

Abstract
Submicroscopic copy-number imbalances contribute significantly to the genetic etiology of human disease. Here, we report a novel microduplication hot spot at Xp11.22 identified in six unrelated families with predominantly nonsyndromic XLMR. All duplications segregate with the disease, including the large families MRX17 and MRX31. The minimal, commonly duplicated region contains three genes: RIBC1, HSD17B10, and HUWE1. RIBC1 could be excluded on the basis of its absence of expression in the brain and because it escapes X inactivation in females. For the other genes, expression array and quantitative PCR analysis in patient cell lines compared to controls showed a significant upregulation of HSD17B10 and HUWE1 as well as several important genes in their molecular pathways. Loss-of-function mutations of HSD17B10 have previously been associated with progressive neurological disease and XLMR. The E3 ubiquitin ligase HUWE1 has been implicated in TP53-associated regulation of the neuronal cell cycle. Here, we also report segregating sequence changes of highly conserved residues in HUWE1 in three XLMR families; these changes are possibly associated with the phenotype. Our findings demonstrate that an increased gene dosage of HSD17B10, HUWE1, or both contribute to the etiology of XLMR and suggest that point mutations in HUWE1 are associated with this disease too.
Introduction
Mental retardation (MR) is a nonprogressive cognitive impairment affecting 2%–3% of the Western population.1 So far, point mutations and subtle deletions and insertions have been shown to represent only a proportion (<40%) of genetic causes underlying X-linked mental retardation (XLMR).2,3 Microarray-comparative genomic hybridization (array-CGH) is a powerful tool for the identification of submicroscopic deletions and duplications, which would otherwise escape detection.4 Array-CGH screening of MR patients have been reported with several different array platforms. The majority of these studies have used full-genome 1 Mb resolution arrays,5–7 but currently, disease-specific targeted arrays have become popular and have been reported for the analysis of thousands of patients with multiple congenital anomalies (MCAs) including MR.8,9 In both cases, copy-number differences of regions not represented on the array will be missed. The excess of male to female patients with MR (ratio of 1.3/1) points to a genetic etiology on the X chromosome. Although many X-linked genes have been identified in the last decade, still in greater than 50% of XLMR families, the genetic cause remains unidentified.3
We have screened a subset of 300 presumable X-linked families by X chromosome-specific array-CGH and identified five families with overlapping microduplications at Xp11.22. Subsequent screening of additional patients by qPCR identified one more positive family. The common duplicated region contains two candidate genes, the hydroxysteroid dehydrogenase HSD17B10 (also known as HADH2, MHBD, or ABAD) and the E3 ubiquitin ligase HUWE1 (also known as UREB1), both of which showed overexpression in the affected individuals. Because a splice-site mutation in HSD17B10 (MIM 300256) has already been reported in XLMR,10 and because we identified three sequence changes of conserved residues in HUWE1 in three XLMR families, we propose that the pathways of both genes contribute to normal cognitive development.
Subjects and Methods
Patients
As a result of a large international collaborative effort led by the European MRX Consortium (EuroMRX), the International GOLD Program (IGOLD), and individual collaborations, we have collected DNA from a large set of well-characterized families with XLMR. These include syndromic as well as nonsyndromic forms of MR. For probands of all families, cytogenetic and FMR1 CGG expansion analyses were normal. The screening protocols were approved by the appropriate Institutional Review Board of the University Hospitals, and informed consent was obtained from the parents of the affected patients. Genomic DNA from patients as well as from healthy controls was isolated from peripheral blood according to standard procedures and stored at 4°C.
X Array-CGH and qPCR Analysis
Full-coverage X chromosome array-CGH was performed essentially as described elsewhere.11 Data normalization was performed against the median of the spot ratios of all clones. Cy5/Cy3 ratios for each clone were plotted in log2 scale (y axis) relative to the position on the X chromosome (x axis). Clones with log2 ratios outside the −0.3 to 0.3 interval were considered aberrant. Delineation of the extent of the duplications and screening for additional duplications was done by real-time relative quantitation (qPCR) with Sybr-green as described previously.12 qPCR primers were designed with the PrimerExpress software (Applied Biosystems, Foster City, CA) and are provided upon request. All samples were run in duplicate, and data were analyzed with the SDS software v1.2.3 of the instrument and further analyzed in Excel with the comparative ddCt method (Sequence Detection System, bulletin #2; Applied Biosystems).
FISH Analysis
Analysis of the position and orientation of the duplication was performed by standard fluorescent in situ hybridization (FISH) on metaphase-chromosome spreads or interphase chromosomes of patients and controls. DOP-PCR products of the genomic BAC clones, RP6-29D12 (53.34 Mb) and RP1-154P24 (53.66 Mb), were labeled with the direct labeling kit (Invitrogen, Paisley, UK) for SpectrumOrange and SpectrumGreen (Abbott Molecular, Des Plaines, IL), respectively as described elsewhere.7 After hybridization according to standard protocols, signals were visualized by digital-imaging microscopy with Cytovision capturing software (Applied Imaging, Santa Clara, CA).
cDNA Expression Analysis by qPCR
Total RNA was extracted from white blood cells or Epstein Barr Virus-transformed peripheral blood lymphocytes (EBV-PBLs) as described elsewhere.12 Real-time quantitation was performed in Leuven (Belgium) with the SYBR-green method on 50 ng cDNA with the gene-specific primers (provided upon request) as described elsewhere.12 The housekeeping genes ACTB and HPRT were used for normalization. In Adelaide (Australia), expression levels of each gene were normalized to ACTB expression in the same sample and calculated with the relative standard curve method. Standard curves were prepared from equally pooled control cDNAs at 10-, 102-, 103-, 104-, and 105-fold dilutions for each primer pair. Each reaction well contained 2 μl cDNA at a 10-fold dilution, 50 pmol of each primer, and 1× SYBR green PCR Master Mix (Applied Biosystems). Reactions were carried out with a 7300 Real-time PCR System (Applied Biosystems). Purity of the PCR products was determined by a melt-curve analysis, and data analysis was done with SDS Software v.1.2.2 (Applied Biosystems).
Mutations Screening
Sequence analysis of the HSD17B10 and HUWE1 genes was performed on the 250 probands in the IGOLD project as described elsewhere.13 This cohort includes 44 Australian families that were analyzed here by X array-CGH including MRX17, MRX31, and A057 but not A119. In addition, all 737 VEGA genes were checked for mutations in MRX17, MRX31, and A057.
