

Abstract
Mutations in the gene encoding human copper-zinc superoxide dismutase (SOD1) cause a dominant form of the progressive neurodegenerative disease amyotrophic lateral sclerosis. Transgenic mice expressing the human G85R SOD1 variant develop paralytic symptoms concomitant with the appearance of SOD1-enriched proteinaceous inclusions in their neural tissues. The process(es) through which misfolding or aggregation of G85R SOD1 induces motor neuron toxicity is not understood. Here we present structures of the human G85R SOD1 variant determined by single crystal x-ray diffraction. Alterations in structure of the metal-binding loop elements relative to the wild type enzyme suggest a molecular basis for the metal ion deficiency of the G85R SOD1 protein observed in the central nervous system of transgenic mice and in purified recombinant G85R SOD1. These findings support the notion that metal-deficient and/or disulfide-reduced mutant SOD1 species contribute to toxicity in SOD1-linked amyotrophic lateral sclerosis.
Superoxide (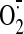 ) is
a reactive byproduct of mitochondrial respiration and fatty acid oxidation
that can damage critical cellular components. To protect against such damage,
cells express antioxidant enzymes such as copper-zinc superoxide dismutase
(SOD1)5
(1), catalase
(2), and glutathione peroxidase
(3) that together act to
convert superoxide to molecular oxygen and water using redox-active metal
cofactors. SOD1 comprises between 0.1 and 2.0% of the detergent-soluble
protein in spinal cord and brain
(4,
5), and this abundance
presumably protects against the plentiful superoxide generated by these
metabolically active (respiring) tissues. However, expression of SOD1 variants
can be harmful if the molecule functions abnormally as demonstrated by
dominantly inherited mutations in the human SOD1 gene that give rise
to familial forms of the fatal neurodegenerative disease amyotrophic lateral
sclerosis (ALS) (6,
7).
) is
a reactive byproduct of mitochondrial respiration and fatty acid oxidation
that can damage critical cellular components. To protect against such damage,
cells express antioxidant enzymes such as copper-zinc superoxide dismutase
(SOD1)5
(1), catalase
(2), and glutathione peroxidase
(3) that together act to
convert superoxide to molecular oxygen and water using redox-active metal
cofactors. SOD1 comprises between 0.1 and 2.0% of the detergent-soluble
protein in spinal cord and brain
(4,
5), and this abundance
presumably protects against the plentiful superoxide generated by these
metabolically active (respiring) tissues. However, expression of SOD1 variants
can be harmful if the molecule functions abnormally as demonstrated by
dominantly inherited mutations in the human SOD1 gene that give rise
to familial forms of the fatal neurodegenerative disease amyotrophic lateral
sclerosis (ALS) (6,
7).
Seminal studies in transgenic mice have established that pathogenic SOD1 proteins elicit motor neuron dysfunction through the acquisition of a deleterious property and not a loss of enzymatic function (8-10). Proteinaceous inclusions enriched in SOD1 are observed in cell culture model systems, ALS-SOD1 transgenic mice, and in familial ALS patients, suggesting that SOD1-linked ALS pathology may be related to protein misfolding or aggregation (for reviews, see Refs. 11-13). However, the molecular mechanisms underlying SOD1 toxicity to motor neurons remain unknown, and it remains to be clarified whether the observed inclusions are causal or symptomatic of motor neuron dysfunction.
Since the discovery of the SOD1-ALS link nearly 15 years ago, the pathogenic human SOD1 variant in which Arg is substituted for Gly at position 85 (G85R) has been frequently studied. When expressed in transgenic mice, G85R SOD1 causes rapidly progressive motor neuron degeneration while remaining at low levels in the soluble fraction of spinal cord extracts (14-16). Visible SOD1-containing inclusions first become apparent in astrocytes with the onset of paralytic symptoms in these mice and steadily increase in abundance as the disease progresses. As the disease nears end stage, inclusions enriched in SOD1 also begin to appear in the motor neurons. Once initiated in these animals, however, motor neuron loss proceeds synchronously with approximately half degenerating over a 2-week period (14).
Accumulating evidence strongly suggests that the G85R substitution weakens metal ion binding and destabilizes the molecule relative to wild type when both are metal-free (17). Purified recombinant G85R SOD1 proteins are always observed to be metal-deficient on a per polypeptide basis relative to the wild type enzyme expressed in the same system (12, 18-20). In addition, recent studies in G85R-overexpressing mice reveal that much of the G85R SOD1 protein found in the soluble fraction of spinal cord lysates is uncharged with copper, disulfide-reduced, and monomeric (15, 16, 21).
Here we present high resolution structures of the human G85R variant determined by single crystal x-ray diffraction. The structures suggest a molecular basis for alterations in metal binding and provide a platform upon which the relationship between metal deficiency and SOD1 toxicity can be expanded.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification—The DNA fragments encoding mutant G85R human SOD1 were generated by PCR and were ligated into the YEP351-hSOD plasmid, which directs the expression of the protein under the control of its own promoter. Human G85R SOD1 was expressed in the EGy118 strain of Saccharomyces cerevisiae that lacks the endogenous yeast sod1 gene. Protein coming from the University of Texas group was obtained as described previously (22). For protein generated by the California State University Long Beach group, the procedure was generally the same except that the lysis buffer did not contain EDTA and a linear gradient rather than a step gradient was used to elute the protein from the phenyl-Sepharose column. An additional column step (DEAE-Sephacryl, linear gradient 0.25 mm potassium phosphate, pH 7.0 to 250 mm potassium phosphate, pH 7.0) was also used prior to the gel filtration column, which was run in 50 mm sodium phosphate, 50 mm sodium chloride, pH 7.0.
Crystallization, Data Collection, Structure Solution, and Refinement—All crystals were grown using the hanging drop vapor diffusion method. For crystals grown in the United States, pure G85R SOD1 at 26 mg/ml in 2.25 mm potassium phosphate, 80 mm NaCl, pH 7.0, was mixed with an equal volume of reservoir solution containing 2.4 m sodium malonate, pH 7.0. Crystals grew within 1 week at room temperature in space groups I212121 (structure I) and P212121 (structure II) in the same drop and contain two and one dimeric G85R protein molecules per asymmetric unit, respectively. Suitable crystals were soaked in a cryoprotectant solution comprised of 15% ethylene glycol (glycerol) in the appropriate mother liquor before flash cooling in the cryostream. Native x-ray data were collected at the Argonne National Laboratory Advanced Photon Source 19BM (structure I) and on a Rigaku FR-D rotating copper anode x-ray generator equipped with an R-AXIS high throughput crystallography imaging plate system (structure II), respectively. The resolution was assessed as 1.95 Å (structure I) and 1.9 Å (structure II). The data were indexed, scaled, and merged using HKL2000 (23).
