

Abstract
The fragile histidine triad (FHIT) gene is frequently inactivated in various types of tumours. However, the system-wide pathology caused by FHIT inactivation has not been examined in detail. Here we demonstrate that Fhit gene knockout mice develop tumours in the lymphoid tissue, liver, uterus, testis, forestomach and small intestine, together with structural abnormalities in the small intestinal mucosa. These results suggest that Fhit plays important roles in systemic tumour suppression and in the integrity of mucosal structure of the intestines.
Keywords: fragile histidine triad gene (FHIT), tumour suppressor gene, intestinal polyps, intestinal lesions, Apc, β-catenin
The fragile histidine triad (FHIT) gene has been identified as a candidate tumour suppressor gene localized in FRA3B, the most sensitive common fragile site, at chromosome 3p14.2 (Ohta et al, 1996). Chromosomal deletion of the FHIT-containing locus or inactivation of FHIT is frequently observed in various types of cancers (for a review, see Huebner and Croce, 2003), consistent with a tumour suppressor function in a variety of organs. The Fhit protein carries a proapoptotic activity through a caspase-dependent pathway in human cancer cells, which may contribute to the tumour suppressor activity (Ji et al, 1999; Ishii et al, 2001; Roz et al, 2002).
Previously, we have demonstrated that Fhit mutant mice develop tumours spontaneously in lymphatic tissues, sebaceous glands, liver, stomach, colonic submucosa, uterus, skin, salivary glands, and parathyroid glands (Zanesi et al, 2001). Moreover, Fhit knockout mice are susceptible to chemical carcinogen-induced tumour formation in the forestomach (Fong et al, 2000), which is reversed by adenoviral transduction of the human FHIT gene (Dumon et al, 2001). These results are consistent with the tumour suppressor function of FHIT. To further characterise tissue types affected by inactivation of Fhit, including nontumorous lesions, we have constructed a second mouse strain with a targeted Fhit gene, and conducted a thorough pathological analysis. These Fhit mutant mice develop tumours in a variety of tissues, including intestinal adenomatous polyps, and show abnormal tissue building of intestinal mucosa, suggesting avenues for further study of Fhit function in normal tissues.
MATERIALS AND METHODS
All in vivo experiments were carried out with ethical committee approval and met the standards required by the UKCCCR guidelines (Workman et al, 1998). The mouse Fhit gene-targeting vector was constructed to replace exon 5, containing the translation initiation codon, with a pGK-Neo-bpA cassette. Homologous recombination in ES cells (RW4) was confirmed by genomic Southern hybridisation (data not shown). Germline transmitted F1 (+/−) mice were intercrossed to obtain Fhit (+/+), (+/−) and (−/−) progeny. Both males and females of each genotype from 12 to 20 months of age were used for thorough pathological examinations. Tissue samples were first examined under a dissection microscope. Then, all tissues were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 16 h at 4°C, embedded in paraffin wax, sectioned at 4 μm thickness and stained with H&E. For immunostaining, sections were preincubated with 3% BSA/10% goat serum in PBS for 2 h and incubated with anti-β-catenin antibody (Sigma Chemical Co., St Louis, MO, USA) at 5000-fold dilution or with c-Kit antibody (Santa Cruz, CA, USA) at 400-fold. Signals were visualised using the Vectastain Elite Kit (Vector Laboratories, Burlingame, CA, USA). For mutation analysis and microsatellite instability (MSI) analysis, DNA samples were extracted from paraffin-embedded sections using DEXPAT (TaKaRa, Japan). Extracted DNA samples were subjected to PCR in 11 contiguous fragments spanning nucleotides 2750–4830 of exon 15 of the Apc gene, where germline and somatic mutations were frequently detected. Amplified DNA samples were sequenced directly with the respective primers using the BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems; Rotkreuz, Switzerland) and ABI Prism 377 DNA Sequencing System (Applied Biosystems). For MSI analysis, four primer sets, D1Mit4, D6Mit59, D9Mit67 and D10Mit2 (http://www.informatics.jax.org), were used. DNA samples extracted from polyps were amplified by PCR. After denaturation (100°C, 5 min), PCR products were loaded onto 5% Long Ranger Gel (TaKaRa) in 8 M urea, and electrophoresed at 150 V for 40 min. The dried gel was scanned in a BAS-1800 (Fujifilm, Japan).
