

Abstract
Liver-enriched nuclear receptors (NRs) collectively function as metabolic and toxicological ‘sensors’ that mediate liver-specific gene-activation in mammals. NR-mediated gene-environment interaction regulates important steps in the hepatic uptake, metabolism and excretion of glucose, fatty acids, lipoproteins, cholesterol, bile acids, and xenobiotics. Hence, liver-enriched NRs play pivotal roles in the overall control of energy homeostasis in mammals. While it is well-recognized that ligand-binding is the primary mechanism behind activation of NRs, recent research is revealing that multiple signal transduction pathways modulate NR-function in liver. The interface between specific signal transduction pathways and NRs helps to determine their overall responsiveness to various environmental and physiological stimuli. In general, phosphorylation of hepatic NRs regulates multiple biological parameters including their transactivation capacity, DNA-binding, sub-cellular location, capacity to interact with protein cofactors, and protein stability. Certain pathological conditions including inflammation, morbid obesity, hyperlipidemia, atherosclerosis, insulin resistance, and type-2 diabetes are known to modulate selected signal transduction pathways in liver. This review will focus upon recent insights regarding the molecular mechanisms that comprise the interface between disease-mediated activation of hepatic signal transduction pathways and liver-enriched NRs. This review will also highlight the exciting opportunities presented by this new knowledge to develop novel molecular and pharmaceutical strategies for combating these increasingly prevalent human diseases.
Introduction
General NR Structure and Function
NRs are one of the largest groups of transcription factors with 48 members in the human genome that regulate diverse biological processes including metabolism, homeostasis, development and reproduction 1. The activity of many NRs is controlled by the binding of small lipophilic molecules such as hormones, fatty acids, bile acids and oxysterols, and xenobiotics.
All members of the NR superfamily share several conserved structural domains that are essential for receptor function 2. The C-terminal region encompasses the ligand binding domain (LBD) and includes a region termed activation function 2 (AF-2), which is an important site for co-activator protein-binding. Binding of ligand induces a conformational change that creates a new surface for the recruitment of co-activator proteins in the AF-2 region 3. The LBD is connected to a DNA binding domain (DBD) by a hinge region (H) which contains a nuclear localization signal. The DBD is highly conserved and contains two alpha helices and two zinc fingers that are involved in the specificity of response-element-recognition and in receptor dimerization. Most liver-enriched NRs are active as dimers, functioning either as homodimers, or as heterodimers with retinoid x receptor (RXR) 4. Vertebrate RXR includes at least three distinct genes (RXRα, RXRβ, and RXRγ) which give rise to a large number of protein products through differential promoter usage and alternative splicing.
The N-terminal region of NRs is highly variable in sequence and length, but all contain a region termed activation function 1 (AF-1) that acts independently of ligand 5. The AF-1 domain contains many consensus phosphorylation sites and is therefore, the target of multiple kinases. Although most of the phosphorylation sites identified in NRs are located in the N-terminal domain, many receptors have at lease one phosphorylation site in the H region, and there are limited reports of sites located in the LBD and DBD. In addition, there are likely many yet to be identified phosphorylation sites in NRs.
Intracellular Localization
Most NRs are constitutively localized in the nucleus, however, the major proportion of steroid receptors and other a few other exceptional receptors may be located in the cytoplasm in the absence of ligand. Nuclear localization of receptors is mainly regulated by protein-protein interactions such as dimerization with RXRs or co-regulator proteins 6. In the cytoplasm, NRs are bound to heat shock proteins and this association prevents receptor transportation through the nuclear pores and thus sequesters NRs from binding to DNA 7. In the nucleus, ligand-mediated activation of NRs causes redistribution of the receptor to chromatin. Recent evidence which will be discussed in more detail has suggested that nuclear localization of some NRs is a cell signaling- and phosphorylation-dependent event.
Co-regulator Proteins
The full activity of NRs depends on a large number of co-regulator proteins that do not bind to DNA directly, but have a pronounced effect on the outcome of gene expression 3. In general, non-liganded NRs form a complex with co-repressor proteins which inhibit transcriptional activity, often through the recruitment of other cofactor proteins that contain histone deacetylase (HDAC) activity. HDACs alter chromatin structure by promoting chromatin compaction, thus rendering enhancer regions of genes less accessible to the necessary basal transcriptional machinery. Activation of NRs by ligand-binding or through phosphorylation induces a conformational change which results in the dissociation of the co-repressor multiprotein complexes and subsequent recruitment of co-activator protein complexes that enhance the rate of gene transcription, often thought the recruitment multiprotein complexes containing histone acetyltransferase (HAT) activity. Co-regulator proteins thus provide a second level of specificity in the modulation of gene expression by NRs. Most NR-co-activator proteins identified to date preferentially interact with NRs through the C-terminal AF-2 domain via an -LXXLL-motif, which constitutes a prototypical NR-interaction motif. However, in contrast to most co-activator proteins, the peroxisome proliferator activated receptor gamma co-activator 1 alpha ((PPARGC1a/PGC-1α) interacts not only with the AF-2 region of NRs, but also with the H region of the selected liver-enriched NRs 8. In addition to NRs, it has also been shown that the intrinsic and recruited enzymatic activities of several NR-associated co-factor proteins are regulated by phosphorylation in a dynamic manner in response to specific signal transduction pathways, and this will be discussed later in this review in more detail.
Liver-Enriched NRs as Targets of Signal Transduction Pathways
The majority of amino acid residues (serine and threonine) identified as being regulated-phosphorylation sites lay within the N-terminal region of NRs that correspond to consensus sites for proline-dependent kinases such as cyclin-dependent kinases (CDK) 9, 10 and MAP kinases 11. For some NRs, such as progesterone receptor (PR) which contains at least 13 sites, phosphorylation of the N-terminus is quite complex. However, other NRs such as perioxisome proliferator activated receptors (PPARs) contain only one or two phosphorylation sites in the N-terminus 12.
NRs can be phosphorylated constitutively in the absence of ligand, or in response to ligand-mediated activation. Other NRs can be phosphorylated independently of ligand in response to cellular signaling events by MAPKs. For example, growth factors, stress, cytokines, and other signals activate several serine kinase cascade pathways that activate different MAP kinases (Erk, JNK, or p38 MAPK) which can enter the nucleus and phosphorylate NRs. The N-terminal domains of PR 13, 14, ERα 15, 16, ERβ 17, 18, AR 19, PPARs 20, 21, and RARγ 22, 23 have all been reported to be substrates of p42/p44 or p38 MAPK, and that of RXRα is phosphorylated by JNK. In addition to MAPK sites, the N-terminus of many NRs also contains consensus sites for Akt kinase, or PKB, a kinase critical for cell survival and proliferation 24, 25. Activated Akt negatively regulates downstream MAP kinases 26, and upon nuclear translocation phosphorylates specific NRs including ERα 27 and AR 28.
In addition to the N-terminal domain, the LBD and DBD are also targets for protein kinases. Phosphorylation of the LBD can involve the same proline dependent kinases. For example, serine residues contained within the LBD of RXRs are targeted by the stress activated protein kinase- JNK 29, 30. Phosphorylation by other kinases such as tyrosine kinases for ERα 31, 32 and RXRα 30 or PKA for RAR 33 is also common. Evidence for phosphorylation of the DBD involves either PKA for ERα 34 or PKC for RARα 35 and VDR 36, 37.
While signal transduction pathways and phosphorylation regulate most, if not all NRs in multiple tissue types, this review will focus on the regulation of non-steroid NRs expressed in a liver-enriched manner.
NR2A1, HNF-4α
Hepatic nuclear factor-4α (HNF-4α) is a NR expressed mainly in liver, intestine, and kidney and is critical for development and liver specific gene expression 38. HNF-4α has been implicated in the regulation of many genes in liver such as Cyp7a139, the constitutive androstane receptor (CAR) 40, and genes involved in glucose transport and glycolysis 41. Although typically thought of as an orphan receptor, HNF-4α has been shown to be activated by fatty acyl-CoA thioesters 42. In addition, the transcriptional activity of HNF-4α is regulated by phosphorylation of serine, threonine, and tyrosine residues. Phosphorylation of HNF-4α is required for DNA binding and appropriate subnuclear localization. Violett et al. indentified a PKA consensus phosphorylation site in the DBD of HNF-4α and report that HNF-4α is directly phosphorylated by PKA. PKA-mediated phosphorylation of wild-type HNF-4α strongly repressed the binding affinity and transcriptional activity of the receptor based on gel-shift assays and reporter gene analysis 43. On the other hand, phosphorylation of specific serine and threonine residues in HNF-4α alters its tertiary structure, which increases the affinity and specificity of DNA-binding in COS-7 cells 44. In addition to alterations in DNA-binding, tyrosine phosphorylation is required for appropriate sub-nuclear localization and transactivation activity of HNF4α as evidenced by immunofluorescence, electron microscopy, and reporter gene assay using genistein treatment to inhibit tyrosine phosphorylation 45.
It has also been recently shown that p38 kinase phosphorylates HNF-4α at S158 increasing its interaction with co-activator PC4, DNA-binding, and transactivation activity in the presence of IL-1β and hydrogen peroxide 46. In addition, inhibition of p38 kinase activity diminishes HNF-4α nuclear protein levels and its phosphorylation, rendering a less stable protein. Induction of p38 kinase by insulin results in an increase of HNF4α protein and Cyp7a1 gene expression in primary rat hepatocytes, thus providing a functional link between HNF-4α phosphorylation and bile acid synthesis 47. Since HNF-4α has been shown to activate multiple genes and interacts with multiple transcription factors and co-regulators in liver such as COUP-TF's, CBP, GRIP-1 SRC-1, p300 and PGC-1α 40, 48, 49, further study is required to determine the mechanism by which phosphorylation of HNF-4α modulates protein-protein interactions and differential gene expression.
The NR1C Subfamily- PPARs
The three perioxisome proliferators activated receptor (PPAR) isotypes, PPAR α, β, and γ, form a subfamily of NRs that are mainly involved in lipid and glucose homeostasis, control of inflammation and wound healing, and regulation of food intake and body weight 50, 51. PPARα is expressed in metabolically active tissues including liver, kidney, heart, skeletal muscle and brown fat. PPARγ is expressed to a high extent in adipose tissue with lower amounts present in kidney, liver, and skeletal and smooth muscle 52. Fatty acids and fatty acid derivatives are endogenous ligands for PPARs and induce PPAR-dependent gene activation. In addition, PPARs are very important therapeutic targets for the treatment of hyperlipidemia and type-2-diabetes. The hypolipidemic fibrates were the first known synthetic ligands of PPARα, while thiazolidinediones are the best characterized PPARγ ligands used in the treatment of type-2-diabetes 53. Therefore, understanding the mechanisms that regulate PPAR activity is crucial for effective therapeutic treatment of metabolic diseases.
Insulin treatment enhances PPARα activity via phosphorylation of S12 and S21 by p42/p44 MAP kinase, but represses PPARγ activity via phosphorylation of S112 20, 21. Furthermore, phosphorylation of the N-terminal domain of PPARγ has been shown to decrease PPARγ activity. For example, PDGF treatment decreases PPARγ transcriptional activity in reporter gene assays, and in vivo labeling experiments demonstrated that PPARγ undergoes EGF-stimulated MAPK-dependent phosphorylation at S82 54. Further studies indicate that PPARγ activity is decreased through phosphorylation of the N-terminus at S84 by ERK2 and JNK via TNF4α and EGF stimulation 55. In contrast, one study reports an increase in transactivation of PPARγ via insulin-stimulated ERK2 phosphorylation in CHO cells 56. Differential modulation of PPAR activity by phosphorylation is likely due to the relationships between specific kinases, serine residues, ligands, receptor isoforms in specific cell types. For example, ERK2 and JNK, but not p38 MAP kinase can phosphorylate PPARγ on S84 55, while PPARα is a substrate for both ERK2 and p38 MAP kinase in a ligand-dependent manner 57. In addition, phosphorylation of PPARα by PKA in transient reporter gene assays was shown to have different effects depending on which promoter was used experimentally 58.