The structures were determined via molecular replacement using MOLREP (24) with the G37R SOD1 structure (Protein Data Bank code 1AZV (25)) as the search model. The positioned models were refined using the maximum likelihood method implemented in REFMAC5 (26) as part of the CCP4 program suite and rebuilt interactively using SIGMAA maps (27) in the program O (28). Five percent of the data were set aside (randomly as a function of resolution) to calculate a free R value (29). No stereochemical restraints were applied to metal-ligand distances or bond angles. A bulk solvent correction was applied, and solvent molecules were added using ARP/WARP (30).
Anomalous diffraction data for structures I and II were collected at two different wavelengths. The first data set was collected at a wavelength of 1.28 Å, at the zinc absorption edge and above the copper absorption edge, giving anomalous data with copper f″= 3.4 e and zinc f″= 3.9 e. The second data set was collected at a wavelength of 1.38 Å, at the copper and below the zinc absorption edges, giving anomalous data with copper f″= 3.9 e and zinc f″= 0.6 e. Both anomalous data sets were collected at beam line 8.2.1 at the Advanced Light Source (Lawrence Berkeley National Laboratory) and were used to exploit differential changes in the anomalous scattering of copper and zinc (see below).
For crystals grown in the United Kingdom, G85R SOD1 at 15 mg/ml in 2.25 mm sodium phosphate buffer, pH 7.0, 100 mm sodium chloride was mixed with an equal volume of reservoir solution containing 20% (w/v) polyethylene glycol 3350, 0.2 m sodium thiocyanate. Crystals grew within 3 weeks in space groups P212121 (structure III) and P21 (structures IV and V) in the same drop and contain one dimeric G85R SOD1 molecule per asymmetric unit. Suitable crystals were soaked in a cryoprotectant solution comprised of reservoir solution with a 25% (v/v) final glycerol concentration before flash cooling in the cryostream. Diffraction data were collected at 100 K on an Area Detector Systems Corp. Quantum 4 charged couple device detector at station 14.1 (structure III), station 9.6 (structure IV), and station 10.1 using an MAR225 detector (structure V) of the Synchrotron Radiation Source, Daresbury Laboratory.
The structures were determined using MOLREP with the wild type human SOD1 dimer (31) as the search model. The models were refined using the maximum likelihood method implemented in REFMAC5 as part of the CCP4 program suite and rebuilt interactively using SIGMAA maps in the program O. Five percent of the data were set aside from the refinement to calculate a free R value. No stereochemical restraints were applied to metal-ligand distances or bond angles. A bulk solvent correction was applied, and solvent molecules were added using ARP/WARP.
Anomalous diffraction data for structures III and IV were collected at two different wavelengths. The first data set was collected at a wavelength of 1.2 Å, above both the zinc and copper absorption edges, giving anomalous data with copper f″= 2.7 e and zinc f″= 3.5 e. The second data set was collected at a wavelength of 1.33 Å, between the copper and zinc absorption edges, giving anomalous data with copper f″= 3.6 e and zinc f″= 0.6 e. Both anomalous data sets were collected at beam line 10.1, Synchrotron Radiation Source, Daresbury Laboratory and were used to exploit differential changes in the anomalous scattering of copper and zinc (see “Results”).
For those metal-binding sites containing mixtures of copper and zinc ions, different ratios of copper and zinc occupancies (0.7/0.3, 0.6/0.4, etc.) were input prior to crystallographic refinement followed by visual inspection of electron density maps resulting from the refinement process. The atomic displacement parameters (B-factors) of the metal ions were compared with those of the liganding atoms coming from histidine and/or aspartic acid residues as described previously (31).
Figure Preparation—Figs. 1, 2, 3 and 4 were created with the program PyMol.
FIGURE 1.
Structural comparison of human wild type and pathogenic G85R SOD1. A, wild type human SOD1. The Greek key β-barrel is shown in yellow. The electrostatic and zinc loop elements are shown in orange and blue, respectively. The disulfide loop and β-strand 8 that comprise the bulk of the dimer interface are shown in green. The disulfide bond is shown in red. Copper and zinc ions are represented as cyan and gray spheres, respectively. The relationship between the two subunits of the dimer is indicated. B, identification of copper and zinc ions in G85R SOD1 structure I, subunit A (see Table 2) using x-rays tuned to the copper and zinc absorption edges. X-rays tuned to the copper edge do not promote anomalous scattering by zinc and thereby act as the discriminator between the two metal ions. Copper does absorb slightly at the zinc edge. The green electron density is calculated from the copper edge, and the red electron density is calculated from the zinc edge. Note that the green electron density is only found around the metal in the copper site, indicating that it is copper and that there is no copper in the zinc site. The small amount of red electron density around the metal in the copper site comes from the slight anomalous scattering by copper at the zinc edge. Occupancies are estimated by comparing the thermal parameters of the metal ions and their liganding atoms. Fully occupied metal-binding sites should have thermal parameters for metal and ligand atoms that are very similar. The copper edge and zinc edge anomalous difference Fourier electron density maps are contoured at 5σ. C, human G85R SOD1 structure II, subunits E and F (see Table 2). The color coding is as in A except that the Arg85 side chains are shown as light pink and blue tubes. Zinc is bound to the copper site in the right subunit F, and the open circle indicates the position zinc would be if it were bound to the zinc site of protein as in A. The electrostatic and zinc loop elements are not shown because they are disordered in the crystal structure.
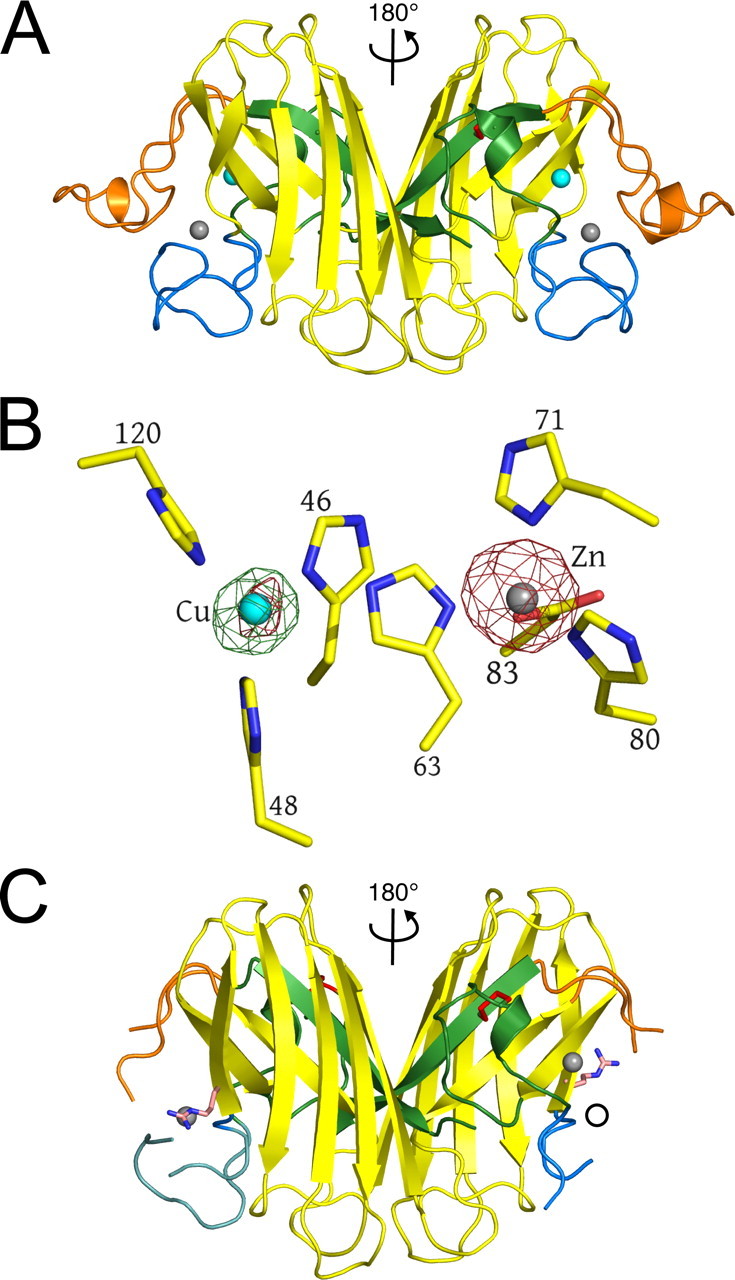
FIGURE 2.
