

Abstract
Although dendritic cells (DCs) play an important role in mediating protection against influenza virus, the precise role of lung DC subsets, such as CD11b− and CD11b+ conventional DCs or plasmacytoid DCs (pDCs), in different lung compartments is currently unknown. Early after intranasal infection, tracheal CD11b−CD11chi DCs migrated to the mediastinal lymph nodes (MLNs), acquiring co-stimulatory molecules in the process. This emigration from the lung was followed by an accumulation of CD11b+CD11chi DCs in the trachea and lung interstitium. In the MLNs, the CD11b+ DCs contained abundant viral nucleoprotein (NP), but these cells failed to present antigen to CD4 or CD8 T cells, whereas resident CD11b−CD8α+ DCs presented to CD8 cells, and migratory CD11b−CD8α− DCs presented to CD4 and CD8 T cells. When lung CD11chi DCs and macrophages or langerin+CD11b−CD11chi DCs were depleted using either CD11c–diphtheria toxin receptor (DTR) or langerin-DTR mice, the development of virus-specific CD8+ T cells was severely delayed, which correlated with increased clinical severity and a delayed viral clearance. 120G8+ CD11cint pDCs also accumulated in the lung and LNs carrying viral NP, but in their absence, there was no effect on viral clearance or clinical severity. Rather, in pDC-depleted mice, there was a reduction in antiviral antibody production after lung clearance of the virus. This suggests that multiple DCs are endowed with different tasks in mediating protection against influenza virus.
Influenza type A is a cytolytic virus that causes acute respiratory infection, of which the clinical outcome can vary greatly. The way in which the innate and adaptive immune systems initially recognize and deal with replicating virus could be decisive in determining the outcome of infection, as this might heavily influence the kinetics of viral clearance (1–3). Little is known about the initial recognition event of influenza virus by the lung immune system in vivo.
In immediate response to viral infection, innate defense mechanisms consist of high level production of type I IFNs by infected epithelial cells, alveolar macrophages, and natural IFN-producing cells (also known as preplasmacytoid DCs [pre-pDCs]), as well as the recruitment of conventional DCs (cDCs), neutrophils, and NK cells (4, 5). By expressing a wide array of microbial pattern recognition receptors shared with cells of the innate immune response, and at the same time displaying the potential to process and present antigen to naive T cells, DCs bridge innate and adaptive immunity. After recognizing foreign antigen in the periphery of the body, DCs migrate via afferent lymphatics into the draining LNs, where they can induce antigen-specific protective CD8 CTL responses, as well as CD4 T helper cells that enforce cellular and humoral immunity (6).
In the lung, DCs are situated in immediate proximity to the respiratory epithelial cells, where they form an elaborate network that rapidly reacts to all kinds of foreign antigens and inflammatory stimuli, including respiratory viruses (7–10). Different DC subsets can be found in the lung, each with a functional specialization (11–15). In the mouse, all DC subsets express the integrin CD11c, and subsets are further defined based on the expression of the marker CD11b, as well as anatomical location (11, 13, 16). The trachea and large conducting airways have a well-developed network of intraepithelial DCs, even in steady-state conditions (17, 18). These cells have been shown to express langerin and CD103 while lacking expression of CD11b (13, 10, 15). In the submucosa of the conducting airways, CD103−CD11b+CD11c+ cDCs can be found, particularly under conditions of inflammation, and these cells might be particularly suited for priming and restimulating effector CD4 cells in the lung in response to protein antigens (19, 20, 15). The lung interstitium that is accessible by enzymatic digestion also contains CD11b+ and CD11b− DCs that access the alveolar lumen and migrate to the mediastinal LNs (MLNs) (14, 11). In the nearby alveolar lumen, CD11chi alveolar macrophages control the function of these interstitial DCs (21). pDCs are CD11b−CD11cint cells expressing SiglecH and bone marrow stromal antigen 2 (recognized by the mAbs mouse pDC antigen 1 [mPDCA-1] or 120G8) (22, 23). In the lungs, pDCs are predominantly found in the lung interstitium and produce large amounts of IFN-α in response to triggering by CpG motifs or viral infection ex vivo (24, 5). Under particular conditions, lung pDCs can prevent immunopathology in response to inhalation of harmless antigens or in response to respiratory syncitial virus (24–26).
Several investigators have studied the involvement of APCs such as DCs, macrophages, and B cells in mediating protective immunity to influenza virus (27). It was previously shown that a mouse-adapted strain of influenza virus induced the in vivo maturation of CD11c+ DCs in the lung (28) and their migration to the MLNs (29, 8, 2). DCs isolated ex vivo from the MLNs of influenza-infected mice presented viral nucleoprotein (NP)-derived peptides to virus-specific CD8 T cells in vitro for at least 10 d, a peak occurring ∼72 h after infection (27, 29–32). In humans, both monocyte-derived DCs and pDCs were shown to be capable of activating already primed influenza-specific T cells in vitro (33, 34) and upon adoptive transfer in vivo (35).