There are multiple mechanisms by which phosphorylation of PPARs modulates their activity. Transfection studies have suggested that phosphorylation of the N-terminus induces the dissociation of co-repressor proteins such as NCoR from PPARα 21. In a similar fashion, phosphorylation of the N-terminus of PPARα increases co-activation by PGC-1α 57. Phosphorylation can also enhance DNA-binding of PPARs, as is the case with PPARα phosphorylation by PKA 58. PPARs may also be modulated through kinase cascades that up-regulate their own expression as shown by the PKC-dependent upregulation of PPARα gene expression 59.
MAPKs (ERK1, ERK2, p38, and JNK), PKA, and PKC are the three kinase families that have been implicated in the phosphorylation of PPARα and γ. Activation of these signaling pathways and phosphorylation of PPARs could affect the endogenous and therapeutic function of PPARs. For example stress or fasting may activate PKA signaling which phosphorylates PPARα and enhances its activity through the recruitment of PGC-1α and may affect the function of PPARα as a drug target.
NR1I2, PXR
Pregnane x receptor (PXR) is a master-regulator of xenobiotic-inducible cytochrome-p450 (CYP) gene expression in liver. The CYPs identified as PXR target-genes encode enzymes responsible for the oxidative metabolism of over 60% of clinically prescribed drugs. In addition, several studies have shown that PXR regulates other genes involved in the metabolism of xenobiotic and endobiotic compounds such as glutathione S-transferases, sulfotransferases, and UDP-glucuronosyltransferases 60-63. PXR also regulates the expression of the drug-transporter genes Oatp2, Mdr1, Mrp2, and Mrp3 64-66. PXR is a promiscuous receptor activated by a wide variety of compounds including synthetic and endogenous steroids, bile acids, and a variety of drugs and natural compounds 67. In this manner, the modulation of PXR activity by ligands and/or signaling pathways represents the basis for an important class of drug-drug interactions.
Drug-inducible CYP gene expression is known to be responsive to cytokine, PKC, and PKA signaling pathways, however the exact mechanism by which these pathways intersect with PXR is unknown. For example, a significant reduction in the hepatic expression of Mdr1 and Mrp3 genes were seen in endotoxin treated mice. Similarly, IL-6-treated mice displayed a 40-70% reduction in the mRNA levels of all Mdr isoforms 68. Inflammatory cytokines inhibit the inducible expression of Oatp2 during intrahepatic cholestasis 69. It has also been shown using primanry cultures of human hepatocytes that IL-6 markedly decreases the expression of PXR and its close cousin constitutive androstane receptor (CAR). IL-6 also decreases both rifampicin- and phenobarbital-mediated induction of CYP3A and CYP2B gene expression 70.
Recent evidence has demonstrated crosstalk between PXR and NF-κB signaling pathways. NF-κB activation by LPS and TNF4α repressed PXR association with the CYP3A promoter by disrupting the association of the PXR-RXR protein complex 71. In addition, PXR activation inhibited the activity of NF-κB and the expression of its target genes. This inhibition was shown to be PXR-dependent and was potentiated by PXR ligands in vitro and in vivo 72. PXR activation has also been shown to alleviate the symptoms of inflammatory bowel disease (IBS). Studies using IBS and PXR knockout mouse models have shown that PXR agonist treatment decreased the expression of NF-κB target gene expression in a PXR-dependent manner 73.
In addition to cytokine signaling, CYP3A gene expression is also modulated by PKA and PKC signaling pathways. Co-treatment of primary cultures of rat hepatocytes with phenobarbital and cyclic AMP analogs and PKA activators clearly results in cyclic AMP-associated inhibition of CYP gene expression 74. However, treatment with the andenlyl cyclase activator forskolin and its non-PKA-activating analog 1,9 dideoxyforskolin both resulted in the stimulation of CYP3A gene expression 75. Ding and Staudinger have shown that both forskolin and 1,9 dideoxyforskolin induce CYP3A expression in primary mouse hepatocytes by functioning as PXR agonists. In addition, activation of PKA signaling potentiated PXR-mediated induction of CYP3A expression and increased the strength of PXR-co-activator protein interactions in mammalian 2-hybrid reporter gene assays. Further kinase assays show that PXR can be a substrate for PKA in vitro, suggesting a potential mechanism for PKA-mediated modulation of CYP3A gene expression 76. It is also of interest that while PKA activation potentiates the expression of CYP3A in mouse hepatocytes, it is a repressive signal in both human and rat hepatocyte cultures (our unpublished data). This suggests a species-specific effect for the modulation of CYP3A by PKA signaling. Differential phosphorylation may be a possible factor the species-specific responses to PKA signaling. In addition, activation of PKC signaling dramatically represses PXR activity in reporter gene assays and in hepatocytes by increasing the strength of interaction between PXR and the co-repressor NCoR, and by abolishing the ligand-dependent interaction between PXR and co-activator SRC-1 77.
NR1I3, CAR
Similar to PXR, the NR CAR was first classified as a xenobiotic-sensing transcription factor that regulates numerous hepatic genes in response to a large group of xenobiotics and endobiotics. CAR was originally found to regulate the transcription of genes encoding the CYP2B subfamily 78. In addition to CYP2B, CAR also regulates the expression of multiple drug and hormone metabolizing enzymes and transporter proteins such as CYP3A, CYP2C, glutathione S-transferases, sulfotransferases, and UDP-glucuronosyltransferases, Oatp2, Mrp2 and Mrp3 79. Interestingly, treatment of wild type and CAR knockout mice with phenobarbital, the prototypical CAR activator, both induces and represses certain hepatic genes in a CAR-dependent manner suggesting that CAR has diverse roles as both a positive and negative regular of hepatic gene expression in response to phenobarbital 80. As the function of CAR has expanded, so has interest in the deciphering the molecular mechanism of its activation by drugs.
It is well documented that the phenobarbital-mediated induction of CYP2B genes in cultured hepatocytes is responsive to several serine/threonine protein kinases and phosphatases. However, the mechanism by which these signaling pathways interact with CAR remains poorly understood. For example, activation of PKA signaling negatively impacts the induction of CYP2B expression in primary cultures of rat hepatocytes 74, and co-treatment with phosphatase inhibitors further potentiates the repressive effects of PKA signaling 81. Serine/threonine-specific protein phosphatases PP1 and PP2A have a positive role in the induction of CYP2B 82. Pustylnyak et al. further reports that rats treated with inhibitors of Ca(2+)/calmodulin-dependent kinase inhibitors exhibited increased gene expression of both CAR and CYP2B, while rats treated with the protein phosphatase PP1 and PP2A inhibitor okadiac acid exhibited the opposite effect 83.
In the absence of a ligand or activator, CAR is sequestered in the cytoplasm where it forms a complex with Hsp90 and cytoplasmic CAR retention protein (CCRP). In response to phenobarbital the complex recruits protein phosphatase 2A before translocation of CAR to the nucleus. The protein phosphatase inhibitor okadiac acid represses phenobarbital-induced nuclear translocation of CAR 84. In addition, dephosphorylation of S202 in mouse CAR is required for its nuclear translocation 85. The signaling pathway involved in the phosphorylation of S202 remains unknown. Unlike most NRs, CAR translocates to the nucleus without directly binding phenobarbital 86. Taken together, these data indicate that the phosphorylation status of CAR is intimately involved its cyptoplasmic retention and nuclear translocation.
Recent evidence shows that epidermal growth factor (EGF) represses phenobarbital-mediated activation of CAR-dependent transcription 87, and that the MEK inhibitor U0126 increases the phenobarbital mediated induction of CYP2B in primary rat heptocytes 88. In addition, hepatocyte growth factor (HGF) treatment represses induction of cyp2b10 by phenobarbital in primary mouse hepatocytes. HGF treatment increased the phosphorylation of ERK1/2, thus decreasing the nuclear translocation of CAR 89, however, the exact mechanism by which this occurs is unknown. In addition to MEK/ERK signaling, AMP-activated protein kinase (AMPK) has been suggested to activate CAR in hepatocytes 90. Additional studies using AMPK knockout mice demonstrate that that although AMPK does not regulate the phenobarbital-induced translocation of CAR, it may be involved in the activation of CAR in the nucleus 91. Shindo et al., reported that activation of AMPK resulted in nuclear accumulation of CAR but was not sufficient to induce CYP2B gene expression 92. Additional studies suggest that phenobarbital targets LKB1 for the activation of AMPK 93, adding a proximal target to the elusive sequence of events by which phenobarbital activates transcription of CYP2B. While AMPK appears to be an activating signal for phenobarbital-mediated induction of CYP2B, MEK/ERK seems to be repressive. Further study into the signal-related mechanisms of CAR activation is required to determine the effect that these pathways might have on the phosphorylation status of CAR or CAR-interacting proteins.
NR1H3, LXR
Liver x receptors (LXRα and LXRβ) have emerged as important regulators of cholesterol metabolism and transport, lipid metabolism, glucose homeostasis and inflammation 94. LXRα is primary expressed in liver, macrophages, and adipose tissue, while LXRβ is more ubiquitously expressed 67. LXR-activating ligands include several oxysterols and 6α-hydroxy bile acids 95, 96. Since the discovery of LXRs, multiple LXR-target genes that are involved in cholesterol and lipid homeostasis have been identified. These include CYP7A1, the rate limiting enzyme in the classical pathway of bile acid synthesis, ATP-binding cassette (ABC) transporters, lipoproteins such as apolipoprotein E (ApoE), lipoprotein lipase (LPL), and lipogenic proteins such as sterol response element binding protein (SREBP)-1C and fatty acid synthase (FAS) 97-100. Although early reports emphasized the role of LXR in cholesterol homeostasis, recent studies suggest that LXR negatively regulates gluconeogenesis 101 and inflammatory responses 102, 103.
LXR has been shown to exist as a phosphoprotein in HEK293 cells. Mutational analysis and metabolic labeling indicate that LXR is constitutively phosphorylated at S198 in the hinge region of the receptor at a MAPK consensus site 104. However, the biological significance of this phosphorylation event has yet to be elucidated.
Early studies demonstrate that PKA/PKC modulators such as prostaglandin E2, phorbol esters, 8-bromo-cyclic AMP, and forskolin enhanced the induction of reporter genes by LXR ligands 105. These experiments suggest that transactivation by ligand-activated LXR may be further modulated through kinase signaling. PKA can directly phosphorylate LXR, and has been reported to both increase and decrease transactivation depending on the experimental conditions 106-108. In primary cultures of rat hepatocytes, activation of PKA repressed LXR-mediated SREBP-1C gene expression. Direct phosphorylation of LXR by PKA in vitro and in vivo at two PKA consensus sites (S195, S196 and S290, S291) located in the LBD was required for trans-repression. PKA-mediated phosphorylation of LXR impaired DNA-binding through the disruption of LXR/RXR dimerization, and decreased transcriptional activity by inhibiting the recruitment of the co-activator protein- SRC-1, and enhancing the recruitment of co-repressor protein- NCoR 108. On the other hand, Tamura et al. have demonstrated that PKA signaling can increase LXR transactivation in reporter gene assays conducted in remal As4.1 mouse cell lines 106.
In addition to inducing genes involved in cholesterol and glucose homeostasis, LXR reciprocally represses a set of inflammatory genes including iNOS, COX-2 and IL-6, MMP-9 after bacterial, LPD, TNFα or IL-1β stimulation 102, 103. Importantly, LXR agonists reduce inflammation in vivo. The mechanism by which LXR represses inflammatory genes is not well understood. No LXREs have been identified on the promoters of the repressed genes. In addition to possible competition for co-regulator proteins, recent evidence suggests that inhibition of the NF-κB pathway is involved likely through trans-repression of NF-kB in the nucleus 109. In a recent study of trans-repression of the iNOS promoter, SUMOylation of PPARγ was identified as a mechanism of repression 110. It remains to be determined the extent to which post-translational modification of LXR is involved in LXR-mediated trans-repression of inflammatory genes.
