 was due to HA-receptor mediated cellular uptake, they recently synthesized a fluorescently labeled PTX-HA-FITC drug conjugate (scheme 20) by step wise addition of PTX-NHS ester (90) and FITC to the HA-ADH adduct (100), which enabled them to measure the cellular uptake by using flow cytometry.102 In these experiments, the labeled drug conjugate 102 was found to be strongly absorbed by the tumors cells, SK-OV-3, HBL-100, HCT-116, that express the HA receptors, as indicated by a dose dependent shift in the fluorescent peak in FACS. No fluorescence binding or shift was noticed when 102 was treated with NIH-3-T-3 cells, clearly indicates that PTX-conjugated to HA was selectively binding to and taken by HA receptor positive cells. These results further confirm the selective cytotoxicity results obtained by non-fluorescently labeled PTX-HA conjugate.
Additionally, in a competition experiment, the drug conjugate 101 did not show activity against cancer cells which were pre incubated (2 h) with 100-fold excess high molecular weight HA, where as the same drug conjugate together with 100 fold HA, or PTX alone with 100 fold excess HA showed cytotoxicity. These experiments conclude the receptor mediated binding of PTX-HA conjugate to the tumor cells and leave the notion that high molecular weight HA has slow binding properties compared to low molecular weight HA, because simultaneous addition of HA with free PTX and or PTX-HA did not protect the cells. Also in other experiment a synthesized low molecular weight HA has been rapidly absorbed by the tumor cells.100
In this study they have also carried out how an active drug (PTX) could be released after HA-PTX taken up by the cells. PTX-HA was incubated in 3 different cell culture medias (cell culture media with HASe enzyme, esterases, human plasma). All these media released PTX from the PTX-HA conjugate, among which esterase’s media released the PTX at a faster rate compared to others, suggesting that when HA-mediated cellular uptake has been achieved the active drug could be release hydrolytically from the conjugate inside the cells by enzymatic hydrolysis. The results from this study are encouraging and raise the hope for better chemotherapeutics.
3.8. Fatty acid as taxoid drug targeting agent
As pointed out in the introduction, rapidly proliferating cancer cells require more energy than their normal cells and consequently consume increased amounts of primary metabolites including natural fatty acids as biochemical precursors.103 Most human diets contain a variety of fatty acids and these are categorized as saturated fatty acids, monounsaturated fatty acids and polyunsaturated fatty acids (PUFAs). PUFAs considered as essential fatty acids because human body cannot synthesize them and they are not found adequately in most diets, so they must be provided as supplements. These PUFAs were subdivided into several classes based on location of the first double bond from ω-methyl group located at opposite end to the carboxylic acid (Table 11).104 Essential PUFAs such as linolenic acid (LNA), docosahexaenoic acids (DHA) were previously associated with increased risk of several types of cancers such as breast, colon and prostate. Also reported was that ω-6 PUFAs enhance the tumorigenisis and metastasis in animals by several mechanisms of action, where as ω-3 PUFAs inhibit the growth of initiated cancer cells. 105 All these properties suggest that PUFAs could serve as tumor marking agents and could selectively deliver cytotoxic drugs to the tumor environment. In this regard attempts have already been directed toward targeting cytotoxic drugs such as 4′-dimethyldeoxypodophyllotoxin while other side taxoids drugs are being explored and the results seem very encouraging.106,107,108
Table 11.
Representative examples of fatty acids
| Class | Name | Structure of fatty acid | No. of carbons (double bonds) | Subclass |
|---|---|---|---|---|
| Saturated fatty acids | Lauric acid |
|
12 | |
| Palmitic acid |
|
16 | ||
| Stearic acid |
|
18 | ||
| Mono Unsaturated fatty acids | Oleic acid |
|
18 (1) | ω-9 |
| Linoleic acid (LA) |
|
18 (2) | ω-6 | |
| Poly unsaturated fatty acids | Linolenic acid (LNA) |
|
18 (3) | ω -3 |
| Eicosapentanoic acid |
|
20 (5) | ω -3 | |
| Docosahexaenoic acid (DHA) |
|
22 (6) | ω -3 |
Bradley et al. have synthesized a PTX-DHA conjugate (103) in a single step reaction between PTX and docosahexaenoic acid using DCC as a catalyst and formulated in 10% Cremophor EL-P/10% ethanol/80% normal saline.107
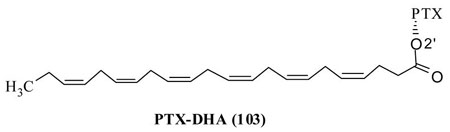
Two very important points are worth mentioning here. The PTX is a hydrophobic molecule and found to be a very good substrate for P-glycoprotein (Pgp) efflux pumps, which is why was inactive against several resistant (MDR) cancer cells. Conjugation of a fatty acid to PTX could makes the drug a much better substrate for efflux pumps hence, the therapeutic effects could be limited. PTX is already found to be insoluble in aqueous solvent and conjugation with a fatty acid could make it much less soluble in aqueous media therefore the conjugate must be formulated in a vehicle which may develop undesirable side effects.
PTX-DHA conjugate 103 was tested against NCI’s 56 human tumor cell lines which are derived from human leukemias, melanomas, lung, colon, ovarian, renal and breast tumors. Results indicated that the conjugate 103 showed at least 10-orders of magnitude lower cytotoxic than PTX. PTX-DHA conjugate (103) showed very interesting biochemical properties including no microtubule assembly activity (in vitro) at 10μM concentration, however, microtubule bundle formation when SKOV3 cells treated with 5μM concentration of 103. Very interestingly, it was found to be a relatively (4 times) weaker substrate for Pgp-efflux pumps, compared to PTX. The PTX-DHA conjugate has also increased the maximum tolerated dose to 4.5, 3.6, 2.9 times compared to PTX for mice, rats, dogs respectively.
In vivo experiments on female mice inoculated with M109 lung tumor, the PTX at optimum dose (OD) (20 mg/kg × 5 days) has shown some tumor growth inhibition but tumors continued to grow as in control experiment after 10 days and also PTX developed severe hind-limb paralysis. However, the PTX-DHA (103) at OD (120 mg/kg) eliminated all the tumors for 60 days on 10/10 mice and did not show any hind-limb paralysis. One point to be noted here the activities of PTX-DHA mentioned here are only at higher doses, since at equimolar concentration to PTX (i. e. 27.4 mg/kg) the conjugate 103 did not eliminate any tumors. In the same study it was investigated that free DHA was also lethal to CD2F1 mice at >60 mg/kg (i.v. for 5 days) and a ethyl ester of DHA was non toxic.
