

Abstract
Müllerian inhibiting substance (MIS) causes regression of the fetal Müllerian duct on binding a heteromeric complex of types I and II cell-surface receptors in the fetal urogenital ridge. The MIS type II receptor (MISRII), which provides specificity for MIS, is also expressed in the adult testis, ovary, and uterus. The rat MISRII promoter was cloned to study the molecular mechanisms underlying its temporal and cell-specific expression. The 1.6-kilobase (kb) promoter contained no recognizable TATA or CAAT box, but there was a consensus Sp1 site upstream of the transcription initiation site. Two binding sites for the orphan nuclear receptor steroidogenic factor-1 (SF-1) are occupied in vitro by using nuclear extracts from R2C cells, an MIS-responsive rat Leydig cell line that expresses endogenous MISRII, with differing affinities, indicating that the distal SF-1 site is bound more avidly than is the proximal SF-1 site. R2C cells transfected with MISRII promoter/luciferase reporter constructs show a 12-fold induction with the 1.6-kb fragment and deletion of sequences upstream of −282-bp lowered luciferase expression to one-third. Mutation of both SF-1 sites greatly inhibited luciferase expression, whereas mutation of either site alone resulted in continuing activation by endogenous SF-1, indicating redundancy. In vitro binding and transcriptional analyses suggest that a proximal potential Smad-responsive element and an uncharacterized element also contribute to activation of the MISRII gene. R2C cells and MISRII promoter regulation can now be used to uncover endogenous transcription factors responsible for receptor expression or repression.
The testis determining factor, SRY, initiates a cascade of signaling events that leads to differentiation of the indifferent fetal gonad into the testis, which in turn leads to the production of at least two hormones with distinct roles essential for normal male reproductive tract development, Müllerian inhibiting substance (MIS; also known as anti-Müllerian hormone), and testosterone (1). MIS, which is produced by Sertoli cells of the fetal testes before the seminiferous tubules become morphologically distinct, is responsible for regression of the Müllerian ducts, the anlagen of the uterus, Fallopian tubes, and upper vagina. Testosterone, which is produced by Leydig cells, induces differentiation of the Wolffian duct into the epididymis, vas deferens, and seminal vesicles and virilizes the external genitalia (2–5). Adult testes and ovaries also produce MIS, albeit at much lower levels, but its role in the adult gonad is still being evaluated. Although the orphan nuclear receptor steroidogenic factor-1 (SF-1) appears to be required for MIS expression, several SF-1-interacting factors are also purportedly involved, and the exact molecular interactions defining how SF-1 regulates MIS transcription remain to be clarified (6–9).
MIS is a member of the transforming growth factor-β (TGF-β) family of hormones (10, 11) involved in the regulation of growth and differentiation. Other members of this family include the activins, bone morphogenetic proteins, inhibins, Drosophila decapentaplegic, Xenopus Vg1, and a large number of growth and differentiation factors. The bioactive carboxyl-terminal fragments of these hormones bind as dimers to a heteromeric complex of type I and type II single transmembrane serine/threonine kinase receptors. When ligand bound, the type II receptor component, which supplies specificity for the ligand on its own or in conjunction with the type I receptor, recruits and phosphorylates its type I receptor partner. The type I receptor then initiates subsequent downstream signaling that, among other pathways, involves direct activation of subsets of ligand-specific Smads (12).
The MIS type II receptor (MISRII) cDNA was first isolated from a rat Sertoli cell cDNA library during a search for androgen-regulated genes, but its expression was subsequently found to be androgen independent (13). The MISRII cDNA was also isolated by homology with other members of the TGF-β family of type II receptors by library screening (14) and by using a PCR approach with degenerate oligonucleotide primers (15). The mRNA corresponding to the MISRII was localized to the Müllerian duct at the time when the Müllerian duct undergoes regression and to Sertoli cells and granulosa cells of both embryonic and adult gonads (15). Sertoli cells and granulosa cells, which surround the germ cells, also produce the MIS ligand, suggesting that MIS is an autocrine factor in the fetal male gonad and in the adult male and female gonad.
Deletion of the MISRII in mice resulted in a phenocopy of the MIS-deficient male mice, including Leydig cell hyperplasia, and provided definitive proof that the cloned receptor was essential for MIS signal transduction (16). Coupled with the observation that mice chronically overexpressing a human MIS transgene had a male pseudohermaphrodite phenotype (17) characterized by feminized external genitalia, underdeveloped internal male reproductive tract structures, and low serum testosterone, it was apparent that MIS was also affecting Leydig cell function (18, 19). The direct effect of MIS on Leydig cells was shown in subsequent studies with primary Leydig cells, in which steroidogenesis was inhibited by MIS (20, 21). We have demonstrated by transfection studies in a mouse Leydig cell line, which expresses the MISRII, that testosterone synthesis was inhibited by the effect of MIS on the transcription of P450c17, the enzyme that catalyzes the conversion of progesterone to androstenedione, the immediate precursor of testosterone (22). These experiments, coupled with the unequivocal demonstration of type II receptor mRNA in primary Leydig cells (22), indicate that MIS, which is produced by the Sertoli cells of the testis, affects Leydig cell function directly in a paracrine fashion via its specific receptor.
We have initiated studies of the temporal and tissue-specific expression of the MISRII to understand the varied roles played by MIS in both the adult and the embryo. Here we report the isolation of a rat MISRII genomic clone containing approximately 1.6-kilobase (kb) 5′ flanking DNA and analyses of the transcriptional regulation of the MISRII promoter in rat Leydig cells, which normally express the MISRII.
Materials and Methods
General Materials and Experimental Procedures.
32P-radiolabeled nucleotides were purchased from New England Nuclear. Sequencing was done by primer walking and cycle sequencing with reagents from Amersham Pharmacia, and DNA sequence analysis was done with macvector software (Oxford Molecular Group, Campbell, CA). Pyrostase thermostable polymerase was purchased from Molecular Genetic Resources (Tampa, FL). Female fetal calf serum was from Aires Scientific/Biologos (Richardson, TX). Antibody to the SF-1 DNA-binding domain was purchased from Upstate Biotechnology (Lake Placid, NY). Unless otherwise noted, standard recombinant DNA techniques were used (23, 24).