Although these studies certainly suggest the strong involvement of DCs in mediating protective immunity to influenza A virus, direct description of the functional in vivo involvement of the now well-described DC subsets in the different compartments of the lung during early infection is lacking. In this paper, using eight-color flow cytometry, we have carefully studied the kinetics of the reaction of different DC subsets and alveolar macrophages in various lung compartments to intranasal (i.n.) infection with influenza virus X-31 (H3N2), focusing particularly on CD11b+ and CD11b− subsets, as well as on pDCs, describing which subsets carry viral NP to the nodes, and carefully dissecting which subsets present antigen to specific CD8 and CD4 T cells. Using genetic cell-specific targeting techniques, CD11chi DCs and macrophages or langerin+CD11chi DCs could be depleted in CD11c–diphtheria toxin receptor (DTR) or langerin-DTR mice (36, 37), and pDCs could be depleted using mAbs. Collectively, our results demonstrate that different DC subsets perform specialized tasks during the primary encounter with influenza virus in the lung.
RESULTS
Lung DC subtypes and alveolar macrophages respond to influenza virus infection differentially
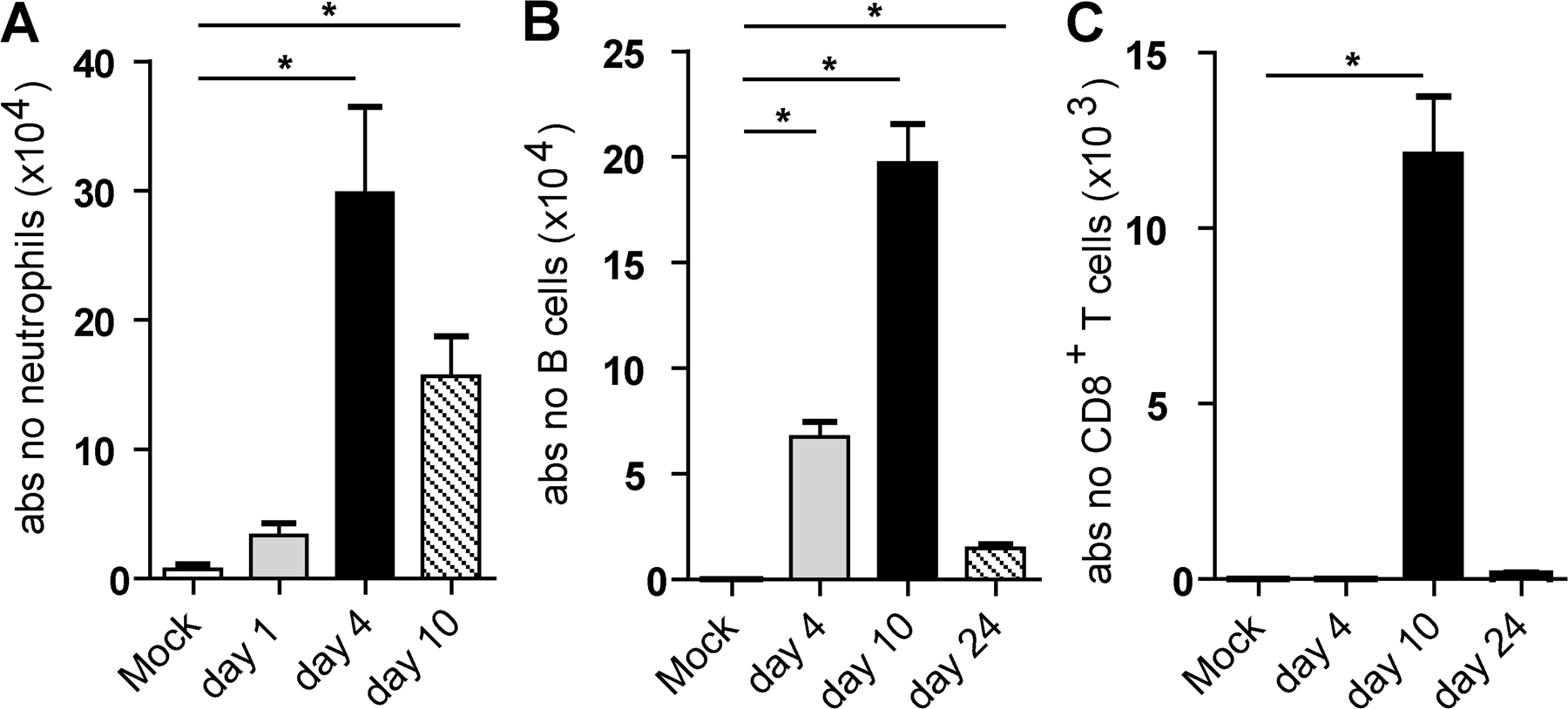
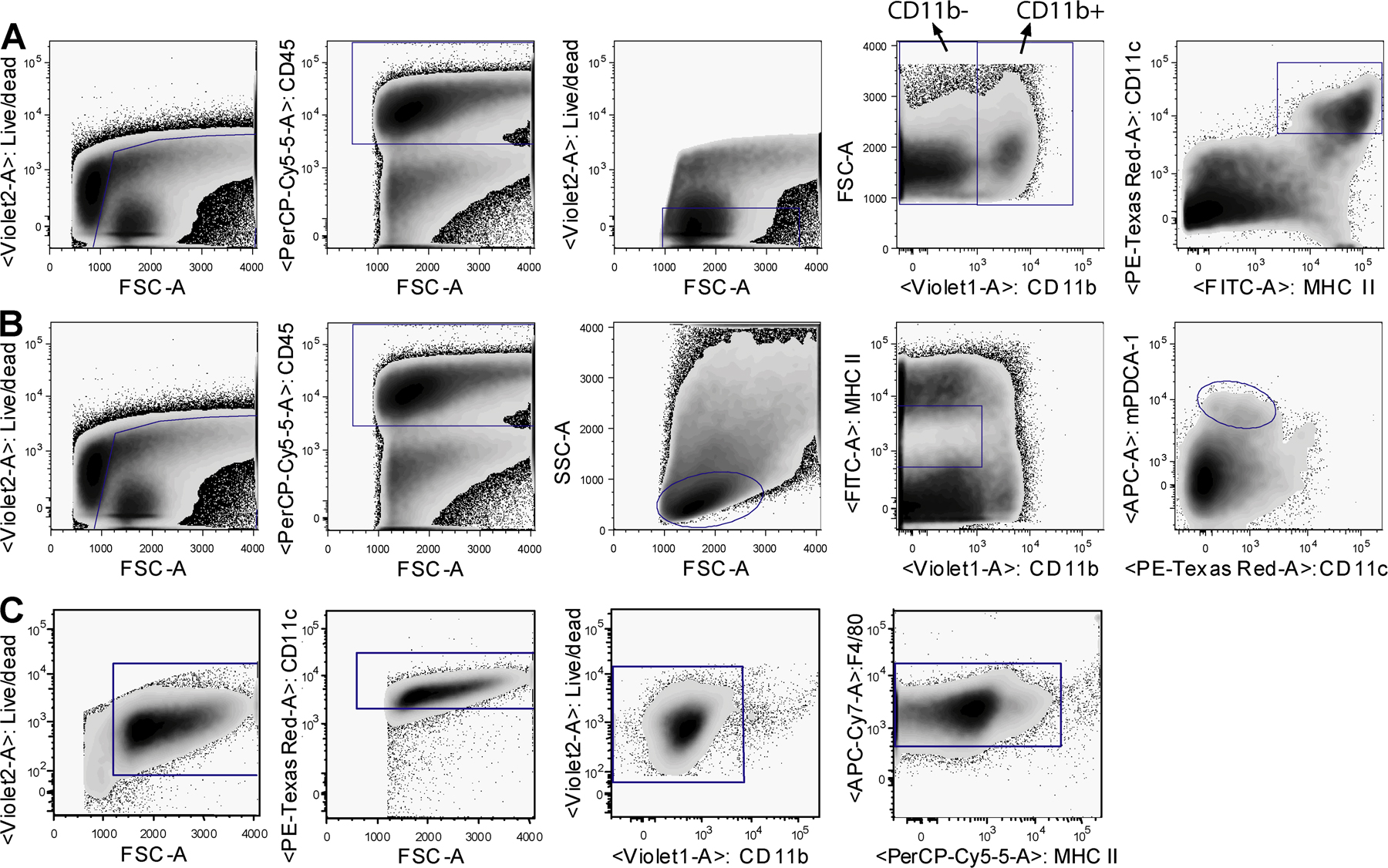
As expected after i.n. inoculation with 5 × 105 tissue culture ID50 influenza A virus X-31 (H3N2), there was induction of vigorous innate and adaptive immune responses in the lung. The innate cellular immune response was already present 1 day postinfection (dpi) and peaked at 4 dpi, exemplified by a strong increase in neutrophils. B lymphocytes (CD19+) were present in the lung by day 4 and peaked around day 10. Total CD8+ cells in the lung peaked around day 10 (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20071365/DC1). To investigate the number of DC subsets and macrophages after influenza virus infection, enzymatically digested lungs (from which the large conducting airways were dissected) were analyzed at different time points after infection by eight-color flow cytometry. Digested lung cells were first gated for live leukocytes and expression of the panleukocyte marker CD45. cDCs were characterized as low fluorescent cells, with a CD45hi, MHCIIhi CD11chi phenotype (Fig. S2 A), and further subdivided into CD11b+ or CD11b− cells (11, 15, 30, 13). As previously described, pDCs were small, low autofluorescent cells expressing a CD45hi, CD11blo, MHCint CD11cint mPDCA-1+ phenotype (Fig. S2 B) (24). Alveolar macrophages were identified in the bronchoalveolar lavage (BAL) fluid as highly fluorescent cells with a CD11chi, CD11blo, MHCIIlo, F4/80hi phenotype (Fig. S2 C). The use of CD11c and CD11b expression in combination with the autofluorescence typically seen in alveolar macrophages adequately discriminated alveolar DCs from macrophages, as previously described (38, 39, 11).
In the mock situation, 65% of CD11chi DCs were CD11b− and 35% were CD11b+. After infection with influenza virus X-31, a significant increase in both CD11b+ (Fig. 1 A) and CD11b− (Fig. 1 B) lung DCs was seen from 2 dpi, and these DC subsets remained increased up to at least 10 dpi. The DCs expressed high levels of MHCII and up-regulated maturation markers such as CD86 (Fig. 1, A and B), CD40, CD80, and intercellular adhesion molecule 1 (not depicted). The increase in the expression level of these markers peaked shortly after inoculation (2 d) and gradually returned back to the levels seen in mock-infected mice (Fig. 1, A and B, bar graphs). In contrast, the number of pDCs peaked shortly after infection (2 dpi) and then returned to baseline conditions (Fig. 1 C). This increased recruitment of pDCs was accompanied by a distinct but very transient up-regulation of maturation markers (CD86 is shown). Alveolar macrophages were studied in the BAL fluid accessible by BAL. This compartment contains a large amount of resident alveolar macrophages in mock situation. After influenza virus infection, there was a nonsignificant trend of increase in the alveolar macrophage number at 4 dpi (Fig. 1 D), accompanied by a temporary increase in CD86 and a more continual increase in MHCII expression (mean fluorescence intensity [MFI] of 820 in mock and 2,769 at 10 dpi).
Figure 1.

Number and surface phenotype of mouse lung CD11b+ and CD11b− DCs, pDCs, and alveolar macrophages after influenza infection. Populations were gated as shown in Fig. S2 (available at http://www.jem.org/cgi/content/full/jem.20071365/DC1) and as indicated by the gates in the left column of each panel. (A) CD11b+ DCs significantly increased after infection and remained increased up to 10 dpi (left). CD86 expression was plotted in histograms (right), with MFI shown in the graph (bottom). (B) The increase in CD11b− DCs is demonstrated in flow cytometric plots (left), with absolute numbers shown in the graph (bottom). CD86 expression was up-regulated and plotted as MFI (C, bottom). pDCs increased significantly at 2 dpi and then returned to baseline. Recruitment of pDCs was accompanied by up-regulation of CD86 (right). (D) Alveolar macrophages slightly increased in number, but CD86 expression was not increased. Gray histograms represent isotype controls and were measured at 4 dpi. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from at least three separate experiments. *, P < 0.05; **, P < 0.01.