The G85R mutation site and the copper- and zinc-binding sites. The wild type protein is shown in yellow. A, three of the four conformations observed in the 10 unique subunits of the four crystal structures are shown in light blue, green, and pink, respectively (see text). Copper and zinc ions are represented as cyan and gray spheres, respectively. The dual hydrogen bonds formed by Asp124 to the copper ligand His46 and the zinc ligand His71 as well as the hydrogen bonding network between Pro74, Arg79, and Asp101 are shown as dotted lines. B, the image is the same as in A except rotated ∼90° around the horizontal and vertical axes in the plane of the page. The electrostatic loop has been removed for clarity. C, the location of the three proline residues of the zinc loop (see text). The disulfide loop (residues 50-62), a substructure of the zinc loop, is shown in green. The remainder of the zinc loop containing the zinc-binding ligands, residues 63-83, is shown in blue. Pro62, Pro66, and Pro74 in the zinc-bound conformation of the zinc loop are shown in magenta. The altered position of the five-membered ring of Pro74 and the Arg85 side chain are shown in orange. The hydrogen bond normally found between the carbonyl oxygen of Pro74 and the guanidinium group of Arg79 is disrupted. D, structural state four (see text) found in G85R subunits C, E, and F (see Table 2). The absence of electron density around the α carbon and the Arg85 side chain indicates that this residue is conformationally mobile, sampling many positions. This movement is correlated with both zinc deficiency in the zinc site and disorder of the zinc and electrostatic loop elements.
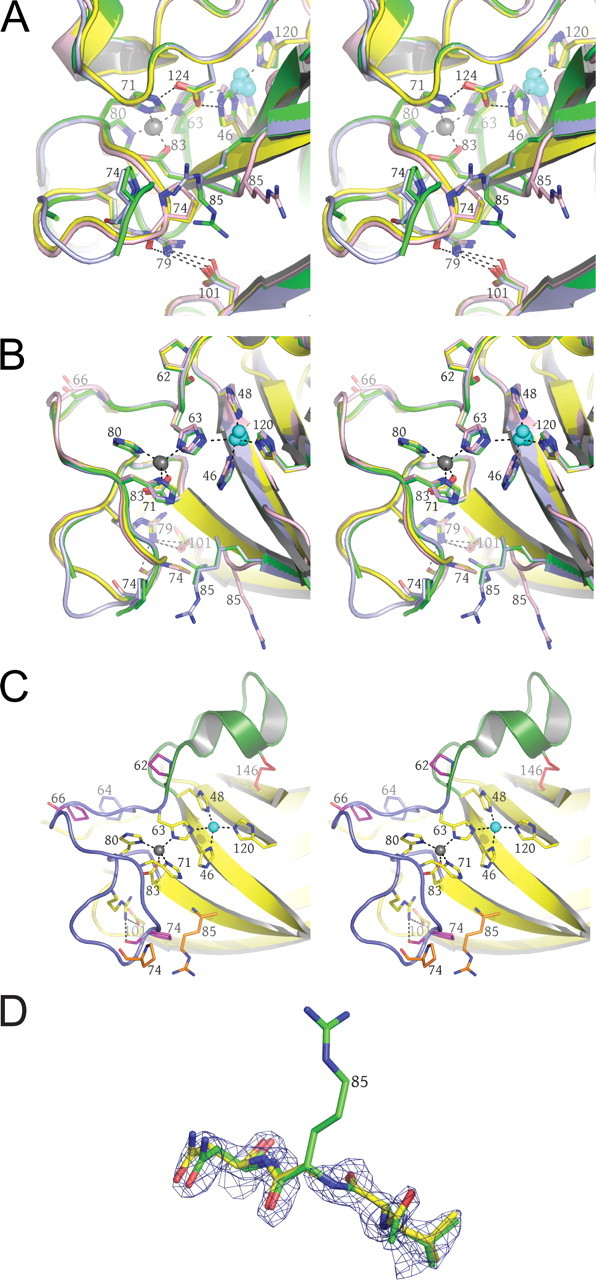
FIGURE 3.
Surface zinc-binding site observed in structures IV and V. A, the bridging zinc ion between two G85R SOD1 dimers is shown as a gray sphere. Residues coming from a subunit of one dimer are shown in yellow, and those coming from another dimer are shown in pink. The thiocyanate anions are labeled. The SIGMAA electron density is contoured at 1.5σ. B, the bridging zinc ions link each SOD1 dimer to other dimers through a 2-fold rotation and a translation of one subunit. The bridging zinc ion and its ligands are shown in red. These interactions are propagated throughout the entire crystal along the b axis. The zinc ion in the zinc-binding site is shown as a blue sphere where it caps the helix dipole at the C terminus of the short α-helix in the electrostatic loop.
FIGURE 4.

A novel water molecule gains access to the active site area in the three subunits with a displaced 85-86 peptide bond (see also Table 2 and the text). The wild type enzyme is shown in yellow, and the G85R SOD1 mutant is shown in pink. The water molecule gaining access to zinc site in the G85R structure is shown as a green sphere.