Exon-Expression Array
RNA was extracted from established cell lines of two patients from MRX17, one from MRX31, one from A057, and five unrelated and unaffected individuals as described above. Pooled samples of patients and controls were sent to the Australian Genome Research Facility for labeling and hybridization onto Human Genome 1.0 ST arrays (Affymetrix, Santa Clara, CA). Background correction and normalization was performed with robust multiarray averaging (RMA) with GC correction.14 Expression values were determined for each probe set by median polish. Expression values at the gene level were determined as the median expression across all probe sets in the gene. Statistical significance between affected individuals with the duplication and unaffected controls for both gene level and alternative splicing was determined by one-way ANOVA. All data manipulation for exon arrays was performed with Partek Genomics Suite V6.3beta-6.07.0613 software (Partek, St. Louis, MI). Pathways that may be affected by the duplications were identified with the assistance of the Database for Annotation, Visualization and Integrated Discovery (DAVID) 2007.15
Metabolic Profiling
The determination of both acylcarnitines and amino acids were performed on a Turbo V ion source (T = 100°C) of an Applied Biosystems/MDS-SCIEX API4000 triple quadropole tandem mass spectrometer (MS/MS) instrument (Concord, Ontario, Canada). Samples were infused into the MS/MS at a flow of mobile phase (100 μl/min) with an Agilent 1100 HPLC system (Waldbronn, Germany). We derivatized Whole blood-spot samples with butanolic-HCl to form butyl-esters, the acylcarnitines were determined by precursor scan of 85.1 amu, and the amino acids were determined by neutral total loss of 102 amu. The levels of each acylcarnitine and amino acid in μmol/l whole blood were determined against the respective deuterated stable isotope with the Analyst software, Chemoview (Applied Biosystems) and were compared against a normal age-matched population.
Western Blotting
Proteins were extracted by triturating LCL in radioimmunoprecipitation assay (RIPA) buffer (65.3 mM Tris [pH 7.4], 150 mM NaCl, 1% Nonidet P40, 1 mM NaVO3, 1 mM NaF, and 1× protease-inhibitor cocktail [Sigma, Australia]). A total of 10 μg of each extract was separated on a 4%–12% gradient polyacrylamide gel and transferred to nitrocellulose membrane. The membrane was blocked with 5% skim milk and 0.1% normal goat serum and probed with a mouse monoclonal antibody specific to human p53 at 1:1000 dilution (DO-7, Novacastra, UK) and a goat anti-mouse IgG conjugated to HRP secondary antibody at 1:1000 dilution (Dako, Glostrup, Denmark). Bands were visualized by autoradiography after exposure to enhanced chemiluminescent detection reagents (GE Healthcare). An identical gel was stained with colloidal Coomassie brilliant blue G250 so that total protein loading in each lane could be shown.
Results
Clinical Details of the Six Families with Microduplications at Xp11.22
Family FAM3
The three sons, III-1, III-2, and III-3 (Figure 1 for pictures and Figure 2A for pedigree) of Finnish origin present with similar clinical findings of moderate mental retardation, unique dysmorphic faces, a remarkable speech difficulty with hypotonic open mouth, severe dysarthria, and only one to two word expressions, normal stature, and OFC, short attention span, and happy disposition. The youngest son (III-3) has a surgically repaired submucous cleft palate. All boys have satisfactory self-help skills. The mother (II-1) shares similar dysmorphic features with her sons. She is successfully employed as a cleaning lady and has normal intelligence. The sons showed normal results on subtelomeric FISH of the X chromosome, sequencing of MECP2, and the mutational hot spot region of XNP. MRI brain imaging in the youngest son showed normal structure and myelination.
Figure 1.
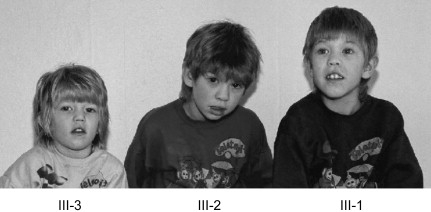
Pictures of the Three Affected Individuals of FAM3 with a Duplication at Xp11.22
Facial features of the sons (from left to right; III-3, III-2, and III-1). Note hypertelorism, short and upslanting palpebral fissures, epicanthi, lateral flare of eyebrows, broad nose, simple protruding ears, open mouth, and thick lips.
Figure 2.
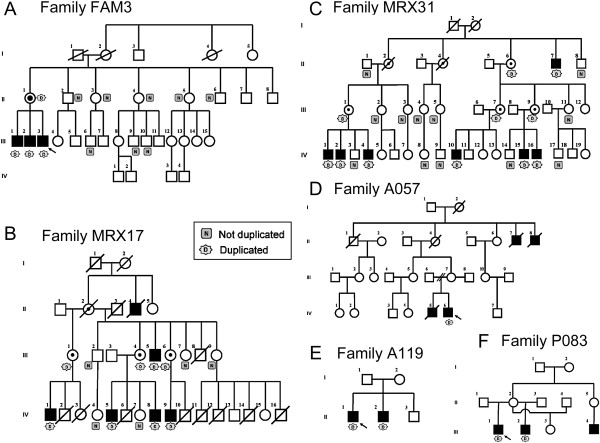
Pedigrees of Families with a Microduplication at Xp11.22
(A) FAM3, (B) MRX17, (C) MRX31, (D) A057, (E) A119, and (F) P083. Family members that could be tested for the duplication are indicated as N (do not carry the duplication) or D (carry the duplication).
Families MRX17 and MRX31
Families MRX17 and MRX31 were published earlier16,17 but were revisited; normal growth parameters and absence of dysmorphic features were confirmed. The extended pedigrees of both large families are shown in Figures 2B and 2C. For family MRX31, the phenotype of the more recently identified affected boys is also classified as mild retardation with nondysmorphic features.
Family A057
The probands were two retarded brothers (IV-5 and IV-6 in Figure 2D) with two affected maternal great uncles (II-7 and II-8). Both probands were born at term after normal deliveries; IV-5 (born in 1947) had a normal birth weight (3.7 kg), but that of IV-6 (born in 1948) was a little low (2.7 kg) and was ascribed to poor maternal nutrition. Both boys appeared normal except for minor hypospadias in IV-6. Both had normal motor development, and concern was only raised because of speech delay by the age of 3 years. Both were hyperactive and were admitted to long-term residential care at ages 8 and 11 years. As adults, no dysmorphic facial features were recognized, and heights (170 cm) were normal; the head circumference of IV-5 was normal (55.5 cm), and that of IV-6 (52 cm) was ∼3 SDs below the mean. Both had limited speech, needed some help with the activities of daily living, and were regarded as having moderate intellectual handicap. No particular health problems were recorded until IV-5, at the age of 55, was found to have the nephrotic syndrome shown by renal biopsy to be due to amyloidosis, type A-A. He died from heart failure soon after. Both maternal uncles had been in long-term institutional care; II-7 died in his twenties from a “clot on the brain.” II-8 became demented and died of “enterocolitis” at age 69.