Response of DC subsets in the large conducting airways to influenza virus infection
To examine the DC network in the large conducting airways, we stained in vivo fixed and permeabilized tracheal whole mounts with an mAb to MHCII, revealing the presence of a highly developed network of DCs and demonstrating delicate dendritic processes in between bronchial epithelial cells (Fig. 2 A). After infection, there was an increase in MHCII+ cells at 2 dpi, and cells had a more rounded appearance, making it difficult to quantify DC density. At 4 dpi, there was a strong decrease in the density of cells staining for MHCII, followed by a restoration to baseline density and morphology by 10 dpi. To better quantify these changes, eight-color flow cytometry was performed on tracheal digestions, demonstrating considerable heterogeneity in DCs at various time points after infection (Fig. 2 B). In the mock situation, all CD45+ MHCII+ cells expressed CD11c, identifying them as DCs. The majority of these cells (∼75%) were CD11b− and expressed the mucosal αE integrin marker CD103 (Fig. 2 C). At 2 dpi, this subset was depleted from the tracheal digest and a majority of cells now expressed CD11b but lacked CD103, which was most consistent with the phenotype of a freshly recruited monocyte-derived DC (Fig. 2, B and C). A temporary increase in the expression level of CD86 was found on these CD11b+ DCs at day 2 (Fig. 2 B). At 10 dpi, the subset balance between CD11b+ and CD11b− DCs was almost restored to baseline, and cells again showed a highly dendritic morphology and CD86 expression returned to baseline levels. As previously reported (11), we detected a minor percentage (0.35% of live tracheal CD45+ leukocytes) of mPDCA-1+ pDCs in tracheal digests of mock-infected mice (Fig. 2 D). Again, pDCs increased after influenza virus infection and temporarily up-regulated their expression of co-stimulatory molecules (Fig. 2 E).
Figure 2.

DC subtypes in tracheal tissue after influenza infection. (A) Tracheal whole mount sections stained for MHCII expression were performed at various days after infection. Bar, 35 μm. (B) Flow cytometric analysis of DCs in tracheal cell suspensions stained for CD45, CD11c, CD11b, F4/80, and CD103. CD11c+ cells contained two subsets, one CD11b− and the other CD11b+. Histograms represent CD86 expression on both subsets. The dotted lines indicate MFI in a mock situation. (C) Absolute numbers of the two subsets after influenza infection expressed as mean values ± SEM. *, P < 0.05; **, P < 0.01. Histograms represent CD103 expression on both subsets. (D) pDCs were identified as CD11cintPDCA-1+ cells representing a minor percentage of CD45+ leukocytes in tracheal cell suspensions and only temporarily expressing CD86 (histogram). (E) Absolute number of pDCs at various days after infection expressed as mean values ± SEM. **, P < 0.01.
Influenza virus infection increases DC subsets in MLNs
Upon recognition of antigen, lung DCs are known to migrate to the MLNs. We followed the kinetics of increase and expression of maturation markers on various DC subsets within this node. cDC subsets were CD11c+MHCII+ cells (Fig. 3 A) that were further discriminated based on expression of the myeloid marker CD11b (Fig. 3 B). After infection, there was an increase in the absolute amount of CD11c+MHCII+CD11b− and CD11c+MHCII+CD11b+ DCs starting at 2 dpi, and the increase was more pronounced in the CD11b+ subset. Within the CD11b−CD11chi cells, there was a clear increase in cells coexpressing langerin and CD103 (30 × 104 cells at 4 dpi vs. 2 × 104 cells in mock-infected mice). Belz et al. previously suggested that a resident subset of CD11b−CD8α+ DCs was responsible for generating virus-specific CD8 CTLs, whereas the population of CD11b−CD11c+ cells transported antigen from the periphery (30). Therefore, cells were further discriminated based on CD8α (Fig. 3 C). Compared with mock-infected mice, at 4 dpi there was a generalized increase in all CD8α− subsets (CD11b− and CD11b+), whereas the resident CD8α+CD11b− DCs were not increased. After infection, CD11c+MHCII+ cells had higher levels of the CD86 maturation marker compared with mock-infected mice, which was consistent with their potential to prime CD8 T cell responses (Fig. 3 D), and this was found in all DC subsets.
Figure 3.

DC subsets in MLNs after influenza virus infection. (A) Kinetics of CD11c+MHCII+ DCs demonstrating an almost fourfold increase after infection. Percentages of living cells are shown. (B) Expression of CD11b by CD11c+ DCs gated in A. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from separate experiments. (C) CD8α expression on different DC subsets was demonstrated in combination with CD11b, with percentages of the different populations plotted in the graph. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from separate experiments. *, P < 0.05. (D) CD86 expression on different MLN subsets. The gray histogram represents isotype control, the dotted histogram represents mock controls, and the black histogram represents infected animals. (E) Sorted DC subsets were stained for intracellular NP. Uninfected cells stain dull red because of Evan's blue in the solution, and the green fluorescence indicates NP. Bar, 20 μm. These images are representative of one experiment out of at least five performed. (F) Flow cytometric analysis of the total detectable amount of intracellular NP in DC subsets as a percentage of total live LN cells per DC subset. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from two separate experiments. (G) Plots represent CFSE-labeled T cell proliferation 4 d after co-culture with the various sorted DC populations obtained from pooled LNs of 20 influenza-infected mice. (top) CD8+ T cells and (bottom) CD4+ T cell proliferation are shown. Numbers (top left corners) represent the percentage of cells recruited into cell division. Cell sortings with co-culture were performed three times each, and the results shown are representative plots of these experiments.
To test the antigen-presenting potential of the various DC subsets in the MLNs, mice were infected with influenza virus encoding either the immunodominant OVA257-264 Kb–restricted MHCI epitope recognized by the OT-I TCR-transgenic (Tg) strain (40) or carrying the OVA323-339 MHCII epitope recognized by the OT-II TCR-Tg strain (41), allowing us to probe presentation of sorted lung DC subsets to naive OVA-specific CD8 and CD4 T cells directly ex vivo. To have an indication of the uptake of viral antigen or virally infected apoptotic cells, preparations of sorted DC subsets from the MLNs were also stained for the presence of viral NP using a specific antibody (Fig. 3 E), and confirmed using eight-color flow cytometric staining on permeabilized cells (Fig. 3 F). Viral NP was found, particularly in the CD11b+CD8α− subset as well as abundantly in the pDC subset, and was practically absent from the CD11b− subsets (Fig. 3 F). When DC subsets were sorted and co-cultured with OVA-specific OTI cells (Fig. 3 G, top) or OTII cells (Fig. 3 G, bottom), the CD11b−CD8α− subset presented antigen to both CD8 and CD4 T cells, whereas the CD11b−CD8α+ resident DCs presented exclusively to CD8 cells. Despite the fact that these cells had seen viral antigens (Fig. 3, E and F, NP staining), the CD11b+ DCs and pDCs did not present antigen to naive CD4 or CD8 T cells. As a control, OT-I or OT-II T cells incubated with total MLN DCs obtained from mice infected with the virus containing the OVA-MHCII or –MHCI epitope, respectively, failed to proliferate.
Conditional depletion of lung CD11chi cells aggravates features of infection
DCs are extremely potent APCs that are uniquely suited to prime naive T cells. To study the immune response against influenza virus infection with and without CD11chi DCs, CD11c-DTR mice were treated with DT intratracheally (i.t.) 1 d before infection with influenza virus X-31. By this localized treatment, lung CD11chi DCs (both CD11b+ and CD11b−) and alveolar macrophages were efficiently depleted from the lungs (Fig. 4 A). In the trachea, the localized treatment with DT led to a reduction of CD11b+ DCs, but not to depletion of CD11b− DCs (Fig. S3 A, available at http://www.jem.org/cgi/content/full/jem.20071365/DC1) or pDCs (not depicted). In the MLNs, all DC subsets were partially depleted after DT treatment, with the biggest depletion occurring in the resident CD11b−CD8α+ DCs (Fig. S3 B) (19, 37). On the contrary, CD11chi cells in the spleen or nondraining LNs were not affected (unpublished data) (19, 37). At different time points after infection, the clinical immune response and viral replication were analyzed. First, clinical severity of infection was determined by measuring body weight. In general, mice show a maximal 10% weight loss after a mild X-31 infection and rapidly regain weight once the virus has been cleared from the lungs. When CD11c-DTR Tg mice were treated with DT before infection, the clinical severity of infection dramatically increased and mice lost up to 20% of their weight, representing a sublethal infection. This weight loss could not be attributed to DT treatment, as the control group, which was treated with DT before mock infection, did not lose weight (Fig. 4 B). As weight loss might be related to the initial severity of infection or failure of the immune system to clear infectious particles from the lung, we next studied the generation of an efficient immune response in CD11chi–depleted mice. For efficient lung clearance of a primary influenza virus infection, CD8+ CTLs play an important role. We therefore measured the number of NP366-374 peptide/H-2Db–specific CTL cells using tetramer reagents and correlated their numbers to viral clearance. In DT-treated and -infected animals, the number of virus-specific CTLs was significantly reduced in the lung, spleen (Fig. 4 C), and MLN (not depicted). In addition to the measurement of CTL, CD3-stimulated IFN-γ production by CD4 and CD8 T cells from MLNs was determined and dramatically suppressed in the absence of DCs (Fig. 4 D).