MIS Type II Receptor Promoter Cloning and S1 Analysis.
A rat genomic library in λEMBL3 was purchased from CLONTECH and screened with a radiolabeled 40-mer designed from the 5′ end of the rat MISRII DNA (15). One of the isolated clones contained an insert of approximately 15 kb, from which a XbaI 1.6-kb promoter fragment was subcloned into pBluescript (Promega) for sequencing. The S1 single-stranded DNA probe was made from an XbaI/SacI fragment of the promoter cloned into M13 that was annealed to an oligonucleotide primer (5′-TGCCGCAGGAAGCAGTGCCA-3′) complementary to a region in the first exon for primer extension with the Klenow fragment of DNA polymerase and [α-32P] dCTP. The radiolabeled product was digested with EcoNI, denatured, and gel purified. RNA (20 μg) was heated to 65°C for 10 min and hybridized overnight with 5 × 104 cpm of S1 probe in a total volume of 50 μl hybridization solution that contained 40 mM piperazine-N,N′-bis[2-ethanesulfonic acid], pH 6.4, 1 mM EDTA, and 400 mM NaCl. S1 nuclease digestion was carried out the next day at room temperature for 1 h by adding 250 μl S1 digestion buffer [250 mM NaCl/20 mM K acetate, pH 4.5/1 mM Zn(SO4)2/200 units of S1 nuclease (Boehringer Mannheim)]. The reaction was stopped by adding 100 μl S1 Stop buffer (3 M ammonium acetate/2.5 mM EDTA/2 mg/ml yeast tRNA), ethanol precipitated, and resuspended in formamide with dyes for electrophoresis in a 7 M urea/6% polyacrylamide sequencing gel alongside a sequencing reaction done with the same primer used to make the S1 probe.
Transfection of R2C Cells.
R2C adult rat Leydig tumor cells (ATCC CCL97) were cultured in Ham’s F-10 supplemented with 15% horse serum/2.5% female fetal bovine serum/20 mM Hepes, pH 7.2/100 units/ml penicillin/100 μg/ml streptomycin and were transfected by calcium phosphate coprecipitation. Promoter activity was assayed by using the Dual-Luciferase Reporter Assay System (Promega), in which the cells were cotransfected with the pRL-TK Renilla luciferase construct to control for transfection efficiency. On day 0, 2 × 105 cells were plated in each well of a six-well culture plate and cultured in 2 ml of growth medium. On day 2, the growth medium was changed and the transfection initiated in triplicate for each construct with 5 μg total DNA. On day 3, the precipitate was washed off once with Hank’s balanced salt solution, and growth medium was added. Cells were assayed for luciferase activity on day 4 according to the supplied protocol for the Dual-Luciferase System, with the exception that firefly and Renilla luciferase data were collected on separate aliquots of the same sample of cellular lysate.
Preparation of Chimeric MRII Promoter/Luc Reporter Constructs.
A chimeric rat MISRII promoter/luciferase reporter construct was created by subcloning the 1.575-kb genomic fragment isolated above into pGL3-Basic (Promega). Shorter fragments were amplified by PCR with pyrostase [see Table 1 in the supplemental material (see www.pnas.org) for primers used] and inserted directly into pCRII or pCRII-TOPO (Invitrogen) for blue/white selection and sequencing. Appropriate clones were digested with SacI-XhoI, yielding fragments containing approximately 48 bp of vector sequence at the 5′ end of the promoter fragment, which were inserted into similarly digested pGL3-Basic. Constructs with mutations in the SF-1 were made by PCR with the mutant SF-1 primers shown in supplemental Table 1. Sequencing of all constructs was done to ensure the fidelity of the thermostable polymerase.
Nuclear Extract Preparation.
Nuclear extracts were prepared according to methods first described by Dignam et al. (25). Cells (6 × 107) were washed twice with Hank’s balanced salt solution and then resuspended in 1 ml Triton Lysis Buffer (9 mM Tris, pH7.5/135 mM NaCl/0.9 mM MgCl2/freshly added 0.5 mM DTT/0.5 mM PMSF). Cells were lysed for 5 min on ice, and nuclei were separated by centrifugation at 800 × g for 5 min in a refrigerated microcentrifuge and resuspended in 1 ml Dignam’s Buffer C (25% glycerol/20 mM Hepes, pH 7.9/0.42 M NaCl/1.5 mM MgCl2/0.2 mM EDTA/0.5 mM DTT/0.5 mM PMSF). Nuclear proteins were extracted by rocking at 4°C for 45 min and centrifuged at 13,800 × g for 15 min. The supernatant was dialyzed at 4°C against 250–500 volumes of modified Shapiro’s Buffer D (20 mM Hepes, pH 7.9/20% glycerol/100 mM KCl/0.2 mM EDTA/0.2 mM EGTA/2 mM DTT/0.5 mM PMSF), and any precipitated proteins were removed by another 15-min centrifugation at 13,800 × g. Protein concentration was determined by Bradford Assay (26), and the extracts were stored at −70°C.
DNase I Footprinting.
Radiolabeled DNA probes for footprinting analysis were constructed by PCR amplification of fragments of pB2, the 1.575-kb promoter plasmid. Oligonucleotides [pp6, pp5, and pp1; see supplemental Table 1 (www.pnas.org)] were phosphorylated with [γ-32P]ATP and T4 polynucleotide kinase and were used as 5′ primers for amplification by PCR to create the promoter fragments of interest with the 3′ primer XhoLuc (supplemental Table 1; see www.pnas.org). Probes were then PAGE purified. The binding of protein to DNA occurred in a total volume of 100 μl containing 50 μl 2× binding buffer (50 mM Tris, pH 8/100 mM KCl/12.5 mM MgCl2/1 mM EDTA/20% glycerol/1 mM DTT)/0.1 μg poly(dA/dT)/3 × 104 cpm-labeled probe/varying quantities of nuclear extract. The volumes were adjusted to 100 μl with Shapiro’s Buffer D. Binding was allowed for 15 min on ice, after which 100 μl of a solution containing 5 mM CaCl2/10 mM MgCl2 was added, followed by 1.25 μg DNase I. Digestion was done for 1 min 45 sec and was terminated with 300 μl stop solution (200 mM NaCl/30 mM EDTA/1% SDS/100 μg/ml yeast tRNA). The resultant digestion products were phenol and chloroform extracted and ethanol precipitated. The pellet was dissolved in 10 μl 98% formamide loading solution and electrophoresed on a 6% polyacrylamide denaturing sequencing gel alongside sequencing reactions primed with the same oligonucleotide. The gels were dried and exposed to x-ray film overnight with Kodak HE intensifying screens at −70°C.