Family A119
This family consists of two affected brothers (II-1 and II-2 in Figure 2E) from a mother (I-2) who was adopted. Both boys were slow to pass their motor milestones and speech development. Both had febrile seizures, but II-2 continued to have generalized seizures well controlled by valproate. In childhood, attention-deficit and hyperactivity disorders were diagnosed in both individuals, and they were treated with stimulants. When first seen in adolescence, they were off stimulant medication, and no significant behavioral problems were reported. They were of average stature with a muscular build and normal head circumferences (56 cm). There were no significant dysmorphic features, although the older brother had synophris and a broad nasal root. Both were friendly and sociable. They were regarded as having mild-to-moderate intellectual disability. Further investigations included a CT scan on one brother and MRI on the other, which were normal.
Family P083
The proband (III-1 in Figure 2F) suffers from mild MR with minor additional clinical features: moderate macroorchidism, synophris, and diastema between the incisor teeth and delayed tendon reflexes. He was born after normal pregnancy with birth weight (3.1 kg) and length (51 cm) and with head circumference (34 cm) within normal ranges. He had severe speech delay and some behavioral problems thought to be due to lack of affection and stimulation. His current weight (77 kg), length (1.84 m), and head circumference (60 cm) at the age of 19 years are within the normal range. The results of a neurological examination were all normal. His mother was described as “marginal,” and several other male (his half-brother and maternal nephew) and female (his half sister from a third father) family members had normal-to-borderline IQ or psychomotor retardation.
Clinical Features of the Three Families with Missense Mutations in HUWE1
Family A323
This large family, with linkage to the pericentromeric region of the X chromosome including the HUWE1 gene, has been reported previously.18 In brief, all affected males in the family were nondysmorphic and had macrocephaly and moderate MR. All heterozygous females were also macrocephalic and had significant degrees of learning difficulty. The pedigree is shown in Figure 3A.
Figure 3.
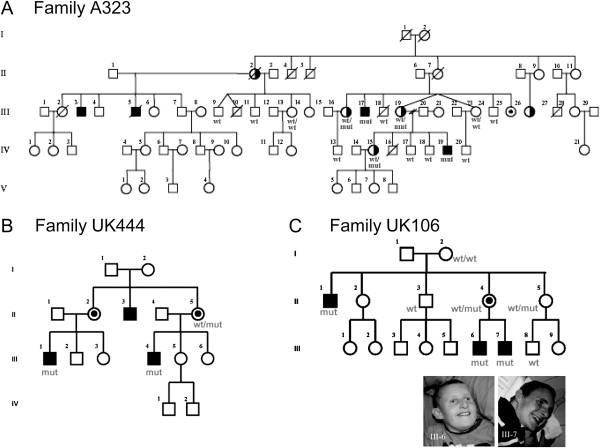
Pedigrees of Families with a Sequence Change in HUWE1
(A) A323, (B) UK444, and (C) UK106. Additional family members that were screened are indicated on the pedigrees as WT or WT/WT (do not carry the likely mutation), mut (carry the mutation), or WT/mut (heterozygous for the mutation). For the large family A323, the HUWE1 gene is located within the linked interval.18 Female I.2 of family UK106 most probably is germline mosaic. Pictures of the affected individual III-6 and III-7 of family UK106 are shown.
Family UK444
The three affected males presented with moderate mental retardation (Figure 3B). III-4 is 38 years old and lives at home with his parents. He works in a sheltered workshop in town and travels to work independently. He is able to read and uses the internet extensively. He has normal language but has occasional problems with articulation. His growth parameters are as follows: height, 1.60 m (0.4–2nd centile); weight, 53 kg (0.4–2nd centile); and head circumference, 56 cm (50th centile). As a child, his speech and language development was delayed. His motor development was normal, although he was clumsy and had frequent falls and poor hand-eye coordination. He was fully toilet trained at 2 1/2 years. He had an operation for strabismus performed at 6 years with some success. He attended mainstream primary school but went to a special school for children with moderate learning disability at the age of 11. He had little sense of danger. He is not dysmorphic, although he has a long face and a pointed chin. His cousin III-1 shows similar characteristics, including the degree of disability. He is slightly less outgoing and sociable and is less confident.
Family UK106
The affected males in this family have severe to profound mental retardation. The brothers III-6 and III-7 (Figure 3C) are looked after at home. The boys are currently aged 17 and 12 years, respectively. Neither of the boys have any speech nor language but communicate their basic needs with objects of reference. The elder boy is very shy, although he makes good eye contact when interacting with familiar people. He has a flexed flexion deformity of the knees but is able to walk. He needs to eat mashed or soft food because of difficulties with chewing. He wears incontinence pads day and night because he has no sense of toileting. His height is on the ninth centile and his weight is on the 0.4th centile, whereas his head circumference is on the 50th centile. The younger brother is physically more active at 12 years and has complete absence of eye contact even within the family. He walks and does not have knee contractures. He has long-standing eczema and requires medication for constipation. The affected uncle (II-1) lives in a residential home with one-to-one care. He walks little because of knee contractures and prefers to move around on his knees. Otherwise, he is similar to his nephews. He has no understandable language and has had problems with constipation. He is dry by day and night if regularly taken to the toilet. He feeds himself with a spoon.
X Array-CGH Screen
Upon initial screening of 200 probands (68 from XLMR families of the EuroMRX Consortium, 124 from small families of the Clinical Hospital of Leuven, and 8 from Finnish origin) with idiopathic XLMR by X chromosome-specific array-CGH, we identified a small duplication at Xp11.22 in the Finnish family FAM3 (Figure 4A). Subsequently, we screened probands from 100 Australian families. Four families from this set—MRX17, MRX31, A057 (Figures 4B–4D), and A119—were found to have duplications of the same Xp11.22 region. Affected males from the last four families presented with mild-to-moderate MR without additional dysmorphic features. Family FAM3 presented with a unique dysmorphic syndrome that has not been reported up to now.
Figure 4.