Further in vivo results of PTX-DHA (103) are very promising. When it was tested on H-29 human colon carcinoma implanted into BALB/c-nu/nu mice, it showed superior activity with 40% complete responses and 60% partial responses, where as PTX showed no response. To verify the toxicity of PTX-DHA was due to PUFA mediated targeting, they have conducted several pharmacokinetic studies of tumors, plasma and muscle of M109 tumor bearing mice. They concluded that PUFA actually targeted the drug to tumors. The PTX-DHA (103) has shown several improved biochemical properties and entered in to phase II clinical study, with trade name ‘Taxoprexin ®’ for several types of cancer.109
Recently Ojima et al., have also reported the synthesis of several PUFA-taxoid conjugates.108 Their main purpose was to target the taxoid drugs to resistant (MDR) cancer cells. Though 103 proved to be a weaker substrate for Pgp-efflux pumps, there is a chance that, once PTX has released slowly from the conjugate, it will be captured and eliminated by efflux pumps. For this reason they have selected to use highly active taxoid (38) showing activity against resistant tumor cells (which express Pgp-efflux pumps). They have also investigated other PUFAs including DHA which already produced efficacious results.107 The synthesis of fatty acid conjugates was carried out by coupling between taxoid and a PUFA using DIC and DMAP as catalysts. Scheme 21 shows only conjugates with improved performance. In this study they have also resynthesized PTX-DHA (103) for comparison studies.
Scheme 21.
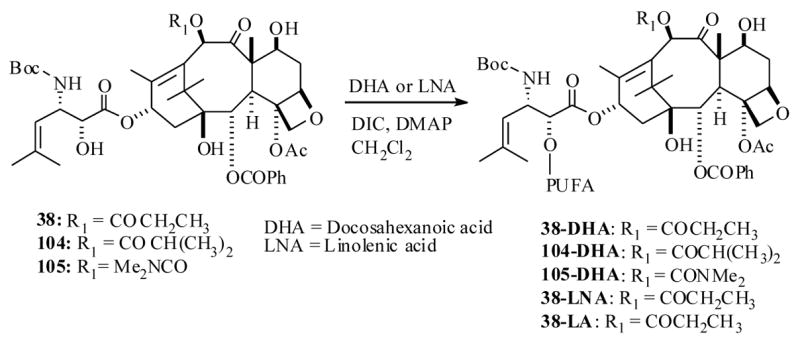
The taxoid-fatty acid conjugates were tested against drug sensitive human ovarian tumor xenograft A121 (Pgp negative) cell line and drug resistant human colon tumor xenograft DLD-1(Pgp positive) in SUID mice. The 38-DHA conjugate showed complete tumor regression of the DLD-1 tumor in 5/5 mice at 80 mg/kg dose administered at 3 day interval starting from day five after tumor implant. It also showed 187 days of growth delay time where as PTX-DHA conjugate (103), as expected, was ineffective against the same tumor xenograft. Likewise 38-DHA showed delayed tumor growth for more than 186 days and complete tumor regression in 5/5 mice of drug sensitive A121 xenograft at 30 mg/kg × 3 doses. Similar results were obtained with 105-DHA, where as PTX-DHA in this case cured only 2/5 mice and tumors appeared after 150 days in 3/5 mice. The relatively low efficacies of PTX-DHA compared to previous study can be attributed to relatively low dose used in the latter study as well as low cytotoxicity of PTX compared to taxoid 38.
Prompted by the results of taxoid-DHA conjugates, they have also investigated the other PUFA conjugates for the activity. Notably is the 38-LNA conjugate tested against drug resistant human colon xenograft DLD-1 and results showed that it caused complete tumor regression in 2/5 mice tested with tumor growth delayed up to more than 109 days. Authors indicated that 38-LNA exhibited overall better activity compared to 38-DHA conjugate, despite some toxicity noted with 38-LNA. Moreover 38-LA conjugate synthesized in this study did not show fruitful results. Clearly the results obtained from the above two studies, that have utilized fatty acids to transport taxoid drugs to tumor cells are very promising and could lead to better and alternative chemotherapeutics compared to parent taxoid drugs.
Owing to time and space constrains, the literature cited in this review article is not exhaustive, so the reader is referred to a review article published with a broader scope, during the preparation of this article110 and also a perspective article appeared during the revised stage of this manuscript.111
4. Conclusions
Significant efforts have already appeared to improve the aqueous solubility and stability of the taxoid drugs in plasma and also circumvent toxic side effects poised by them. Such efforts have resulted in two different sets of protocols as categorized in this article; they are prodrug and drug targeting methods. In many studies both of these methods led to taxoid derivatives with significantly enhanced biochemical properties. As outlined at the end of every section several drug conjugates demonstrated potential success in normal taxoid chemotherapy. Noteworthy are the antibody and fatty acid mediated drug targeting protocols already entered in to the clinical trials not only for the taxoid drug, but also for several other cytotoxic drugs such as doxobubicin.
The designed linkers that couple the drug and tumor marking agent to form a drug conjugate so far belong to a restricted subclass. Very importantly, the self-immolating linker strategy, which was originally designed by Carl et al.112 in the early 80’s, has now been carefully applied in several cases, however the approaches still warrant some new classes of linkers that would increase the stability of the drug in the plasma and liberate the active taxoid drug readily upon reaching the conjugate to the tumor site and or by the tumor microenvironment such as low pH. This was often the limitation encountered by some of the drug conjugates described in this article.
We strongly believe that any anticancer drug or potential anticancer agent that ultimately operates by cellular death mechanism is not fully devoid of side effects due to non-selective toxicity. Therefore prodrug and drug targeting protocols reviewed in this article will play a pivotal role in future generations of anti-cancer agents that either belongs to taxoid class (tubulin binders such as epothilones, discodermolide, eluetherobins and peloruside etc) or other classes. There is little doubt that the successful protocols described in this article, eventually, could be applied to newly discovered drugs.
Fig 2.