RESULTS
Structures of G85R SOD1 and Metal Ion Binding—Fig. 1A shows a ribbon diagram of the dimeric human wild type holoenzyme (Protein Data Bank code 1HL5 (32)). Each SOD1 subunit folds as an eight-stranded Greek key β-barrel, binds one copper and one zinc ion, and harbors an intrasubunit disulfide bond (33). Two lengthy loop elements protrude from the β-barrel to form the walls of the active site and are intimately involved in metal binding. The first, termed the “zinc loop” (loop IV, residues 50-83) houses most of the zinc-binding ligands. The second, termed the “electrostatic loop” (loop VI, residues 121-142), acts as a “lid” by covering the metal-binding sites. The electrostatic loop also contains charged amino acid residues that help guide the negatively charged substrate to the active site entrance while simultaneously excluding larger non-substrate ions (34, 35). A third, smaller loop, the “disulfide loop” (a substructure of loop IV, residues 50-62), is covalently linked to the β-barrel through a disulfide bond between Cys57 and Cys146. The disulfide loop and the eighth strand of the β-barrel from each subunit interact reciprocally and comprise the bulk of the SOD1 dimer interface (22) (Fig. 1A).
Five x-ray crystal structures of the human familial ALS G85R mutant were determined at resolutions ranging from 1.3 to 1.9 Å. Table 1 summarizes the data collection and refinement statistics for these structures, designated I-V. All sites where copper or zinc ions were bound to the protein in these crystal forms were identified by measuring diffraction data using x-radiation of wavelength (energy) tuned to the copper and zinc absorption edges. The scattering of x-rays is altered by a metal ion at its absorption edge (anomalous scattering), and this property causes the metal to be illuminated in an electron density map, revealing both its location and an estimate of the amount (occupancy) bound at each site (see “Experimental Procedures”). The energy of x-rays tuned to the copper edge does not illuminate zinc, providing a means to discriminate between the two metal ions. Supplemental Table 1 shows the x-ray data collection statistics at the copper and zinc absorption edges from different G85R crystals. Fig. 1B shows a representative electron density map calculated using these edge data. The rightmost columns in Table 2 summarize the results of these metal analyses.
TABLE 1.
X-ray diffraction data and refinement statistics for the G85R SOD1 structures I-V
R.m.s., root mean square; pdb, Protein Data Bank.
| Structure I | Structure II | Structure III | Structure IV | Structure V | |
| pdb code 3CQP | pdb code 3CQQ | pdb code 2VR6 | pdb code 2VR7 | pdb code 2VR8 | |
| Data Collection | |||||
| Space Group | I212121 | P212121 | P212121 | P21 | P21 |
| Cell Dimensions | |||||
| a, b, c (Å) | 73.5, 116.8, 147.8 | 40.0, 58.4, 104.5 | 35.3, 73.8, 111.4 | 36.8, 56.4, 75.0 | 34.2, 56.7, 74.7 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 103.0, 90.0 | 90.0, 102.1, 90.0 |
| Resolution (Å)* | 50.0-1.95 (2.02-1.95) | 50.0-1.9 (1.97-1.90) | 50.0-1.3 (1.35-1.3) | 50.0-1.58 (1.64-1.58) | 50.0-1.36 (1.41-1.36) |
| Rsym (on I) (%) | 6.2 (36.9) | 6.0 (51.1) | 6.6 (58.6) | 5.1 (8.7) | 4.3(33.3) |
| I/σI | 23.4(4.4) | 32.8(5.1) | 21.8(2.0) | 28.4(8.2) | 25.6(2.26) |
| Completeness (%) | 99.4 (99.6) | 100 (100) | 96.6 (85.5) | 93.5(73.5) | 91.0(70.5) |
| Redundancy | 5.3(5.1) | 6.8(7.0) | 5.2(2.9) | 3.9(2.7) | 3.4(2.2) |
| Refinement | |||||
| Resolution (Å) | 50.0-1.95 | 37.3-1.9 | 20.0-1.3 | 20.0-1.58 | 30-1.36 |
| No. reflections | 43,936 | 18,971 | 66,380 | 36,731 | 52,158 |
| Rwork/Rfree(%) | 18.3/22.3 | 19.2/24.0 | 13.2/18.1 | 13.5/17.4 | 11.2/15.3 |
| Number of atoms | |||||
| Protein | 4277 | 1899 | 2377 | 2244 | 2296 |
| Metal ions | 7 | 3 | 6 | 8 | 8 |
| Water | 344 | 146 | 779 | 624 | 520 |
| B- factors | |||||
| Protein | 25.5 | 30.6 | 11.99 | 11.98 | 12.24 |
| Metal ions | 34.7 | 24.3 | 8.7 | 12.53 | 11.37 |
| Water | 30.8 | 37.4 | 30.41 | 32.19 | 33.52 |
| R.m.s deviations | |||||
| Bond lengths (Å) | 0.009 | 0.013 | 0.017 | 0.017 | 0.017 |
| Bond angles (°) | 1.178 | 1.392 | 1.771 | 1.671 | 1.770 |
The number in parentheses is for the highest resolution.
TABLE 2.
Summary of structural observations in the four different crystal systems
|
Structure and subunit |
Arg85 backbone position |
Pro74 ring position |
Disordered residues |
Metal content |
||
|---|---|---|---|---|---|---|
| Copper site | Zinc site | External zinc site | ||||
| I212121 (I) | ||||||
| A | Wild type | Flipped | 1.0 copper | 1.0 zinc | ||
| B | Wild type | Flipped | 76-78 | 1.0 copper | 0.5 zinc | |
| C | Disordered | Disordered | 67-80, 127-139 | 1.0 zinc | Empty | |
|
D
|
Wild type
|
Flipped
|
1.0 copper
|
1.0 zinc
|
||
| P212121 (II) | ||||||
| E | Disordered | Disordered | 73-79, 126-139 | 1.0 zinc | 0.4 zinc | |
|
F
|
Disordered
|
Disordered
|
66-80, 124-139
|
1.0 zinc
|
Empty
|
|
| P212121 (III) | ||||||
| G | Rotated | Wild type | 0.2 copper/0.8 zinc | 1.0 zinc | ||
|
H
|
Wild type
|
Flipped
|
0.2 copper/0.8 zinc
|
1.0 zinc
|
||
| P21 (IV) | ||||||
| I | Rotated | Wild type | 0.3 copper/0.7 zinc | 1.0 zinc | 1.0 zinc | |
|
J
|
Rotated
|
Wild type
|
0.3 copper/0.7 zinc
|
1.0 zinc
|
1.0 zinc
|
|
| P21 (V) | ||||||
| K | 0.25 copper/0.35 zinc | 0.6 zinc | 0.7 zinc | |||
| L | 0.2 copper/0.4 zinc | 0.6 zinc | 0.7 zinc | |||
As reflected in Table 2, only two of 12 G85R SOD1 subunits (A and D of structure I) bind both copper and zinc normally at 1 eq each per subunit. Nine of the 12 G85R SOD1 subunits examined in this study are copper-deficient to varying degrees. Two subunits (subunit C of structure I and subunit F of structure II) reveal a complete absence of metal ions in the zinc site but contain exclusively zinc in the copper site. Six G85R subunits contain a mixture of copper and zinc bound to the copper site. These differences in metal binding by G85R SOD1 cause unique but defined alterations in their structures, which in turn are selected by the crystal form in which they pack. As shown in Fig. 1C and Table 2, those proteins that have lost zinc ions from the zinc-binding site demonstrate extreme conformational disorder in the electrostatic and zinc loop elements that protrude from the β-barrel.