RESULTS AND DISCUSSION
Fhit (+/−) and (−/−) mice were viable, fertile and clinically normal up to 12 months of age. However, upon necropsy, tumours and abnormal lesions were found in mice older than 12 months, consistent with observations of another Fhit mutant strain (Fong et al, 2000). Although several types of tumours developed spontaneously even in the age-matched wild-type littermates, overall incidences of tumours and abnormal lesions were significantly higher in both Fhit (+/−) and (−/−) mice than in the wild-type littermates (60, 77 and 30% in Fhit (+/−), (−/−) and (+/+) mice, respectively; Table 1 ). In both Fhit (+/−) and (−/−) mice, lymphoid malignancies including B-cell lymphoma and lymphocyte infiltrations, hemangioma and adenoma of the liver, and uterine leiomyoma were found, which is consistent with the previous report (Zanesi et al, 2001). Gastrointestinal stromal tumour (GIST) was described in the other Fhit mutant strain. Here, we have confirmed the diagnosis by immunohistochemistry for c-Kit protein (Figure 1A inset). In the present study, several additional types of tumours were detected, such as Leydig cell tumour, endometrial hyperplasia and hepatocellular carcinoma (Table 1). However, the last two tumours were observed also in Fhit (+/+) mice, and no increase was found in incidence in Fhit mutant mice for either tumour type. One Leydig cell tumour was observed in one of 90 Fhit (+/−) mice (Figure 1B), although the number is too small to draw a statistical link with Fhit deficiency, and none was found in (−/−) animals.
Table 1. Tumour and abnormality incidence and tumour spectrum.
|
Histological tumour types |
|||||||
|---|---|---|---|---|---|---|---|
| Genotype | na | Age (months) | Incidence of tumours and/or abnormal lesions (% of animals) | Intestinal lesions (% of animals) | Forestomach papilloma | Lymphoid malignancies | Other neoplasms |
| Fhit (+/+) | 26 | 12–20 | 8 (30%) | 0 | 3 (12%) | 3 (12%) | 1 hepatocellular carcinoma |
| 2 lymphomas (1 B-cell lymphoma) | 1 periosis hepatis | ||||||
| 1 lymphocyte infiltration | 1 endometrial hyperplasia | ||||||
| Fhit (+/–) | 90 | 12–20 | 54 (60%)b | 19 (21%) | 22 (24%) | 23 (26%) | 1 hepatocellular carcinoma |
| 6 small intestinal polyps | 18 lymphomas (16 B-cell lymphomas) | 1 hepatocellular adenoma | |||||
| 5 fused villi | 5 lymphocyte infiltrations | 1 liver hemangioma | |||||
| 8 swollen crypts | 2 endometrial hyperplasia | ||||||
| 1 uterine leiomyoma | |||||||
| 1 lung adenoma | |||||||
| 1 Leydig cell tumour | |||||||
| 1 dermoid cyst | |||||||
| Fhit (–/–) | 26 | 12–20 | 20 (77%)b | 7c (27%) | 6 (23%) | 11 (42%) | 2 hepatocellular carcinomas |
| 4 small intestinal polyps | 9 lymphomas (6 B-cell lymphomas) | 2 endometrial hyperplasia | |||||
| 3 fused villi | 2 lymphocyte infiltrations | 1 GISTd | |||||
| 1 swollen crypts | |||||||
Number of animals.
P<0.05 compared to wild-type controls by chi-squared test.
One of these mice contained a small intestinal polyp and a fused villi. The other mice contained only one lesion per mouse.