Figure 4.

Infection parameters after conditional depletion of CD11chi cells in a CD11c-DTR Tg mouse model. CD11c-DTR Tg mice received an i.t. injection of DT on day −1, followed by X-31 i.n. infection. (A) Efficient depletion of lung CD11chi cells by DT treatment compared with PBS treatment. (right) Plots demonstrate flow cytometry data, and numbers indicate the percentage of live cells within each gate. (B) Body weight after influenza infection. A weight loss of 20% represents a sublethal infection. (C) Virus-specific CTL response in spleen and lung as measured by Flupeptide/H-2Db tetramer (TM) staining. (D) IFN-γ levels in supernatants of MLN cell cultures restimulated with anti-CD3 antibody. (E) Viral titers measured in lung tissue after influenza X-31 infection. Virus is normally completely cleared at 8 dpi. nd, nondetectable. (F) IFN-α levels in BAL fluid. (G) Adoptive transfer of wild-type DCs and alveolar macrophages into DT-treated CD11c-DTR Tg mice before viral infection. CD11c-DTR Tg mice were either treated with PBS or DT, and received either DCs or macrophages before influenza infection. Numbers of Flupeptide/H-2Db–specific CTLs in spleen suspensions (left) and viral titer in lung tissue (right) are shown. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from at least two separate experiments. *, P < 0.05; **, P < 0.01.
As induction of virus-specific CTL responses is necessary for viral clearance from the lung, we measured viral titers in the lungs. In this mild infection model, virus is normally cleared completely at 8 dpi (Fig. 4 E) (42). However, in the absence of lung CD11chi cells and mediastinal CD8α+CD11chi cells, the virus had not been cleared at this time.
A delay in viral clearance could be caused by a direct defect in CTL priming but could also result from a less efficient innate response consisting mainly of type I IFN production. IFN-α has been described as an important antiviral cytokine that is mainly produced by epithelial cells and alveolar macrophages, but also by pDCs and even myeloid DCs after influenza infection in vitro (3, 5). We therefore determined IFN-α production in the BAL fluid at different time points after infection in CD11chi–depleted mice. In vivo, IFN-α was produced in the lungs after X-31 infection, but levels were not affected by CD11chi cell depletion (Fig. 4 F).
The data in this paragraph show that lung CD11chi cells are required for an efficient immune response against influenza virus infection. However, as CD11c is also highly expressed by alveolar macrophages and these cells are also depleted by local i.t. administration of DT, we performed adoptive transfer reconstitution experiments (19, 37). Tg mice depleted of CD11chi cells therefore received i.t. unpulsed wild-type CD11chi DCs or alveolar macrophages at the moment of infection. DCs were capable of restoring the CTL response (Fig. 4 G), and as a consequence, viral clearance from the lung was complete by day 8 (Fig. 4 G, right). Alveolar macrophages were not capable of restoring antiviral immunity or viral clearance. These experiments demonstrated that CD11chi DCs are sufficient for inducing an adequate immune response against influenza virus infection.
Depletion of lung langerin+ DCs aggravates infection parameters
The conditional depletion of CD11chi cells in CD11c-DTR mice did not allow us to address the contribution of tracheal CD11b−CD11chi DCs, as neither tracheal CD11b-CD11chi DCs nor their CD11b− progeny in the MLNs were depleted in CD11c-DTR mice. Therefore, we performed experiments in langerin-DTR mice. When DT was administered i.t. to these mice, there was a strong reduction in CD11b−CD11chi DCs in the trachea, whereas tracheal CD11b+ DCs were unaffected. In the lungs, CD11chi DCs were depleted, whereas alveolar macrophages were unaffected. The CD8α+CD11b− resident MLN cDC subset does not express langerin, and consequently, lung administration of DT to these mice only led to a reduction in lung-derived CD11b−CD8α− migratory DCs (Fig. S3). Depletion of langerin+ DCs resulted in a severe weight loss in the mice until 8 dpi (Fig. 5 B), and this was correlated with a significant decrease in CTL response (Fig. 5 C) and a deficient viral clearance at 8 dpi (Fig. 5 D).
Figure 5.

Effect of conditional depletion of langerin+ DCs cells during influenza infection. Langerin-DTR Tg mice received an i.t. injection of DT on day −1, followed by X-31 i.n. infection. (A) Efficient depletion of MHCII+CD11c DCs in lung after DT treatment. Numbers indicate percentages of live cells within the gate. (B) Body weight after influenza infection. A weight loss of >20% represents a sublethal infection. (C) Virus-specific CTL response in spleen and lung measured by Flupeptide/H-2Db tetramer (TM) staining. (D) Viral titers measured in lung tissue after influenza X-31 infection. Virus is normally cleared completely at 8 dpi. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from at least two separate experiments. *, P < 0.05; **, P < 0.01. nd, nondetectable.
Depletion of pDCs did not alter the course of infection
The experiments using CD11c-DTR and langerin-DTR mice mainly depleted CD11chi cells, whereas CD11cint pDCs are globally not affected by this targeting strategy (43). To additionally address the role of pDCs, we performed experiments in which pDCs were depleted by injection of the 120G8 mAb (22). Using this antibody, an effective depletion of pDCs to <10% of baseline numbers was achieved in the lung (Fig. 6 A), as measured using antibodies directed against both bone marrow stromal antigen and SiglecH (24). Surprisingly, this efficient depletion of pDCs did not affect any of the infection parameters and virus was cleared efficiently by 8 dpi (Fig. 6, B–E). As pDCs have been previously described as the main IFN-α–producing cells in response to a viral infection (44), it was similarly striking that there was no reduction in the protein level of IFN-α in the BAL fluid or in the mRNA level for IFN-α in lung tissue after the depletion of pDCs (Fig. 6 F).
Figure 6.

Infection parameters after influenza in mice depleted of pDCs. Mice received three i.p. injections of the depleting mAb 120G8 before infection with influenza on day 0. (A) Efficient depletion of lung pDCs after 3 d of 120G8 i.p. treatment compared with PBS treatment. (B) Body weight after infection. (C) Virus-specific CTL responses in spleen and lung cell suspensions, as measured by Flupeptide/H-2Db tetramer staining. (D) IFN-γ levels in supernatants of MLN cell cultures restimulated with anti-CD3 antibody. (E) Viral titers in lung after influenza infection. (F) IFN-α levels in BAL fluid. The values are representative of five mice per group and are expressed as mean ± SEM. Similar results were obtained from at least two separate experiments. *, P < 0.05; **, P < 0.01.
The production of hemagglutinin (HA)-specific antibodies depends on pDCs and not on CD11chi DCs
The induction of antiviral CD8 T cell responses is only one aspect of adaptive immunity to influenza. We also measured the production of serum HA-specific antibodies at 8 and 28 dpi by measuring HA inhibition (HI) titers. Although there was no effect on the induction of CD8 responses after 120G8-mediated pDC depletion, virus-specific antibody titers measured at both time points were significantly reduced (Fig. 7 A). We found no evidence for 120G8 staining on lung B cells, making it unlikely that this would be a depleting effect of 120G8 on B cells directly (unpublished data). Antibody responses were maintained in mice depleted of CD11chi DCs by DT treatment of CD11c-DTR mice 1 d before infection in CD11c-DTR mice (Fig. 7 B).
Figure 7.

Virus-specific serum antibodies of influenza virus–infected mice after DC subset depletion. (A) Virus-specific antibodies in the serum of C57BL/6 mice after i.p. treatment with 120G8 depleting antibody or PBS, or (B) in the serum of CD11c-DTR mice after i.t. treatment with DT or PBS, measured at 8 dpi and 1 mo after infection. The values are representative of at least five mice per group and are expressed as geometric mean titer (GMT) of HI ± SEM. The differences in HI titer can be explained by the background of the mice. CD11c-DTR mice were F1 (BALB/c × C57BL/6) animals and developed less high viral and HI titers than the pure C57BL/6 mice that were used in 120G8 depletion experiments. Similar results were obtained from at least two separate experiments. *, P < 0.05; **, P < 0.01.