NR5A2, LRH-1
The orphan NR liver receptor homolog 1 (LRH-1) functions to regulate the expression of a number of genes involved in bile acid homeostasis and other liver functions. Unlike the majority of NRs that function as dimers, LRH-1 binds as a monomer to an extended NR half site in the promoter its target genes 111. LRH-1 is an important regulator of CYP7A1 gene expression 112, 113. LRH-1 also regulates the expression of other genes involved in cholesterol and bile acid homeostasis including CYP8B1, MRP3, cholesterol ester transfer protein (CETP), apical sodium-dependent bile acid transporter (ASBT), and apolipoprotein A1 60, 114-119.
No ligands for LRH-1 have yet been identified, and the mechanisms that modulate its activity are still unclear. Lee et al. have shown that treatment with PMA increases LRH-1 activation in Hela cells. This response is blocked by the ERK1/2 inhibitor U0126. Mutation analysis confirm that phosphorylation of LRH-1 at S238 and S243 in the H domain stimulates LRH-1 transactivation 120. In contrast, activation of stress-activated protein kinase (JNK) pathways is associated with inhibitory effects on the LRH-1 target CYP7A1; however the role of LRH-1 in this pathway is unclear 121, 122.
Treatment of HepG2 cells with the inflammatory cytokine- TNFα, a potent activator of the JNK pathway, increased the expression of LRH-1 and MRP3, and also increased LRH-1-binding to the MRP3 promotor 114. Krylova et al., have suggested that phosphatidylinositols, major intracellular signaling molecules, bind to LRH-1, linking phospholipids signaling and gene expression 123. However the function role of phosphatidylinositols as modulators of LRH-1 function remains unknown.
NR1H4, FXR
Farnesoid x receptor (FXR) is a NR expressed in liver, intestine, kidney and adipose tissue. FXR has emerged as a key player involved in the maintenance of cholesterol and bile acid homeostasis through its regulation of the expression of genes involved in the synthesis, uptake, and excretion of bile acids 124. An important breakthrough in the FXR field was the discovery that FXR is directly activated by several bile acids including chenodeoxycholic acid (CDCA), lithocholic acid (LCA), and deoxycholic acid (DCA) 125-127. Studies with FXR knockout mice revealed that a number of genes involved in cholesterol homeostasis are also regulated by FXR including Cyp7a1, Cyp8b1, intestinal bile acid binding protein (I-BABP), canalicular bile salt excretory pump (BSEP), phospholipids transfer protein (PLTP), and the hepatic basolateral transporter NTCP 128. Additional studies reveal that FXR induces expression of the NR- SHP. Increased SHP then represses Cyp7a1 transcription by inhibiting the activity of LRH-1, which is a positive regulator of the Cyp7a1 promoter 112.
Although there is no evidence at this time for direct phosphorylation of FXR, the expression and activity of FXR are modulated by signal transduction pathways. For example, FXR is thought to modulate insulin signaling. FXR expression is reduced in streptozotocin (STZ)-induced diabetic rat models, and administration of insulin restores FXR mRNA to normal levels 129. Additional support for this concept comes from the observation that glucose reduces FXR expression in the liver 129, while activation of FXR by the synthetic agonist GW4064 reduced plasma glucose levels in mice 130. Furthermore, loss of FXR disrupts normal glucose homeostasis and leads to the development of insulin resistance in FXR knockout mice 131-133. FXR may also play a role in regulating glucose and lipid metabolism during alterations in nutritional status. A recent study reports that FXR expression is induced in mouse liver in response to fasting, a condition during which PKA signaling is enhanced 134.
Bile acids that are FXR agonist have been shown to activate multiple signal transduction pathways. Treatment with taurocholic acid results in activation of the JNK pathway 135, and deoxycholic acid treatment activates the Raf-1/MEK/ERK signaling cascade in primary rat hepatocytes 136. In addition, treatment of HepG2 cells with bile acids results in the activation of PKC, and treatment with PKC inhibitors reduces the bile acid-mediated repression of Cyp7a1 gene expression 137. While there is evidence for multiple bile acid-responsive pathways, future research goals include elucidation of the effects that kinase activation has on the phosphorylation status and functional activity of FXR.
NR0B2, SHP
The small heterodimer partner (SHP) is an atypical orphan member of the NR superfamily in that it lacks the conserved DBD. SHP was isolated in a yeast 2-hybrid screen based on its ability to dimerize with other NRs 138. It is expressed mainly in liver, small intestine, spleen, heart and pancreas 139. SHP interacts with a variety of NRs in liver including PPARα 140, LRH-1 112, 141, LXR 142, and HNF4α 143. SHP acts as a direct transcriptional repressor and inhibits the activity of most NRs with which it interacts 144. Two notable exceptions are that SHP enhances the transcriptional activation of PPARα 145, and PPARγ 146 under certain conditions.
It has been shown that SHP expression is regulated by the JNK pathway. Gupta et al. provide evidence that bile acids rapidly down-regulate CYP7A1 transcription via activation of the JNK/c-Jun pathway, and that SHP is a direct target-gene of activated c-Jun 135. Over-expression of c-Jun resulted in increased SHP promoter activity, whereas mutation of the c-Jun response element in the SHP promoter abolished activation induction of reporter gene expression under the control of the SHP promoter. This study provides an alternative mechanism for bile acid-mediated induction of SHP expression that is independent of FXR.
NRs and FGFs
Recent evidence has uncovered several novel NR-dependent mechanisms involving fibroblast growth factors (FGFs). This recent thrust of research has created a new paradigm that particular FGFs function as metabolic hormones and act through yet to be described signal transduction cascades to elicit specific physiological responses. FGFs function in processes such as development and wound healing. However, three member of the FGF family, FGF19 (FGF15 in mouse), FGF21, and FGF23 have recently emerged as novel metabolic hormones.
As mentioned earlier in this review, activation of FXR by bile acids down-regulates Cyp7a1 gene expression in mice through an indirect mechanism involving the induction and activation of the negative regulator- SHP. Recently, an additional FXR-dependent mechanism involving FGF19 has been described. FGF19 binds to its cell surface receptor, FGFR4, and increases JNK-dependent signaling 147. In primary cultures of human hepatocytes, FXR activation induces expression of FGF19. FGF19 then modulates bile acid biosynthesis by reducing the expression of CYP7A1 through a JNK-dependent pathway without affecting SHP expression 148. It has also been shown that over-expression of FGF19, using either a transgenic approach or with chronic FGF19 treatment, improves insulin sensitivity and glucose homeostasis in diet-induced obese mice, in part through increased metabolic rate and fatty acid oxidation 149, 150. Conversely, activation of the JNK signaling pathway in the liver has been shown to increase insulin resistance 151, however there is currently no explanation for this discrepancy.
FGFR4 deficient mice exhibit reduced JNK activity, an increased bile acid pool, and enhanced expression of CYP7A1 152. On the other hand, transgenic mice expressing constitutively active FGFR4 exhibit increased JNK activity, a reduced bile acid pool, and reduced expression of CYP7A1 153. The expression of FGF15, the mouse ortholog of FGF19, is induced by FXR activation in the small intestine, but not in the liver. FGF15 expression then represses CYP7A1 in liver through a mechanism that involves FGFR4 and SHP 154. In addition, mice lacking FGF15 have increased hepatic Cyp7a1 expression and activity corresponding to increased bile acid excretion 154. Taken together, these studies define FGF19 in humans and FGF15 in mice as pivotal components of a novel signaling pathway that cooperates with FXR and SHP to maintain bile acid homeostasis.
PPARα, a fatty acid-activated NR, regulates the utilization of fat during the starvation response. Recently, a PPARα-dependent role for FGF 21 in the adaptive response to starvation has been described. FGF21 has been observed to have a variety of beneficial effects on metabolic parameters. Treatment of obese and leptin-deficient mice with FGF21 decreases serum glucose and triglyceride concentrations, and increases insulin sensitivity and glucose clearance. Moreover, mice that over-expressed FGF21 are resistant to diet induced obesity 155. Similar results were observed in FGF21 treatment of diabetic rhesus monkeys 156. While theses studies show that the administration of FGF21 has important metabolic effects, recent studies have provided insight to the physiological role of FGF21. Inagaki et al. and Badman et al., show that FGF21 expression in the liver of fasted mice is induced following activation of PPARα 157, 158. Adenoviral knockdown of endogenous FGF21resulted in fatty liver, increased serum triglyceride levels, and decreased serum ketone levels in mice fed a low carbohydrate, high fat diet 157. This was associated with the decreased expression of fatty acid oxidizing enzymes and key enzymes in ketone body production that are known PPARα-target genes 157. In transgenic mice over-expressing FGF21, ketogenesis and ketone body concentration in serum was increased several fold. Interestingly, recombinant FGF21 treatment rescued defective ketone body production in f PPARα knockout mice 158. Over-expression of FGF21 in mice also produced decreased body temperature and locomotor activity during fasting and increased lipolysis in white adipose tissue 158. These studies make it evident that FGF21 signaling collaborates with PPARα, and together they function in liver as master regulators of energy balance. The precise molecular mechanisms that are downstream of these FGF signaling pathways in liver remain to be elucidated.
Co-Regulator Proteins as Targets of Signal Transduction Pathways
Interaction of NRs with co-regulator proteins provides a second level of regulation in target gene activation. The association of co-regulator proteins with NRs is clearly controlled at the level of ligand binding. In addition, the activation of cell signaling events and/or protein kinases directly regulates the association of NRs with co-regulator proteins. Numerous examples sited above illustrate how phosphorylation of NRs can result in increased or decreased strength of interaction between the receptor and co-activator or co-repressor multiprotein complexes. Realization that the specificity and activity of co-regulator proteins may also be regulated by signal transduction and phosphorylation is relatively new concept for which much less is known.
NCoR and SMRT Co-repressor Proteins
The most extensively characterized co-repressor proteins for NRs are NR co-repressor (NCoR) and silencing-mediator for retinoid and thyroid hormone receptors (SMRT) 159, 160. NCoR and SMRT interact with and mediate the repression of overlapping sets of NRs. NCoR and SMRT do not have intrinsic enzymatic activity; instead they have conserved modular domains that interact with HDACs. These co-repressor proteins can bind to NRs at their conserved C-terminal receptor interacting domain (RID) in the presence or absence of ligand and are regulated by a variety of signal transduction pathways 161.
It had been previously observed that activation of tyrosine kinases negatively regulates the interaction between transcription factors and SMRT 162. Further studies reveal that phosphorylation of SMRT in the C-terminal RID by the MAP kinase-kinase MEK-1 and MEK-1 kinase (MEKK-1) inhibits the interaction between SMRT and NRs 163. In addition, introduction of MEK-1 and MEKK-1 signaling into transfected cells led to the redistribution of SMRT from the nucleus to the perinucleus or cytoplasm 163. In contrast, phosphorylation of SMRT by casein kinase 2 (CK2) on S1492 stabilizes SMRT-NR interactions 164. Therefore, different signaling pathways can modulate different transcriptional outcomes via SMRT phosphorylation.
In contrast to SMRT, NCoR is refractory to MEKK1 phosphorylation, does not release from NR partners and does not change its sub-cellular distribution in response to MEKK1 signaling 165. These results indicate that the closely related SMRT and NCoR are regulated by distinct kinase signaling pathways. Although NCoR is fully refractory to MEKK-1 signaling, it is partially inhibited by EGF receptor signaling, indicating that NCoR may respond to an as yet undefined secondary pathway activated by EGF signaling 165. Recent evidence suggests that this differential response may be determined by alternative mRNA splicing of SMRT and NCoR 166. NCoR has been shown to be phosphorylated by Akt at S401, leading to the reversal of NCoR-mediated repression and nuclear export of NCoR. However, SMRT possesses and alanine residue at position 401 and is resistant to the actions of Akt 167.