Gastrointestinal stromal tumour.
Figure 1.
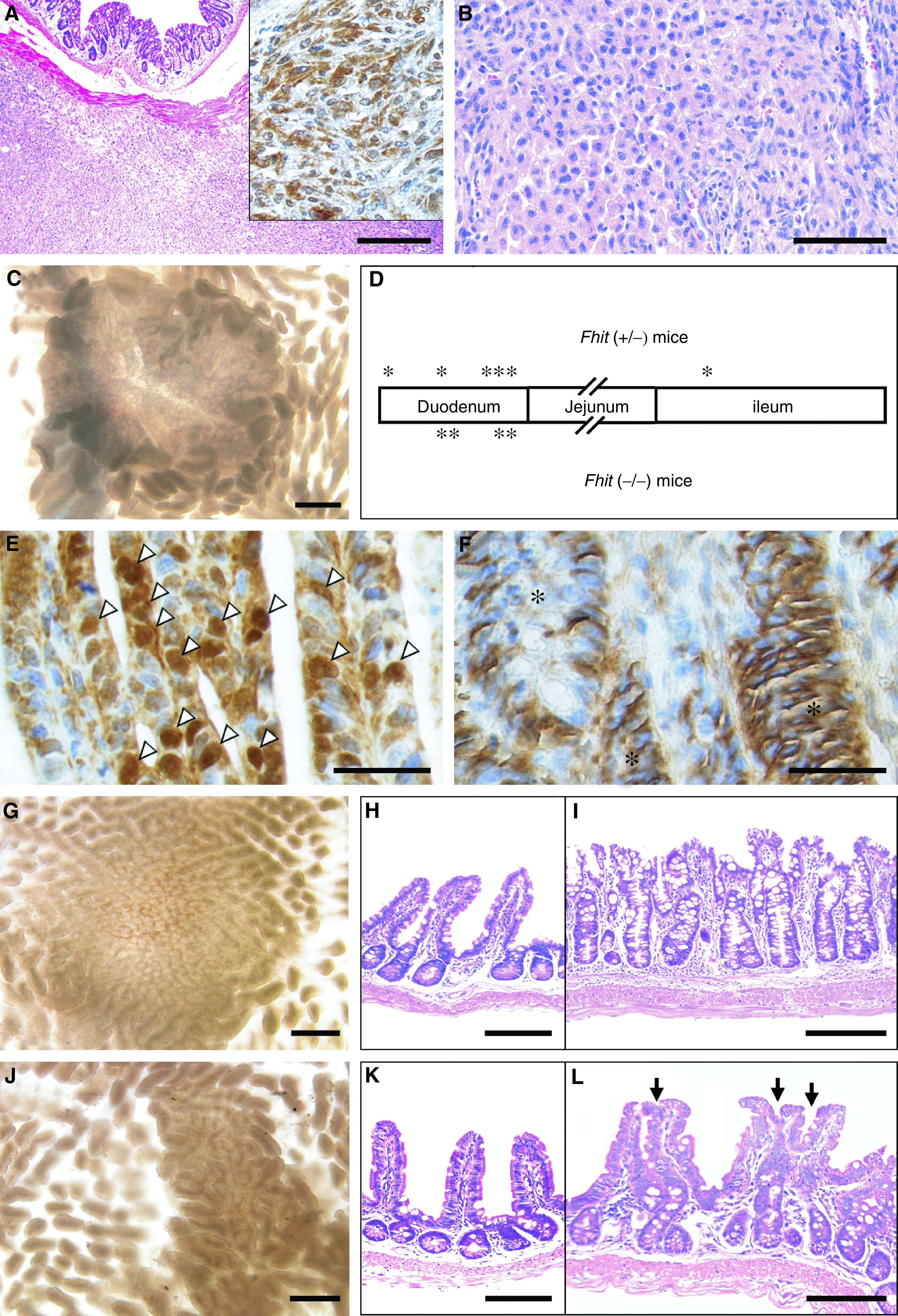
Histology of tumours and abnormal intestinal mucosa in Fhit-deficient mice. (A) Gastrointestinal stromal tumour in a female Fhit (−/−) mouse at 20 months of age. Inset, immunostaining with an anti-c-Kit antibody. (B) Leydig cell tumour in a male Fhit (+/−) mouse at 15 months. (C) Dissection micrograph of a duodenal polyp in a Fhit (−/−) mouse at 15 months. (D) Schematic distribution of the small intestinal polyps. Asterisks indicate relative locations of the polyps. (E, F) Immunostaining for β-catenin in polyps of a Fhit (+/−) mouse at 12 months, and Fhit (−/−) at 15 months, respectively. Note the nuclear accumulation of β-catenin (white arrowheads) in (E) and basolateral staining (asterisks) in (F). (G, J) Dissection micrographs of swollen crypts (ileum) in a Fhit (+/−) mouse at 20 months, and fused villi (duodenum) in a Fhit (+/−) at 20 months, respectively. (H, K) Normal ileal mucosa adjacent to the lesions shown in (I, L), respectively. (I, L) Histological sections of ileal swollen crypts in a Fhit (+/−) mouse at 20 months, and fused villi in a Fhit (+/−) at 20 months, respectively. Bars in: (A) 500 μm; (B) 100 μm; (C) 250 μm; (E, F) 50 μm; (G, J) 250 μm; (H, I, K, L) 200 μm.
Interestingly, some adenomatous polyps were found in the small intestine of Fhit mutant mice (Figure 1C, D), whereas these polyps have never been observed in wild-type littermates (Table 1). Although the incidence of polyp development in Fhit (−/−) was higher (15.4%) than that in Fhit (+/−) mice (6.7%), the polyp multiplicity was not different in the two genotypes. These results suggest that tumours develop in Fhit (+/−) mice because of Fhit haploinsufficiency, either directly or through an indirect mechanism.