DISCUSSION
Belz et al. have elegantly described that after influenza infection, both CD11b−CD8α+ resident MLN DCs and the CD11b−CD8α− lung-derived migratory DC subset presented antigen to naive CD8+ T cells ex vivo (30, 32). However, these authors have not described in detail where exactly in the lung these CD11b− migratory DCs originated from and whether any of the DCs would also present to CD4+ T cells, and they have also not studied in detail the precise contribution of inflammatory CD11b+ DCs or pDCs that are recruited to inflamed lungs.
Our studies on the various anatomical lung compartments suggest that a predominant source of CD11b− DCs arriving in the MLNs are from the network of highly dendritic-shaped CD103+ intraepithelial DCs that lines the large conducting airways (Fig. 1) (18, 13). There was a marked decrease in this subset of highly dendriform tracheal CD11b−CD103+ DCs at 2 dpi, at a time that the langerin+CD103+CD11b−CD8α− DCs started to accumulate in the MLNs. The disappearance of CD11b−CD11c+ tracheal DCs was accompanied by a new influx of CD11b+CD11c+ DCs into the trachea (present as more rounded cells at day 2 after infection on whole mounts) and into the lung interstitium. These cells most likely differentiate from recruited Ly6C+ monocytes that give rise to inflammatory-type DCs (16, 45), or that arose from local proliferation and differentiation of a myeloid precursor population that also generates alveolar macrophages (46). An identical subset of CD11b+CD11c+ DCs was found to be increased in the MLNs from days 2 to 4 onwards, again suggesting migration from the lungs to the MLN.
It was striking that there were several subsets of DCs found to be recruited and/or activated in the MLNs after influenza infection, strongly suggesting the division of labor between various APCs. Recently, several papers have shown that the cross presentation of exogenous harmless or viral antigen to CD8+ lymphocytes or presentation of exogenous antigen to CD4 T cells is a mutually exclusive function of CD8α+ or CD8α− DC subsets, respectively (47–49). We therefore performed a head-to-head comparison of the potential to present viral antigen to naive CD4 or CD8 T cells, taking advantage of influenza virus encoding either the MHCI or MHCII immunodominant OVA epitope.
As previously described by Belz et al., CD8α+CD11b− resident DCs presented viral antigen to naive CD8 cells, supported by the fact that these cells had up-regulated co-stimulatory molecules. Strikingly, the population of airway-derived CD11b−CD8α− DCs also up-regulated co-stimulatory molecules and presented not only to CD8 T cells but also to CD4 T cells. This suggests that processing for and presentation on both MHCI and MHCII molecules can occur in a single cell population in vivo in the lung, contrary to what was shown for the spleen or lymph nodes (30). Vermaelen et al. also previously demonstrated that antigen presentation of harmless antigen to naive CD4 T cells in the MLNs was an exclusive function of a migratory DC population (50). Despite the fact that both CD8α+ and CD8α− CD11b− DCs presented antigen to naive T cells, the strength of viral NP staining was not abundant. This could be explained by the fact that viral NP was digested in these subsets as part of an antigen-processing step, leading to a loss of immunoreactivity toward the NP-specific antibody. Alternatively, Belz et al. previously suggested that resident CD8α+ cells acquired the antigen from other migratory APCs, proposed as the CD11b−CD8α− lung-derived DCs, but this was never directly demonstrated (30). Our data on viral NP staining suggest that the CD11b+ DCs as well as the pDCs were abundantly positive for viral antigen in the MLNs and, therefore, could be the most important source for providing viral antigens to resident CD8α+ DCs.
Another striking observation was the fact that the CD11b+CD11c+ subset found to be increased in the trachea, lung, and MLNs after influenza infection hardly presented any viral antigen to CD4 or CD8 T cells. Clearly, the CD11b+ DCs had seen viral antigens (either directly or through phagocytosis of virally infected apoptotic epithelial cells) as they carried an abundant amount of viral NP in their cytoplasm. The absence of APC function by this subset is in striking contrast to the situation when harmless noninflammatory antigen is inhaled (15). What could be the purpose of recruitment of a different DC subset, in addition to CD11b− DCs, if these cells do not present antigen to naive T cells? First, as these cells have been shown to massively produce inflammatory chemokines, CD11b+ DCs might be crucial in attracting effector CD4 and CD8 cells that have been generated in the LNs back to the lung and trachea, where they would mediate effector function (51). Second, CD11b+ DCs might have direct innate antiviral activity by producing TNF-α and inducible NO synthase–dependent NO, analogous to the situation seen with Listera monocytogenes infection (52, 53). Third, recruited CD11b+ DCs might also stimulate the innate antiviral activity of NK cells. The important function of inflammatory DCs in initiating the innate response is supported by the fact that influenza produces the nonstructural protein 1, with a specific aim to subvert the innate immune function of the CD11b+ DC subset (54, 3). Future studies in our laboratory will have to address the direct or indirect innate and adaptive functions of inflammatory CD11b+ DCs in influenza infection.
Another way to study the function of lung DC subsets is to deplete them using various cell-specific genetic targeting strategies using expression of the DTR under control of a specific promoter and administration of the DT via the airways (19). In CD11c-DTR mice, there is a predominant depletion of lung CD11chi DCs, of tracheal CD11b+CD11chi DCs, and of resident MLN CD8α+CD11chi DCs. Because CD11b−CD11chi migratory tracheal DCs are not depleted in these mice, we consequently observed no decrease in MLN CD11b−CD8α− DCs. In CD11c-DTR mice given DT, we noticed that generation of virus-specific CD8+ CTLs and production of effector cytokines (IFN-γ) by MLN cells was severely diminished. Based on our antigen presentation studies and the previous work of Belz et al. (30), we propose that this is caused by depletion of the resident CD8α+CD11chi DCs or to the depletion of chemokine-producing lung CD11b+ DCs. CD11chi alveolar macrophages are also depleted in CD11c-DTR mice, although it is unlikely that this contributed to a decrease in antiviral immunity, as adoptive transfer of wild-type DCs but not macrophages restored immunity.
To address the specific role of migratory tracheal CD11b− DCs, we performed experiments in langerin-DTR mice (36). Langerin was found to be particularly present on mucosal CD11b−CD103+ DCs in the lung (13), a population of cells that strictly relies on CCR7 to migrate to the MLNs (15). In accordance, we also found increased numbers of langerin+CD103+CD11b−CD11c+ DCs in the MLNs after influenza infection. After i.t. administration of DT, there was a selective depletion of CD11b−CD8α− migratory DCs in the trachea and MLNs, while the CD8α+ resident LN population or alveolar macrophages were unaffected. In the absence of the migratory langerin+ CD11b− DC population, influenza ran a particularly severe course. Strikingly, these data also suggest that the langerin+ DCs of the lung are much more immunogenic than their langerin+ Langerhans cell counterparts in the skin (55, 36, 56).
Finally, we also addressed the in vivo function of lung pDCs, previously known as natural IFN-α–producing cells (44). In humans, these cells produce copious amounts of IFN-α when exposed to influenza virus in vitro (57) and have been shown to stimulate already-primed influenza-specific CD4 and CD8 T cells (34). It has therefore been suggested that upon proper stimulation, pDCs develop into bona fide APCs that primarily stimulate antiviral immune response (58). To address the antigen-presenting function of pDCs, we sorted pDCs from the MLNs of infected mice and found no evidence for presentation to either CD4 or CD8 T cells ex vivo, despite the fact that the cells contained copious amounts of intracellular viral NP. Previous studies with other respiratory viruses have suggested that pDCs have a more immunoregulatory role, which is crucial for preventing excessive immune activation and immunopathology (12, 24, 25). Although pDCs were attracted to the lungs and tracheal wall during influenza infection, their confirmed depletion using 120G8 antibody did not affect viral titers, generation of virus-specific T cells, or the severity of infection, arguing against a predominant role for pDCs as APCs, immunoregulatory cells, or innate immune cells in this mild infection model. It was similarly striking to see that there was no effect on IFN-α production when pDCs were effectively depleted. Infections in epithelial surfaces that bathe in IFN-α (produced by epithelial cells) might be less dependent on IFN-α production by pDCs. Kumagai et al. recently demonstrated that during lung infection with RNA viruses, alveolar macrophages are a predominant source of type I IFN, and that pDCs only start producing IFN-α when macrophages are depleted from the lungs (5). The only effect of treatment with the 120G8 antibody was a significant reduction of the titer of virus-specific HI antibodies at 8 and 28 dpi, signifying a possible role for pDCs in stimulating humoral antiviral immunity, as previously suggested (59). Although we found no evidence for 120G8 staining on lung B cells after influenza infection, treatment with 120G8 antibody could also deplete plasma cells directly and in this way reduce HI antibody titers. Therefore, we are awaiting more specific (e.g., genetic) pDC targeting strategies to address this point further. In conclusion, our paper demonstrates a division of labor between different DC subsets during pulmonary influenza infection, knowledge that might be used for the design of better influenza vaccines and could increase our understanding of why particular strains of influenza are more pathogenic than others.