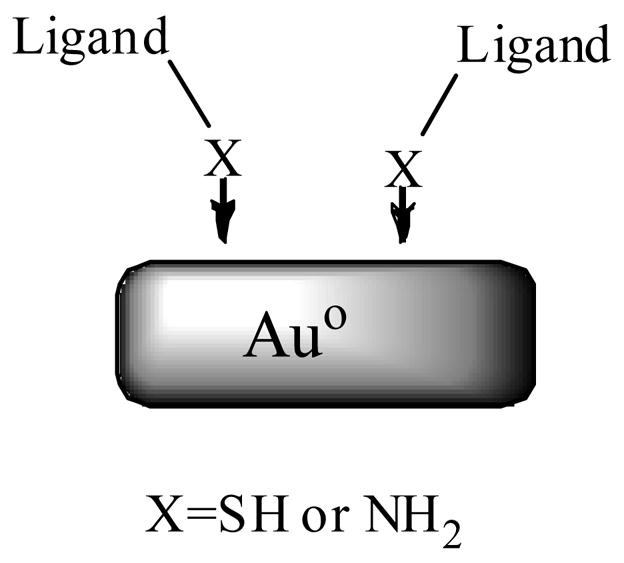
Table 3.
Half lives in DTT buffered solution (pH 7), in vitro cytotoxicity of the PTX and dithio-PTX prodrugs 17a–c against L2987 lung carcinoma cells
| Compound | Half life of prodrug to release PTX in DTT buffer (min) | IC50 (μM) of the compound | IC50 (μM) of the compound + DTT |
|---|---|---|---|
| PTX | 0.2 | ||
| 17a | 12 | 5.7 | 0.2 |
| 17b | 60 | 10.1 | 0.4 |
| 17c | 25 | 130.0 | 2.3 |
| 18 | 4 | 6.2 | 0.2 |
Table 5.
In vitro cytotoxicity of PTX and drug conjugates 31–34.
| Compound | IC50 (nM) | IC50 (nM) | ||
|---|---|---|---|---|
| A2780 | PC-3 | MDA-MB-231 | MCF-7 | |
| ER-β-(−) | ER-β-(+) | ER-α-(−) | ER-α-(+) | |
| PTX | 25 | 77 | 4.5 | 4.9 |
| 31 | 180 | 73 | 22 | 39 |
| 32 | 680 | 40 | 51 | 62 |
| 33 | 8300 | 120 | 2200 | 1600 |
| 34 | 1900 | 68 | 304 | 103 |
Table 6.
In vitro cytotoxicities of taxoid derivatives 43–46 and immunoconjugate 47
| Taxoid | IC50 (nM) | |
|---|---|---|
| A431 | A549 | |
| 38 | 0.09 | 0.1 |
| 43 | 2.0 | ND |
| 46 | 3.0 | 0.9 |
| 45 | 0.5 | 0.8 |
| 44 | >3.0 | ND |
| 47-KS78 | 1.5 | ND |
Acknowledgments
Author sincerely expresses his gratitude to Prof. David G. I. Kingston for encouragement and for reading the proof of this manuscript. Author also thanks Drs. J. P. Snyder, A. Sun, S Kotha, N. Sreenivasachary, P. G. Steel and G. L. and David Krupadanam for their support. Thanks are due to Ms. J. Sorrells for early proof read of this article. Author is grateful to Dr. Dennis C. Liotta, Emory University for financial support.
Abbreviations
- EEDQ
2-Ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline
- DCC
N,N′-Dicyclohexylcarbodiimide
- DMAP
4-N,N-Dimethylaminopyridine
- DIC
DIPC, N,N′-Diisopropylcarbodiimide
- EDCI
N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride
- PEG
poly(ethylene glycol)
- DMF
N,N-Dimethylformamide
- DMSO
Dimethyl sulfoxide
- TBAF
Tetrabutyl ammonium fluoride
- TBSCl
tert-butyldimethylsilyl chloride
- TIPSCl
Triisopropylsilyl chloride
Biography
Dr. Thota Ganesh was born in the village Ananthasagar (India) and obtained his M. Sc. and Ph. D degrees from Osmania University, Hyderabad, India. After his postdoctoral studies on synthetic methodologies and total synthesis of natural products at Indian Institute of Technology-Bombay (India) and then at University of Durham (UK), he joined Virginia Polytechnic Institute and State University (Virginia Tech) as a senior post doctoral fellow then promoted to research scientist. As a research scientist at VT, he carried out the research on anti-cancer drug Taxol® and potential anti-cancer agents epothilones. In the program he designed and synthesized several Taxol analogs that kill cancer cells more effectively than Taxol, and his most recent investigations have focused on the synthesis of epothilone analogs and study their interaction with tubulin receptor and the total synthesis of natural products isolated from marine organisms. Dr. Ganesh is now working as senior research scientist in medicinal chemistry core at Emory University. His research interests; drug delivery to cancer cells, develop small molecules inhibitors for novel protein targets, rational drug design.

Footnotes
This article is dedicated to Dr. David G. I. Kingston for his outstanding contributions in natural products and paclitaxel research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowinsky EK. Ann Rev Med. 1997;48:353–374. doi: 10.1146/annurev.med.48.1.353. [DOI] [PubMed] [Google Scholar]
- 2.Crown J, O’Leary M. The Lancet. 2000;355:1176–1178. doi: 10.1016/S0140-6736(00)02074-2. [DOI] [PubMed] [Google Scholar]
- 3.For leading references see: Wang TH, Wang HS, Soong YK. Cancer. 2000;88:2619–2628. doi: 10.1002/1097-0142(20000601)88:11<2619::aid-cncr26>3.0.co;2-j.Blagosklonny MV, Fojo T. Int J Cancer. 1999;83:151–156. doi: 10.1002/(sici)1097-0215(19991008)83:2<151::aid-ijc1>3.0.co;2-5.Jordan MA, Toso RJ, Thrower D, Wilson L. Proc Natl Acad Sci USA. 1993;90:9552–9556. doi: 10.1073/pnas.90.20.9552.Soger PK, Dobles M, Tournebize R, Hyman AA. Curr Opin Cell Biol. 1997;9:807–814. doi: 10.1016/s0955-0674(97)80081-6.
- 4.For seminal reports please see: Schiff PB, Fant J, Horwitz SB. Nature. 1979;277:665–667. doi: 10.1038/277665a0.Horwitz SB. Trends Pharmacol Sci. 1992;13:134–136. doi: 10.1016/0165-6147(92)90048-b.