Gel-Shift Assay.
Oligonucleotides SF1–250 A, SF1–250 B, SF1–200 A, and SF1–200 B (see supplemental Table 1) were diluted to 10 μM in 0.1 M NaCl. Equimolar amounts of each complementary strand were heated to 80°C and allowed to cool to room temperature. Ten picomoles of these annealed oligonucleotides was then phosphorylated by using T4 polynucleotide kinase and [γ-32P]ATP. After phenol/chloroform extraction, the aqueous layer was further purified over a Kodak NuClean D25 column. cpm (105) was combined with 1 μg nuclear extract in 1.5 μl 10×binding buffer (66.7 mM Hepes, pH 7.9/50 mM DTT/10 mM EDTA) with 4.5 μg BSA/0.1 μl 5M NaCl/0.8 μg poly(dI⋅dC). A total volume of 15 μl was obtained by adding sufficient Shapiro’s Buffer D. Binding was done for 30 min at room temperature. The entire reaction was run on a 4% 40:1 acrylamide/bisacrylamide nondenaturing gel, which was dried and exposed to x-ray film overnight with intensifying screens at −70°C. Quantitation, when done, was with a Molecular Dynamics PhosphorImager.
Results and Discussion
Rat MIS Type II Receptor Promoter Cloning and Sequence Analysis.
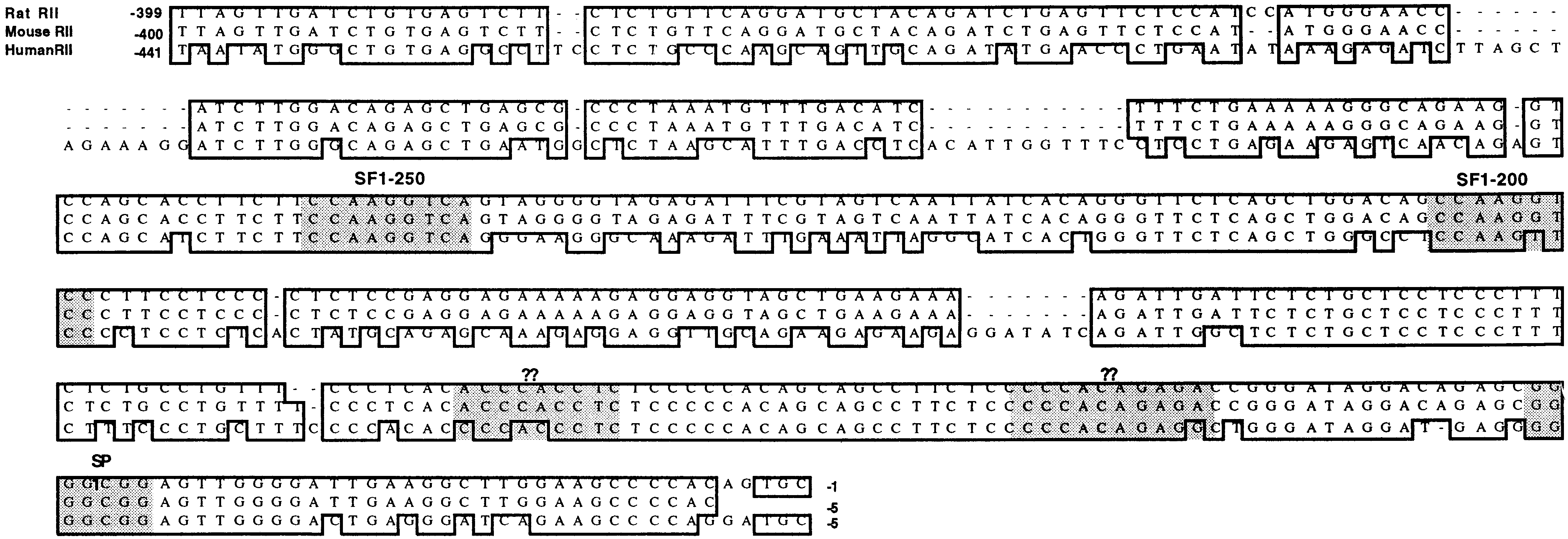
To define the molecular mechanisms governing the MIS-mediated effects on the Müllerian duct in the urogenital ridge and on regulation of steroidogenesis in Leydig cells, we have studied the regulation of MISRII expression. We isolated a 15-kb rat MISRII genomic clone from which we have subcloned and sequenced a 1.6-kb fragment which corresponds to the 5′ flanking region of the first exon (see Fig. 1). Sequence analysis showed approximately 150-bp inverted Alu-family repeats, which could be involved in DNA secondary structure and regulatory variations during evolution (27). Alignment of the Alu repeats underlined in Fig. 1A is shown in Fig. 1B. After subcloning and sequence analysis, it was necessary to determine the transcriptional start site of the mRNA (Fig. 1A, arrow). This was done with a radiolabeled single-stranded DNA probe, followed by solution hybridization to RNA from 28-day-old rat testes and S1 nuclease digestion analysis (see Fig. 2). Subsequent studies have been done by using 5′ rapid amplification of cDNA ends to confirm that the 5′ end is identical to that seen in the cDNA and the genomic fragment up to the transcription initiation site found by S1 nuclease (data not shown).
Figure 1.