Finally, since SMRT and NCoR exist in co-repressor multiprotein complexes, their activity may be affected by activation of signaling cascades that result in the phosphorylation of an HDAC, or other proteins in the complex. For example, phosphorylation of HDAC4 by ERK1 and ERK2 enhances its nuclear accumulation, whereas phosphorylation of HDAC1 and HDAC2 alters their interactions with co-repressor complexes 168-170. IL-1β has been reported to inhibit NCoR through an indirect pathway resulting in the MEKK-1 phosphorylation of a TAB2 subunit present in a subset of NCoR-HDAC3 complexes, whereas SMRT is resistant to this pathway 171.
p160/SRC Co-activator Proteins
Steroid receptor co-activators (SRC) proteins are widely expressed and co-activate most NRs as well and many general transcription factors. The C-terminal region of SRC contains HAT activity, albeit relatively weak 172, 173. Furthermore, SRCs recruit other co-activator proteins such as CBP/p300 and pCAF to a larger multiprotein complex that participates in chromatin remodeling 174. There are three member of the SRC family, all of which contain conserved centrally located –LXXLL- motifs that are responsible for ligand-dependent interaction with NR through the AF-2 domain 175.
Seven phosphorylation sites for SRC-1 and six for SRC-3 have been identified 176, 177. All seven of the sites identified in SRC-1 contained consensus-phosphorylation sequences for serine/threonine-proline directed kinases, and two contained perfect consensus sequences for the MAPK family and are phosphorylated by ERK-2. Phosphorylation of SRCs can be induced by a variety of environmental stimuli including EGF, cyclic AMP, cytokines, and steroid hormones 176-180. In addition, the phosphorylation of SRCs induced by these agents is required for optimal co-activator activity. For example, ERKs can phosphorylate SRC-2 at S736 and treatment of cells with EGF increases the transcriptional activity of GAL4-GRIP1 181. In addition, SRC-1 phosphorylation at S1185 and T1179 is induced by cyclic AMP, and phosphorylation at these sites enhances the ligand-dependent and –independent activity of PR 179. PKA did not phosphorylate these sites in vitro, but blockage of PKA activity in COS-1 cells prevented cyclic AMP mediated phosphorylation of these sites 179. This phosphorylation event was also shown to be required for the interaction of SRC with pCAF or CBP 179. In a similar manner, EGF-stimulated phosphorylation of SRC-3 by MAPK stimulates the recruitment of p300 and enhances ligand-dependent ER activity 178. Phosphorylation of SRC-3 was shown to selectively affect the interactions with NRs, NF-κB, and CBP 177. These data suggest that the phosphorylation of SRCs seems to be involved in the regulation of protein-protein interactions, however, it remains to be seen whether phosphorylation can affect other aspects of SRC-function.
All three SRCs contain both redundant and distinct functions which may be modulated by the signal transduction pathways that interact with SRCs and result in their phosphorylation. For example, SRC-3 contains several distinct patterns of phosphorylation. Phosphoylation of all six sites of SRC-3 is shown to be induced by estrogen and androgen hormones and is required for co-activation of estrogen and androgen receptors 177. However, phosphorylation of only five of the six sites is induced by TNFα and is required for co-activation of NF-κB 177. Further evidence shows that SRC-3, but not SRC-1 was co-purified in complex with IκB kinase (IKK), and consequently, phosphorylation of SRC-3, but not SRC-1 is enhanced in response to TNFα stimulation 180. Thus, it appears that phosphorylation provides a molecular basis that determines the ability of SRCs to distinguish among various transcription factor families, and helps to provide specific responses to various upstream signaling pathways.
In addition to SRC, phosphorylation of other proteins in the co-activator complex can modulate transactivation potential of NRs. CBP/p300can be phosphorylated in vivo and participates in cyclic AMP-regulated gene expression 182. Kinase activities are also found to be associated with CBP/p300. Activation of cellular Ras with insulin treatment stimulated the recruitment of S6 kinase pp90RSK to CBP 183. Binding of pp90RSK to CBP repressed the transcription of cyclic AMP-responsive genes via CREB 183. CBP also contains a signal-regulated transcriptional activation domain that is controlled by calcium/calmodulin-dependent protein kinase IV and by cyclic AMP 184. Signal transduction pathways may also influence acetyltransferase activities and substrate preferences. An example of this is best illustrated with the POU homeodomain transcription factor Pit-1. Pit-1 function requires CBP/p300 and pCAF and is positively regulated by cyclic AMP and MAPK signal transduction pathways 185. Interestingly, stimulation of Pit-1 activity by cyclic AMP requires the intrinsic HAT activity of CBP, whereas stimulation of Pit-1 activity by the MAP kinase pathway requires the HAT activity of pCAF 185. It is thus plausible that activation of different signaling pathways could influence the group of co-activators that are required for NR mediated transactivation.
The PGC Family of Co-integrator Proteins
There are three members of the PPARγ co-activator (PGC) family, PGC-1α, PGC-1β, and PGC-1 related co-activator (PRC). However, PGC-1α is the most extensively characterized member of the family. Like many protein cofactors, PGC1α co-activates multiple NRs. PGC-1α is selectively expressed mainly in skeletal muscle, cardiac muscle, white fat, and liver 186. PGC-1α binds to NR-LBDs with high affinity and, similar to SRCs, contains a triplet –LXXLL- motif for binding to NRs through their AF-2 domains. PGC-1α does not have intrinsic HAT activity, and serves as a molecular scaffold that recruits additional factors such as CBP/p300 187. The physiological role of PGC-1α has been well characterized as a master regulator of energy homeostasis in fat, liver and muscle 188. Specifically in liver, PGC1-α plays a prominent role in the regulation of genes involved in energy metabolism and glucose homeostasis. PGC-1α is induced in liver by fasting and up-regulates the expression of key genes that participate in gluconeogenesis 189, fatty acid oxidation 190, and bile acid synthesis 191.
PGC-1α interacts with a multitude of signaling pathways that affect both its expression and/or phosphorylation status. Agents that increase cyclic AMP signaling such glucagon, catecholamines, and glucocorticoids induce PGC-1α expression in liver 189. This cyclic AMP/PKA-dependent induction of PGC-1α is mediated by phosphorylation and activation of the transcription factor CREB which directly regulates the PGC-1α promoter 192. On the other hand, LKB1/AMPK signaling appears to regulate the repression PGC-1α gene expression. In LKB1 deficient liver, transducer of regulated CREB activity 2 (TORC2), a transcriptional co-activator of CREB, was de-phosphorylated and entered the nucleus, driving expression of PGC-1α 193.
In insulin stimulated skeletal muscle, PGC-1α gene expression is down-regulated by Akt-mediated phosphorylation and nuclear exclusion of FOXO1 194. In addition to insulin, obesity and saturated fatty acids decrease PGC-1α gene expression and function via p38 MAPK-dependent transcriptional pathways 195. Moreover, palmitate, a common saturated fatty acid, reduces PGC-1α expression in skeletal muscle through a mechanism involving MAPK-ERK and NF-κB activation 196.
It is worth noting that the most prominent PGC-1α post-translational modification in terms of control of its activity and physiological output is acetylation 197, 198. Methylation can also enhance PGC-1α activity 199. However, this review will focus on the effect of phosphorylation of PGC-1α. Cytokines such as IL-1α, IL-1β, and TNFα have been shown to activate the transcriptional activity of PGC-1α in muscle through direct phosphorylation by p38 MAPK resulting in increased stability and half-life and activation of PGC-1α protein 200. p38 MAPK phosphorylates PGC-1α at three residues (T262, S265, and T298) that occur in a region previously shown to interact with NRs, however, it remains to be seen whether phosphorylation of PGC-1α affects NR docking. Further studies performed in primary hepatocytes confirm that PGC-1α phosphorylation by p38 MAPK is necessary for free fatty acid induced activation of PEPCK, a PGC-1α-target gene involved in gluconeogenesis 201. The precise mechanism by which p38 MAPK-mediated phosphorylation of PGC-1α alters the amount and activity of PGC-1α will likely provide important physiological, and perhaps therapeutic, insight. Kralli's group has also shown that activation of p38 MAPK leads to and increase in PGC-1α activity. They propose that a repressor binds to the PGC-1α –LXXLL- motif and that the interaction is terminated upon activation of p38 MAPK 202. This suggests a model where the repressor and NRs compete to recruit PGC-1α to an inactive or active state, and that cellular signaling such as ligand or kinase signaling can shift the equilibrium between the two states.
In addition to p38 MAPK, two recent reports show that PGC-1α is phosphorylated by AMPK and Akt/PKB. AMPK activation in muscle increases the expression of genes required for glucose uptake, fatty acid oxidation, and mitochondrial biogenesis. Using primary muscle cells and PGC-1α knockout mice, Jager et al. demonstrated that the effect of AMPK mediated gene expression is dependent on PGC-1α function. Furthermore, AMPK phosphorylates PGC-1α at T177 and S583, which is required for PGC-1α dependent induction of the PGC-1a promoter 203. In liver, the mechanism by which insulin regulates lipid synthesis and degradation are largely unknown. Insulin treatment, through protein kinase Akt2/protein kinase B (PKB) resulted in the phosphorylation and inhibition of PGC-1α 204. Akt phosphorylates PGC-1α at S570 which prevents the recruitment of PGC-1α to its target promoters 204. Repression of PGC-1α activity by phosphorylation impairs its ability to promote gluconeogenesis and fatty acid oxidation in liver. PGC-1α has an additional role in the regulation of this pathway. PGC-1α co-activates PPARα in the expression of tribbles homolog TRB-3, a fasting inducible inhibiter of Akt/PKB 205. This mechanism by which insulin signaling regulates PGC-1α activity could provide insight into alternative drug targets for the treatment of type-2-diabetes.
Theraputic Obstacles and Opportunities
NRs control many aspects of biology including development, reproduction, and homeostasis through target gene activation. The ability to modulate by their activity using fat-soluble molecules makes them extremely attractive drug targets. As our understanding of NR signaling increases, so does our appreciation of the complexity of their regulation. It is possible that management of diseases in the future will include therapies that not only target NRs, but also co-regulator proteins and signaling pathways that are critical in the modulation of their function.
PPARs are the targets of some commonly used drugs in the treatment of hyperlipidemia and type-2-diabetes. Activation of PPARα by fibrates causes the up-regulation of genes involved in the β-oxidation of fatty acids. This results in the decreased synthesis of triglycerides and decreased LDL secreation by the liver 67. Glitazones such as rosiglitazone and pioglitazone are PPARγ agonists. PPARγ is known to regulate glucose homeostasis and adipogenesis, making it an attractive target for the treatment of type-2-diabetes. However, recent evidence has indicated an increased risk of heart attacks with rosiglitazone (marketed as Avandia) and the FDA released a safety alert on the drug in May 2007 206. Further research surrounding the signaling events and co-regulator proteins that affect PPARγ activity in multiple tissues may be useful in separating the therapeutic effects from the toxic effects of drugs like rosiglitazone.
One therapeutic challenge and opportunity in development of drugs that target NRs are selective therapeutic modulators (SRMs). SRMs are NR ligands that exhibit agonistic or antagonistic activity in a cell- or tissue-dependent manner. The classic SRM is tamoxifen, which can selectively activate or inhibit estrogen receptor and is commonly used in the treatment of breast cancer. Tamoxifen exhibits agonist (estrogen-like) activity in uterus and antagonist (anti-estrogen-like) activity in breast 207. SRM-induced alterations in the conformation of NRs may affect the ability of the receptor to bind to co-regulators or to be phosphorylated. The expression profile of specific co-activator proteins and co-repressor proteins in a given cell type may affect the relative agonist –vs-antagonist activity of SRMs. However, as evidenced in this review, it is likely that cellular signaling events contribute to SRM activity due to altered activation, binding, and localization of co-regulator proteins, as well as NRs. Increased understanding of the effect of cellular signaling on NRs and their co-regulator proteins has the potential to aid in the process of discovery of novel SRMs and the development of new and more effective drug therapeutics.
Most NRs regulate a myriad of target genes that control multiple processes. One of the challenges in designing NR agonists is separating the desired therapeutic effects from the undesirable side effects. For example, the functional ability of LXRs to promote reverse cholesterol transport, improve glucose tolerance, and alleviate inflammation makes them attractive drug targets for the treatment of metabolic and inflammatory diseases. However, the finding that first generation synthetic ligands of LXR increase hepatic lipogenesis and plasma triglyceride levels is a therapeutic obstacle that needs to be overcome 208. The increase in hepatic lipogenesis has been attributed to the LXR mediated induction of SREBP-1c, therefore an agonist designed to increase reverse cholesterol transport but not to induce SREBP-1c may be a more effective therapy. Loss of LXR results in the increased expression of ABCA1 and decreased expression of SREBP-1c suggesting that LXRs interact differentially with the transcriptional machinery on either promoter. Better understanding of the differential mechanisms and signaling pathways that interface with LXR during activation of specific target genes may provide insight in to the design of a selective agonist or may present new drug targets.