- 5.Brown DT. ch. 11. In: Itokawa H, Lee K-H, editors. Taxus: The Genus Taxus. Taylor and Francis; London: 2003. pp. 387–435. [Google Scholar]
- 6.Thayer AM. Chem & Eng News. 2000;78(45):20. [Google Scholar]
- 7.(a) Rigas J. The Oncologist. 2004;9:16–22. doi: 10.1634/theoncologist.9-suppl_2-16. [DOI] [PubMed] [Google Scholar]; (b) Nabholtz J-M, Tonkin K, Smylie M, Au H-J, Lindsay M-A, Mackey J. Expert Opin Pharmacother. 2000;1:187–206. doi: 10.1517/14656566.1.2.187. [DOI] [PubMed] [Google Scholar]; (c) Miller KD, Sledge GW., Jr Cancer Invest. 1999;17:121–136. [PubMed] [Google Scholar]
- 8.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 9.ter Haar E, Kowalski RJ, Hamel E, Lin CM, Longley RE, Gunasekera SP, Rosenkranz HS, Day BW. Biochemistry. 1996;35:243–250. doi: 10.1021/bi9515127. [DOI] [PubMed] [Google Scholar]
- 10.Long BH, Carboni JM, Wasserman AJ, Cornell LA, Casazza AM, Jensen PR, Lindel T, Fenical W, Fairchild CR. Cancer Res. 1998;58:1111–1115. [PubMed] [Google Scholar]
- 11.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Cancer Res. 1999;59:653–660. [PubMed] [Google Scholar]
- 12.Hood KA, West LM, Rouwé B, Northcote PT, Berridge MV, Wakefield SJ, Miller JH. Cancer Res. 2002;62:3356–3360. [PubMed] [Google Scholar]
- 13.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JTM, Fojo T, Poruchynsky MS. J Biol Chem. 1997;272:17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]; (b) Chou TC, Zhang XG, Harris CR, Kuduk SD, Balog A, Savin KA, Bertino JR, Danishefsky SJ. Proc Natl Acad Sci USA. 1998;95:15798–15802. doi: 10.1073/pnas.95.26.15798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma A, Mayhew E, Bolcsak L, Cavanaugh C, Harmon P, Janoff A, Bernacki RJ. Int J Cancer. 1997;71:103–107. doi: 10.1002/(sici)1097-0215(19970328)71:1<103::aid-ijc17>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 15.Szebeni J, Muggia FM, Alving CR. J Natl Cancer Inst. 1998;90:300–306. doi: 10.1093/jnci/90.4.300. [DOI] [PubMed] [Google Scholar]
- 16.Crown J, O’Leary M, Ooi WS. The Oncologist. 2004;9:24–32. doi: 10.1634/theoncologist.9-suppl_2-24. [DOI] [PubMed] [Google Scholar]
- 17.(a) Nicolau KC, Riemer C, Kerr MA, Rideout D, Wrasidlo W. Nature. 1993;364:464–466. doi: 10.1038/364464a0. [DOI] [PubMed] [Google Scholar]; (b) Nicolaou KC, Guy RK, Pitsinos EN, Wrasidlo W. Angew Chem, Int Ed Engl. 1994;33:1583–1587. [Google Scholar]; (c) Seligson AL, Terry RC, Bressi JC, Douglass JG, III, Sovak M. Anti-cancer Drugs. 2001;12:305–313. doi: 10.1097/00001813-200104000-00002. [DOI] [PubMed] [Google Scholar]
- 18.(a) Khmelnitsky YL, Budde C, Arnold MJ, Usyatinsky A, Clark DS, Dordick JS. J Am Chem Soc. 1997;119:11554–11555. [Google Scholar]; (b) Hayashi Y, Skwarczynski M, Hamada Y, Sohma Y, Kimura Y, Kiso Y. J Med Chem. 2003;46:3782–3784. doi: 10.1021/jm034112n. [DOI] [PubMed] [Google Scholar]; (c) Vyas DM, Wong H, Crosswell AR, Casazza AM, Knipe JO, Mamber SW, Doyle TW. Bioorg Med Chem Lett. 1993;3:1357–1360. [Google Scholar]
- 19.Michaud LB, Valero V, Hortobagyi G. Drug Safety. 2000;23:401–428. doi: 10.2165/00002018-200023050-00005. [DOI] [PubMed] [Google Scholar]
- 20.Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Nature Medicine. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]
- 21.(a) Greenwald RB, Gilbert CW, Pendri A, Conover CD, Xia J, Martinez A. J Med Chem. 1996;39:424–431. doi: 10.1021/jm950475e. [DOI] [PubMed] [Google Scholar]; (b) Zhao Z, Kingston DGI, Crosswell AR. J Nat Prod. 1991;54:1607–1611. [Google Scholar]; (c) Deutsch HM, Glinski JA, Hernandez M, Haugwitz RD, Narayanan VL, Suffness M, Zalkow LH. J Med Chem. 1989;32:788–792. doi: 10.1021/jm00124a011. [DOI] [PubMed] [Google Scholar]; (d) Mathew AE, Mejillano MR, Nath JP, Himes RH, Stella VJ. J Med Chem. 1992;35:145–151. doi: 10.1021/jm00079a019. and references cited in 17 and 18. [DOI] [PubMed] [Google Scholar]
- 22.Kingston DGI, Jagtap PG, Yuan H, Samala L. Prog Chem Org Nat Prod. 2002;84:53–225. doi: 10.1007/978-3-7091-6160-9_2. [DOI] [PubMed] [Google Scholar]
- 23.Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E. Proc Natl Acad Sci USA. 2001;98:5312–5316. doi: 10.1073/pnas.051309398. and references cited there in. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Please see references 26, 50, 62, 81 cited in this article.