Mutations in the gene encoding Apc or β-catenin result in intestinal adenomatous polyposis through Wnt signalling activation (Oshima et al, 1995; Harada et al, 1999). To examine whether the Wnt pathway was activated in the intestinal polyps of Fhit-deficient mice, we localised β-catenin in nine polyp tissues from both Fhit (+/−) and Fhit (−/−) mice by immunohistochemistry. β-Catenin had accumulated in the nucleus in two polyps of Fhit (+/−) mice, indicating Wnt activation in these adenoma cells (Figure 1E). β-Catenin was localised to the basolateral side of adenoma cells in the other seven polyps (Figure 1F). By sequence analysis of the Apc gene of the two nuclear β-catenin-positive polyps, we further found a nonsense mutation in the Apc gene at codon 1055 (wt: GAA (Glu) → mutant: TAA (STOP)) in one polyp. This mutation may explain the cause of at least a fraction of the polyps, although other genes may be mutated giving rise to other adenomas. As the intestines were not inspected in detail in the other Fhit knockout strain, it is conceivable that similar intestinal lesions existed at low frequencies. These results suggest that an insufficient Fhit level can induce Apc gene mutations, which is consistent with the enhanced survival and mutation frequency of Fhit-deficient cells after UVC or mitomycin C damage (Ottey et al, 2004). In contrast, MSI was not observed in genomic DNAs from Fhit mutant mouse tails or tumours (Fong et al, 2000). In the present study, we also examined MSI in cells from two intestinal polyps, using four different sets of markers, and confirmed no MSI in these samples (data not shown). Accordingly, it is possible that inactivation of Fhit results in tumorigenesis through induction of mutations in tumour suppressor genes.
On the other hand, morphological abnormalities in the small intestine, such as swollen crypts (goblet cell hyperplasia, Figure 1G, H) and fused villi (aggregated villi, Figure 1I, J), were observed in both Fhit (+/−) and (−/−) mice at similar incidences. Such lesions were not found in the age-matched wild-type mice. Histologically, these lesions consisted of differentiated epithelial cells without any signs of dysplasia. Thus, it is conceivable that Fhit expression is necessary also for the maintenance of normal intestinal architecture, in addition to suppressing tumorigenesis.