Understanding of the signaling mechanisms that interface with NRs could also be useful in modulating the effect of a receptor without directly targeting it, or in the development of therapeutic molecules that only induce specific NR-target genes. There are multiple areas of potential therapeutic usefulness for FXR modulators such as cholestatic disorders, fatty liver disorder, or metabolic and inflammatory diseases. However, activation of FXR induces a complex physiological response that may lead to undesirable side effects in addition to the beneficial response. For example, the use of an FXR agonist in the regulation of glucose homeostasis may also result in the inhibition of bile acid synthesis and impact cholesterol excretion. Therefore, the identification of selective bile acid receptor modulators (SBARMs) may be necessary to target specific groups of genes modulated by FXR. Additionally, the identification of how signaling pathways intersect with and modulate FXR may provide additional therapeutic opportunities that don't target FXR itself. For example, FGF19 or FGFR4 may prove to have interesting therapeutic potential in cholesterol and bile acid regulation.
The xenobiotic receptors PXR and CAR may have useful implications in the treatment of cholestatic liver disease. However, it has been hypothesized that unwanted activation of PXR is responsible for nearly 60% of all drug-drug interactions. Due to their promiscuous nature PXR and CAR are capable of modulating a number of genes in response to many different ligands. PXR and CAR activation by a specific drug results in the in the increased metabolism of not only that drug, but other drugs that may be in the system as well. In order for PXR and CAR to be effective therapeutic targets, the activation of a potential therapeutic-target gene must be separated from the activation of genes involved in drug metabolism. A better understanding of the co-regulator proteins and signaling pathways that interface with PXR and CAR may provide alternative drug therapies toward that end. In addition, pharmaceutical companies commonly screen for PXR activation by drug candidates in rodent and human species in order to avoid future drug-drug interactions. However, there is a significant species-specific response of PXR- and CAR-target genes with respect to activating ligands and signaling pathways. Understanding the signaling pathways that affect these two receptors may also be useful in the development of more accurate activation assays in order to predict and prevent unwanted and potentially lethal drug-drug interactions.
Conclusion
It is clear that multiple signaling pathways and phosphorylation events affect NR-mediated signaling. They modulate protein-protein interactions, sub-cellular localization, DNA-binding, protein stability, and transactivation capacity. The situation is further complicated by the fact that many NR cofactor proteins are themselves modulated by signaling pathways and phosphorylation events that affect their intrinsic and recruited enzymatic activities. Further investigation into the role of cell signaling pathways in NR-mediated transcription, and into signaling pathway crosstalk will be necessary to fully understand the functional implication of these signaling events. In addition, further characterization of these processes will likely lead to the development of novel and selective therapeutic molecules for a multitude of indications.
Figure 1. Activation of signaling pathways modulates nuclear receptor transcriptional activity.
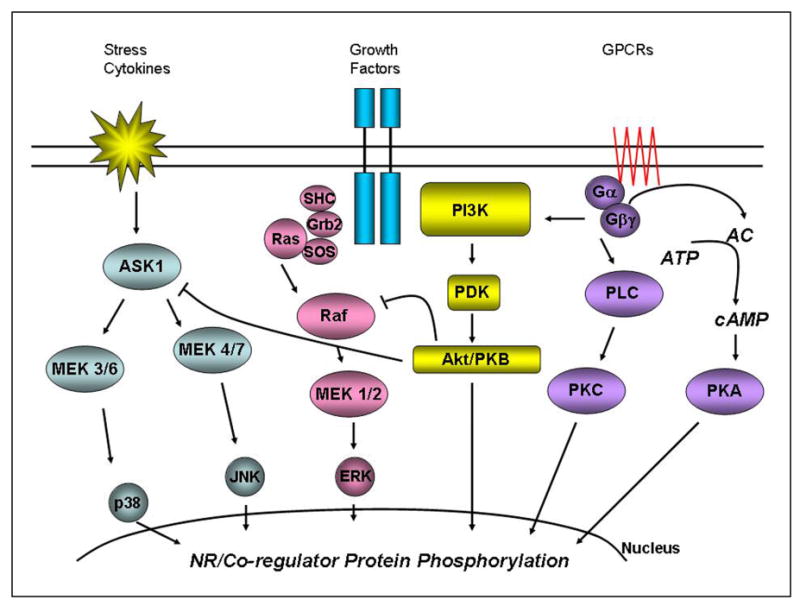
Activation of MAPK signaling cascades (p38, JNK, and ERK), FGF signaling, and GPCR signaling results in phosphorylation-dependent modulation of NR activity. Signaling pathways and phosphorylation events affect nuclear receptors or nuclear receptor cofactors through the modulation of protein–protein interactions, subcellular localization, DNA-binding, protein stability, and transactivation capacity. The interface between signal transduction pathways and NRs is critical in the responsiveness of the system to environmental and physiological stimuli.
References
- 1.Maglich JM, Sluder A, Guan X, Shi Y, McKee DD, Carrick K, Kamdar K, Willson TM, Moore JT. Genome Biol. 2001;2(8):29.1–29.7. doi: 10.1186/gb-2001-2-8-research0029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar R, Johnson BH, Thompson EB. Essays Biochem. 2004;40:27–39. doi: 10.1042/bse0400027. [DOI] [PubMed] [Google Scholar]
- 3.Glass CK, Rosenfeld MG. Genes Dev. 2000;14(2):121–41. [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Evans RM. Cell. 1995;83(6):841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Nagpal S, Friant S, Nakshatri H, Chambon P. Embo J. 1993;12(6):2349–60. doi: 10.1002/j.1460-2075.1993.tb05889.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumann CT, Maruvada P, Hager GL, Yen PM. J Biol Chem. 2001;276(14):11237–45. doi: 10.1074/jbc.M011112200. [DOI] [PubMed] [Google Scholar]
- 7.DeFranco DB, Ramakrishnan C, Tang Y. J Steroid Biochem Mol Biol. 1998;65(16):51–8. doi: 10.1016/s0960-0760(97)00177-5. [DOI] [PubMed] [Google Scholar]
- 8.Tcherepanova I, Puigserver P, Norris JD, Spiegelman BM, McDonnell DP. J Biol Chem. 2000;275(21):16302–8. doi: 10.1074/jbc.M001364200. [DOI] [PubMed] [Google Scholar]
- 9.Morgan DO. Annu Rev Cell Dev Biol. 1997;13:261–91. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 10.Morgan DO. Nature. 1995;374(6518):131–4. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 11.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Endocr Rev. 2001;22(2):153–83. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 12.Rochette-Egly C. Cell Signal. 2003;15(4):355–66. doi: 10.1016/s0898-6568(02)00115-8. [DOI] [PubMed] [Google Scholar]
- 13.Lange CA, Shen T, Horwitz KB. Proc Natl Acad Sci U S A. 2000;97(3):1032–7. doi: 10.1073/pnas.97.3.1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen T, Horwitz KB, Lange CA. Mol Cell Biol. 2001;21(18):6122–31. doi: 10.1128/MCB.21.18.6122-6131.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bunone G, Briand PA, Miksicek RJ, Picard D. Embo J. 1996;15(9):2174–83. [PMC free article] [PubMed] [Google Scholar]
- 16.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, Metzger D, Chambon P. Science. 1995;270(5241):1491–4. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 17.Driggers PH, Segars JH, Rubino DM. J Biol Chem. 2001;276(50):46792–7. doi: 10.1074/jbc.M106927200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tremblay A, Tremblay GB, Labrie F, Giguere V. Mol Cell. 1999;3(4):513–9. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 19.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. Proc Natl Acad Sci U S A. 1999;96(10):5458–63. doi: 10.1073/pnas.96.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu E, Kim JB, Sarraf P, Spiegelman BM. Science. 1996;274(5295):2100–3. doi: 10.1126/science.274.5295.2100. [DOI] [PubMed] [Google Scholar]
- 21.Juge-Aubry CE, Hammar E, Siegrist-Kaiser C, Pernin A, Takeshita A, Chin WW, Burger AG, Meier CA. J Biol Chem. 1999;274(15):10505–10. doi: 10.1074/jbc.274.15.10505. [DOI] [PubMed] [Google Scholar]
- 22.Gianni M, Bauer A, Garattini E, Chambon P, Rochette-Egly C. Embo J. 2002;21(14):3760–9. doi: 10.1093/emboj/cdf374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gianni M, Kopf E, Bastien J, Oulad-Abdelghani M, Garattini E, Chambon P, Rochette-Egly C. J Biol Chem. 2002;277(28):24859–62. doi: 10.1074/jbc.C200230200. [DOI] [PubMed] [Google Scholar]
- 24.Alessi DR, Cohen P. Curr Opin Genet Dev. 1998;8(1):55–62. doi: 10.1016/s0959-437x(98)80062-2. [DOI] [PubMed] [Google Scholar]
- 25.Scheid MP, Woodgett JR. Nat Rev Mol Cell Biol. 2001;2(10):760–8. doi: 10.1038/35096067. [DOI] [PubMed] [Google Scholar]
- 26.Datta SR, Brunet A, Greenberg ME. Genes Dev. 1999;13(22):2905–27. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 27.Campbell RA, Bhat-Nakshatri P, Patel NM, Constantinidou D, Ali S, Nakshatri H. J Biol Chem. 2001;276(13):9817–24. doi: 10.1074/jbc.M010840200. [DOI] [PubMed] [Google Scholar]
- 28.Lin HK, Yeh S, Kang HY, Chang C. Proc Natl Acad Sci U S A. 2001;98(13):7200–5. doi: 10.1073/pnas.121173298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam-Stitah S, Penna L, Chambon P, Rochette-Egly C. J Biol Chem. 1999;274(27):18932–41. doi: 10.1074/jbc.274.27.18932. [DOI] [PubMed] [Google Scholar]
- 30.Lee HY, Suh YA, Robinson MJ, Clifford JL, Hong WK, Woodgett JR, Cobb MH, Mangelsdorf DJ, Kurie JM. J Biol Chem. 2000;275(41):32193–9. doi: 10.1074/jbc.M005490200. [DOI] [PubMed] [Google Scholar]
- 31.Arnold SF, Obourn JD, Jaffe H, Notides AC. Mol Endocrinol. 1995;9(1):24–33. doi: 10.1210/mend.9.1.7539106. [DOI] [PubMed] [Google Scholar]
- 32.Migliaccio A, Pagano M, Auricchio F. Oncogene. 1993;8(8):2183–91. [PubMed] [Google Scholar]
- 33.Rochette-Egly C, Oulad-Abdelghani M, Staub A, Pfister V, Scheuer I, Chambon P, Gaub MP. Mol Endocrinol. 1995;9(7):860–71. doi: 10.1210/mend.9.7.7476969. [DOI] [PubMed] [Google Scholar]
- 34.Chen D, Pace PE, Coombes RC, Ali S. Mol Cell Biol. 1999;19(2):1002–15. doi: 10.1128/mcb.19.2.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Delmotte MH, Tahayato A, Formstecher P, Lefebvre P. J Biol Chem. 1999;274(53):38225–31. doi: 10.1074/jbc.274.53.38225. [DOI] [PubMed] [Google Scholar]