- 25.Hayashi Y, Skwarczynski M, Hamada Y, Sohma Y, Kimura T, Kiso Y. J Med Chem. 2003;46:3782–3784. doi: 10.1021/jm034112n. [DOI] [PubMed] [Google Scholar]
- 26.Damen EWP, Weigerinck PHG, Braamer L, Sperling D, de Vos D, Scheeren HW. Bioorg Med Chem. 2000;8:427–432. doi: 10.1016/s0968-0896(99)00301-6. [DOI] [PubMed] [Google Scholar]
- 27.Jones MC, Leroux JC. Eur J Pharma Biopharma. 1999;48:101–111. doi: 10.1016/s0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 28.(a) Brigger I, Dubernet C, Couvreur P. Adv Drug Deliv Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]; (b) Visaria RK, Griffin RJ, Williams BW, Ebbini ES, Paciotti GF, Song CW, Bischof JC. Mol Can Ther. 2006;5:1014–1020. doi: 10.1158/1535-7163.MCT-05-0381. [DOI] [PubMed] [Google Scholar]
- 29.Esfand R, Tomalia DA. Drug Discovery Today. 2001;6:427–436. doi: 10.1016/s1359-6446(01)01757-3. [DOI] [PubMed] [Google Scholar]
- 30.Koshkina NV, Waldrep JC, Roberts LE, Golunski E, Melton S, Knight V. Clin Cancer Res. 2001;7:3258–3262. [PubMed] [Google Scholar]
- 31.Medina OP, Zhu Y, Kairemo K. Curr Pharma Design. 2004;10:2981–2989. doi: 10.2174/1381612043383467. [DOI] [PubMed] [Google Scholar]
- 32.Klibanove A, Maruyama K, Torchillin VP, Huang L. FEBS Lett. 1990;268:235–237. doi: 10.1016/0014-5793(90)81016-h. [DOI] [PubMed] [Google Scholar]
- 33.(a) Oku N, Namba Y, Okada S. Biochim Biophys Acta. 1992;1126:255–260. doi: 10.1016/0005-2760(92)90238-q. [DOI] [PubMed] [Google Scholar]; (b) Allen TM, Hansen C, Martin F, Redeman C, Yau-Yong A. Biochim Biophys Acta. 1991;1066:29–36. doi: 10.1016/0005-2736(91)90246-5. [DOI] [PubMed] [Google Scholar]
- 34.Koshkina NV, Waldrep JC, Knight V. Curr Cancer Drug Targets. 2003;3:251–264. doi: 10.2174/1568009033481930. [DOI] [PubMed] [Google Scholar]
- 35.Knight V, Koshkina NV, Waldrep JC, Giovanella BC, Gilbert BE. Cancer Chemother Pharmacol. 1999;44:177–186. doi: 10.1007/s002800050965. [DOI] [PubMed] [Google Scholar]
- 36.Zakharian TY, Seryshev A, Sitharaman B, Gilbert BE, Knight V, Wilson LJ. J Am Chem Soc. 2005;127:12508–12509. doi: 10.1021/ja0546525. [DOI] [PubMed] [Google Scholar]
- 37.Wouters BG, Weppler SA, Koritzinisky M, Landuyt W, Nuyts S, Thyes J, Chiu RK, Lambin P. Eur J Cancer. 2002;38:240–257. doi: 10.1016/s0959-8049(01)00361-6. and references cited their in. [DOI] [PubMed] [Google Scholar]
- 38.Damen EWP, Nevalainen TJ, van den Bergh TJM, de Groot FMH, Scheeren HW. Bioorg Med Chem. 2002;10:71–77. doi: 10.1016/s0968-0896(01)00235-8. [DOI] [PubMed] [Google Scholar]
- 39.Vrudhula VM, MacMaster JF, Li Z, Kerr DE, Senter PD. Bioorg Med Chem Lett. 2002;12:3591–3594. doi: 10.1016/s0960-894x(02)00784-9. [DOI] [PubMed] [Google Scholar]
- 40.Volpert OV, Ward WF, Lingen MW, Chesler L, Solt DB, Johnson MD, Molteni A, Polverini PJ, Bouck NP. J Clin Invest. 1996;98:671–679. doi: 10.1172/JCI118838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.(a) Yamashita JC, Ogawa M. Int J Oncol. 1997;10:807–813. doi: 10.3892/ijo.10.4.807. [DOI] [PubMed] [Google Scholar]; (b) Reuning U, Magdolen V, Wilhelm O, Fischer K, Lutz V, Graeff H, Schmitt M. Int J Oncol. 1998;13:893–906. doi: 10.3892/ijo.13.5.893. [DOI] [PubMed] [Google Scholar]
- 42.(a) Eisenbrand G, Lauck-Birkel S, Tang WC. Synthesis. 1996:1246–1258. [Google Scholar]; (b) Carl PL, Chakravarty PK, Katzenellenbogen JA, Weber MJ. Proc Natl Acad Sci USA. 1980;77:2224–2228. doi: 10.1073/pnas.77.4.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) de Groot FMH, de Bart ACW, Verheijen JH, Scheeren HW. J Med Chem. 1999;42:5277–5283. doi: 10.1021/jm9910472. [DOI] [PubMed] [Google Scholar]
- 43.de Groot FMH, van Berkom LWA, Scheeren HW. J Med Chem. 2000;43:3093–3102. doi: 10.1021/jm0009078. [DOI] [PubMed] [Google Scholar]
- 44.Ueda Y, Wong H, Matiskella JD, Mikkilinani AB, Farina V, Fairchild C, Rose WC, Member SW, Long BH, kerns EH, Casazza AM, Vyas DM. Bioorg Med Chem Lett. 1994;4:1861–1864. [Google Scholar]
- 45.Osborne CK. Brest Cancer Res Treat. 1998:227–238. doi: 10.1023/a:1006132427948. [DOI] [PubMed] [Google Scholar]
- 46.Park WC, Jordan C. Trends Mol Med. 2002;8:82–88. doi: 10.1016/s1471-4914(02)02282-7. [DOI] [PubMed] [Google Scholar]
- 47.Bianchi E, Cohen RL, Thot AT, Todd RF, III, Mizukami IF, Lawrence DA, Ljung BM, Shuman MA, Smith HS. Cancer Res. 1994;54:861–864. [PubMed] [Google Scholar]