Rat MIS type II receptor promoter sequence. (A) The sequence of a genomic fragment containing approximately 1.6 kb of the MISRII was subcloned from a 15-kb clone, which was isolated from λEMBL3 rat library and sequenced on both strands. Potential SF-1-binding sites are shown boxed, as are GATA and Sp1 sites. The boxed sequences labeled CAGA and ?? indicate DNase I footprinted regions (see Fig. 4) that are potential binding sites for Smads and some unknown factor, respectively. The arrow indicates the transcription initiation site found by S1 analysis; the first exon is shown with predicted amino acids underneath, and the first intron is shown with uncapitalized letters. The bases are negatively numbered on the right-hand side from the transcription initiation site. The 5′ ends of the promoter fragments used in the luciferase experiments are bracketed ([) and indicated with their names in bold. The 3′ end for all the promoter fragments is indicated by #. These sequences have been submitted to GenBank under the accession number AF092445. Inverted repeats with Alu-like sequences were observed by sequence analysis at −1336 to −1486 and −476 to 576 and are shown underlined in A and aligned in B.
Figure 2.

Transcription initiation site of the Rat MIS type II receptor. The transcriptional start site of the rat MISRII mRNA was mapped by S1 nuclease digestion. RNA from the indicated sources (rat testes, liver, liver poly(A)+, and yeast) was hybridized overnight in solution with a single-stranded radiolabeled DNA probe. The resulting hybrids were digested with S1 nuclease and analyzed by denaturing gel electrophoresis. The band observed indicates the length of the probe protected from digestion by the hybridized mRNA. Sequencing was done with the same primer as that used for making the S1 probe and electrophoresed alongside the S1-digested fragment to indicate the exact nucleotides for transcription initiation.
In addition to showing that the genomic fragment was a functional promoter, identification of the transcriptional start site allows us to determine where the proximal promoter for the rat type II receptor gene ended and the 5′-untranslated end of the mRNA began. The promoter sequence has no recognizable TATA or CCAAT boxes in the vicinity of the transcription start site, which has also been observed in the promoter region of the human MIS (28) as well as numerous other genes (29), including the human MISRII promoter (30). The TATA box-binding protein is a member of the TFIID transcription complex that binds to a TATAAA consensus sequence 25–30 bases upstream of the transcription initiation site and recruits the general transcriptional machinery. In the absence of a TATA box, an Initiator element with a consensus YYA+1NT/ATYY can effectively nucleate preinitiation complex assembly by multiple Initiator-binding proteins by a less well understood mechanism (29). The MISRII promoter does have a pyrimidine-rich transcription initiation site, GTGC+1CCTGT, which does not conform to the consensus Initiator. A lack of canonical TATA or CCAAT sites was common to the promoter regions of rat, mouse, and human MISRIIs [shown in supplemental Fig. 7 (see www.pnas.org)]. Homology between mouse and rat MISRII promoters was nearly 100% and approximately 80% between rodent and human in the proximal 150 bases, but less so further from the transcriptional start site.
Scanning for transcription factor binding sites that were conserved revealed that consensus elements were found for Sp1 at nucleotides −37 to −44 in the rat. Sp1 belongs to a family of ubiquitous transcription factors that binds to G+C-rich regions of many promoters and cooperates with other DNA-binding proteins such as SF-1 and the CAAT-box/enhancer-binding protein to promote transcription from TATA-less and initiator-less promoters (31–33). When activated, Sp1 can also enhance transcription of the TGF-β type II receptor in breast cancer cells and is required for the early response of α2(I) collagen to TGF-β (34).
Footprinting analysis of the region immediately upstream of the transcription initiation site (Fig. 3A) was done to determine whether the site for Sp1 was protected from DNase I digestion by protein in R2C nuclear extract. The Sp1 site was protected from DNase I digestion as shown in Fig. 4 and is probably critical for transcriptional activation from the MISRII promoter. Although the precise role for Sp1, the first of a family of proteins that bind to G+C-rich DNA, in regulating the MISRII is yet to be determined, we suspect that the ubiquitous transcription factor might be involved in nucleating the preinitiation complex, as it has been shown to do in other TATA-less promoters (33, 35, 36) and might interact with transacting factors binding to upstream SF-1 sites (37). Detailed studies need to be done to determine whether Sp1 functions to recruit the transcriptional machinery to the MISRII promoter in the absence of a TATA box or initiator region and how it interacts with other factors to initiate transcription, as has been observed in other genes.
Figure 3.

R2C nuclear proteins occupy cis-elements in the MIS type II receptor promoter. DNase I footprinting was done with 32P-labeled fragments PCR of the rat MISRII promoter incubated with 25, 50, and 75 μg of protein and digested with DNase I. The digested products were electrophoresed on an acrylamide gel alongside a sequencing reaction (not shown) performed with the same oligonucleotide primer to precisely map the footprinted nucleotides in the fragment. The lines indicate the DNA fragment, and the open boxes indicate the footprinted region. The wedges above the digests indicate increasing amounts of nuclear protein. The nucleotides protected from DNase I digestion are indicated in Fig. 1. (A) Fragment made from the proximal 144 bases of the MISRII promoter shows footprints in the GC box or SP1 site and to previously unidentified sites (nucleotides are shown in Fig. 1) which are labeled with a question mark. The GATA site is indicated where there appears to be increased digestion. (B) Fragments made with the distal SF-1 site (SF-1 250) and with the proximal SF-1 site (SF-1 200) are shown with their corresponding footprints. (C) The proximal SF-1 probe did not contain the distal SF-1 site.
Figure 4.

Electrophoretic mobility of SF-1 sites in the MIS type II receptor promoter are altered when incubated with R2C nuclear extracts. Gel-shift assays were done as described in Materials and Methods with annealed complimentary oligonucleotides corresponding to the footprinted SF-1 sites. 32P-labeled oligonucleotides were incubated in the absence (Free) or presence of 1-μg R2C nuclear protein alone (NE) or with indicated 50-fold molar excess unlabeled competitor or antibody. Unbound DNA probes are indicated with the arrow. (A) The distal SF-1 site (SF1–250) is shown competed with unlabeled specific (SF1–250), nonspecific (SP1), and mutated SF-1 site (SF1–250*) and the proximal SF-1 site (SF1–200). Antibody to the SF-1 DNA-binding domain was also added to the R2C nuclear extract before addition of probe (αSF-1-pre). (B) The proximal SF-1 site (SF1–200) is shown competed with unlabeled specific (SF1–200), nonspecific (SP1), mutated SF-1 site (SF1–200*), and the distal SF-1 site (SF1–250). Antibody to the SF-1 DNA-binding domain was also added to the R2C nuclear extract before addition of probe (αSF-1-pre).