- 36.Hsieh JC, Jurutka PW, Galligan MA, Terpening CM, Haussler CA, Samuels DS, Shimizu Y, Shimizu N, Haussler MR. Proc Natl Acad Sci U S A. 1991;88(20):9315–9. doi: 10.1073/pnas.88.20.9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hsieh JC, Jurutka PW, Nakajima S, Galligan MA, Haussler CA, Shimizu Y, Shimizu N, Whitfield GK, Haussler MR. J Biol Chem. 1993;268(20):15118–26. [PubMed] [Google Scholar]
- 38.Hayhurst GP, Lee YH, Lambert G, Ward JM, Gonzalez FJ. Mol Cell Biol. 2001;21(4):1393–403. doi: 10.1128/MCB.21.4.1393-1403.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stroup D, Chiang JY. J Lipid Res. 2000;41(1):1–11. [PubMed] [Google Scholar]
- 40.Ding X, Lichti K, Kim I, Gonzalez FJ, Staudinger JL. J Biol Chem. 2006;281(36):26540–51. doi: 10.1074/jbc.M600931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stoffel M, Duncan SA. Proc Natl Acad Sci U S A. 1997;94(24):13209–14. doi: 10.1073/pnas.94.24.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wan YJ, Cai Y, Cowan C, Magee TR. Cancer Lett. 2000;154(1):19–27. doi: 10.1016/s0304-3835(00)00341-4. [DOI] [PubMed] [Google Scholar]
- 43.Viollet B, Kahn A, Raymondjean M. Mol Cell Biol. 1997;17(8):4208–19. doi: 10.1128/mcb.17.8.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang G, Nepomuceno L, Yang Q, Sladek FM. Arch Biochem Biophys. 1997;340(1):1–9. doi: 10.1006/abbi.1997.9914. [DOI] [PubMed] [Google Scholar]
- 45.Ktistaki E, Ktistakis NT, Papadogeorgaki E, Talianidis I. Proc Natl Acad Sci U S A. 1995;92(21):9876–80. doi: 10.1073/pnas.92.21.9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo H, Gao C, Mi Z, Zhang J, Kuo PC. J Biochem (Tokyo) 2007;141(5):635–40. doi: 10.1093/jb/mvm066. [DOI] [PubMed] [Google Scholar]
- 47.Xu Z, Tavares-Sanchez OL, Li Q, Fernando J, Rodriguez CM, Studer EJ, Pandak WM, Hylemon PB, Gil G. J Biol Chem. 2007 doi: 10.1074/jbc.M611481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang JC, Stafford JM, Granner DK. J Biol Chem. 1998;273(47):30847–50. doi: 10.1074/jbc.273.47.30847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshida E, Aratani S, Itou H, Miyagishi M, Takiguchi M, Osumu T, Murakami K, Fukamizu A. Biochem Biophys Res Commun. 1997;241(3):664–9. doi: 10.1006/bbrc.1997.7871. [DOI] [PubMed] [Google Scholar]
- 50.Desvergne B, Wahli W. Endocr Rev. 1999;20(5):649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 51.Escher P, Wahli W. Mutat Res. 2000;448(2):121–38. doi: 10.1016/s0027-5107(99)00231-6. [DOI] [PubMed] [Google Scholar]
- 52.Auboeuf D, Rieusset J, Fajas L, Vallier P, Frering V, Riou JP, Staels B, Auwerx J, Laville M, Vidal H. Diabetes. 1997;46(8):1319–27. doi: 10.2337/diab.46.8.1319. [DOI] [PubMed] [Google Scholar]
- 53.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. J Biol Chem. 1995;270(22):12953–6. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- 54.Camp HS, Tafuri SR. J Biol Chem. 1997;272(16):10811–6. doi: 10.1074/jbc.272.16.10811. [DOI] [PubMed] [Google Scholar]
- 55.Adams M, Reginato MJ, Shao D, Lazar MA, Chatterjee VK. J Biol Chem. 1997;272(8):5128–32. doi: 10.1074/jbc.272.8.5128. [DOI] [PubMed] [Google Scholar]
- 56.Zhang B, Berger J, Zhou G, Elbrecht A, Biswas S, White-Carrington S, Szalkowski D, Moller DE. J Biol Chem. 1996;271(50):31771–4. doi: 10.1074/jbc.271.50.31771. [DOI] [PubMed] [Google Scholar]
- 57.Barger PM, Browning AC, Garner AN, Kelly DP. J Biol Chem. 2001;276(48):44495–501. doi: 10.1074/jbc.M105945200. [DOI] [PubMed] [Google Scholar]
- 58.Lazennec G, Canaple L, Saugy D, Wahli W. Mol Endocrinol. 2000;14(12):1962–75. doi: 10.1210/mend.14.12.0575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yaacob NS, Norazmi MN, Gibson GG, Kass GE. Toxicol Lett. 2001;125(13):133–41. doi: 10.1016/s0378-4274(01)00433-7. [DOI] [PubMed] [Google Scholar]
- 60.Chen F, Ma L, Dawson PA, Sinal CJ, Sehayek E, Gonzalez FJ, Breslow J, Ananthanarayanan M, Shneider BL. J Biol Chem. 2003;278(22):19909–16. doi: 10.1074/jbc.M207903200. [DOI] [PubMed] [Google Scholar]
- 61.Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. Mol Pharmacol. 2002;62(3):638–46. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- 62.Sonoda J, Xie W, Rosenfeld JM, Barwick JL, Guzelian PS, Evans RM. Proc Natl Acad Sci U S A. 2002;99(21):13801–6. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei P, Zhang J, Dowhan DH, Han Y, Moore DD. Pharmacogenomics J. 2002;2(2):117–26. doi: 10.1038/sj.tpj.6500087. [DOI] [PubMed] [Google Scholar]
- 64.Geick A, Eichelbaum M, Burk O. J Biol Chem. 2001;276(18):14581–7. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 65.Kast HR, Goodwin B, Tarr PT, Jones SA, Anisfeld AM, Stoltz CM, Tontonoz P, Kliewer S, Willson TM, Edwards PA. J Biol Chem. 2002;277(4):2908–15. doi: 10.1074/jbc.M109326200. [DOI] [PubMed] [Google Scholar]
- 66.Staudinger JL, Madan A, Carol KM, Parkinson A. Drug Metab Dispos. 2003;31(5):523–7. doi: 10.1124/dmd.31.5.523. [DOI] [PubMed] [Google Scholar]
- 67.Francis GA, Fayard E, Picard F, Auwerx J. Annu Rev Physiol. 2003;65:261–311. doi: 10.1146/annurev.physiol.65.092101.142528. [DOI] [PubMed] [Google Scholar]
- 68.Hartmann G, Kim H, Piquette-Miller M. Int Immunopharmacol. 2001;1(2):189–99. doi: 10.1016/s0162-3109(00)00271-x. [DOI] [PubMed] [Google Scholar]
- 69.Hartmann G, Cheung AK, Piquette-Miller M. J Pharmacol Exp Ther. 2002;303(1):273–81. doi: 10.1124/jpet.102.039404. [DOI] [PubMed] [Google Scholar]
- 70.Pascussi JM, Gerbal-Chaloin S, Pichard-Garcia L, Daujat M, Fabre JM, Maurel P, Vilarem MJ. Biochem Biophys Res Commun. 2000;274(3):707–13. doi: 10.1006/bbrc.2000.3219. [DOI] [PubMed] [Google Scholar]
- 71.Gu X, Ke S, Liu D, Sheng T, Thomas PE, Rabson AB, Gallo MA, Xie W, Tian Y. J Biol Chem. 2006;281(26):17882–9. doi: 10.1074/jbc.M601302200. [DOI] [PubMed] [Google Scholar]
- 72.Zhou C, Tabb MM, Nelson EL, Grun F, Verma S, Sadatrafiei A, Lin M, Mallick S, Forman BM, Thummel KE, Blumberg B. J Clin Invest. 2006;116(8):2280–2289. doi: 10.1172/JCI26283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shah YM, Ma X, Morimura K, Kim I, Gonzalez FJ. Am J Physiol Gastrointest Liver Physiol. 2007;292(4):G1114–22. doi: 10.1152/ajpgi.00528.2006. [DOI] [PubMed] [Google Scholar]
- 74.Sidhu JS, Omiecinski CJ. J Biol Chem. 1995;270(21):12762–73. doi: 10.1074/jbc.270.21.12762. [DOI] [PubMed] [Google Scholar]
- 75.Sidhu JS, Omiecinski CJ. J Pharmacol Exp Ther. 1996;276(1):238–45. [PubMed] [Google Scholar]
- 76.Ding X, Staudinger JL. J Pharmacol Exp Ther. 2005;312(2):849–56. doi: 10.1124/jpet.104.076331. [DOI] [PubMed] [Google Scholar]
- 77.Ding X, Staudinger JL. Biochem Pharmacol. 2005;69(5):867–73. doi: 10.1016/j.bcp.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 78.Honkakoski P, Zelko I, Sueyoshi T, Negishi M. Mol Cell Biol. 1998;18(10):5652–8. doi: 10.1128/mcb.18.10.5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pascussi JM, Dvorak Z, Gerbal-Chaloin S, Assenat E, Maurel P, Vilarem MJ. Drug Metab Rev. 2003;35(4):255–68. doi: 10.1081/dmr-120026394. [DOI] [PubMed] [Google Scholar]
- 80.Ueda A, Hamadeh HK, Webb HK, Yamamoto Y, Sueyoshi T, Afshari CA, Lehmann JM, Negishi M. Mol Pharmacol. 2002;61(1):1–6. doi: 10.1124/mol.61.1.1. [DOI] [PubMed] [Google Scholar]
- 81.Sidhu JS, Omiecinski CJ. J Pharmacol Exp Ther. 1997;282(2):1122–9. [PubMed] [Google Scholar]
- 82.Joannard F, Galisteo M, Corcos L, Guillouzo A, Lagadic-Gossmann D. Cell Biol Toxicol. 2000;16(5):325–37. doi: 10.1023/a:1026702615125. [DOI] [PubMed] [Google Scholar]
- 83.Pustylnyak VO, Gulyaeva LF, Lyakhovich VV. Toxicology. 2005;216(23):147–53. doi: 10.1016/j.tox.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 84.Kawamoto T, Sueyoshi T, Zelko I, Moore R, Washburn K, Negishi M. Mol Cell Biol. 1999;19(9):6318–22. doi: 10.1128/mcb.19.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hosseinpour F, Moore R, Negishi M, Sueyoshi T. Mol Pharmacol. 2006;69(4):1095–102. doi: 10.1124/mol.105.019505. [DOI] [PubMed] [Google Scholar]
- 86.Swales K, Negishi M. Mol Endocrinol. 2004;18(7):1589–98. doi: 10.1210/me.2003-0397. [DOI] [PubMed] [Google Scholar]
- 87.Bauer D, Wolfram N, Kahl GF, Hirsch-Ernst KI. Mol Pharmacol. 2004;65(1):172–80. doi: 10.1124/mol.65.1.172. [DOI] [PubMed] [Google Scholar]
- 88.Joannard F, Rissel M, Gilot D, Anderson A, Orfila-Lefeuvre L, Guillouzo A, Atfi A, Lagadic-Gossmann D. Toxicol Lett. 2006;161(1):61–72. doi: 10.1016/j.toxlet.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 89.Koike C, Moore R, Negishi M. Mol Pharmacol. 2007;71(5):1217–21. doi: 10.1124/mol.107.034538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rencurel F, Stenhouse A, Hawley SA, Friedberg T, Hardie DG, Sutherland C, Wolf CR. J Biol Chem. 2005;280(6):4367–73. doi: 10.1074/jbc.M412711200. [DOI] [PubMed] [Google Scholar]
- 91.Rencurel F, Foretz M, Kaufmann MR, Stroka D, Looser R, Leclerc I, da Silva Xavier G, Rutter GA, Viollet B, Meyer UA. Mol Pharmacol. 2006;70(6):1925–34. doi: 10.1124/mol.106.029421. [DOI] [PubMed] [Google Scholar]
- 92.Shindo S, Numazawa S, Yoshida T. Biochem J. 2007;401(3):735–41. doi: 10.1042/BJ20061238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Blattler SM, Rencurel F, Kaufmann MR, Meyer UA. Proc Natl Acad Sci U S A. 2007;104(3):1045–50. doi: 10.1073/pnas.0610216104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beaven SW, Tontonoz P. Annu Rev Med. 2006;57:313–29. doi: 10.1146/annurev.med.57.121304.131428. [DOI] [PubMed] [Google Scholar]
- 95.Janowski BA, Willy PJ, Devi TR, Falck JR, Mangelsdorf DJ. Nature. 1996;383(6602):728–31. doi: 10.1038/383728a0. [DOI] [PubMed] [Google Scholar]
- 96.Song C, Liao S. Endocrinology. 2000;141(11):4180–4. doi: 10.1210/endo.141.11.7772. [DOI] [PubMed] [Google Scholar]