MATERIALS AND METHODS
Mice.
6–8-wk-old C57BL/6 mice were purchased from Harlan. The generation and screening of CD11c-DTR Tg mice has been previously described (37). Male BALB/c background CD11c-DTR Tg (H2-Dd) mice were crossed to C57BL/6 (H2-Db), to obtain F1 progeny, to allow detection of the H2-Db tetramer. CD11chi cells were depleted in CD11c-DTR × C57BL/6 Tg mice by i.t. injection of 50 ng DT, a dose previously determined by titration (15). pDCs were depleted by i.p. injection of pDC-selective depleting 120G8 antibody (22). All experiments were approved by an independent animal ethics committee of Erasmus Medical Centre Rotterdam.
Influenza virus infection.
Influenza virus X-31 (Medical Research Council) was inoculated in the allantoic cavity of 11-d-old embryonated chicken eggs. The allantoic fluid was harvested after 2 d. Infectious virus titers were determined in Madin-Darby canine kidney (MDCK) cells (NBL-2; American Type Culture Collection), as described previously (60). Virus titers were obtained at days 4 and 8 after infection. Lungs were stored at −70°C. Lungs were homogenized with a homogenizer (Polytron; Kinematica AG) in infection medium (Eagle's minimal essential medium), 0.3% BSA (fraction V), 4 μg/ml trypsin, 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 0.15% NaHCO3, 20 mM Hepes, and 0.1 mM of nonessential amino acids. 10-fold serial dilutions of these samples were used 8-fold to determine the virus titers in MDCK cells, as described previously (60). For antigen presentation assays of LN DCs, mice were infected with WSN influenza virus encoding the OVA257-264 Kb–restricted MHCI epitope in neuraminidase (40), and with X-31 influenza virus encoding the OVA323-339 MHCII epitope in HA of the virus (41). The OVA viruses were provided by R. Webby (St. Jude Children's Research Hospital, Memphis, TN).
Flow cytometry.
For detection and phenotyping of DCs at days 2, 4, and 10 after infection, single-cell suspensions of MLNs and lung samples were prepared as described previously (24). Trachea was digested in collagenase solution for 1 h at 37°C to promote the release of DCs. Cells were subsequently stained with mAbs directed against MHCII FITC, intracellular NP FITC, CD86 PE, CD11c PE Texas red, CD45 PECy5, CD103 PECy7, intracellular langerin APC, mPDCA-1 APC, F4/80 APC Cy7, CD11b Pacific blue, and a fixable live/dead marker in Aqua (Invitrogen). Acquisition of eight to nine color samples was performed on a cytometer (FACSAria) equipped with FACSDiva software (both from BD Biosciences). The final analysis and graphical output were performed using FlowJo software (Tree Star, Inc.). Cell sorting of DC subsets from MLNs was performed on a FACSAria. The purity of sorted populations was >95%.
Immunofluorescence on sorted DC subsets.
Sorted cell populations were spotted on microscope slides, dried, and fixed in acetone. Subsequently, the slides were incubated with FITC-labeled influenza A NP-specific antibody (IMAGEN influenza virus; Dako) at 37°C for 15 min. The slides were washed twice with PBS and once with distilled water, dried, and embedded into a glycerol–PBS solution (Citifluor; UKC Chemlab). Uninfected cells stain dull red because of the Evan's blue in the solution. Green and yellow signals indicate the presence of NP. Fluorescence was scored using a fluorescence microscope (LSM 510; Carl Zeiss, Inc.).
Analysis of T cell proliferation.
OT-1 and OT-2 Tg T cells were isolated from spleens and LNs of respective mice, enriched by MACS cell sorting with anti-CD8 or -CD4 antibodies according to manufacturer's protocol (Miltenyi Biotec), and labeled with CFSE (61). Sorted DC subsets were co-cultured with T cells in a v-bottom plate at a 1:10 ratio for 4 d. T cell divisions were measured by flow cytometry. The percentage of cells recruited into each cell division was calculated by dividing the number of individual cells by CFSE content, as previously described (61), using the formula 100 × {1 − [n0/(n0 + n1/2 + n2/4 + n3/8 + n4/16 + n5/32 + n6/64 + n7/128)] } to correct for the multiplying effect of division.
Tracheal whole mount staining.
Animals were anesthetized with a lethal dose of Nembutal (Sanofi) and perfused in vivo with 1% paraformaldehyde fixative in PBS, pH 7.4, for 2 min through the ascending aorta (18). Tracheas were removed, opened by a midline incision, and pinned flat in silicone-coated Petri dishes. After permeabilization in PBS containing 0.3% Triton X-100 (Sigma-Aldrich), tissues were preblocked with rabbit serum, incubated with M5/114 mAb (rat IgG2, anti–I-A, and I-Eb,d,k; Boehringer Mannheim) for 36 h at room temperature, washed, and incubated with peroxidase-conjugated rabbit anti–rat IgG, followed by incubation for 20 min in 0.05% diaminobenzidine in Tris-buffered saline, pH 7.6. Tissues were dehydrated in serial alcohol steps and cleared in toluene. The entire trachea was mounted in DPX mounting medium.
Detection of virus-specific CTL by tetramer-staining.
Single-cell suspensions of lung and spleen samples were prepared as described previously (24). Red blood cells were removed using erythrocyte lysis buffer (Roche). The cells were washed with 0.5% BSA in PBS and stained for 20 min at room temperature with the following antibodies: CD3e-PerCP and CD8b.2-FITC (BD Biosciences), TO-PRO 3–APC (Invitrogen), and PE-labeled H-2Db tetramer with the NP366–374 epitope ASNENMETM (Sanquin Research) (62).
Effector cytokine production.
On day 4 after infection, single-cell suspensions of lung draining LNs were prepared. Cells were cultured in RPMI 1640 medium with 5% FCS at a concentration of 2 × 106 cells/ml in the presence of either 1 μg/ml of plate-bound anti-CD3 (BD Biosciences) or medium alone and incubated at 37°C. After 4 d, supernatants were collected and stored at −20°C until ELISA for IFN-γ (BD Biosciences) was performed. IFN-α ELISA (PBL Biomedical Laboratories) was performed on BAL fluid on several days after infection. Quantitative RT-PCR for IFN-α was performed on homogenized lungs on day 4 after infection (Assay-On-Demand; Applied Biosystems).
Generation of bone marrow–derived DCs.
Bone marrow cells were cultured for 9 d in DC culture medium (RPMI 1640 containing glutamax-I; Invitrogen) supplemented with 5% (vol/vol) FCS (Sigma-Aldrich), 50 μM 2-ME (Sigma-Aldrich), 50 μg/ml gentamicin (Invitrogen), and 20 ng/ml recombinant mouse GM-CSF, as previously described (15).
Adoptive transfer.
Adoptive transfer reconstitution experiments were performed in DT-treated CD11c-DTR × C57BL/6 mice. At the moment of infection, they were treated i.t. with 2 × 106 bone marrow–derived DCs and 2 × 105 alveolar macrophages, as previously described (19).
Detection of virus-specific antibodies in serum.
After treatment with cholera filtrate and heat inactivation at 56°C, the serum samples were tested for the presence of anti-HA antibodies. For this purpose, an HI assay was used according to the standard protocol of 1% turkey erythrocytes and 4 HA U of H3N2 influenza virus (63).
Statistical analysis.
All experiments were performed using 5–10 animals per group. The difference between groups was calculated using the Mann-Whitney U test for unpaired data (Prism software version 4.0; GraphPad Software, Inc.). Differences were considered significant when P < 0.05.
Online supplemental material.
Fig. S1 shows time kinetics of neutrophils, B cells, and CD8+ T cells in the lung after influenza infection. Fig. S2 shows the gating strategy for myeloid DCs (A), pDCs (B), and alveolar macrophages (C). Fig. S3 shows the depletion of DC subsets in the tracheas (A) and MLNs (B) in CD11c-DTR (left) and langerin-DTR (right) mice. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20071365/DC1.
Supplementary Material
Acknowledgments
C.H. GeurtsvanKessel is supported by a Viral Genomics (VIRGO) grant from the Netherlands Organisation for Scientific Research. L..S. van Rijt is supported by an Erasmus University Rotterdam fellowship, and B.N. Lambrecht is supported by VIDI and VIRGO grants from the Netherlands Organisation for Scientific Research, as well as by an Odysseus Grant from the Flemish government. M. Kool is supported by a grant from the Dutch Asthma foundation. B.E. Clausen is the recipient of a VIDI grant and a career development grant from the Landsteiner Foundation for Blood Transfusion Research.