The crystals used to determine structures IV and V were harvested from the same crystallization condition, but the x-ray diffraction data for structure V were collected approximately 1 year after the data for structure IV. Interestingly structure V is metal-deficient relative to structure IV in all metal-binding sites, strongly suggesting metal ion loss as a function of time. This metal loss phenomenon is never observed in similar experiments performed using wild type human SOD1 crystals.
The G85R Substitution—Four distinct conformational states are observed at the G85R mutation sites. Fig. 2 shows these conformational states superimposed on the corresponding region of the wild type enzyme. The observed conformations of the regions local to the G85R substitution site are correlated with the metal binding properties in each subunit listed in Table 2.
A Surface Binding Site for Zinc—Fig. 3A shows a view of 1.6-Å resolution electron density superimposed on the molecular model coming from structure IV, revealing a zinc ion bridging two G85R subunits from different dimers. His110 from one G85R dimer and Glu24 from another together with two thiocyanate anions coordinate zinc in a tetrahedral geometry. The metal is verified to be zinc through the anomalous scattering methods described above. Fig. 3B shows that this surface zinc-binding site effectively creates tightly linked G85R SOD1 layers where the yellow and green dimers are related to the pink dimers by a 180° rotation and a translation of one SOD1 subunit.
DISCUSSION
Copper and Zinc Deficiency in G85R SOD1—When expressed in sod1-/ccs+ yeast, nine of the 12 human G85R SOD1 subunits are found to contain at least some copper in the copper-binding site (Table 2). This observation apparently conflicts with reports that the human G85R SOD1 variant and its G86R mouse analog are inactive (10, 36). In those studies, however, the G85R SOD1 proteins were exposed to EDTA during non-denaturing gel electrophoresis prior to an enzymatic activity assay, and this treatment could conceivably remove weakly bound copper from the protein. Copper binding, even by a small subset of G85R SOD1 subunits, would be sufficient to explain the ability of this mutant to rescue the oxygen-sensitive phenotype of SOD1 knock-out yeast (18), and the results presented here are consistent with this observation. Six of 12 G85R subunits (B, C, E, F, K, and L) are partially or completely devoid of zinc ion in the zinc-binding site. Structures IV and V reveal that G85R SOD1 crystals grown from the same crystallization experiment tend to lose their zinc over time.
The G85R Mutation Alters Structural Elements Harboring Zinc Ligands—The apparent decreased affinity for zinc likely comes from the proximity of the G85R substitution to the zinc-binding site, which consists of amino acids His63, His71, His80, and Asp83. In four subunits (A, B, D, and H), the backbone atoms of Arg85 superimpose with the backbone atoms of Gly85 of the wild type enzyme. For steric reasons, this wild type-like backbone position dictates that the Arg85 side chain must clash with Pro74 of the zinc loop, driving its five-membered ring ∼90° from its wild type conformation. This disruption in the conformation of the zinc loop occurs only two residues from the zinc ligand His71. As shown in Fig. 2, A and B, the steric clash of Arg85 with Pro74 also disrupts the hydrogen bond normally found between the guanidinium nitrogen of Arg79 and the carbonyl oxygen of Pro74, a hydrogen bond that, together with the hydrogen bond between Arg79 and Asp101, tethers the zinc loop to the β-barrel.
The zinc loop (including its substructure, the disulfide loop) is predicted to be conformationally dynamic in metal-free and disulfide-reduced nascent SOD1 (37). The zinc loop contains three proline residues, Pro62, Pro66, and Pro74, which are all in the trans configuration when zinc is bound to the zinc site. As shown in Fig. 2C, Pro62 sits N-terminal to His63, which simultaneously coordinates copper and zinc in the Cu(II) form of the enzyme. Pro66 is positioned between the copper- and zinc-binding sites where its five-membered ring stacks with the side chain of Phe64, thereby creating a bend in the zinc loop and making space for His80 to act as a zinc ligand. As described above, Pro74 occupies a key location where the zinc loop circles back upon itself before being tethered to the β-barrel via hydrogen bonds.
Proline isomerization is known to be a relatively slow process that can impede protein folding by “trapping” one or more proline residues in the non-native isomer (38). A cis conformation of Pro62, Pro66, or Pro74 would induce a zinc loop conformation where one or more of the zinc ligands would be out of position to bind zinc ion. The zinc loop would have to subsequently unfold, the cis-proline would have to isomerize, and the zinc loop would have to refold. The presence of the Arg85 side chain that can clash with Pro74 could hinder or delay the attainment of the zinc-bound conformation of the zinc loop.
Table 2 shows that the majority of G85R subunits that are zinc-deficient or zinc-free (subunits B, C, E, and F) demonstrate disorder in zinc loop residues, strongly suggesting that zinc binding and loop mobility are related phenomena. Similar zinc and electrostatic loop disorder has been observed previously in other zinc-deficient pathogenic SOD1 proteins (39, 40). Recent molecular dynamics simulations (41) and isothermal titration calorimetry studies (42) have demonstrated that, in the absence of copper, the binding of zinc alone in the zinc site can order these protective loop elements.
Three subunits (G, I, and J) retain the wild type conformation of Pro74 but instead reveal a rotation of the plane of the Arg85-Asn86 peptide bond by ∼60°, permitting the Arg85 side chain and its carbonyl oxygen access to bulk solvent. This distortion in the protein backbone occurs only two residues from the zinc ligand Asp83. In these cases, a novel water molecule gains access to the β-barrel interior as shown in Fig. 4 and forms hydrogen bonds to the amide nitrogen of Phe45 (adjacent to copper ligand His46) and the side chain of Asp124. In the wild type enzyme, the carbonyl oxygen of Gly85 is hydrogen-bonded only to the amide nitrogen of Phe45 as part of the hydrogen bonding network between β-strands 4 and 5, and the disruption of this network due to the Arg85-Asn86 peptide bond rotation weakens the interaction of these β-strands.
Asp124 of the electrostatic loop accepts hydrogen bonds simultaneously from the nonliganding imidazole nitrogen atoms of the zinc ligand His71 and the copper ligand His46 and is critical to proper zinc binding by the enzyme. D124N and D124G mutants are severely zinc-deficient even when dialyzed extensively against 0.5 m ZnCl2 at neutral pH for extended periods (43). The pathogenic SOD1 mutant D124V is quite similar to G85R subunits C and F in that it contains no metal in zinc-binding site but does contain zinc bound to the copper site.6 It is unknown whether the novel water molecule affects the folding of the molecule or the stability of the metal-binding sites through its non-native interaction with Asp124. Figs. 2A and 3B show that when zinc binds to the zinc-binding site it caps the negatively charged dipole formed by the short helix consisting of residues 132-137. Thus, the binding of zinc to the zinc-binding site appears to be critical to the ordering of both the zinc and electrostatic loop elements, and it is therefore not entirely surprising that these loop elements are disordered in zinc-deficient G85R SOD1.