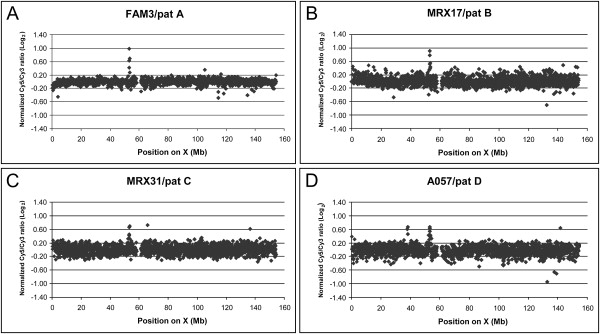
X Chromosome-Specific Array-CGH Plots Obtained for Probands of Four Families Revealing the Duplications at Xp11.22
These families are (A) FAM3, (B) MRX17, (C) MRX31, and (D) A057. DNA from two unrelated MR patients were differentially labeled and cohybridized onto the X array. The log2 normalized intensity ratios of the Cy5 (patient with duplication) and Cy3 (unrelated MR patient, named pat A–D) signals is plotted (y axis) against the position on the X chromosome (in Mb), from Xpter to Xqter (x axis). The duplications are visible as clones with aberrant ratios >0.3 at ∼53 Mb. For A057, an additional polymorphic duplication is visible as three aberrant clones at 38 Mb. Other clones outside the normal interval (log2 ratio of −0.3 to 0.3) are polymorphic clones or outliers.
Array-CGH data revealed aberrant Cy5/Cy3 log2 ratios for different but overlapping sets of clones indicating varying sizes of these duplications in the different families (indicated by green arrows in Figure 5).
Figure 5.

Schematic Representation of the Xp11.22 Region Showing the Duplications
(A) Indicated from top to bottom: the location of the qPCR primer sets (1–20), genomic BAC or PAC clones, and annotated genes present in the interval composed of the six duplications. This region is represented for each family by the arrows indicating the genomic clones from the X array with their corresponding log2 ratios. Clones with aberrant ratios are in green; those with ratios in the normal interval are open. For each family, the mapped duplicated region, defined by qPCR, is given as solid blue bars above these clones, indicating that the primer sets within these bars yielded relative copy numbers of ∼2.0 whereas the primer pairs outside (indicated with the gray bars) gave values ∼1.0. Proximal and distal breakpoints must lie between these bars (no bars).
(B) For P083, only qPCR data are shown. The maximal common duplicated region is boxed (broken red line) and entirely contains the genes RIBC1, HSD17B10, and HUWE1.
Mapping of the Duplications by qPCR
Subsequent qPCR analysis with 20 primer sets from the region refined the position as well as the extent of individual duplications (blue bars in Figure 5A). The duplications varied in size from 0.4 to 0.8 Mb with a common minimal overlapping region at 53.3–53.7 Mb as based on the Ensembl view release 43, Feb 2007. This critical region includes four annotated genes: SMC1A, RIBC1, HSD17B10, and HUWE1, as well as the microRNAs mir-98 and let-7f-2, both situated within the HUWE1 gene. So far, information on these specific microRNA subtypes is very scarce, and a role in brain development still needs to be investigated. Because SMC1A and RIBC1 escape X inactivation19 and thus are present at a double dosage in females compared to males, it is unlikely that they are significantly involved in the observed phenotype in these families. The larger duplications in families MRX17, A057, and A119 also harbor the known MRX gene SMCX/JARID1C20 (MIM 314690), whereas GPR173 and TSPYL2 are the additional genes duplicated in families A057 and A119. The largest duplication in family A119 also harbors TMEM29 and potentially SPANXN5 and some genes of the XAGE and SSX clusters (Figure 5A). As far as we could test, all duplications segregate with the disease in the respective families (Figures 2A–2E). For the previously published and mapped families (MRX17 and MRX31), the duplication resides within the completely overlapping linked intervals (LOD > 2.0) of 42 Mb and 23 Mb, respectively.
Screening for Additional Duplications
qPCR screening with the primers designed for the HSD17B10 gene did not reveal any duplication in 350 X chromosomes from control samples of Finnish, Australian, and Belgian origin. In addition, the Database for Genomic Variants from The Center for Applied Genomics (build 36; March 2006) and the Human Genome Structural Variation Project (assembly May 2004) do not report this variation. Next, we screened from the EuroMRX Consortium 50 XLMR individuals that were not yet analyzed by array-CGH and 30 XLMR probands linked to Xp11.2, collected by the Greenwood Genetics Center and part of the IGOLD study, via qPCR by using HSD17B10 and HUWE1 primer sets. One additional positive family, P083, present in the EuroMRX panel, was identified. Fine mapping revealed a 320 kb duplication (Figure 5B), which involved RIBC1, HSD17B10, and HUWE1 but only part of the SMC1A gene. The proband of P083 suffers from mild MR with minor additional clinical features. qPCR on DNA of the affected half-brother demonstrated that he also carries this duplication (Figure 2F). No other family members were available for analysis.
FISH Analysis
In order to determine the location and orientation of the duplicated region, we performed FISH on metaphase and interphase chromosomes from affected individuals of families MRX17, MRX31, and A057, for which a cell line was available. Two differentially labeled BAC clones within the common duplicated region revealed single fluorescent signals at Xp11 on metaphase chromosomes (Figure 6A), demonstrating that the duplicated regions in the respective families are located very close and most probably next to each other. A tandem arrangement was deduced from the color order of fluorescent signals on interphase chromosomes, which was red (RP6-29D12), green (RP1-154P24), red (RP6-29D12), and green (RP1-154P24) for all three families (Figure 6B).
Figure 6.
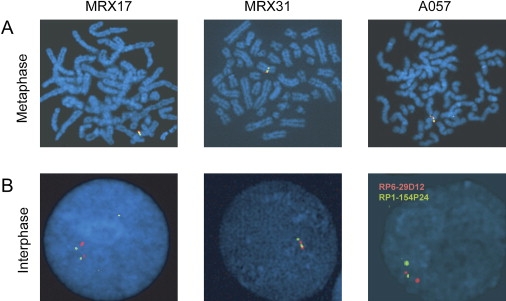
Representative FISH Data on Chromosomes from Probands of Families MRX17, MRX31, and A057
(A) Labeled DNA of RP6-29D12 (red) and RP1-154P24 (green) were hybridized to metaphase spreads and show signals for both probes only at Xp11.
(B) On interphase chromosomes, alternating red-green-red-green signals are indicative of a tandem duplication event.