- 48.(a) Purohit A, Wyatt J, Hynd G, Wright J, Shafey AE, Swamy N, Ray R, Jones JB. Tetrahedron Lett. 2001;42:8579–8582. [Google Scholar]; (b) Jones GB, Huber RS, Mathews JE, Li A. Tetrahedron Lett. 1996;37:3643–3646. [Google Scholar]; (c) Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Bioorg Med Chem Lett. 1999;9:1233–1238. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]; (d) Swamy N, James DA, Mohr SC, Hanson RN, Ray R. Bioorg Med Chem. 2002;10:3237–3243. doi: 10.1016/s0968-0896(02)00242-0. [DOI] [PubMed] [Google Scholar]
- 49.Rink SM, Yarema KJ, Solomaon MS, Paige LA, Rebek T, Essigman JM, Croy RG. Proc Natl Acad Sci USA. 1996;93:15063–15068. doi: 10.1073/pnas.93.26.15063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu C, Strobl JS, Bane S, Schilling J, McCracken M, Chatterjee SK, Rahim-Bata R, Kingston DGI. J Nat Prod. 20004;67:152–159. doi: 10.1021/np030296x. [DOI] [PubMed] [Google Scholar]
- 51.(a) Kuduk SD, Harris CR, Zheng FF, Sepp-Lorenzino L, Ouerfelli Q, Rosen N, Danishefsky SJ. Bioorg Med Chem Lett. 2000;10:1303–1306. doi: 10.1016/s0960-894x(00)00208-0. [DOI] [PubMed] [Google Scholar]; (b) James DA, Swamy N, Paz N, Hanson RN, Ray R. Bioorg Med Chem Lett. 1999;9:2379–2384. doi: 10.1016/s0960-894x(99)00390-x. [DOI] [PubMed] [Google Scholar]
- 52.Chari RVJ. Adv Drug Deliv Rev. 1998;31:89–104. doi: 10.1016/s0169-409x(97)00095-1. [DOI] [PubMed] [Google Scholar]
- 53.Robert D. Cancer Invest. 2001;19:833–841. doi: 10.1081/cnv-100107745. [DOI] [PubMed] [Google Scholar]
- 54.Sunada H, Magun BE, Mendelsohn J, MacLeod CL. Proc Natl Acd Sci USA. 1986;83:3825–3829. doi: 10.1073/pnas.83.11.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bender H, Takahashi H, Adachi K, Belser P, Liang SH, Prewett M, Schrappe M, Sutter A, Rodeck U, Herlyn D. Can Res. 1992;52:121–126. [PubMed] [Google Scholar]
- 56.Liu C, Tadayoni BM, Bourret LA, Mattocks KM, Derr SM, Widdison WC, Kedersha NL, Ariniello PD, Goldmacher VS, Lambert JM, Blättler WA, Chari RVJ. Proc Natl Acd Sci USA. 1996;93:8618–8623. doi: 10.1073/pnas.93.16.8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chari RV, Jackel KA, Bourret LA, Derr SM, Tadayoni BM, Mattocks KM, Shah SA, Liu C, Blattler WA, Goldmacher VS. Cancer Res. 1995;55:4079–4084. [PubMed] [Google Scholar]
- 58.Saragvi HU, Gehring K. Trends Pharmacol Sci. 2000;21:93–98. doi: 10.1016/s0165-6147(99)01444-3. [DOI] [PubMed] [Google Scholar]
- 59.LeSauter L, Maliartchouk S, LeJuene H, Saragovi HU. J Nuerosci. 1996;16:1308–1316. doi: 10.1523/JNEUROSCI.16-04-01308.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chandler CE, Parsons LM, Hosang M, Shooter EM. J Biol Chem. 1984;259:6882–6889. [PubMed] [Google Scholar]
- 61.Guillemard V, Saragovi HU. Cancer Res. 2001;61:694–699. [PubMed] [Google Scholar]
- 62.Ojima I, Geng X, Wu X, Qu C, Borella CP, Xie H, Wilhelm SD, Leece BA, Bartle LM, Goldmacher VS, Chari RVJ. J Med Chem. 2002;45:5620–5623. doi: 10.1021/jm025540g. [DOI] [PubMed] [Google Scholar]
- 63.Ojima I, Kuduk SD, Pera P, Veith JM, Bernacki RJ. J Med Chem. 1997;40:279–285. doi: 10.1021/jm9606711. [DOI] [PubMed] [Google Scholar]
- 64.(a) Arteaga CL. The Oncologist. 2002;7(suppl 4):31–39. doi: 10.1634/theoncologist.7-suppl_4-31. and references cited there in. [DOI] [PubMed] [Google Scholar]; (b) Salomon DS, Brandt R, Ciardiello F, Normanno N. Crit Rev Oncol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 65.Bagshawe K. Brit J Cancer. 1987;56:531–532. doi: 10.1038/bjc.1987.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.(a) Bagshawe K. Drug Dev Res. 1995;34:220–230. [Google Scholar]; (b) Melton R, Knox R, Conner T. Drugs of the Future. 1996;21:167–181. [Google Scholar]
- 67.(a) Melton RG, Scherwood RF. J Natl Can Inst. 1996;88:153–165. doi: 10.1093/jnci/88.3-4.153. and references cited there in. [DOI] [PubMed] [Google Scholar]; (b) Jung M. Mini Rev Med Chem. 2001;1:399–407. doi: 10.2174/1389557013406747. and references cited there in. [DOI] [PubMed] [Google Scholar]; (c) Dubowchik GM, Mosure K, Knipe JO, Firestone RA. Bioorg Med Chem Lett. 1998;8:3347–3352. doi: 10.1016/s0960-894x(98)00610-6. [DOI] [PubMed] [Google Scholar]; (d) Dubowchik GM, Firestone RA. Bioorg Med Chem Lett. 1998;8:3341–3356. doi: 10.1016/s0960-894x(98)00609-x. [DOI] [PubMed] [Google Scholar]; (e) Leenders RGG, Damen EWP, Bijsterveld EJA, Scheeren HW, Houba PHJ, van der Meulen-Muileman IH, Boven E, Haisma HJ. Bioorg Med Chem. 1999;7:1597–1610. doi: 10.1016/s0968-0896(99)00095-4. [DOI] [PubMed] [Google Scholar]; (f) Haenseler E, Esswein A, Vitols KS, Montejano Y, Mueller BM, Reisfeld RA, Huennekens FM. Biochemistry. 1992;31:891–897. doi: 10.1021/bi00118a035. [DOI] [PubMed] [Google Scholar]