External Metal Ion-binding Site—As shown in Fig. 3, His110 from one G85R dimer and Glu24 from another together with two thiocyanate anions coordinate zinc in a tetrahedral geometry, creating tightly linked G85R SOD1 layers. Although zinc has not been observed to bind at this location in any wild type SOD1 structure determined to date, zinc is observed to bridge dimers through His110 and Glu77 in the crystal structure of the H46R/H48Q double copper site SOD1 variant, which contains an ablated copper-binding site (Protein Data Bank code 2NNX (44)). It is worth noting that Cys111 sits immediately adjacent to His110, providing another potential metal-binding ligand in the vicinity. It is currently unknown whether the external zinc-binding site observed in this study is relevant to pathogenesis, although metal ions such as copper and zinc are abundant in brain and spinal tissue and are postulated to play a role as bridging entities in aggregates found in a range of proteins that cause human neurodegenerative diseases, including Alzheimer peptides (45) and prion proteins (Ref. 46; for a review, see Ref. 47).
The G85R Substitution and Copper Chaperone for SOD1 (CCS)—Recent studies have strongly suggested that the CCS catalyzes the formation of the disulfide bond concomitant with copper ion delivery to the SOD1 protein (48, 49). The binding of zinc to the zinc site is thought to be a prerequisite for copper charging of SOD1 by CCS, and it is tempting to speculate that G85R SOD1 subunits C and F could not acquire copper via CCS due to their failure to meet this prerequisite. The observation that zinc is bound to the copper site in these same subunits suggests that zinc was, at least at some point in time, available to the protein but was unable to bind to an impaired or weakened zinc-binding site. The decreased affinity of G85R SOD1 for zinc may enrich a population of G85R SOD1 subunits that is not competent to interact efficiently with CCS through its SOD1-like domain. It is also possible that the G85R substitution might directly interfere with the protein-protein interactions in the recognition of nascent SOD1 by CCS, which would result in a copper-depleted, disulfide-reduced population of molecules consistent with the observations of Marklund and co-workers (15, 16) in the central nervous system of transgenic mice expressing the pathogenic G85R SOD1 variant.
Metal Deficiency and Pathogenic G85R SOD1 Aggregation—Conformational disorder in the zinc and electrostatic loop elements in zinc-deficient pathogenic human SOD1 proteins is known to result in loss of coverage of “edge strands” of the β-barrel, which can in turn lead to non-native SOD1-SOD1 protein-protein interactions that result in the formation of linear amyloid-like and helical tube-like filamentous arrays (39, 40). Recent studies have revealed that the metal-free, disulfide-reduced wild type SOD1 protein exists as a monomer (22, 50, 51). Metal-deficient and disulfide-reduced SOD1 proteins are predicted to lead to an increased propensity for monomerization and aggregation (for a review, see Ref. 52). For example, a monomeric SOD1 intermediate was detected in an oxidation model of SOD1 aggregation (53), and the introduction of cysteine residues at the dimer interface with the intention of creating an intersubunit disulfide bond seemed to protect against aggregation of the A4V mutant (54). Fifteen compounds predicted to bind at the SOD1 dimer interface were shown to stabilize the molecule against guanidinium hydrochloride unfolding and to protect against aggregation in vitro (55). More recently, using an antibody that recognizes an epitope normally buried at the SOD1 homodimer interface, Chakrabartty and co-workers (56) established the presence of “misfolded” monomeric SOD1 in the proteinaceous inclusions of transgenic mice expressing the pathogenic SOD1 mutants G37R, G85R, and G93A. Despite ubiquitous expression in these animals, the monomeric pathogenic SOD1 protein was found primarily within degenerating motor neurons.
Differential scanning calorimetry studies have demonstrated that the metal-free, disulfide-reduced wild type SOD1 protein melts at a temperature ∼10 °C lower than does the metal-free disulfide-oxidized enzyme. Pathogenic SOD1 mutants such as A4V and G93A are so destabilized in this “nascent” state that melting transitions cannot be measured (17). This observation is in accordance with the shortened half-lives of human G85R in cultured cells (57, 58). As mentioned, recent studies by Marklund and co-workers (15, 16, 21) revealed that G85R SOD1 showed low steady-state protein levels in mouse central nervous system, lacked enzyme activity, and had no Cys57-Cys146 disulfide bond. However, these mutants were enriched in the central nervous system relative to other organs, suggesting inefficient recognition and degradation of misfolded disulfide-reduced SOD1 in susceptible tissues.
Although there has been much focus on the insoluble SOD1 aggregates in transgenic mouse studies, mounting evidence suggests that they may not be the noxious entity causing paralysis in these animals. Elliott and co-workers (59) crossed a G93A SOD1-overexpressing mouse with a CCS-overexpressing mouse with the surprising result that the progeny mice developed neurological deficits with a mean survival of 36 days compared with 242 days for mice expressing G93A SOD1 alone. Even more surprising was the observation that there were no insoluble aggregates in the neural tissues of the G93A/CCS-overexpressing mice and that the effect of CCS overexpression was to enhance the population of disulfide-reduced and not disulfide-oxidized G93A SOD1 as might be expected (59).7 Together these data seem to support the notion that it may be the soluble, disulfide-reduced pathogenic SOD1 species that are the noxious entity in SOD1-linked ALS, although the precise mechanism of toxicity remains unknown.
In conclusion, the structural studies on the pathogenic G85R SOD1 variant strongly suggest a molecular basis for the metal ion deficiency consistently observed for this protein in in vitro and in vivo studies. This metal deficiency is linked to an increased propensity for the G85R SOD1 protein to exist as a disulfide-reduced species that is increasingly suspect as the noxious agent in SOD1-linked ALS and may arise from a failure of the molecule to mature via normal posttranslational modification by CCS.
Acknowledgments
Support for the X-ray Crystallography Core Laboratory by The University of Texas Health Science Center at San Antonio Executive Research Committee and the San Antonio Cancer Institute is gratefully acknowledged. We are indebted to Dr. Sue Weintraub for mass spectrometry analyses and to our colleagues in the International Consortium on SOD and ALS (ICOSA) for valuable discussions. We also acknowledge support of Science and Technology Facilities Council Daresbury Laboratory throughout the course of this study.
The atomic coordinates and structure factors (codes 3CQP, 3CQQ, 2VR6, 2VR7, and 2VR8) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported, in whole or in part, by National Institutes of Health Grants NS39112 (to P. J. H.), NS44170 (to L. J. H.), and NS049134 (to J. S. V.) from the NINDS and Grant GM28222 (to J. S. V.) from the NIGMS. This work was also supported by the Judith and Jean Pape Adams Charitable Foundation (to P. J. H.), the Motor Neuron Disease Association (to S. S. H.), and the ALS Association (to L. J. H., J. A. C., and J. S. V.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1.