Expression of Duplicated Genes
In silico expression analysis with Unigene at NCBI and immunohistochemistry data from adult-mouse brain sections (Allen Brain Atlas) demonstrated ubiquitous expression of HSD17B10 and HUWE1, with high expression in the hippocampus. In contrast, the expression of RIBC1 was restricted to only a few tissues with barely detectable levels in brain, if any. Accurate qPCR measurement of mRNA levels of HSD17B10 and HUWE1 directly in PBLs or in EBV-transformed PBLs from controls revealed very similar expression levels, indicating a tight regulation of their expression in these cells. Moreover, similar mRNA levels were detected in males and females (data not shown), and such a finding suggests that both genes are subject to X inactivation. Next, we determined their relative expression in RNA extracted from blood in family members from FAM3. A 5-fold upregulation was detected for HSD17B10 (5.05 ± 0.60) and HUWE1 (5.00 ± 0.47) in the two affected males (Figure 7), whereas no significant change was noticed for either gene in the carrier mother (data not shown). Expression analysis in the five other families was done on EBV-PBLs because of the unavailability of fresh blood. Again, a significant increased expression of 2- to 5-fold was detected for HSD17B10 and HUWE1 (Figure 7). We also analyzed the relative mRNA content of SMC1A in all six families. A substantial increase of 2- to 7-fold was detected in all families except for P083 in which no upregulation was found (Figure 7). These data confirm that SMC1A is not entirely duplicated in P083 and, therefore, would probably not account for the observed phenotype.
Figure 7.
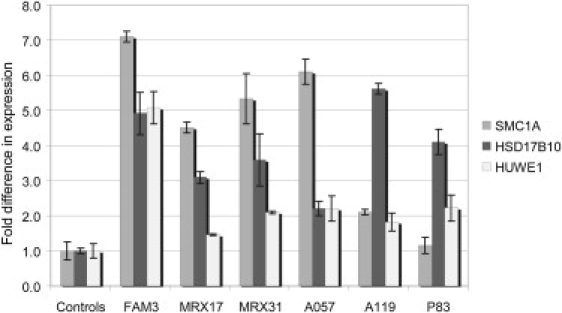
mRNA Expression Analysis of SMC1A, HSD17B10, and HUWE1 in All Six Families with a Duplication at Xp11.22
cDNA was prepared from RNA extracted from blood (controls and FAM3) or EBV-transformed PBL cell lines (controls, MRX17, MRX31, A057, A119, and P083). Compared to controls, all affected individuals showed significantly increased mRNA levels for all three genes except for SMC1A in family P083. Expression was determined by real-time RT-PCR with the comparative ddCt method and normalized to the expression of HPRT and ACTB (FAM3, P083) or with the standard curve method with ACTB for normalization (MRX17, MRX31, A057, and A119). Expression levels are calculated relative to the mean levels obtained in the control samples (fold difference; y axis). Standard deviations of at least two independent experiments are indicated for each bar.
Sequencing of Genes on the X Chromosome
Sequencing of the coding exons of 737 VEGA annotated X chromosome genes in the three families present in the IGOLD study (MRX17, MRX31, and A057) did not reveal any obvious disease-causing mutations, suggesting that a simple mutation is unlikely to account for the observed phenotypes in these families. Moreover, mutation analysis of the genes within the duplication in all probands from 250 XLMR families of the IGOLD project did not detect any potential disease-associated changes in RIBC1 or HSD17B10. However, three HUWE1 missense changes were identified. One of these (12037C > T/R4013W) was found in a large published Australian family (A323) with XLMR and macrocephaly.18 The other two abnormalities (8942G > A/R2981H and 12559C > T/R4187C) were identified in yet-unpublished families UK106 (severe to profound MR with knee contractures) and UK444 (nonsyndromic MR), respectively. All three mutated arginine residues are highly conserved among the animal orthologs of HUWE1 (Figure 8). None of these changes were found in 750 control individuals. Given the conservation and location of these changes within or near known functional domains and cosegregation with the phenotype in the respective families (Figures 3A–3C), as well as their absence from controls, we hypothesize that these sequence changes are deleterious to HUWE1 function.
Figure 8.
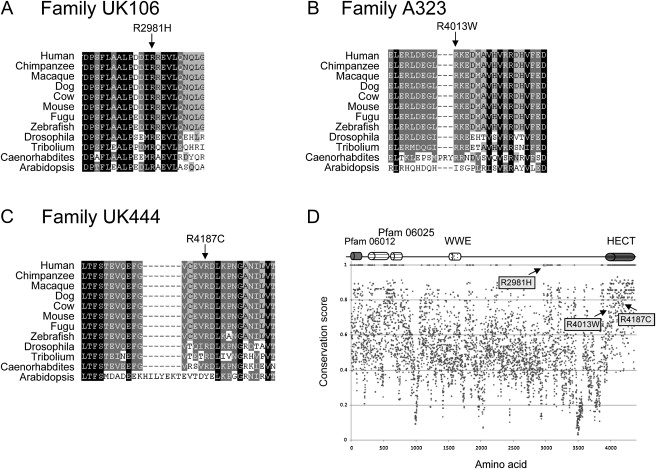
Missense Changes in the HUWE1 Gene Identified in Three XLMR Families
Partial ClustalW alignments of HUWE1 orthologs in the regions surrounding (A) R2981H (UK106), (B) R4013W (A323), and (C) R4187C (UK444) amino acid changes. Whereas the R2981 is invariable across all orthologs, R4013 and R4187 are highly conserved. In (D), the position of these arginine residues within the HUWE1 protein is indicated with the arrows. The cartoon above the conservation plot (Scorecons) indicates domains within the HUWE1 protein. Pfam 06012 (aa 104–374) and Pfam 06025 (aa 424–704 and 762–815) domains are of unknown function, WWE domain (aa 1617–1678) might be involved in the regulation of ubiquitin-mediated proteolysis, and HECT domain (aa 4036–4374) is the catalytic domain of HUWE1 ubiquitin protein ligase.