The authors have no conflicting financial interests.
Abbreviations used: BAL, bronchoalveolar lavage; cDC, conventional DC; dpi, days postinfection; DTR, diphtheria toxin receptor; HA, hemagglutinin; HI, HA inhibition; i.n., intranasal; i.t., intratracheal; MFI, mean fluorescence intensity; MLN, mediastinal lymph node; mPDCA-1, mouse pDC antigen 1; NP, nucleoprotein; pDC, plasmacytoid DC; Tg, transgenic.
G.F. Rimmelzwaan and B.N. Lambrecht contributed equally to this paper.
References
- 1.Eichelberger, M., W. Allan, M. Zijlstra, R. Jaenisch, and P.C. Doherty. 1991. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibility complex-restricted CD8+ T cells. J. Exp. Med. 174:875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Legge, K.L., and T.J. Braciale. 2005. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity. 23:649–659. [DOI] [PubMed] [Google Scholar]
- 3.Pichlmair, A., O. Schulz, C.P. Tan, T.I. Naslund, P. Liljestrom, F. Weber, and C. Reis e Sousa. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science. 314:997–1001. [DOI] [PubMed] [Google Scholar]
- 4.Tecle, T., M.R. White, and K.L. Hartshorn. 2005. Innate immunity to influenza A virus infection. Current Respir. Med. Rev. 1:127–145. [Google Scholar]
- 5.Kumagai, Y., O. Takeuchi, H. Kato, H. Kumar, K. Matsui, E. Morii, K. Aozasa, T. Kawai, and S. Akira. 2007. Alveolar macrophages are the primary interferon-alpha producer in pulmonary infection with RNA viruses. Immunity. 27:240–252. [DOI] [PubMed] [Google Scholar]
- 6.Banchereau, J., and R.M. Steinman. 1998. Dendritic cells and the control of immunity. Nature. 392:245–252. [DOI] [PubMed] [Google Scholar]
- 7.McWilliam, A.S., D.J. Nelson, J.A. Thomas, and P.G. Holt. 1994. Rapid dendritic cell recruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J. Exp. Med. 179:1331–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Legge, K.L., and T.J. Braciale. 2003. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity. 18:265–277. [DOI] [PubMed] [Google Scholar]
- 9.Lambrecht, B.N., J.B. Prins, and H.C. Hoogsteden. 2001. Lung dendritic cells and host immunity to infection. Eur. Respir. J. 18:692–704. [PubMed] [Google Scholar]
- 10.Vermaelen, K., and R. Pauwels. 2005. Pulmonary dendritic cells. Am. J. Respir. Crit. Care Med. 172:530–551. [DOI] [PubMed] [Google Scholar]
- 11.von Garnier, C., L. Filgueira, M. Wikstrom, M. Smith, J.A. Thomas, D.H. Strickland, P.G. Holt, and P.A. Stumbles. 2005. Anatomical location determines the distribution and function of dendritic cells and other APCs in the respiratory tract. J. Immunol. 175:1609–1618. [DOI] [PubMed] [Google Scholar]
- 12.de Heer, H.J., H. Hammad, M. Kool, and B.N. Lambrecht. 2005. Dendritic cell subsets and immune regulation in the lung. Semin. Immunol. 17:295–303. [DOI] [PubMed] [Google Scholar]
- 13.Sung, S.S., S.M. Fu, C.E. Rose Jr., F. Gaskin, S.T. Ju, and S.R. Beaty. 2006. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176:2161–2172. [DOI] [PubMed] [Google Scholar]
- 14.Wikstrom, M.E., and P.A. Stumbles. 2007. Mouse respiratory tract dendritic cell subsets and the immunological fate of inhaled antigens. Immunol. Cell Biol. 85:182–188. [DOI] [PubMed] [Google Scholar]
- 15.del Rio, M.-L., J.-I. Rodriguez-Barbosa, E. Kremmer, and R. Forster. 2007. CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J. Immunol. 178:6861–6866. [DOI] [PubMed] [Google Scholar]
- 16.Shortman, K., and S.H. Naik. 2007. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 7:19–30. [DOI] [PubMed] [Google Scholar]
- 17.Sertl, K., T. Takemura, E. Tschachler, V.J. Ferrans, M.A. Kaliner, and E.M. Shevach. 1986. Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. J. Exp. Med. 163:436–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lambrecht, B.N., B. Salomon, D. Klatzmann, and R.A. Pauwels. 1998. Dendritic cells are required for the development of chronic eosinophilic airway inflammation in response to inhaled antigen in sensitized mice. J. Immunol. 160:4090–4097. [PubMed] [Google Scholar]
- 19.van Rijt, L.S., S. Jung, A. Kleinjan, N. Vos, M. Willart, C. Duez, H.C. Hoogsteden, and B.N. Lambrecht. 2005. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J. Exp. Med. 201:981–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Rijt, L.S., J.B. Prins, V.C. deVries, P.J. Leenen, K. Thielemans, H.C. Hoogsteden, and B.N. Lambrecht. 2002. Allergen-induced accumulation of airway dendritic cells is supported by an increase in CD31hi Ly-6Cneg hematopoietic precursors. Blood. 100:3663–3671. [DOI] [PubMed] [Google Scholar]
- 21.Lambrecht, B.N. 2006. Alveolar macrophage in the driver's seat. Immunity. 24:366–368. [DOI] [PubMed] [Google Scholar]
- 22.Asselin-Paturel, C., G. Brizard, J.J. Pin, F. Briere, and G. Trinchieri. 2003. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J. Immunol. 171:6466–6477. [DOI] [PubMed] [Google Scholar]
- 23.Blasius, A.L., E. Giurisato, M. Cella, R.D. Schreiber, A.S. Shaw, and M. Colonna. 2006. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J. Immunol. 177:3260–3265. [DOI] [PubMed] [Google Scholar]
- 24.de Heer, H.J., H. Hammad, T. Soullie, D. Hijdra, N. Vos, M.A. Willart, H.C. Hoogsteden, and B.N. Lambrecht. 2004. Essential role of lung plasmacytoid dendritic cells in preventing asthmatic reactions to harmless inhaled antigen. J. Exp. Med. 200:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Smit, J.J., B.D. Rudd, and N.W. Lukacs. 2006. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J. Exp. Med. 203:1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang, H., N. Peters, and J. Schwarze. 2006. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J. Immunol. 177:6263–6270. [DOI] [PubMed] [Google Scholar]
- 27.Hamilton-Easton, A., and M. Eichelberger. 1995. Virus-specific antigen presentation by different subsets of cells from lung and mediastinal lymph node tissues of influenza virus-infected mice. J. Virol. 69:6359–6366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto, N., S. Suzuki, A. Shirai, M. Suzuki, M. Nakazawa, Y. Nagashima, and T. Okubo. 2000. Dendritic cells are associated with augmentation of antigen sensitization by influenza A virus infection in mice. Eur. J. Immunol. 30:316–326. [DOI] [PubMed] [Google Scholar]
- 29.Brimnes, M.K., L. Bonifaz, R.M. Steinman, and T.M. Moran. 2003. Influenza virus–induced dendritic cell maturation is associated with the induction of strong T cell immunity to a coadministered, normally nonimmunogenic protein. J. Exp. Med. 198:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belz, G.T., C.M. Smith, L. Kleinert, P. Reading, A. Brooks, K. Shortman, F.R. Carbone, and W.R. Heath. 2004. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc. Natl. Acad. Sci. USA. 101:8670–8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Belz, G.T., L. Zhang, M.D. Lay, F. Kupresanin, and M.P. Davenport. 2007. Killer T cells regulate antigen presentation for early expansion of memory, but not naive, CD8+ T cell. Proc. Natl. Acad. Sci. USA. 104:6341–6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belz, G.T., S. Bedoui, F. Kupresanin, F.R. Carbone, and W.R. Heath. 2007. Minimal activation of memory CD8+ T cell by tissue-derived dendritic cells favors the stimulation of naive CD8+ T cells. Nat. Immunol. 8:1060–1066. [DOI] [PubMed] [Google Scholar]
- 33.Bender, A., L.K. Bui, M.A.V. Feldman, M. Larsson, and N. Bhardwaj. 1995. Inactivated influenza virus, when presented on dendritic cells, elicits human CD8+ cytolytic T cell responses. J. Exp. Med. 182:1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fonteneau, J.F., M. Gilliet, M. Larsson, I. Dasilva, C. Munz, Y.J. Liu, and N. Bhardwaj. 2003. Activation of influenza virus-specific CD4+ and CD8+ T cells: a new role for plasmacytoid dendritic cells in adaptive immunity. Blood. 101:3520–3526. [DOI] [PubMed] [Google Scholar]