- 68.Rodrigues ML, Presta LG, Kotts CE, Wirth C, Mordenti J, Osaka G, Wong WL, Nuijens A, Blackburn B, Carter P. Cancer Res. 1995;55:63–70. [PubMed] [Google Scholar]
- 69.Rodrigues ML, Carter P, Wirth C, Mullins S, Lee A, Blackburn BK. Chem Biol. 1995;2:223–227. doi: 10.1016/1074-5521(95)90272-4. [DOI] [PubMed] [Google Scholar]
- 70.de Bont DBA, Leenders RGG, Haisma HJ, van der Meulen-Muileman I, Scheeren HW. Bioorg Med Chem. 1997;5:405–414. doi: 10.1016/s0968-0896(96)00249-0. [DOI] [PubMed] [Google Scholar]
- 71.(a) Schmidt F, Ungureanu I, Duval R, Pompon A, Monneret C. Eur J Org Chem. 2001:2129–2134. [Google Scholar]; (b) Bouvier E, Thirot S, Schmidt F, Monneret C. Org & Biomol Chem. 2003;1:3343–3352. doi: 10.1039/b306236h. [DOI] [PubMed] [Google Scholar]; (c) Bouvier E, Thirot S, Schmidt F, Monneret C. Bioorg Med Chem. 2004;12:969–977. doi: 10.1016/j.bmc.2003.12.013. [DOI] [PubMed] [Google Scholar]
- 72.Islam R, Tomotsu S, Shah G, Grubb S, Jain W, Sly J. J Biol Chem. 1999;274:23451–23455. doi: 10.1074/jbc.274.33.23451. [DOI] [PubMed] [Google Scholar]
- 73.Saari WS, Schwering JE, Lyle PA, Smith SJ, Engelhardt EL. J Med Chem. 1990;33:97–101. doi: 10.1021/jm00163a016. [DOI] [PubMed] [Google Scholar]
- 74.(a) Toffoli G, Cernigoi C, russo A, gallo A, Bagnoli M, Boiocchi M. Int J cancer. 1997;74:193–198. doi: 10.1002/(sici)1097-0215(19970422)74:2<193::aid-ijc10>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]; (b) Gibbs DD, Theti DS, Wood N, Green M, Raynaud F, Valenti M, Forster MD, Mitchell F, Bavetsias V, Henderson E, Jackman AL. Cancer Res. 2005;65:11721–11728. doi: 10.1158/0008-5472.CAN-05-2034. [DOI] [PubMed] [Google Scholar]
- 75.Antony AC. Blood. 1992;79:2807–2820. [PubMed] [Google Scholar]
- 76.Elnakat H, Ratnam M. Adv Drug Deliv Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 77.McHugh M, Cheng YC. J Biol Chem. 1979;254:11312–113-18. [PubMed] [Google Scholar]
- 78.(a) Leamon CP, Reddy JA. Adv Drug Deliv Rev. 2004;56:1067–1084. doi: 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]; (b) Lu Y, Sega E, Leamon C, Low PS. Adv Drug Deliv Rev. 2004;56:1161–1176. doi: 10.1016/j.addr.2004.01.009. [DOI] [PubMed] [Google Scholar]; (c) Roy EJ, Gawlick U, Orr BA, Kranz DM. Adv Drug Deliv Rev. 2004;56:1219–1231. doi: 10.1016/j.addr.2004.01.006. [DOI] [PubMed] [Google Scholar]; (d) Theti DS, Jackman AL. Clin Can Res. 2004;10:1080–1089. doi: 10.1158/1078-0432.ccr-03-0157. [DOI] [PubMed] [Google Scholar]
- 79.(a) Kamen BA, Capdevila A. Proc Natl Acad Sci USA. 1986;83:5983–5987. doi: 10.1073/pnas.83.16.5983. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Leamon CP, Low PS. Proc Natl Acad Sci USA. 1991;88:5572–5576. doi: 10.1073/pnas.88.13.5572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.(a) Steinburg G, Borch RF. J Med Chem. 2001;44:69–73. doi: 10.1021/jm000306g. [DOI] [PubMed] [Google Scholar]; (b) Aronov O, Horowitz AT, Gabizon A, Gibson D. Bioconjugate Chem. 2003;14:563–574. doi: 10.1021/bc025642l. [DOI] [PubMed] [Google Scholar]
- 81.Lee JW, Lu JY, Low PS, Fuchs PL. Bioorg Med Chem. 2002;10:2397–2414. doi: 10.1016/s0968-0896(02)00019-6. [DOI] [PubMed] [Google Scholar]
- 82.Majoros IJ, Myc A, Thomas T, Mehta CB, Becker JR., Jr Biomacromolecules. 2006;7:572–579. doi: 10.1021/bm0506142. [DOI] [PubMed] [Google Scholar]
- 83.Arap W, Pasqualini R, Ruoslahti E. Science. 1998;279:377–380. doi: 10.1126/science.279.5349.377. and references cited there in. [DOI] [PubMed] [Google Scholar]
- 84.(a) Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, Krajewski S, Lombardo CR, Rao R, Ruoslahti E, Bredesen DE, Pasqualini R. Nature Medicine. 1999;5:1032–1038. doi: 10.1038/12469. [DOI] [PubMed] [Google Scholar]; (b) Ruoslahti E. Nature Rev. 2002;2:83–90. doi: 10.1038/nrc724. [DOI] [PubMed] [Google Scholar]
- 85.(a) Zhou J, Chen J, Zhong R, Mokotoff M, Shultz LD, Ball ED. Clin Can Res. 2006;12:2224–2231. doi: 10.1158/1078-0432.CCR-05-1524. [DOI] [PubMed] [Google Scholar]; (b) Zhou J, Chen J, Mokotoff M, Zhong R, Shultz LD, Ball ED. Clin Can Res. 2003;9:4953–4960. [PubMed] [Google Scholar]
- 86.Safavy A, Raisch KP, Khazaeli MB, Buchsbaum DJ, Bonner JA. J Med Chem. 1999;42:4919–4924. doi: 10.1021/jm990355x. [DOI] [PubMed] [Google Scholar]
- 87.(a) Moody TW, Venugopal R, Hu V, Gozes Y, McDermed J, Leban JJ. Peptides. 1996;17:1337–1343. doi: 10.1016/s0196-9781(96)00195-7. [DOI] [PubMed] [Google Scholar]; (b) Moody TW, Lee M, Kris RM, Bellot G, Bepler G, Oie H, Gazdar AF. J Cell Biochem. 1990;43:139–147. doi: 10.1002/jcb.240430205. [DOI] [PubMed] [Google Scholar]; (c) Moody TW, Zia F, Venugopal R, Fagarasan M, Oie H, Hu V. J Cell Biochem (Suppl) 1996;24:247–256. doi: 10.1002/jcb.240630520. [DOI] [PubMed] [Google Scholar]