Exon-Microarray Analysis
Exon-array analysis on pooled cDNA from MRX17, MRX31, and A057 versus five control males showed 2- to 2.5-fold increased expression of SMC1A, HSD17B10, and HUWE1 (Table S1 available online). In total, 1677 genes were found to be differentially expressed, whereas 2517 genes were identified to be alternatively spliced between the affected individuals and the controls with a p < 0.05 as defined by ANOVA (Tables S1 and S2, respectively). These genes were then analyzed against the known molecular pathways of HSD10 and HUWE1 with the molecular-interaction databases DAVID and KEGG. We noticed for example a downregulation of ACAA1 that metabolizes products downstream of HSD10 in the pathways of fatty acid (Figure S1), and valine, isoleucine, and leucine metabolism (data not shown). We also found alteration in the expression of genes involved in the ubiquitin-mediated protein-degradation pathway, consistent with the known function of HUWE1 (data not shown). Finally, we observed changes in the expression of many cell-cycle genes with a central role for TP53 (P53), consistent with an upregulation of HUWE1 (Figure S2).
Functional Analysis of HSD10 and HUWE1 Activities
Metabolic screening of the dehydrogenase activity of HSD10 in lymphocytes21 will not allow for the detection of this microduplication because we did not find altered levels of a number of acylcarnitines or amino acids associated with the isoleucine degradative pathway in a blood spot of one of the patients from family MRX17, compared to age-matched controls, by using tandem mass spectrometry (MS/MS) (data not shown).
For HUWE1 activity, we analyzed the TP53 mRNA and TP53 protein abundances by RT-qPCR and western blotting, respectively, in EBV-PBLs from affected individuals from families MRX17, MRX31, A057, P083 (duplications), and A323 (missense change) but did not find reduced levels compared to control cell lines (data not shown).
Discussion
X chromosome-specific array-CGH analysis of XLMR families has uncovered several disease-associated as well as polymorphic submicroscopic copy-number changes.11,22 However, for several of these aberrations, the association with the disease is not clear. In our screen of 300 presumed XLMR families, we identified six different but overlapping microduplications at Xp11.22. The proposed causal role for this duplication comes from the fact that all segregate with the disease in the six families, and it was never found in controls (own screen and online databases). The common 0.32 Mb region contains four genes: part of SMC1A, RIBC1, HSD17B10, and HUWE1. The RIB43A domain with coiled coils 1 (RIBC1) gene of unknown function does not seem to be expressed in the brain. The fact that SMC1A is not completely duplicated in P083, as reflected in its normal relative expression profile in a cell line derived from this patient, strongly suggests that SMC1A duplication is unlikely to contribute to the phenotype seen in these families. Mutations in SMC1A have recently been associated with Cornelia de Lange syndrome (MIM 300590).23 This leaves two candidate genes for which a 2-fold increase in expression might be responsible for the cognitive deficit.
The gene 17 β-hydroxysteroid dehydrogenase type 10 (HSD17B10) codes for a 261 amino acid mitochondrial enzyme HSD10 important in isoleucine and branched-chain fatty-acid degradation (reviewed in 24). Interestingly, three missense mutations in this gene have been associated with progressive neurodegenerative disease,25 whereas a silent mutation, which affects alternative splicing of exon 5, has recently been related to a syndromic form of MR with choreoathetosis.10 Additionally, a role in Alzheimer's as well as Parkinson's disease has been suggested through binding with amyloid-β peptide and regulation of dopamine-containing neurons, respectively.26,27 Elevated HSD10 levels were found in hippocampi of Alzheimer's patients.28 It is therefore likely that not only a reduced activity but also an increased dosage of HSD10 might interfere with normal cognition through disturbed metabolism of neurosteroid modulators of GABAA receptors.28 The localization of the HSD10 protein to postsynaptic protein complexes29 further reinforces its likely role in neuronal communication. It is of interest that in family A057, one affected brother died of amyloidosis at the age of 55.
The large HECT, UBA, and WWE domain-containing protein 1 (HUWE1) is an E3 ubiquitin ligase initially found implicated in oncogenesis. In colorectal cancers, overexpression of HUWE1 leads to increased ubiquitination of the tumor suppressor TP53 with its subsequent degradation.30 Because a critical role for TP53 has been implicated in maintaining the balance between the continuous generation of neuroblasts and their elimination through apoptosis,31 reduced levels of TP53 are expected to result in developmental abnormalities in the nervous system. This process has been demonstrated in Tp53-deficient mice in part because of overproduction of neuronal precursors.32 Moreover, ubiquitin and protein degradation have been implicated in neuronal function (reviewed in33), and mutations in several proteins involved in the ubiquitin pathway have been related to MR.13,34–37 Finally, dysfunction of the ubiquitin metabolism is a hallmark in several neurological diseases.38,39
Taken together, our data demonstrate that duplications varying in size from 0.3–0.8 Mb at Xp11.22, which include HSD17B10 and HUWE1, contribute to disturbance of normal cognitive development. The phenotype of our patients with this duplication is nonsyndromic with mild-to-moderate mental handicap in five out of six families, suggesting that this is the group in whom screening is advisable. We do not have as yet an explanation for the syndromic phenotype observed in family FAM3 who carries a rather small duplication. The significant higher relative expression of HUWE1 in this family compared to the others with this duplication (Figure 7) might have affected this peculiar phenotype. Also, the relative high expression levels (4- to 7-fold) observed for SMC1A and HSD17B10 in families with a 2-fold increase in copy number of these genes remains elusive. Whereas there is one mutation of HSD17B10 described in XLMR associated with choreoathotosis,10 the HUWE1 gene had not been implicated in XLMR so far. Our data indicate that HUWE1 point mutations indeed are associated with MR. On the basis of global-expression analysis, which revealed altered mRNA levels for several genes involved in the cell-cycle pathway, we propose that HUWE1 might be the major but unlikely the only contributor to the observed phenotype in those with a duplication. Ultimately, identification of a duplication carrying either one or the other of the candidate genes or of an appropriate animal model will be required to distill the causal gene in this new microduplication hot spot. These findings will provide additional insight into its role in normal brain development.
Other examples of genes for which duplications, missense mutations, and deletions herein result in cognitive phenotypes are PLP1 (MIM 300401), resulting in Pelizaeus-Merzbacher disease, PMP22 (MIM 601097) in Charcot-Marie-Tooth disease, APP (MIM 104760) in Alzheimer disease, and MECP2 (MIM 300005) in severe forms of syndromic MR.12,40–42 HSD17B10, HUWE1, or both seem to join this growing list, and it is expected that several more will follow.
Supplemental Data
Two figures and two tables are available at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Allen Brain Atlas, http://www.brainatlas.org/aba/
Database for Genomic Variants, http://projects.tcag.ca/variation/
Ensembl, http://www.ensembl.org/
EuroMRX, http://euromrx.com/
Human Genome Structural Variation Project, http://humanparalogy.gs.washington.edu/structuralvariation/
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
UniGene, http://www.ncbi.nlm.nih.gov/sites/entrez?db=unigene
Accession Numbers
The GEO accession number for the array data for all six patients reported in this paper is GSE9143.