A GATA-binding site was also detected just upstream of the Sp1 site. The GATA-binding proteins are a family of tissue-specific zinc finger DNA-binding proteins that bind the consensus motif (A/T)GATA(A/G). GATA-1 and GATA-4 mRNA have been detected in rodent gonads and have been implicated in transcriptional activation of the rat Inhibin α subunit in Leydig cells (38) and of the MIS gene in Sertoli cells (39), respectively. In contrast to protection, DNase I footprinting analysis showed (Fig. 3A) that the GATA site had increased access or was hypersensitive to DNase I with added R2C nuclear protein. DNase I hypersensitive sites in chromatin are normally associated with genes that are expressed, and the DNA sequences within the site are required for expression. Interpretation of a DNase I hypersensitive site in the context of in vitro footprinting experiments with radiolabeled DNA fragments is unclear, but, because proteins are binding to the adjacent DNA both 5′ and 3′ of the GATA site, it is possible that the bound proteins are altering the conformation of the DNA such that the GATA site is more accessible to DNase I. It is interesting to note that the GATA site was not gel shifted with nuclear extract (data not shown).
We also observed footprinted regions at −62 to −73 and from −91 to −99 as shown in Fig. 3A and highlighted in Fig. 1, which correspond to the site for DNA-binding proteins reminiscent of a Smad3/4 DNA-binding element (labeled with CAGA) (40, 41) and to that of an unknown DNA-binding protein (labeled with ?), respectively. Because MIS is likely to signal by a mechanism similar to that of other TGF-β family members, and a possible Smad3/4-binding site was observed in the MISRII promoter, we also tested whether MIS could regulate the expression of its own receptor. However, addition of MIS to R2C cells transfected with MIS type II promoter/luciferase reporter construct did not affect luciferase expression (data not shown).
The SF-1-Binding Sites Are Occupied in the MIS Type II Receptor Promoter.
Two SF-1-binding sites were found at positions −250 and −200 (Fig. 1B) and are subsequently referred to as the distal and the proximal SF-1-binding sites, respectively. The orphan nuclear receptor SF-1 was first isolated from adrenocortical cells because of its capacity to act as a key regulator of steroidogenic enzymes. It is expressed in all steroidogenic cells such as Leydig cells in the testis and theca and granulosa cells in the ovary, in addition to the adrenal cortex cells in which it was originally found. Targeted disruption of the SF-1 gene in mice resulted in complete adrenal and gonadal agenesis and death by postnatal day 8, probably from adrenocortical insufficiency. SF-1 binds to a core DNA element (CCAAGGTCA) as a monomer via a zinc finger DNA-binding domain. The distal SF-1-binding site, which perfectly matches the SF-1 consensus sequence, is well conserved in the rodent and human promoters in terms of distance from the transcription start site as well as in identity at the nucleotide level. The proximal SF-1-binding site is less well conserved. This more proximal site (CCAAGGTCC) differs from the consensus at the last nucleotide in the rodent promoter, whereas in the human promoter there is a T in place of the second G.
Because SF-1 is expressed in Leydig cells and has been shown to modulate transcription of the many genes involved in steroidogenesis and gonadogenesis, including MIS, experiments were designed to determine whether the SF-1-binding sites observed in the rat MIS type II promoter are essential for transcriptional regulation of the MISRII gene in Leydig cells. Nuclear protein extracts from the rat Leydig cell tumor line, R2C, were incubated with radiolabeled double-stranded probes prepared from fragments of the MISRII used for DNase I footprinting analysis (Fig. 3). When incubated with lower concentrations of nuclear extract (Fig. 3B), the proximal SF-1 site (SF1–200) was not protected from DNase I digestion, whereas the distal site (SF1-250) was very well protected and showed a very distinct footprinting pattern with a probe that encompassed both SF-1 sites. At higher concentrations of nuclear extract (Fig. 3B), the proximal SF-1 site is protected. In addition, when a footprinting probe was made that did not contain the distal SF-1-binding site, the proximal site was protected at the lower concentrations of nuclear extract (Fig. 3C).
To investigate whether the protein that bound to SF-1 cognate-binding sites detected by DNase I footprinting was indeed SF-1, we performed electrophoretic mobility shift assays with R2C nuclear protein and labeled duplex oligonucleotides corresponding to the SF-1 sites. SF-1 expression in the steroidogenic Leydig cells in the interstitium of the testis as well as in R2C cells, which were derived from the a rat Leydig cell tumor, has been implicated in regulating the expression of steroidogenic enzymes via its cognate binding site (42, 43).
As shown in Fig. 4, retarded complexes were formed when the distal SF-1 site probe (SF1–250, Fig. 4A) and the proximal SF-1 site probe (SF1–200, Fig. 4B) were incubated with R2C nuclear extract. The specificity of the protein-bound DNA complex was examined by competition experiments. In Fig. 4A, addition of 50-fold molar excess unlabeled SF-250 to the reaction mixture was able to greatly diminish the gel-shifted band. In contrast, addition of 50-fold molar excess of either Sp1-containing (Sp1) or mutant SF1-containing (SF1–250*) duplexed oligonucleotides had no effect on the degree of shift observed. Incubation with 50-fold molar excess DNA containing the proximal SF-1 site (SF1–200) was able to diminish the gel shift but appeared not to compete as well as the distal canonical SF-1 site (SF1–250), which correlates with the footprinting results (Fig. 3B). Finally, preincubation with an antibody produced against the DNA-binding domain of SF-1 was also able to diminish the gel-shifted band, proving that the protein was, at a minimum, immunologically related to SF-1 if not SF-1 itself. A similar series of experiments was done with labeled oligonucleotides corresponding to the proximal SF-1 site (SF1–200, Fig. 4B). Unlabeled SF1–200 competed for binding to the SF1–200 probe, but neither the unlabeled Sp1 nor the mutated SF1–200* oligonucleotides was a very effective competitor, and the SF-1 antibody blocked complex formation. Competition for binding of SF-1 to the proximal site probe (SF1–200) with 50 molar excess unlabeled distal SF-1 site probe SF1–250 was complete. Further analysis of the relative affinities of the proximal and distal sites for SF-1 binding was done with increasing amounts of unlabeled competitor and quantitated with a PhosphorImager (supplemental Fig. 8; see www.pnas.org) that clearly show that the distal site binds DNA more avidly than does the proximal site.