- 35.Dhodapkar, M.V., R.M. Steinman, M. Sapp, H. Desai, C. Fossella, J. Krasovsky, S.M. Donahoe, P.R. Dunbar, V. Cerundolo, D.F. Nixon, and N. Bhardwaj. 1999. Rapid generation of broad T-cell immunity in humans after a single injection of mature dendritic cells. J. Clin. Invest. 104:173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bennett, C.L., E. van Rijn, S. Jung, K. Inaba, R.M. Steinman, M.L. Kapsenberg, and B.E. Clausen. 2005. Inducible ablation of mouse Langerhans cells diminishes but fails to abrogate contact hypersensitivity. J. Cell Biol. 169:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jung, S., D. Unutmaz, P. Wong, G. Sano, K. De los Santos, T. Sparwasser, S. Wu, S. Vuthoori, K. Ko, F. Zavala, et al. 2002. In vivo depletion of CD11c(+) dendritic cells abrogates priming of CD8(+) T cells by exogenous cell-associated antigens. Immunity. 17:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vermaelen, K., and R. Pauwels. 2004. Accurate and simple discrimination of mouse pulmonary dendritic cell and macrophage populations by flow cytometry: methodology and new insights. Cytometry A. 61:170–177. [DOI] [PubMed] [Google Scholar]
- 39.van Rijt, L.S., H. Kuipers, N. Vos, D. Hijdra, H.C. Hoogsteden, and B.N. Lambrecht. 2004. A rapid flow cytometric method for determining the cellular composition of bronchoalveolar lavage fluid cells in mouse models of asthma. J. Immunol. Methods. 288:111–121. [DOI] [PubMed] [Google Scholar]
- 40.Topham, D.J., M.R. Castrucci, F.S. Wingo, G.T. Belz, and P.C. Doherty. 2001. The role of antigen in the localization of naive, acutely activated, and memory CD8(+) T cells to the lung during influenza pneumonia. J. Immunol. 167:6983–6990. [DOI] [PubMed] [Google Scholar]
- 41.Thomas, P.G., S.A. Brown, W. Yue, J. So, R.J. Webby, and P.C. Doherty. 2006. An unexpected antibody response to an engineered influenza virus modifies CD8+ T cell responses. Proc. Natl. Acad. Sci. USA. 103:2764–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kreijtz, J.H., R. Bodewes, G. van Amerongen, T. Kuiken, R.A. Fouchier, A.D. Osterhaus, and G.F. Rimmelzwaan. 2007. Primary influenza A virus infection induces cross-protective immunity against a lethal infection with a heterosubtypic virus strain in mice. Vaccine. 25:612–620. [DOI] [PubMed] [Google Scholar]
- 43.Sapoznikov, A., J.A. Fischer, T. Zaft, R. Krauthgamer, A. Dzionek, and S. Jung. 2007. Organ-dependent in vivo priming of naive CD4+, but not CD8+, T cells by plasmacytoid dendritic cells. J. Exp. Med. 204:1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asselin-Paturel, C., A. Boonstra, M. Dalod, I. Durand, N. Yessaad, C. Dezutter-Dambuyant, A. Vicari, A. O'Garra, C. Biron, F. Briere, and G. Trinchieri. 2001. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat. Immunol. 2:1144–1150. [DOI] [PubMed] [Google Scholar]
- 45.Naik, S.H., D. Metcalf, A. van Nieuwenhuijze, I. Wicks, L. Wu, M. O'Keeffe, and K. Shortman. 2006. Intrasplenic steady-state dendritic cell precursors that are distinct from monocytes. Nat. Immunol. 7:663–671. [DOI] [PubMed] [Google Scholar]
- 46.Blusse van Oud Alblas, A., B. van der Linden-Schrever, and R. van Furth. 1981. Origin and kinetics of pulmonary macrophages during an inflammatory reaction induced by intravenous administration of heat-killed bacillus Calmette-Guerin. J. Exp. Med. 154:235–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dudziak, D., A.O. Kamphorst, G.F. Heidkamp, V.R. Buchholz, C. Trumpfheller, S. Yamazaki, C. Cheong, K. Liu, H.W. Lee, C.G. Park, et al. 2007. Differential antigen processing by dendritic cell subsets in vivo. Science. 315:107–111. [DOI] [PubMed] [Google Scholar]
- 48.Allan, R.S., J. Waithman, S. Bedoui, C.M. Jones, J.A. Villadangos, Y. Zhan, A.M. Lew, K. Shortman, W.R. Heath, and F.R. Carbone. 2006. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 25:153–162. [DOI] [PubMed] [Google Scholar]
- 49.Schnorrer, P., G.M. Behrens, N.S. Wilson, J.L. Pooley, C.M. Smith, D. El-Sukkari, G. Davey, F. Kupresanin, M. Li, E. Maraskovsky, et al. 2006. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc. Natl. Acad. Sci. USA. 103:10729–10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vermaelen, K.Y., I. Carro-Muino, B.N. Lambrecht, and R.A. Pauwels. 2001. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J. Exp. Med. 193:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beaty, S.R., C.E. Rose Jr., and S.S. Sung. 2007. Diverse and potent chemokine production by lung CD11bhi dendritic cells in homeostasis and in allergic lung inflammation. J. Immunol. 178:1882–1895. [DOI] [PubMed] [Google Scholar]
- 52.Serbina, N.V., T.P. Salazar-Mather, C.A. Biron, W.A. Kuziel, and E.G. Pamer. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 19:59–70. [DOI] [PubMed] [Google Scholar]
- 53.Rimmelzwaan, G.F., M.M. Baars, P. de Lijster, R.A. Fouchier, and A.D. Osterhaus. 1999. Inhibition of influenza virus replication by nitric oxide. J. Virol. 73:8880–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Diebold, S.S., M. Montoya, H. Unger, L. Alexopoulou, P. Roy, L.E. Haswell, A. Al-Shamkhani, R. Flavell, P. Borrow, and C.R. Sousa. 2003. Viral infection switches non-plasmacytoid dendritic cells into high interferon producers. Nature. 424:324–328. [DOI] [PubMed] [Google Scholar]
- 55.Stoecklinger, A., I. Grieshuber, S. Scheiblhofer, R. Weiss, U. Ritter, A. Kissenpfennig, B. Malissen, N. Romani, F. Koch, F. Ferreira, et al. 2007. Epidermal langerhans cells are dispensable for humoral and cell-mediated immunity elicited by gene gun immunization. J. Immunol. 179:886–893. [DOI] [PubMed] [Google Scholar]
- 56.Kissenpfennig, A., S. Henri, B. Dubois, C. Laplace-Builhe, P. Perrin, N. Romani, C.H. Tripp, P. Douillard, L. Leserman, D. Kaiserlian, et al. 2005. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 22:643–654. [DOI] [PubMed] [Google Scholar]
- 57.Cella, M., F. Facchetti, A. Lanzavecchia, and M. Colonna. 2000. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat. Immunol. 1:305–310. [DOI] [PubMed] [Google Scholar]
- 58.Soumelis, V., and Y.J. Liu. 2006. From plasmacytoid to dendritic cell: morphological and functional switches during plasmacytoid pre-dendritic cell differentiation. Eur. J. Immunol. 36:2286–2292. [DOI] [PubMed] [Google Scholar]
- 59.Jego, G., A.K. Palucka, J.P. Blanck, C. Chalouni, V. Pascual, and J. Banchereau. 2003. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 19:225–234. [DOI] [PubMed] [Google Scholar]
- 60.Rimmelzwaan, G.F., M. Baars, E.C. Claas, and A.D. Osterhaus. 1998. Comparison of RNA hybridization, hemagglutination assay, titration of infectious virus and immunofluorescence as methods for monitoring influenza virus replication in vitro. J. Virol. Methods. 74:57–66. [DOI] [PubMed] [Google Scholar]
- 61.Lambrecht, B.N., R.A. Pauwels, and B. Fazekas De St Groth. 2000. Induction of rapid T cell activation, division, and recirculation by intratracheal injection of dendritic cells in a TCR transgenic model. J. Immunol. 164:2937–2946. [DOI] [PubMed] [Google Scholar]
- 62.Haanen, J.B., M.C. Wolkers, A.M. Kruisbeek, and T.N. Schumacher. 1999. Selective expansion of cross-reactive CD8 positive memory T cells by viral variants. J. Exp. Med. 190:1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Masurel, N., P. Ophof, and P. de Jong. 1981. Antibody response to immunization with influenza A/USSR/77 (H1N1) virus in young individuals primed or unprimed for A/New Jersey/76 (H1N1) virus. J. Hyg. (Lond.). 87:201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}