Acknowledgments
The authors thank all families for their cooperation, Marleen Willems for help with cell culture, Erica Woollatt for chromosome preparation, Marie Shaw for X inactivation studies, and Janice Fletcher for advice on metabolic screening. M.B. is supported by the IWT (Instituut voor Innovatie door Technologie en Wetenschap), Flanders, Belgium. H.V.E. is a postdoctoral researcher of the Fund for Scientific Research-Flanders (FWO-Vlaanderen), Belgium. J.G. is supported by the Australian NH&MRC senior research fellowship 250340 and Program grant 400121. This study was also supported by the European Union grant QLG3-CT-2002-01810 (EuroMRX Consortium), the NIH grant NINDS (NS31564, USA), the Wellcome Trust (UK), the NIHCD grant HD26202, the Neurology Foundation (Helsinki, Finland) and, in part, the South Carolina Department of Disabilities and Special Needs (SCDDSN).
References
- 1.Leonard H., Wen X. The epidemiology of mental retardation: Challenges and opportunities in the new millennium. Ment. Retard. Dev. Disabil. Res. Rev. 2002;8:117–134. doi: 10.1002/mrdd.10031. [DOI] [PubMed] [Google Scholar]
- 2.Gecz J. The molecular basis of intellectual disability: Novel genes with naturally occurring mutations causing altered gene expression in the brain. Front. Biosci. 2004;9:1–7. doi: 10.2741/1199. [DOI] [PubMed] [Google Scholar]
- 3.Ropers H.H. X-linked mental retardation: Many genes for a complex disorder. Curr. Opin. Genet. Dev. 2006;16:260–269. doi: 10.1016/j.gde.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 4.Albertson D.G., Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum. Mol. Genet. 2003;12:R145–R152. doi: 10.1093/hmg/ddg261. [DOI] [PubMed] [Google Scholar]
- 5.Vissers L.E., de Vries B.B., Osoegawa K., Janssen I.M., Feuth T., Choy C.O., Straatman H., van der Vliet W., Huys E.H., Van Rijk A. Array-based comparative genomic hybridization for the genome-wide detection of submicroscopic chromosomal abnormalities. Am. J. Hum. Genet. 2003;73:1261–1270. doi: 10.1086/379977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw-Smith C., Redon R., Rickman L., Rio M., Willatt L., Fiegler H., Firth H., Sanlaville D., Winter R., Colleaux L. Microarray based comparative genomic hybridisation (array-CGH) detects submicroscopic chromosomal deletions and duplications in patients with learning disability/mental retardation and dysmorphic features. J. Med. Genet. 2004;41:241–248. doi: 10.1136/jmg.2003.017731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menten B., Maas N., Thienpont B., Buysse K., Vandesompele J., Melotte C., De Ravel T., Van Vooren S., Balikova I., Backx L. Emerging patterns of cryptic chromosomal imbalance in patients with idiopathic mental retardation and multiple congenital anomalies: A new series of 140 patients and review of published reports. J. Med. Genet. 2006;43:625–633. doi: 10.1136/jmg.2005.039453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shaffer L.G., Kashork C.D., Saleki R., Rorem E., Sundin K., Ballif B.C., Bejjani B.A. Targeted genomic microarray analysis for identification of chromosome abnormalities in 1500 consecutive clinical cases. J. Pediatr. 2006;149:98–102. doi: 10.1016/j.jpeds.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 9.Stankiewicz P., Beaudet A.L. Use of array CGH in the evaluation of dysmorphology, malformations, developmental delay, and idiopathic mental retardation. Curr. Opin. Genet. Dev. 2007;17:182–192. doi: 10.1016/j.gde.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Lenski C., Kooy R.F., Reyniers E., Loessner D., Wanders R.J., Winnepenninckx B., Hellebrand H., Engert S., Schwartz C.E., Meindl A., Ramser J. The reduced expression of the HADH2 protein causes X-linked mental retardation, choreoathetosis, and abnormal behavior. Am. J. Hum. Genet. 2007;80:372–377. doi: 10.1086/511527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Froyen G., Van Esch H., Bauters M., Hollanders K., Frints S.G., Vermeesch J.R., Devriendt K., Fryns J.P., Marynen P. Detection of genomic copy number changes in patients with idiopathic mental retardation by high-resolution X-array-CGH: Important role for increased gene dosage of XLMR genes. Hum. Mutat. 2007;28:1034–1042. doi: 10.1002/humu.20564. [DOI] [PubMed] [Google Scholar]
- 12.Van Esch H., Bauters M., Ignatius J., Jansen M., Raynaud M., Hollanders K., Lugtenberg D., Bienvenu T., Jensen L.R., Gecz J. Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 2005;77:442–453. doi: 10.1086/444549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarpey P.S., Raymond F.L., O'Meara S., Edkins S., Teague J., Butler A., Dicks E., Stevens C., Tofts C., Avis T. Mutations in CUL4B, which encodes a ubiquitin E3 ligase subunit, cause an X-linked mental retardation syndrome associated with aggressive outbursts, seizures, relative macrocephaly, central obesity, hypogonadism, pes cavus, and tremor. Am. J. Hum. Genet. 2007;80:345–352. doi: 10.1086/511134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 15.Kanehisa M., Goto S., Hattori M., Aoki-Kinoshita K.F., Itoh M., Kawashima S., Katayama T., Araki M., Hirakawa M. From genomics to chemical genomics: New developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gedeon A., Kerr B., Mulley J., Turner G. Pericentromeric genes for non-specific X-linked mental retardation (MRX) Am. J. Med. Genet. 1994;51:553–564. doi: 10.1002/ajmg.1320510453. [DOI] [PubMed] [Google Scholar]
- 17.Donnelly A.J., Partington M.W., Ryan A.K., Mulley J.C. Regional localisation of two non-specific X-linked mental retardation genes (MRX30 and MRX31) Am. J. Med. Genet. 1996;64:113–120. doi: 10.1002/(SICI)1096-8628(19960712)64:1<113::AID-AJMG19>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 18.Turner G., Gedeon A., Mulley J. X-linked mental retardation with heterozygous expression and macrocephaly: Pericentromeric gene localization. Am. J. Med. Genet. 1994;51:575–580. doi: 10.1002/ajmg.1320510456. [DOI] [PubMed] [Google Scholar]