Acknowledgments
We thank Dr M Ohta for discussion of the Fhit gene targeting construct and Y Toda for technical advice on immunostaining. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Dumon KR, Ishii H, Fong LY, Zanesi N, Fidanza V, Mancini R, Vecchione A, Baffa R, Trapasso F, During MJ, Huebner K, Croce CM (2001) FHIT gene therapy prevents tumor development in Fhit-deficient mice. Proc Natl Acad Sci USA 98: 3346–3351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong LY, Fidanza V, Zanesi N, Lock LF, Siracusa LD, Mancini R, Siprashvili Z, Ottey M, Martin SE, Druck T, McCue PA, Croce CM, Huebner K (2000) Muir–Torre-like syndrome in Fhit-deficient mice. Proc Natl Acad Sci USA 97: 4742–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N, Tamai Y, Ishikawa T, Sauer B, Takaku K, Oshima M, Taketo MM (1999) Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J 18: 5931–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner K, Croce CM (2003) Cancer and the FRA3B/FHIT fragile locus: it's a HIT. Br J Cancer 88: 1501–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Dumon KR, Vecchione A, Trapasso F, Mimori K, Alder H, Mori M, Sozzi G, Baffa R, Huebner K, Croce CM (2001) Effect of adenoviral transduction of the fragile histidine triad gene into esophageal cancer cells. Cancer Res 61: 1578–1584 [PubMed] [Google Scholar]
- Ji L, Fang B, Yen N, Fong K, Minna JD, Roth JA (1999) Induction of apoptosis and inhibition of tumorigenicity and tumor growth by adenovirus vector-mediated fragile histidine triad (FHIT) gene overexpression. Cancer Res 59: 3333–3339 [PubMed] [Google Scholar]
- Ohta M, Inoue H, Cotticelli MG, Kastury K, Baffa R, Palazzo J, Siprashvili Z, Mori M, McCue P, Druck T, Croce CM, Huebner K (1996) The FHIT gene, spanning the chromosome 3p14.2 fragile site and renal carcinoma-associated t(3;8) breakpoint, is abnormal in digestive tract cancers. Cell 84: 587–597 [DOI] [PubMed] [Google Scholar]
- Oshima M, Sugiyama H, Kitagawa K, Kobayashi M, Itakura C, Taketo M (1995) Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyposis in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA 92: 4482–4486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottey M, Han S-Y, Druck T, Barnoski B, McCorkell KA, Croce CM, Raventos-Suarez C, Fairchild CR, Wang Y, Huebner K (2004) Fhit deficient normal and cancer cells are mitomycin C and UVC resistant. Br J Cancer (in press) [DOI] [PMC free article] [PubMed]
- Roz L, Gramegna M, Ishii H, Croce CM, Sozzi G (2002) Restoration of fragile histidine triad (FHIT) expression induces apoptosis and suppresses tumorigenicity in lung and cervical cancer cell lines. Proc Natl Acad Sci USA 99: 3615–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman P, Twentyman P, Balkwill F, Balman A, Chaplin D, Double J, Embleton J, Newell D, Raymond R, Stables J, Stephens T, Wallace J (1998) United Kingdom Co-ordinating Committee on Cancer Research (UKCCCR) Guidelines for the Welfare of Animals in Experimental Neoplasia (Second Edition). Br J Cancer 77: 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanesi N, Fidanza V, Fong LY, Mancini R, Druck T, Valtieri M, Rudiger T, McCue PA, Croce CM, Huebner K (2001) The tumor spectrum in FHIT-deficient mice. Proc Natl Acad Sci USA 98: 10250–10255 [DOI] [PMC free article] [PubMed] [Google Scholar]