Footnotes
The abbreviations used are: SOD1, copper-zinc superoxide dismutase; ALS, amyotrophic lateral sclerosis; CCS, copper chaperone for SOD1.
S. V. Seetharaman and P. J. Hart, manuscript in preparation.
V. Culotta, personal communication.
References
- 1.McCord, J. M., and Fridovich, I. (1969) J. Biol. Chem. 244 6049-6055 [PubMed] [Google Scholar]
- 2.Deisseroth, A., and Dounce, A. L. (1970) Physiol. Rev. 50 319-375 [DOI] [PubMed] [Google Scholar]
- 3.Little, C., and O'Brien, P. J. (1968) Biochem. Biophys. Res. Commun. 31 145-150 [DOI] [PubMed] [Google Scholar]
- 4.Marklund, S. L. (1984) J. Clin. Investig. 74 1398-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pardo, C. A., Xu, Z., Borchelt, D. R., Price, D. L., Sisodia, S. S., and Cleveland, D. W. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 954-958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deng, H. X., Hentati, A., Tainer, J. A., Iqbal, Z., Cayabyab, A., Hung, W. Y., Getzoff, E. D., Hu, P., Herzfeldt, B., Roos, R. P., Warner, C., Deng, G., Soriano, E., Smyth, C., Parge, H. E., Ahmed, A., Roses, A. D., Hallewell, R. A., Pericak-Vance, M. A., and Siddique, T. (1993) Science 261 1047-1051 [DOI] [PubMed] [Google Scholar]
- 7.Rosen, D. R., Siddique, T., Patterson, D., Figlewicz, D. A., Sapp, P., Hentati, A., Donaldson, D., Goto, J., O'Regan, J. P., Deng, H. X., Rahmani, Z., Krizus, A., McKenna-Yasek, D., Cayabyab, A., Gaston, S. M., Berger, R., Tanzi, R. E., Halperin, J. J., Herzfeldt, B., Van den Bergh, R., Hung, W.-Y., Bird, T., Deng, G., Mulder, D. W., Smyth, C., Laing, N. G., Soriano, E., Pericak-Vance, M. A., Haines, J., Rouleau, G. A., Gusella, J. S., Horvitz, H. R., and Brown, R. H., Jr. (1993) Nature 362 59-62 [DOI] [PubMed] [Google Scholar]
- 8.Reaume, A. G., Elliott, J. L., Hoffman, E. K., Kowall, N. W., Ferrante, R. J., Siwek, D. F., Wilcox, H. M., Flood, D. G., Beal, M. F., Brown, R. H., Jr., Scott, R. W., and Snider, W. D. (1996) Nat. Genet. 13 43-47 [DOI] [PubMed] [Google Scholar]
- 9.Bruijn, L. I., Houseweart, M. K., Kato, S., Anderson, K. L., Anderson, S. D., Ohama, E., Reaume, A. G., Scott, R. W., and Cleveland, D. W. (1998) Science 281 1851-1854 [DOI] [PubMed] [Google Scholar]
- 10.Borchelt, D. R., Lee, M. K., Slunt, H. S., Guarnieri, M., Xu, Z. S., Wong, P. C., Brown, R. H., Jr., Price, D. L., Sisodia, S. S., and Cleveland, D. W. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 8292-8296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bruijn, L. I., Miller, T. M., and Cleveland, D. W. (2004) Annu. Rev. Neurosci. 27 723-749 [DOI] [PubMed] [Google Scholar]
- 12.Valentine, J. S., Doucette, P. A., and Potter, S. Z. (2005) Annu. Rev. Biochem. 74 563-593 [DOI] [PubMed] [Google Scholar]
- 13.Hart, P. J. (2006) Curr. Opin. Chem. Biol. 10 131-138 [DOI] [PubMed] [Google Scholar]
- 14.Bruijn, L. I., Becher, M. W., Lee, M. K., Anderson, K. L., Jenkins, N. A., Copeland, N. G., Sisodia, S. S., Rothstein, J. D., Borchelt, D. R., Price, D. L., and Cleveland, D. W. (1997) Neuron 18 327-338 [DOI] [PubMed] [Google Scholar]
- 15.Jonsson, P. A., Graffmo, K. S., Andersen, P. M., Brannstrom, T., Lindberg, M., Oliveberg, M., and Marklund, S. L. (2006) Brain 129 451-464 [DOI] [PubMed] [Google Scholar]
- 16.Zetterstrom, P., Stewart, H. G., Bergemalm, D., Jonsson, P. A., Graffmo, K. S., Andersen, P. M., Brannstrom, T., Oliveberg, M., and Marklund, S. L. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 14157-14162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez, J. A., Shaw, B. F., Durazo, A., Sohn, S. H., Doucette, P. A., Nersissian, A. M., Faull, K. F., Eggers, D. K., Tiwari, A., Hayward, L. J., and Valentine, J. S. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 10516-10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Corson, L. B., Strain, J. J., Culotta, V. C., and Cleveland, D. W. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 6361-6366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ratovitski, T., Corson, L. B., Strain, J., Wong, P., Cleveland, D. W., Culotta, V. C., and Borchelt, D. R. (1999) Hum. Mol. Genet. 8 1451-1460 [DOI] [PubMed] [Google Scholar]
- 20.Hayward, L. J., Rodriguez, J. A., Kim, J. W., Tiwari, A., Goto, J. J., Cabelli, D. E., Valentine, J. S., and Brown, R. H., Jr. (2002) J. Biol. Chem. 277 15923-15931 [DOI] [PubMed] [Google Scholar]
- 21.Jonsson, P. A., Ernhill, K., Andersen, P. M., Bergemalm, D., Brannstrom, T., Gredal, O., Nilsson, P., and Marklund, S. L. (2004) Brain 127 73-88 [DOI] [PubMed] [Google Scholar]
- 22.Doucette, P. A., Whitson, L. J., Cao, X., Schirf, V., Demeler, B., Valentine, J. S., Hansen, J. C., and Hart, P. J. (2004) J. Biol. Chem. 279 54558-54566 [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski, Z., and Minor, W. (1997) Methods in Enzymol. 276 306-326 [DOI] [PubMed] [Google Scholar]
- 24.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1025 [Google Scholar]
- 25.Hart, P. J., Liu, H., Pellegrini, M., Nersissian, A. M., Gralla, E. B., Valentine, J. S., and Eisenberg, D. (1998) Protein Sci. 7 545-555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy, L. M., Strange, R. W., and Hasnain, S. S. (1997) Structure (Lond.) 5 371-379 [DOI] [PubMed] [Google Scholar]
- 27.Read, R. J. (1986) Acta Crystallogr. Sect. A 42 140-149 [Google Scholar]
- 28.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard, M. (1991) Acta Crystallogr. Sect. A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 29.Brünger, A. T., and Rice, L. M. (1997) Methods Enzymol. 277 243-269 [DOI] [PubMed] [Google Scholar]