- 97.Chiang JY, Kimmel R, Stroup D. Gene. 2001;262(12):257–65. doi: 10.1016/s0378-1119(00)00518-7. [DOI] [PubMed] [Google Scholar]
- 98.Costet P, Luo Y, Wang N, Tall AR. J Biol Chem. 2000;275(36):28240–5. doi: 10.1074/jbc.M003337200. [DOI] [PubMed] [Google Scholar]
- 99.Mak PA, Laffitte BA, Desrumaux C, Joseph SB, Curtiss LK, Mangelsdorf DJ, Tontonoz P, Edwards PA. J Biol Chem. 2002;277(35):31900–8. doi: 10.1074/jbc.M202993200. [DOI] [PubMed] [Google Scholar]
- 100.Ulven SM, Dalen KT, Gustafsson JA, Nebb HI. J Lipid Res. 2004;45(11):2052–62. doi: 10.1194/jlr.M400119-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.Cao G, Liang Y, Broderick CL, Oldham BA, Beyer TP, Schmidt RJ, Zhang Y, Stayrook KR, Suen C, Otto KA, Miller AR, Dai J, Foxworthy P, Gao H, Ryan TP, Jiang XC, Burris TP, Eacho PI, Etgen GJ. J Biol Chem. 2003;278(2):1131–6. doi: 10.1074/jbc.M210208200. [DOI] [PubMed] [Google Scholar]
- 102.Castrillo A, Joseph SB, Marathe C, Mangelsdorf DJ, Tontonoz P. J Biol Chem. 2003;278(12):10443–9. doi: 10.1074/jbc.M213071200. [DOI] [PubMed] [Google Scholar]
- 103.Joseph SB, Castrillo A, Laffitte BA, Mangelsdorf DJ, Tontonoz P. Nat Med. 2003;9(2):213–9. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 104.Chen M, Bradley MN, Beaven SW, Tontonoz P. FEBS Lett. 2006;580(20):4835–41. doi: 10.1016/j.febslet.2006.07.074. [DOI] [PubMed] [Google Scholar]
- 105.Huang CJ, Feltkamp D, Nilsson S, Gustafsson JA. Biochem Biophys Res Commun. 1998;243(3):657–63. doi: 10.1006/bbrc.1998.8152. [DOI] [PubMed] [Google Scholar]
- 106.Tamura K, Chen YE, Horiuchi M, Chen Q, Daviet L, Yang Z, Lopez-Ilasaca M, Mu H, Pratt RE, Dzau VJ. Proc Natl Acad Sci U S A. 2000;97(15):8513–8. doi: 10.1073/pnas.100519097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tamura K, Chen YE, Tanaka Y, Sakai M, Tsurumi Y, Koide Y, Kihara M, Pratt RE, Horiuchi M, Umemura S, Dzau VJ. Mol Cell Endocrinol. 2004;224(12):11–20. doi: 10.1016/j.mce.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 108.Yamamoto T, Shimano H, Inoue N, Nakagawa Y, Matsuzaka T, Takahashi A, Yahagi N, Sone H, Suzuki H, Toyoshima H, Yamada N. J Biol Chem. 2007;282(16):11687–95. doi: 10.1074/jbc.M611911200. [DOI] [PubMed] [Google Scholar]
- 109.Terasaka N, Hiroshima A, Ariga A, Honzumi S, Koieyama T, Inaba T, Fujiwara T. Febs J. 2005;272(6):1546–56. doi: 10.1111/j.1742-4658.2005.04599.x. [DOI] [PubMed] [Google Scholar]
- 110.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. Nature. 2005;437(7059):759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lu TT, Repa JJ, Mangelsdorf DJ. J Biol Chem. 2001;276(41):37735–8. doi: 10.1074/jbc.R100035200. [DOI] [PubMed] [Google Scholar]
- 112.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, Wilson JG, Lewis MC, Roth ME, Maloney PR, Willson TM, Kliewer SA. Mol Cell. 2000;6(3):517–26. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 113.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Mol Cell. 2000;6(3):507–15. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 114.Bohan A, Chen WS, Denson LA, Held MA, Boyer JL. J Biol Chem. 2003;278(38):36688–98. doi: 10.1074/jbc.M304011200. [DOI] [PubMed] [Google Scholar]
- 115.del Castillo-Olivares A, Gil G. J Biol Chem. 2000;275(23):17793–9. doi: 10.1074/jbc.M000996200. [DOI] [PubMed] [Google Scholar]
- 116.Delerive P, Galardi CM, Bisi JE, Nicodeme E, Goodwin B. Mol Endocrinol. 2004;18(10):2378–87. doi: 10.1210/me.2004-0132. [DOI] [PubMed] [Google Scholar]
- 117.Inokuchi A, Hinoshita E, Iwamoto Y, Kohno K, Kuwano M, Uchiumi T. J Biol Chem. 2001;276(50):46822–9. doi: 10.1074/jbc.M104612200. [DOI] [PubMed] [Google Scholar]
- 118.Luo Y, Liang CP, Tall AR. J Biol Chem. 2001;276(27):24767–73. doi: 10.1074/jbc.M100912200. [DOI] [PubMed] [Google Scholar]
- 119.Schoonjans K, Annicotte JS, Huby T, Botrugno OA, Fayard E, Ueda Y, Chapman J, Auwerx J. EMBO Rep. 2002;3(12):1181–7. doi: 10.1093/embo-reports/kvf238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lee YK, Choi YH, Chua S, Park YJ, Moore DD. J Biol Chem. 2006;281(12):7850–5. doi: 10.1074/jbc.M509115200. [DOI] [PubMed] [Google Scholar]
- 121.Li T, Jahan A, Chiang JY. Hepatology. 2006;43(6):1202–10. doi: 10.1002/hep.21183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stroup D. Front Biosci. 2005;10:1678–92. doi: 10.2741/1652. [DOI] [PubMed] [Google Scholar]
- 123.Krylova IN, Sablin EP, Moore J, Xu RX, Waitt GM, MacKay JA, Juzumiene D, Bynum JM, Madauss K, Montana V, Lebedeva L, Suzawa M, Williams JD, Williams SP, Guy RK, Thornton JW, Fletterick RJ, Willson TM, Ingraham HA. Cell. 2005;120(3):343–55. doi: 10.1016/j.cell.2005.01.024. [DOI] [PubMed] [Google Scholar]
- 124.Fiorucci S, Rizzo G, Donini A, Distrutti E, Santucci L. Trends Mol Med. 2007;13(7):298–309. doi: 10.1016/j.molmed.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 125.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Science. 1999;284(5418):1362–5. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 126.Parks DJ, Blanchard SG, Bledsoe RK, Chandra G, Consler TG, Kliewer SA, Stimmel JB, Willson TM, Zavacki AM, Moore DD, Lehmann JM. Science. 1999;284(5418):1365–8. doi: 10.1126/science.284.5418.1365. [DOI] [PubMed] [Google Scholar]
- 127.Wang H, Chen J, Hollister K, Sowers LC, Forman BM. Mol Cell. 1999;3(5):543–53. doi: 10.1016/s1097-2765(00)80348-2. [DOI] [PubMed] [Google Scholar]
- 128.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Cell. 2000;102(6):731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 129.Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, Kuipers F, Staels B. Diabetes. 2004;53(4):890–8. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 130.Zhang Y, Lee FY, Barrera G, Lee H, Vales C, Gonzalez FJ, Willson TM, Edwards PA. Proc Natl Acad Sci U S A. 2006;103(4):1006–11. doi: 10.1073/pnas.0506982103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, Kuipers F, Staels B. J Biol Chem. 2006;281(16):11039–49. doi: 10.1074/jbc.M510258200. [DOI] [PubMed] [Google Scholar]
- 132.Duran-Sandoval D, Cariou B, Percevault F, Hennuyer N, Grefhorst A, van Dijk TH, Gonzalez FJ, Fruchart JC, Kuipers F, Staels B. J Biol Chem. 2005;280(33):29971–9. doi: 10.1074/jbc.M501931200. [DOI] [PubMed] [Google Scholar]
- 133.Ma K, Saha PK, Chan L, Moore DD. J Clin Invest. 2006;116(4):1102–9. doi: 10.1172/JCI25604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang Y, Castellani LW, Sinal CJ, Gonzalez FJ, Edwards PA. Genes Dev. 2004;18(2):157–69. doi: 10.1101/gad.1138104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gupta S, Stravitz RT, Dent P, Hylemon PB. J Biol Chem. 2001;276(19):15816–22. doi: 10.1074/jbc.M010878200. [DOI] [PubMed] [Google Scholar]
- 136.Rao YP, Studer EJ, Stravitz RT, Gupta S, Qiao L, Dent P, Hylemon PB. Hepatology. 2002;35(2):307–14. doi: 10.1053/jhep.2002.31104. [DOI] [PubMed] [Google Scholar]
- 137.Stravitz RT, Vlahcevic ZR, Gurley EC, Hylemon PB. J Lipid Res. 1995;36(6):1359–69. [PubMed] [Google Scholar]
- 138.Seol W, Choi HS, Moore DD. Science. 1996;272(5266):1336–9. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 139.Lee HK, Lee YK, Park SH, Kim YS, Park SH, Lee JW, Kwon HB, Soh J, Moore DD, Choi HS. J Biol Chem. 1998;273(23):14398–402. doi: 10.1074/jbc.273.23.14398. [DOI] [PubMed] [Google Scholar]
- 140.Masuda N, Yasumo H, Tamura T, Hashiguchi N, Furusawa T, Tsukamoto T, Sadano H, Osumi T. Biochim Biophys Acta. 1997;1350(1):27–32. doi: 10.1016/s0167-4781(96)00196-0. [DOI] [PubMed] [Google Scholar]
- 141.Lee YK, Moore DD. J Biol Chem. 2002;277(4):2463–7. doi: 10.1074/jbc.M105161200. [DOI] [PubMed] [Google Scholar]
- 142.Brendel C, Schoonjans K, Botrugno OA, Treuter E, Auwerx J. Mol Endocrinol. 2002;16(9):2065–76. doi: 10.1210/me.2001-0194. [DOI] [PubMed] [Google Scholar]
- 143.Lee YK, Dell H, Dowhan DH, Hadzopoulou-Cladaras M, Moore DD. Mol Cell Biol. 2000;20(1):187–95. doi: 10.1128/mcb.20.1.187-195.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Seol W, Chung M, Moore DD. Mol Cell Biol. 1997;17(12):7126–31. doi: 10.1128/mcb.17.12.7126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kassam A, Capone JP, Rachubinski RA. Mol Cell Endocrinol. 2001;176(12):49–56. doi: 10.1016/s0303-7207(01)00475-0. [DOI] [PubMed] [Google Scholar]
- 146.Nishizawa H, Yamagata K, Shimomura I, Takahashi M, Kuriyama H, Kishida K, Hotta K, Nagaretani H, Maeda N, Matsuda M, Kihara S, Nakamura T, Nishigori H, Tomura H, Moore DD, Takeda J, Funahashi T, Matsuzawa Y. J Biol Chem. 2002;277(2):1586–92. doi: 10.1074/jbc.M104301200. [DOI] [PubMed] [Google Scholar]
- 147.Xie MH, Holcomb I, Deuel B, Dowd P, Huang A, Vagts A, Foster J, Liang J, Brush J, Gu Q, Hillan K, Goddard A, Gurney AL. Cytokine. 1999;11(10):729–35. doi: 10.1006/cyto.1999.0485. [DOI] [PubMed] [Google Scholar]
- 148.Holt JA, Luo G, Billin AN, Bisi J, McNeill YY, Kozarsky KF, Donahee M, Wang DY, Mansfield TA, Kliewer SA, Goodwin B, Jones SA. Genes Dev. 2003;17(13):1581–91. doi: 10.1101/gad.1083503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fu L, John LM, Adams SH, Yu XX, Tomlinson E, Renz M, Williams PM, Soriano R, Corpuz R, Moffat B, Vandlen R, Simmons L, Foster J, Stephan JP, Tsai SP, Stewart TA. Endocrinology. 2004;145(6):2594–603. doi: 10.1210/en.2003-1671. [DOI] [PubMed] [Google Scholar]
- 150.Tomlinson E, Fu L, John L, Hultgren B, Huang X, Renz M, Stephan JP, Tsai SP, Powell-Braxton L, French D, Stewart TA. Endocrinology. 2002;143(5):1741–7. doi: 10.1210/endo.143.5.8850. [DOI] [PubMed] [Google Scholar]