- 19.Carrel L., Willard H.F. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- 20.Jensen L.R., Amende M., Gurok U., Moser B., Gimmel V., Tzschach A., Janecke A.R., Tariverdian G., Chelly J., Fryns J.P. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poll-The B.T., Wanders R.J., Ruiter J.P., Ofman R., Majoie C.B., Barth P.G., Duran M. Spastic diplegia and periventricular white matter abnormalities in 2-methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency, a defect of isoleucine metabolism: Differential diagnosis with hypoxic-ischemic brain diseases. Mol. Genet. Metab. 2004;81:295–299. doi: 10.1016/j.ymgme.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 22.Lugtenberg D., Veltman J.A., van Bokhoven H. High-resolution genomic microarrays for X-linked mental retardation. Genet. Med. 2007;9:560–565. doi: 10.1097/gim.0b013e318149e647. [DOI] [PubMed] [Google Scholar]
- 23.Musio A., Selicorni A., Focarelli M.L., Gervasini C., Milani D., Russo S., Vezzoni P., Larizza L. X-linked Cornelia de Lange syndrome owing to SMC1L1 mutations. Nat. Genet. 2006;38:528–530. doi: 10.1038/ng1779. [DOI] [PubMed] [Google Scholar]
- 24.Korman S.H. Inborn errors of isoleucine degradation: A review. Mol. Genet. Metab. 2006;89:289–299. doi: 10.1016/j.ymgme.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Ofman R., Ruiter J.P., Feenstra M., Duran M., Poll-The B.T., Zschocke J., Ensenauer R., Lehnert W., Sass J.O., Sperl W., Wanders R.J. 2-Methyl-3-hydroxybutyryl-CoA dehydrogenase deficiency is caused by mutations in the HADH2 gene. Am. J. Hum. Genet. 2003;72:1300–1307. doi: 10.1086/375116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tieu K., Perier C., Vila M., Caspersen C., Zhang H.P., Teismann P., Jackson-Lewis V., Stern D.M., Yan S.D., Przedborski S. L-3-hydroxyacyl-CoA dehydrogenase II protects in a model of Parkinson's disease. Ann. Neurol. 2004;56:51–60. doi: 10.1002/ana.20133. [DOI] [PubMed] [Google Scholar]
- 27.Yang S.Y., He X.Y., Schulz H. 3-Hydroxyacyl-CoA dehydrogenase and short chain 3-hydroxyacyl-CoA dehydrogenase in human health and disease. FEBS J. 2005;272:4874–4883. doi: 10.1111/j.1742-4658.2005.04911.x. [DOI] [PubMed] [Google Scholar]
- 28.Yang S.Y., He X.Y., Miller D. HSD17B10: A gene involved in cognitive function through metabolism of isoleucine and neuroactive steroids. Mol. Genet. Metab. 2007;92:36–42. doi: 10.1016/j.ymgme.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Laumonnier F., Cuthbert P.C., Grant S.G. The role of neuronal complexes in human X-linked brain diseases. Am. J. Hum. Genet. 2007;80:205–220. doi: 10.1086/511441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoon S.Y., Lee Y., Kim J.H., Chung A.S., Joo J.H., Kim C.N., Kim N.S., Choe I.S., Kim J.W. Over-expression of human UREB1 in colorectal cancer: HECT domain of human UREB1 inhibits the activity of tumor suppressor p53 protein. Biochem. Biophys. Res. Commun. 2005;326:7–17. doi: 10.1016/j.bbrc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 31.Medrano S., Scrable H. Maintaining appearances–the role of p53 in adult neurogenesis. Biochem. Biophys. Res. Commun. 2005;331:828–833. doi: 10.1016/j.bbrc.2005.03.194. [DOI] [PubMed] [Google Scholar]
- 32.Armstrong J.F., Kaufman M.H., Harrison D.J., Clarke A.R. High-frequency developmental abnormalities in p53-deficient mice. Curr. Biol. 1995;5:931–936. doi: 10.1016/s0960-9822(95)00183-7. [DOI] [PubMed] [Google Scholar]
- 33.Yi J.J., Ehlers M.D. Emerging roles for ubiquitin and protein degradation in neuronal function. Pharmacol. Rev. 2007;59:14–39. doi: 10.1124/pr.59.1.4. [DOI] [PubMed] [Google Scholar]
- 34.Nascimento R.M., Otto P.A., de Brouwer A.P., Vianna-Morgante A.M. UBE2A, which encodes a ubiquitin-conjugating enzyme, is mutated in a novel X-linked mental retardation syndrome. Am. J. Hum. Genet. 2006;79:549–555. doi: 10.1086/507047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zenker M., Mayerle J., Lerch M.M., Tagariello A., Zerres K., Durie P.R., Beier M., Hulskamp G., Guzman C., Rehder H. Deficiency of UBR1, a ubiquitin ligase of the N-end rule pathway, causes pancreatic dysfunction, malformations and mental retardation (Johanson-Blizzard syndrome) Nat. Genet. 2005;37:1345–1350. doi: 10.1038/ng1681. [DOI] [PubMed] [Google Scholar]
- 36.Matsuura T., Sutcliffe J.S., Fang P., Galjaard R.J., Jiang Y.H., Benton C.S., Rommens J.M., Beaudet A.L. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 37.Kishino T., Lalande M., Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 38.Ross C.A., Pickart C.M. The ubiquitin-proteasome pathway in Parkinson's disease and other neurodegenerative diseases. Trends Cell Biol. 2004;14:703–711. doi: 10.1016/j.tcb.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 39.Bennett E.J., Shaler T.A., Woodman B., Ryu K.Y., Zaitseva T.S., Becker C.H., Bates G.P., Schulman H., Kopito R.R. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- 40.Rovelet-Lecrux A., Hannequin D., Raux G., Le Meur N., Laquerriere A., Vital A., Dumanchin C., Feuillette S., Brice A., Vercelletto M. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- 41.Inoue K. PLP1-related inherited dysmyelinating disorders: Pelizaeus-Merzbacher disease and spastic paraplegia type 2. Neurogenetics. 2005;6:1–16. doi: 10.1007/s10048-004-0207-y. [DOI] [PubMed] [Google Scholar]
- 42.Saifi G.M., Szigeti K., Snipes G.J., Garcia C.A., Lupski J.R. Molecular mechanisms, diagnosis, and rational approaches to management of and therapy for Charcot-Marie-Tooth disease and related peripheral neuropathies. J. Investig. Med. 2003;51:261–283. doi: 10.1136/jim-51-05-14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.