- 30.Lamzin, V. S., Perrakis, A., and Wilson, K. S. (2001) in International Tables for Crystallography (Rossmann, M. G., and Arnold, E., eds) pp. 720-722, Kluwer Academic Publishers, Dordrecht, The Netherlands
- 31.Strange, R. W., Antonyuk, S. V., Hough, M. A., Doucette, P. A., Valentine, J. S., and Hasnain, S. S. (2006) J. Mol. Biol. 356 1152-1162 [DOI] [PubMed] [Google Scholar]
- 32.Strange, R. W., Antonyuk, S., Hough, M. A., Doucette, P. A., Rodriguez, J. A., Hart, P. J., Hayward, L. J., Valentine, J. S., and Hasnain, S. S. (2003) J. Mol. Biol. 328 877-891 [DOI] [PubMed] [Google Scholar]
- 33.Tainer, J. A., Getzoff, E. D., Beem, K. M., Richardson, J. S., and Richardson, D. C. (1982) J. Mol. Biol. 160 181-217 [DOI] [PubMed] [Google Scholar]
- 34.Getzoff, E. D., Tainer, J. A., Weiner, P. K., Kollman, P. A., Richardson, J. S., and Richardson, D. C. (1983) Nature 306 287-290 [DOI] [PubMed] [Google Scholar]
- 35.Bertini, I., Mangani, S., and Viezzoli, M. S. (1998) Adv. Inorg. Chem. 45 127-251 [Google Scholar]
- 36.Kiaei, M., Bush, A. I., Morrison, B. M., Morrison, J. H., Cherny, R. A., Volitakis, I., Beal, M. F., and Gordon, J. W. (2004) J. Neurosci. 24 7945-7950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banci, L., Bertini, I., Cramaro, F., Del Conte, R., and Viezzoli, M. S. (2003) Biochemistry 42 9543-9553 [DOI] [PubMed] [Google Scholar]
- 38.Brandts, J. F., Halvorson, H. R., and Brennan, M. (1975) Biochemistry 14 4953-4963 [DOI] [PubMed] [Google Scholar]
- 39.Elam, J. S., Malek, K., Rodriguez, J. A., Doucette, P. A., Taylor, A. B., Hayward, L. J., Cabelli, D. E., Valentine, J. S., and Hart, P. J. (2003) J. Biol. Chem. 278 21032-21039 [DOI] [PubMed] [Google Scholar]
- 40.Antonyuk, S., Elam, J. S., Hough, M. A., Strange, R. W., Doucette, P. A., Rodriguez, J. A., Hayward, L. J., Valentine, J. S., Hart, P. J., and Hasnain, S. S. (2005) Protein Sci. 14 1201-1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strange, R. W., Yong, C. W., Smith, W., and Hasnain, S. S. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 10040-10044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Potter, S. Z., Zhu, H., Shaw, B. F., Rodriguez, J. A., Doucette, P. A., Sohn, S. H., Durazo, A., Faull, K. F., Gralla, E. B., Nersissian, A. M., and Valentine, J. S. (2007) J. Am. Chem. Soc. 129 4575-4583 [DOI] [PubMed] [Google Scholar]
- 43.Banci, L., Bertini, I., Cabelli, D. E., Hallewell, R. A., Tung, J. W., and Viezzoli, M. S. (1991) Eur. J. Biochem. 196 123-128 [DOI] [PubMed] [Google Scholar]
- 44.Wang, J., Caruano-Yzermans, A., Rodriguez, A., Scheurmann, J. P., Slunt, H. H., Cao, X., Gitlin, J., Hart, P. J., and Borchelt, D. R. (2007) J. Biol. Chem. 282 345-352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curtain, C. C., Ali, F., Volitakis, I., Cherny, R. A., Norton, R. S., Beyreuther, K., Barrow, C. J., Masters, C. L., Bush, A. I., and Barnham, K. J. (2001) J. Biol. Chem. 276 20466-20473 [DOI] [PubMed] [Google Scholar]
- 46.Jobling, M. F., Huang, X., Stewart, L. R., Barnham, K. J., Curtain, C., Volitakis, I., Perugini, M., White, A. R., Cherny, R. A., Masters, C. L., Barrow, C. J., Collins, S. J., Bush, A. I., and Cappai, R. (2001) Biochemistry 40 8073-8084 [DOI] [PubMed] [Google Scholar]
- 47.Bush, A. I. (2000) Curr. Opin. Chem. Biol. 4 184-191 [DOI] [PubMed] [Google Scholar]
- 48.Furukawa, Y., Torres, A. S., and O'Halloran, T. V. (2004) EMBO J. 23 2872-2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown, N. M., Torres, A. S., Doan, P. E., and O'Halloran, T. V. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 5518-5523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lindberg, M. J., Normark, J., Holmgren, A., and Oliveberg, M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 15893-15898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnesano, F., Banci, L., Bertini, I., Martinelli, M., Furukawa, Y., and O'Halloran, T. V. (2004) J. Biol. Chem. 279 47998-48003 [DOI] [PubMed] [Google Scholar]
- 52.Galaleldeen, A., and Hart, P. J. (2006) in Protein Misfolding and Conformational Diseases (Uversky, V., and Fink, A. L., eds) pp. 327-344, Springer, New York
- 53.Rakhit, R., Crow, J. P., Lepock, J. R., Kondejewski, L. H., Cashman, N. R., and Chakrabartty, A. (2004) J. Biol. Chem. 279 15499-15504 [DOI] [PubMed] [Google Scholar]
- 54.Ray, S. S., Nowak, R. J., Strokovich, K., Brown, R. H., Jr., Walz, T., and Lansbury, P. T., Jr. (2004) Biochemistry 43 4899-4905 [DOI] [PubMed] [Google Scholar]
- 55.Ray, S. S., Nowak, R. J., Brown, R. H., Jr., and Lansbury, P. T., Jr. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 3639-3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rakhit, R., Robertson, J., Vande Velde, C., Horne, P., Ruth, D. M., Griffin, J., Cleveland, D. W., Cashman, N. R., and Chakrabartty, A. (2007) Nat. Med. 13 754-759 [DOI] [PubMed] [Google Scholar]
- 57.Borchelt, D. R., Guarnieri, M., Wong, P. C., Lee, M. K., Slunt, H. S., Xu, Z. S., Sisodia, S. S., Price, D. L., and Cleveland, D. W. (1995) J. Biol. Chem. 270 3234-3238 [DOI] [PubMed] [Google Scholar]
- 58.Johnston, J. A., Dalton, M. J., Gurney, M. E., and Kopito, R. R. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 12571-12576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Son, M., Puttaparthi, K., Kawamata, H., Rajendran, B., Boyer, P. J., Manfredi, G., and Elliott, J. L. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 6072-6077 [DOI] [PMC free article] [PubMed] [Google Scholar]