- 151.Nakatani Y, Kaneto H, Kawamori D, Hatazaki M, Miyatsuka T, Matsuoka TA, Kajimoto Y, Matsuhisa M, Yamasaki Y, Hori M. J Biol Chem. 2004;279(44):45803–9. doi: 10.1074/jbc.M406963200. [DOI] [PubMed] [Google Scholar]
- 152.Yu C, Wang F, Kan M, Jin C, Jones RB, Weinstein M, Deng CX, McKeehan WL. J Biol Chem. 2000;275(20):15482–9. doi: 10.1074/jbc.275.20.15482. [DOI] [PubMed] [Google Scholar]
- 153.Yu C, Wang F, Jin C, Huang X, McKeehan WL. J Biol Chem. 2005;280(18):17707–14. doi: 10.1074/jbc.M411771200. [DOI] [PubMed] [Google Scholar]
- 154.Inagaki T, Choi M, Moschetta A, Peng L, Cummins CL, McDonald JG, Luo G, Jones SA, Goodwin B, Richardson JA, Gerard RD, Repa JJ, Mangelsdorf DJ, Kliewer SA. Cell Metab. 2005;2(4):217–25. doi: 10.1016/j.cmet.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 155.Kharitonenkov A, Shiyanova TL, Koester A, Ford AM, Micanovic R, Galbreath EJ, Sandusky GE, Hammond LJ, Moyers JS, Owens RA, Gromada J, Brozinick JT, Hawkins ED, Wroblewski VJ, Li DS, Mehrbod F, Jaskunas SR, Shanafelt AB. J Clin Invest. 2005;115(6):1627–35. doi: 10.1172/JCI23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kharitonenkov A, Wroblewski VJ, Koester A, Chen YF, Clutinger CK, Tigno XT, Hansen BC, Shanafelt AB, Etgen GJ. Endocrinology. 2007;148(2):774–81. doi: 10.1210/en.2006-1168. [DOI] [PubMed] [Google Scholar]
- 157.Badman MK, Pissios P, Kennedy AR, Koukos G, Flier JS, Maratos-Flier E. Cell Metab. 2007;5(6):426–437. doi: 10.1016/j.cmet.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 158.Inagaki T, Dutchak P, Zhao G, Ding X, Gautron L, Parameswara V, Li Y, Goetz R, Mohammadi M, Esser V, Elmquist JK, Gerard RD, Burgess SC, Hammer RE, Mangelsdorf DJ, Kliewer SA. Cell Metab. 2007;5(6):415–25. doi: 10.1016/j.cmet.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 159.Chen JD, Evans RM. Nature. 1995;377(6548):454–7. doi: 10.1038/377454a0. [DOI] [PubMed] [Google Scholar]
- 160.Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B, Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK, et al. Nature. 1995;377(6548):397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 161.Privalsky ML. Annu Rev Physiol. 2004;66:315–60. doi: 10.1146/annurev.physiol.66.032802.155556. [DOI] [PubMed] [Google Scholar]
- 162.Hong SH, Wong CW, Privalsky ML. Mol Endocrinol. 1998;12(8):1161–71. doi: 10.1210/mend.12.8.0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hong SH, Privalsky ML. Mol Cell Biol. 2000;20(17):6612–25. doi: 10.1128/mcb.20.17.6612-6625.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhou Y, Gross W, Hong SH, Privalsky ML. Mol Cell Biochem. 2001;220(12):1–13. doi: 10.1023/a:1011087910699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jonas BA, Privalsky ML. J Biol Chem. 2004;279(52):54676–86. doi: 10.1074/jbc.M410128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jonas BA, Varlakhanova N, Hayakawa F, Goodson M, Privalsky ML. Mol Endocrinol. 2007 doi: 10.1210/me.2007-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hermanson O, Jepsen K, Rosenfeld MG. Nature. 2002;419(6910):934–9. doi: 10.1038/nature01156. [DOI] [PubMed] [Google Scholar]
- 168.Galasinski SC, Louie DF, Gloor KK, Resing KA, Ahn NG. J Biol Chem. 2002;277(4):2579–88. doi: 10.1074/jbc.M107894200. [DOI] [PubMed] [Google Scholar]
- 169.Tsai SC, Seto E. J Biol Chem. 2002;277(35):31826–33. doi: 10.1074/jbc.M204149200. [DOI] [PubMed] [Google Scholar]
- 170.Zhou X, Richon VM, Wang AH, Yang XJ, Rifkind RA, Marks PA. Proc Natl Acad Sci U S A. 2000;97(26):14329–33. doi: 10.1073/pnas.250494697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG. Cell. 2002;110(1):55–67. doi: 10.1016/s0092-8674(02)00809-7. [DOI] [PubMed] [Google Scholar]
- 172.Chen H, Lin RJ, Schiltz RL, Chakravarti D, Nash A, Nagy L, Privalsky ML, Nakatani Y, Evans RM. Cell. 1997;90(3):569–80. doi: 10.1016/s0092-8674(00)80516-4. [DOI] [PubMed] [Google Scholar]
- 173.Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ, O'Malley BW. Nature. 1997;389(6647):194–8. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- 174.Xu J, O'Malley BW. Rev Endocr Metab Disord. 2002;3(3):185–92. doi: 10.1023/a:1020016208071. [DOI] [PubMed] [Google Scholar]
- 175.Heery DM, Kalkhoven E, Hoare S, Parker MG. Nature. 1997;387(6634):733–6. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- 176.Rowan BG, Weigel NL, O'Malley BW. J Biol Chem. 2000;275(6):4475–83. doi: 10.1074/jbc.275.6.4475. [DOI] [PubMed] [Google Scholar]
- 177.Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O'Malley BW. Mol Cell. 2004;15(6):937–49. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 178.Font de Mora J, Brown M. Mol Cell Biol. 2000;20(14):5041–7. doi: 10.1128/mcb.20.14.5041-5047.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rowan BG, Garrison N, Weigel NL, O'Malley BW. Mol Cell Biol. 2000;20(23):8720–30. doi: 10.1128/mcb.20.23.8720-8730.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wu RC, Qin J, Hashimoto Y, Wong J, Xu J, Tsai SY, Tsai MJ, O'Malley BW. Mol Cell Biol. 2002;22(10):3549–61. doi: 10.1128/MCB.22.10.3549-3561.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lopez GN, Turck CW, Schaufele F, Stallcup MR, Kushner PJ. J Biol Chem. 2001;276(25):22177–82. doi: 10.1074/jbc.M010718200. [DOI] [PubMed] [Google Scholar]
- 182.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Nature. 1993;365(6449):855–9. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 183.Nakajima T, Fukamizu A, Takahashi J, Gage FH, Fisher T, Blenis J, Montminy MR. Cell. 1996;86(3):465–74. doi: 10.1016/s0092-8674(00)80119-1. [DOI] [PubMed] [Google Scholar]
- 184.Chawla S, Hardingham GE, Quinn DR, Bading H. Science. 1998;281(5382):1505–9. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- 185.Xu L, Lavinsky RM, Dasen JS, Flynn SE, McInerney EM, Mullen TM, Heinzel T, Szeto D, Korzus E, Kurokawa R, Aggarwal AK, Rose DW, Glass CK, Rosenfeld MG. Nature. 1998;395(6699):301–6. doi: 10.1038/26270. [DOI] [PubMed] [Google Scholar]
- 186.Larrouy D, Vidal H, Andreelli F, Laville M, Langin D. Int J Obes Relat Metab Disord. 1999;23(12):1327–32. doi: 10.1038/sj.ijo.0801106. [DOI] [PubMed] [Google Scholar]
- 187.Puigserver P, Spiegelman BM. Endocr Rev. 2003;24(1):78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 188.Spiegelman BM, Heinrich R. Cell. 2004;119(2):157–67. doi: 10.1016/j.cell.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 189.Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J, Adelmant G, Stafford J, Kahn CR, Granner DK, Newgard CB, Spiegelman BM. Nature. 2001;413(6852):131–8. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 190.Vega RB, Huss JM, Kelly DP. Mol Cell Biol. 2000;20(5):1868–76. doi: 10.1128/mcb.20.5.1868-1876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Shin DJ, Campos JA, Gil G, Osborne TF. J Biol Chem. 2003;278(50):50047–52. doi: 10.1074/jbc.M309736200. [DOI] [PubMed] [Google Scholar]
- 192.Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. Nature. 2001;413(6852):179–83. doi: 10.1038/35093131. [DOI] [PubMed] [Google Scholar]
- 193.Shaw RJ, Lamia KA, Vasquez D, Koo SH, Bardeesy N, Depinho RA, Montminy M, Cantley LC. Science. 2005;310(5754):1642–6. doi: 10.1126/science.1120781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Southgate RJ, Bruce CR, Carey AL, Steinberg GR, Walder K, Monks R, Watt MJ, Hawley JA, Birnbaum MJ, Febbraio MA. Faseb J. 2005;19(14):2072–4. doi: 10.1096/fj.05-3993fje. [DOI] [PubMed] [Google Scholar]
- 195.Crunkhorn S, Dearie F, Mantzoros C, Gami H, da Silva WS, Espinoza D, Faucette R, Barry K, Bianco AC, Patti ME. J Biol Chem. 2007;282(21):15439–50. doi: 10.1074/jbc.M611214200. [DOI] [PubMed] [Google Scholar]
- 196.Coll T, Jove M, Rodriguez-Calvo R, Eyre E, Palomer X, Sanchez RM, Merlos M, Laguna JC, Vazquez-Carrera M. Diabetes. 2006;55(10):2779–87. doi: 10.2337/db05-1494. [DOI] [PubMed] [Google Scholar]
- 197.Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P. Cell Metab. 2006;3(6):429–38. doi: 10.1016/j.cmet.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 198.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nature. 2005;434(7029):113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 199.Teyssier C, Ma H, Emter R, Kralli A, Stallcup MR. Genes Dev. 2005;19(12):1466–73. doi: 10.1101/gad.1295005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, Krauss S, Mootha VK, Lowell BB, Spiegelman BM. Mol Cell. 2001;8(5):971–82. doi: 10.1016/s1097-2765(01)00390-2. [DOI] [PubMed] [Google Scholar]
- 201.Collins QF, Xiong Y, Lupo EG, Jr, Liu HY, Cao W. J Biol Chem. 2006;281(34):24336–44. doi: 10.1074/jbc.M602177200. [DOI] [PubMed] [Google Scholar]
- 202.Knutti D, Kressler D, Kralli A. Proc Natl Acad Sci U S A. 2001;98(17):9713–8. doi: 10.1073/pnas.171184698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jager S, Handschin C, St-Pierre J, Spiegelman BM. Proc Natl Acad Sci U S A. 2007 doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Li X, Monks B, Ge Q, Birnbaum MJ. Nature. 2007;447(7147):1012–6. doi: 10.1038/nature05861. [DOI] [PubMed] [Google Scholar]
- 205.Koo SH, Satoh H, Herzig S, Lee CH, Hedrick S, Kulkarni R, Evans RM, Olefsky J, Montminy M. Nat Med. 2004;10(5):530–4. doi: 10.1038/nm1044. [DOI] [PubMed] [Google Scholar]
- 206.Nissen SE, Wolski K. N Engl J Med. 2007;356(24):2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 207.Smith CL, O'Malley BW. Endocr Rev. 2004;25(1):45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 208.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Genes Dev. 2000;14(22):2831–8. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]