MIS Type II Promoter (1.5 kb) Can Initiate Transcription of a Luciferase Reporter Gene.
Initial studies were done with reporter constructs containing 5′ deletion fragments of the MIS type II promoter driving the expression of the firefly luciferase gene. Rat R2C cells were transfected by calcium phosphate coprecipitation with DNA constructs containing the 1.6-kb fragment of the MISRII promoter and progressively smaller fragments, as shown. All experiments were done with a cotransfected plasmid containing the Renilla luciferase gene driven by the HSV thymidine kinase promoter. Use of Renilla allowed us to normalize MIS promoter-driven luciferase activity for transfection efficiency to ensure that differences in luciferase were caused only by the promoter fragments. To avoid the possibility that poor plasmid quality might skew the results, replicate experiments were also done with different preparations of DNA (n = 4). Additionally, experiments were done in cells that do not express endogenous MISRII (JEG-3, COS, and UGR1 cells) and showed no significant activation of expression of the reporter minigene above that of the minimal promoter fragment, indicating that the 1.6-kb promoter fragment required endogenous factors present in Leydig cells for activation.
In Fig. 5, the relative levels of luciferase expression when compared with the promoterless parent luciferase plasmid are shown along with a schematic representation of the promoter fragments used in this set of experiments. The DNase I footprinted regions in the proximal promoter, which were observed in Fig. 4, are shown to help correlate the luciferase results to possible DNA elements important for transcription. The promoter/reporter construct containing the 1.6-kb fragment of the MIS type II promoter (pFL) drove the expression of luciferase 12-fold over background in this MIS-responsive cell line. When 5′ DNA from the MISRII promoter was deleted to yield the proximal -282 promoter region (p-282), luciferase activity was significantly induced 4-fold over background but one-third when compared with the induction observed with the 1.6-kb pFL. Deletion constructs lacking the distal SF-1 site but still containing the proximal SF-1 site (p-243 and p-208) had roughly 8-fold induction, which is 2-fold greater luciferase activity than p-282, which contains both SF-1 sites.
Figure 5.

Expression of MIS type II receptor promoter/luciferase reporter in R2C cells. The 1.6-kb MISRII promoter and smaller fragments were placed in front of the firefly luciferase gene and used to measure their ability to drive luciferase expression when transfected into R2C cells. The MISRII promoter is schematically drawn above the represented promoter fragments to indicate the footprinted regions of the promoter, which are shown with black boxes and, when recognized by sequence analysis, the sites were labeled with their cognate transcription factor. The numbers shown indicate the number of base pairs from the transcription initiation site, which is indicated with an arrow. The fragments used in the transfection experiments are represented as shaded bars, and the relative size of each is shown with their names. Cells were transfected with the indicated plasmids in triplicate and the average-fold increase over the promoterless parental vector of four different experiments is plotted to the right of each with error bars representing SEM. Luciferase activity from each of the promoter/reporter plasmids was measured and normalized for transfection efficiency with the activity of a cotransfected thymidine kinase promoter/Renilla luciferase reporter.
We have attempted to determine whether MISRII promoter expression can be regulated with increasing exogenous amounts of either native mouse SF-1 expression construct or a construct with a mutant SF-1 lacking the ligand-binding domain. Unlike others, we have been unable to induce luciferase expression by increasing SF-1 (or SF-1 with WT1) in heterologous cell lines such as JEG-3, UGR1, and COS, which lack SF-1 (9). This might be because of a lack of the appropriate ligand or obligatory interacting factor in these cell lines for proper SF-1 activity. It is of considerable practical importance that the MISRII promoter in Leydig cells provides a system in which to identify the components of DNA-binding complex seen in the gel-shift experiments (Fig. 4).
To investigate the relative contributions of the SF-1 sites to the expression of the luciferase reporter, mutations were made in the SF-1-binding sites in the context of the 1.6-kb MISRII promoter fragment and transfected into R2C cells. Luciferase expression of the mutant constructs was normalized to the activity of pFL, the wild-type 1.6-kb MISRII promoter construct (Fig. 6). Mutation of both the distal and the proximal sites in the same construct (DM) resulted in less than 1/10th the luciferase activity observed with pFL. However, mutating the single distal SF-1 site (*250) or proximal SF-1 site (*200) caused luciferase activity that was equal to or greater than that caused by the wild-type promoter. These deletion and mutational analyses with endogenously expressed SF-1 in R2C cells indicate that activation of the MISRII promoter by SF-1 is essential, and that the two sites are redundant. Additionally, luciferase experiments with mutations in the unknown DNA-binding site observed in the footprinting analysis (Fig. 3A) revealed that DNA binding to that site was also required for maximal activity.
Figure 6.

The proximal SF-1 site inhibits transcription of the MIS type II receptor gene. Luciferase experiments were done as described. Mutations were made in the proximal (*200) or distal (*250) SF-1 sites or both (DM) and the unknown site at −91 to −99 (*unk) in the context of the 1.6-kb full-length fragment driving the luciferase reporter. The luciferase activity was normalized to the wild-type pFL and plotted. These experiments were done in triplicate a minimum of four times for each plasmid, and the error bars represent SEM.