- 88.Brigger I, Duberent C, Couvreur P. Adv Drug Deliv Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]; (b) Gao Z, Lukyanov AN, Singhal A, Torchilin VP. Nano Lett. 2002;2:979–982. [Google Scholar]
- 89.Faraday M. Phylos Trans, R Soc London. 1857;14:145–181. [Google Scholar]
- 90.(a) Hengliein A. Langmuir. 1999;15:6738. [Google Scholar]; (b) Selakannan PR, Mandal S, Pasricha R, Adyanthaya SD, Sastry M. Chem Commun. 2002;13:1334–1335. [Google Scholar]
- 91.Shukla R, Bansal V, Chaudhary M, Basu A, Bhonde RR, Sastry M. Langmuir. 2005;21:10644–10654. doi: 10.1021/la0513712. [DOI] [PubMed] [Google Scholar]
- 92.Paciotti GF, Myer L, Weinreich D, Goia D, Pavel N, McLaughlin RE, Tamarkin L. Drug Delivery. 2004;11:169–183. doi: 10.1080/10717540490433895. [DOI] [PubMed] [Google Scholar]
- 93.Paciotti GF, Myer L, Kingston DGI, Ganesh T, Tamarkin L. NSTI-Nanotech. 2005;1:7–10. [Google Scholar]
- 94.Paciotti GF, Kingston DGI, Tamarkin L. Drug Dev Res. 2006;67:47–54. [Google Scholar]
- 95.(a) Kleta R, Gahl WA. Expet Opin Pharmacother. 2004;5:2255–2262. doi: 10.1517/14656566.5.11.2255. [DOI] [PubMed] [Google Scholar]; (b) Khomenko T, Deng X, Jadus MR, Szabo S. Biochem Biophys Res Commun. 2003;309:910–916. doi: 10.1016/j.bbrc.2003.08.092. [DOI] [PubMed] [Google Scholar]
- 96.Toole BP. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. and referenced cited their in. [DOI] [PubMed] [Google Scholar]
- 97.Boregowda RK, Appaiah H, Siddaiah M, Kumarswamy SB, Sunila S, Thimmaiah KN, Mortha K, Toole B, dBanerjee S. J Carcinogenesis. 2006;5:2. doi: 10.1186/1477-3163-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.(a) Udabage L, Browlee GR, Nilsson SK, Brown TJ. Exp Cell Res. 2005;310:205–217. doi: 10.1016/j.yexcr.2005.07.026. [DOI] [PubMed] [Google Scholar]; (b) Serra M, Rabanal RM, Miquel L, Domenzain C, Bassols A. J Com Path. 2004;130:171–180. doi: 10.1016/j.jcpa.2003.10.006. [DOI] [PubMed] [Google Scholar]; (c) Wang C, Thor AD, Moore DH, II, Zhao Y, Kerschmann R, Stern R, Watson PH, Turley EA. Clin Can Res. 1998;4:567–576. [PubMed] [Google Scholar]
- 99.Eliaz RE, Szoka FC., Jr Can Res. 2001;61:2592–2601. [PubMed] [Google Scholar]
- 100.Lio Y, Prestiwitch GD. Bioconjugate Chem. 1999;10:755–763. doi: 10.1021/bc9900338. [DOI] [PubMed] [Google Scholar]
- 101.(a) Iida N, Bourguignon LYWJ. Cell Physiol. 1997;171:152–160. doi: 10.1002/(SICI)1097-4652(199705)171:2<152::AID-JCP5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]; (b) Bourguignon LYW, Zhu H, Chu A, Iida N, Zhang L, Hung MC. J Biol Chem. 1997;272:27913–27918. doi: 10.1074/jbc.272.44.27913. [DOI] [PubMed] [Google Scholar]
- 102.Lio Y, Ziebell R, Prestiwitch GD. Biomacromalecules. 2000;1:208–218. [Google Scholar]
- 103.Sauer LA, Nagel WO, Dauchy RT, Miceli LA, Ausin JE. Can Res. 1986;46:3469–3475. [PubMed] [Google Scholar]; (b) Sauer LA, Stayman JW, III, Dauchy RT. Can Res. 1982;42:4090–4097. [PubMed] [Google Scholar]
- 104.Bartch H, Nair J, Owen RW. Carcinogenesis. 1999;20:2209–2218. doi: 10.1093/carcin/20.12.2209. [DOI] [PubMed] [Google Scholar]
- 105.(a) Boudreau MD, Sohn KH, Rhee SH, Lee SW, Hunt JD, Hwang DH. Can Res. 2001;61:1386–1391. [PubMed] [Google Scholar]; (b) Cave WT., Jr FESEB J. 1991;5:2160–2166. doi: 10.1096/fasebj.5.8.1673664. [DOI] [PubMed] [Google Scholar]; (c) Takahashi M, Przetakiewicz M, Ong A, Borek C, Lowenstein JM. Can Res. 1992;52:154–162. [PubMed] [Google Scholar]
- 106.You YJ, Kim Y, Nam NH, Ahn BZ. Bioorg Med Chem Lett. 2003;13:2629–2632. doi: 10.1016/s0960-894x(03)00558-4. [DOI] [PubMed] [Google Scholar]
- 107.Bradley MO, Webb NL, Anthony FH, Devanesan P, Witman PA, Hemamalini S, Chander MC, Baker SD, He L, Horwitz SB, Swindell CS. Clin Cancer Res. 2001;7:3229–3238. [PubMed] [Google Scholar]
- 108.Kuznetsova L, Chen J, Sun L, Wu X, Pepe A, Veith JM, Pera P, Bernaki RJ, Ojima I. Bioorg Med Chem lett. 2006;16:974–977. doi: 10.1016/j.bmcl.2005.10.089. [DOI] [PubMed] [Google Scholar]
- 109.www.clinicaltrials.gov
- 110.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Bioorg Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 111.Skawarczynski M, Hayashi Y, Kiso Y. J Med Chem. 2006;49:7253–7269. doi: 10.1021/jm0602155. [DOI] [PubMed] [Google Scholar]
- 112.Carl PL, Chakravarthy PK, Katzenellenbogen JA. J Med Chem. 1981;24:479–480. doi: 10.1021/jm00137a001. [DOI] [PubMed] [Google Scholar]