These expression results and those from the in vitro experiments suggest a role for SF-1 in rat MISRII expression that is more complicated than that proposed for the human MISRII in which exogenous SF-1 activates transcription (44). Although the proximal SF-1-binding site in rodent MISRII promoter is well conserved, the human MIS type II promoter appears to contain a less well-conserved more proximal SF-1 site at approximately −200 bp (supplemental Fig. 7; see www.pnas.org). We also expect that expression of the MISRII will be modulated differently depending on the temporal and cell-specific needs for MIS signal transduction. Alternatively, perhaps our different but reproducible results relate to the use of endogenous SF-1 protein in R2C cells that respond to MIS, indicating the presence of functional receptor (22). In comparison, the experiments done with the 1.1-kb human MISRII fragment driving expression of a luciferase reporter were done in heterologous NT2/D1 teratocarcinoma cells cotransfected with an SF-1 overexpression plasmid (44).
Summary and Future Direction
MISRII expression is tightly regulated both developmentally and in a tissue-specific manner. To understand the molecular mechanisms governing regulation of MISRII expression in Leydig cells, we have cloned and characterized the MISRII promoter. Having concluded that we had cloned the transcription initiation site (Fig. 2) and demonstrated that potential transcription factor binding sites are bound by protein extracted from nuclei (Figs. 3 and 4), we studied whether the cloned MISRII genomic fragment was sufficient to promote transcription by generating chimeric minigenes with progressively truncated MISRII promoter fragments driving a luciferase reporter for transient transfection studies in R2C cells (Fig. 5). The deletion causing the most dramatic effect on transcription was that of the distal 1.3 kb, indicating that important regulatory DNA lies further upstream of the proximal promoter that is described in this report. Analysis of this region, in particular the inverted repeats and the unknown DNA-binding regions in the proximal promoter, will form the basis of our future studies of cis-acting elements of the MISRII promoter. Our results also demonstrate the vital role of SF-1 binding to MISRII promoter for transcriptional activation in a system that relies solely on endogenous expression of SF-1.
Expression of the MISRII in the fetal Müllerian duct is critical for normal male reproductive development, but the role(s) of MIS signal transduction in the adult gonad is not yet clear. The receptor is expressed in Sertoli and Leydig cells of the testis as well as in the granulosa cells of the ovary (15). We know MIS regulates expression of steroidogenic enzymes in Leydig cells (22), but neither the molecular mechanisms of enzyme regulation nor that of MISRII expression are well understood. The results reported here provide a foundation for understanding the molecular mechanisms of MISRII expression in Leydig cells and suggest a clear path for further investigation. The Sp1 site and the other DNase I footprinted regions shown in Fig. 3A, one of which might be recognized by Smads and the other totally unknown, will be of particular interest to study in detail in the future.
Supplementary Material
Acknowledgments
We thank Drs. Joel Habener and John Nilson for critically reviewing the manuscript and Drs. Trent Clarke and Xiaohong Liu for helpful advice. We are grateful to Drs. Mishina and Behringer for making the mouse genomic sequence available to us. This work was supported in part by grants from the National Institute for Child Health and Human Development (R01-HD32112 to P.K.D. and F32-HD07954 to J.T.), the National Cancer Institute (R29-CA79459 to J.T.), and the March of Dimes Birth Defects Foundation.
Abbreviations
- MIS
Müllerian inhibiting substance
- MISRII
MIS type II receptor
- SF-1
steroidogenic factor-1
- TGF-β
transforming growth factor-β
- kb
kilobase
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected on April 27, 1999.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. AF092445).
References
- 1.Swain A, Narvaez V, Burgoyne P, Camerino G, Lovell-Badge R. Nature (London) 1998;391:761–767. doi: 10.1038/35799. [DOI] [PubMed] [Google Scholar]
- 2.Jost A. Recent Prog Horm Res. 1953;8:379–418. [Google Scholar]
- 3.Josso N, Cate R L, Picard J Y, Vigier B, di Clemente N, Wilson C, Imbeaud S, Pepinsky R B, Guerrier D, Boussin L, et al. Recent Prog Horm Res. 1993;48:1–59. doi: 10.1016/b978-0-12-571148-7.50005-1. [DOI] [PubMed] [Google Scholar]
- 4.Lee M M, Donahoe P K. Endocr Rev. 1993;14:152–164. doi: 10.1210/edrv-14-2-152. [DOI] [PubMed] [Google Scholar]
- 5.Teixeira J, Donahoe P. J Androl. 1996;17:336–341. [PubMed] [Google Scholar]
- 6.Luo X, Ikeda Y, Parker K L. Cell. 1994;77:481–490. doi: 10.1016/0092-8674(94)90211-9. [DOI] [PubMed] [Google Scholar]
- 7.Parker K L, Schimmer B P. Endocr Rev. 1997;18:361–377. doi: 10.1210/edrv.18.3.0301. [DOI] [PubMed] [Google Scholar]
- 8.de Santa Barbara P, Bonneaud N, Boizet B, Desclozeaux M, Moniot B, Sudbeck P, Scherer G, Poulat F, Berta P. Mol Cell Biol. 1998;18:6653–6665. doi: 10.1128/mcb.18.11.6653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nachtigal M W, Hirokawa Y, Enyeart-VanHouten D L, Flanagan J N, Hammer G D, Ingraham H A. Cell. 1998;93:445–454. doi: 10.1016/s0092-8674(00)81172-1. [DOI] [PubMed] [Google Scholar]
- 10.Cate R L, Mattaliano R J, Hession C, Tizard R, Farber N M, Cheung A, Ninfa E G, Frey A Z, Gash D J, Chow E P, et al. Cell. 1986;45:685–698. doi: 10.1016/0092-8674(86)90783-x. [DOI] [PubMed] [Google Scholar]
- 11.Massague J. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 12.Kretzschmar M, Massague J. Curr Opin Genet Dev. 1998;8:103–111. doi: 10.1016/s0959-437x(98)80069-5. [DOI] [PubMed] [Google Scholar]
- 13.Baarends W M, van Helmond M J L, Post M, van der Schoot P J C M, Hoogerbrugge J W, de Winter J P, Uilenbroek J T J, Karels B, Wilming L G, Meijers J H C, Themmen A P N, et al. Development (Cambridge, UK) 1994;120:189–197. doi: 10.1242/dev.120.1.189. [DOI] [PubMed] [Google Scholar]
- 14.di Clemente N, Wilson C, Faure E, Boussin L, Carmillo P, Tizard R, Picard J-Y, Vigier B, Josso N, Cate R. Mol Endocrinol. 1994;8:1006–1020. doi: 10.1210/mend.8.8.7997230. [DOI] [PubMed] [Google Scholar]
- 15.Teixeira J, He W W, Shah P C, Morikawa N, Lee M M, Catlin E A, Hudson P L, Wing J, Maclaughlin D T, Donahoe P K. Endocrinology. 1996;137:160–165. doi: 10.1210/endo.137.1.8536608. [DOI] [PubMed] [Google Scholar]
- 16.Mishina Y, Rey R, Finegold M J, Matzuk M M, Josso N, Cate R L, Behringer R R. Genes Dev. 1996;10:2577–2587. doi: 10.1101/gad.10.20.2577. [DOI] [PubMed] [Google Scholar]
- 17.Wilson J D, Harrod M J, Goldstein J L, Hemsell D L, MacDonald P C. N Engl J Med. 1974;290:1097–1103. doi: 10.1056/NEJM197405162902001. [DOI] [PubMed] [Google Scholar]
- 18.Behringer R R, Cate R L, Froelick G J, Palmiter R D, Brinster R L. Nature (London) 1990;345:167–170. doi: 10.1038/345167a0. [DOI] [PubMed] [Google Scholar]
- 19.Lyet L, Louis F, Forest M G, Josso N, Behringer R R, Vigier B. Biol Reprod. 1995;52:444–454. doi: 10.1095/biolreprod52.2.444. [DOI] [PubMed] [Google Scholar]
- 20.Racine C, Rey R, Forest M G, Louis F, Ferre A, Huhtaniemi I, Josso N, di Clemente N. Proc Natl Acad Sci USA. 1998;95:594–599. doi: 10.1073/pnas.95.2.594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rouiller-Fabre V, Carmona S, Merhi R A, Cate R, Habert R, Vigier B. Endocrinology. 1998;139:1213–1220. doi: 10.1210/endo.139.3.5785. [DOI] [PubMed] [Google Scholar]
- 22.Teixeira J, Fynn-Thompson E, Payne A H, Donahoe P K. Endocrinology. 1999;140:4732–4738. doi: 10.1210/endo.140.10.7075. [DOI] [PubMed] [Google Scholar]
- 23.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1998. [Google Scholar]
- 24.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 25.Dignam J D, Lebovitz R M, Roeder R G. Nucleic Acids Res. 1983;11:1475–1489. doi: 10.1093/nar/11.5.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 27.Britten R J. Proc Natl Acad Sci USA. 1996;93:9374–9377. doi: 10.1073/pnas.93.18.9374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Haqq C M, King C Y, Donahoe P K, Weiss M A. Proc Natl Acad Sci USA. 1993;90:1097–1101. doi: 10.1073/pnas.90.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smale S T. Biochim Biophys Acta. 1997;1351:73–88. doi: 10.1016/s0167-4781(96)00206-0. [DOI] [PubMed] [Google Scholar]
- 30.Imbeaud S, Faure E, Lamarre I, Mattei M-G, diClemente N, Tizard R, Carre-Eusebe D, Belville C, Tragethon L, Tonkin C, et al. Nat Genet. 1995;11:382–388. doi: 10.1038/ng1295-382. [DOI] [PubMed] [Google Scholar]
- 31.Hagen G, Muller S, Beato M, Suske G. Nucleic Acids Res. 1992;20:5519–5525. doi: 10.1093/nar/20.21.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li N, Seetharam B. J Biol Chem. 1998;273:28170–28177. doi: 10.1074/jbc.273.43.28170. [DOI] [PubMed] [Google Scholar]
- 33.Hu Z Z, Zhuang L, Meng J, Dufau M L. J Biol Chem. 1998;273:26225–26235. doi: 10.1074/jbc.273.40.26225. [DOI] [PubMed] [Google Scholar]
- 34.Greenwel P, Inagaki Y, Hu W, Walsh M, Ramirez F. J Biol Chem. 1997;272:19738–19745. doi: 10.1074/jbc.272.32.19738. [DOI] [PubMed] [Google Scholar]
- 35.Ammanamanchi S, Kim S J, Sun L Z, Brattain M G. J Biol Chem. 1998;273:16527–16534. doi: 10.1074/jbc.273.26.16527. [DOI] [PubMed] [Google Scholar]
- 36.Pugh B F, Tjian R. Cell. 1990;61:1187–1197. doi: 10.1016/0092-8674(90)90683-6. [DOI] [PubMed] [Google Scholar]
- 37.Pugh B F, Tjian R. Genes Dev. 1991;5:1935–1945. doi: 10.1101/gad.5.11.1935. [DOI] [PubMed] [Google Scholar]
- 38.Feng Z M, Wu A Z, Chen C L. Mol Endocrinol. 1998;12:378–390. doi: 10.1210/mend.12.3.0079. [DOI] [PubMed] [Google Scholar]
- 39.Viger R S, Mertineit C, Trasler J M, Nemer M. Development (Cambridge, UK) 1998;125:2665–2675. doi: 10.1242/dev.125.14.2665. [DOI] [PubMed] [Google Scholar]
- 40.Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier J M. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonk L J, Itoh S, Heldin C H, ten Dijke P, Kruijer W. J Biol Chem. 1998;273:21145–21152. doi: 10.1074/jbc.273.33.21145. [DOI] [PubMed] [Google Scholar]
- 42.Lynch J P, Lala D S, Peluso J J, Luo W, Parker K L, White B A. Mol Endocrinol. 1993;7:776–786. doi: 10.1210/mend.7.6.8395654. [DOI] [PubMed] [Google Scholar]
- 43.Ikeda Y, Lala D S, Luo X, Kim E, Moisan M P, Parker K L. Mol Endocrinol. 1993;7:852–860. doi: 10.1210/mend.7.7.8413309. [DOI] [PubMed] [Google Scholar]
- 44.de Santa Barbara P, Moniot B, Poulat F, Boizet B, Berta P. J Biol Chem. 1998;273:29654–29660. doi: 10.1074/jbc.273.45.29654. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}