

Abstract
The “protein folding problem” consists of three closely related puzzles: (a) What is the folding code? (b) What is the folding mechanism? (c) Can we predict the native structure of a protein from its amino acid sequence? Once regarded as a grand challenge, protein folding has seen great progress in recent years. Now, foldable proteins and nonbiological polymers are being designed routinely and moving toward successful applications. The structures of small proteins are now often well predicted by computer methods. And, there is now a testable explanation for how a protein can fold so quickly: A protein solves its large global optimization problem as a series of smaller local optimization problems, growing and assembling the native structure from peptide fragments, local structures first.
Keywords: structure prediction, funnel energy landscapes, CASP, folding code, folding kinetics
INTRODUCTION
The protein folding problem is the question of how a protein’s amino acid sequence dictates its three-dimensional atomic structure. The notion of a folding “problem” first emerged around 1960, with the appearance of the first atomic-resolution protein structures. Some form of internal crystalline regularity was previously expected (117), and α-helices had been anticipated by Linus Pauling and colleagues (180, 181), but the first protein structures—of the globins—had helices that were packed together in unexpected irregular ways. Since then, the protein folding problem has come to be regarded as three different problems: (a) the folding code: the thermodynamic question of what balance of interatomic forces dictates the structure of the protein, for a given amino acid sequence; (b) protein structure prediction: the computational problem of how to predict a protein’s native structure from its amino acid sequence; and (c) the folding process: the kinetics question of what routes or pathways some proteins use to fold so quickly. We focus here only on soluble proteins and not on fibrous or membrane proteins.
THE FOLDING CODE: WHAT BALANCE OF FORCES ENCODES NATIVE STRUCTURES?
Anfinsen’s Thermodynamic Hypothesis
A major milestone in protein science was the thermodynamic hypothesis of Christian Anfinsen and colleagues (3, 92). From his now-famous experiments on ribonuclease, Anfinsen postulated that the native structure of a protein is the thermodynamically stable structure; it depends only on the amino acid sequence and on the conditions of solution, and not on the kinetic folding route. It became widely appreciated that the native structure does not depend on whether the protein was synthesized biologically on a ribosome or with the help of chaperone molecules, or if, instead, the protein was simply refolded as an isolated molecule in a test tube. [There are rare exceptions, however, such as insulin, α-lytic protease (203), and the serpins (227), in which the biologically active form is kinetically trapped.] Two powerful conclusions followed from Anfinsen’s work. First, it enabled the large research enterprise of in vitro protein folding that has come to understand native structures by experiments inside test tubes rather than inside cells. Second, the Anfinsen principle implies a sort of division of labor: Evolution can act to change an amino acid sequence, but the folding equilibrium and kinetics of a given sequence are then matters of physical chemistry.
One Dominant Driving Force or Many Small Ones?
Prior to the mid-1980s, the protein folding code was seen a sum of many different small interactions—such as hydrogen bonds, ion pairs, van der Waals attractions, and water-mediated hydrophobic interactions. A key idea was that the primary sequence encoded secondary structures, which then encoded tertiary structures (4). However, through statistical mechanical modeling, a different view emerged in the 1980s, namely, that there is a dominant component to the folding code, that it is the hydrophobic interaction, that the folding code is distributed both locally and nonlocally in the sequence, and that a protein’s secondary structure is as much a consequence of the tertiary structure as a cause of it (48, 49).
Because native proteins are only 5–10 kcal/mol more stable than their denatured states, it is clear that no type of intermolecular force can be neglected in folding and structure prediction (238). Although it remains challenging to separate in a clean and rigorous way some types of interactions from others, here are some of the main observations. Folding is not likely to be dominated by electrostatic interactions among charged side chains because most proteins have relatively few charged residues; they are concentrated in high-dielectric regions on the protein surface. Protein stabilities tend to be independent of pH (near neutral) and salt concentration, and charge mutations typically lead to small effects on structure and stability. Hydrogen-bonding interactions are important, because essentially all possible hydrogen-bonding interactions are generally satisfied in native structures. Hydrogen bonds among backbone amide and carbonyl groups are key components of all secondary structures, and studies of mutations in different solvents estimate their strengths to be around 1–4 kcal/mol (21, 72) or stronger (5, 46). Similarly, tight packing in proteins implies that van der Waals interactions are important (28).
However, the question of the folding code is whether there is a dominant factor that explains why any two proteins, for example, lysozyme and ribonuclease, have different native structures. This code must be written in the side chains, not in the backbone hydrogen bonding, because it is through the side chains that one protein differs from another. There is considerable evidence that hydrophobic interactions must play a major role in protein folding. (a) Proteins have hydrophobic cores, implying nonpolar amino acids are driven to be sequestered from water. (b) Model compound studies show 1–2 kcal/mol for transferring a hydrophobic side chain from water into oil-like media (234), and there are many of them. (c) Proteins are readily denatured in nonpolar solvents. (d) Sequences that are jumbled and retain only their correct hydrophobic and polar patterning fold to their expected native states (39, 98, 112, 118), in the absence of efforts to design packing, charges, or hydrogen bonding. Hydrophobic and polar patterning also appears to be a key to encoding of amyloid-like fibril structures (236).
What stabilizes secondary structures? Before any protein structure was known, Linus Pauling and colleagues (180, 181) inferred from hydrogen-bonding models that proteins might have α-helices. However, secondary structures are seldom stable on their own in solution. Although different amino acids have different energetic propensities to be in secondary structures (6, 41, 55, 100), there are also many “chameleon” sequences in natural proteins, which are peptide segments that can assume either helical or β conformations depending on their tertiary context (158, 162). Studies of lattice models (25, 29, 51) and tube models (11, 12, 159) have shown that secondary structures in proteins are substantially stabilized by the chain compactness, an indirect consequence of the hydrophobic force to collapse (Figure 1). Like airport security lines, helical and sheet configurations are the only regular ways to pack a linear chain (of people or monomers) into a tight space.
Figure 1.
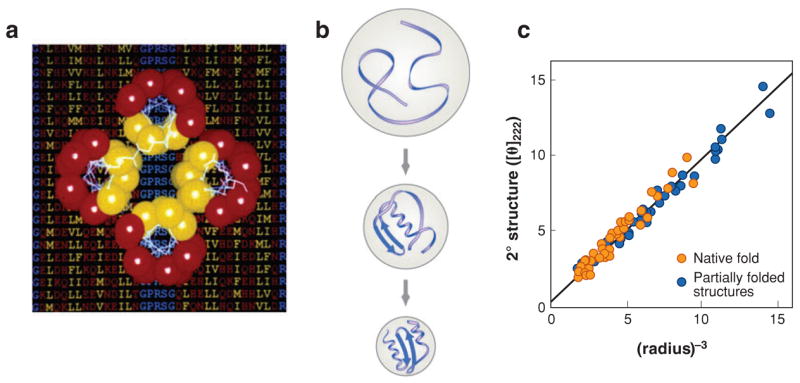
(a) Binary code. Experiments show that a primarily binary hydrophobic-polar code is sufficient to fold helix-bundle proteins (112). Reprinted from Reference 112 with permission from AAAS.
(b) Compactness stabilizes secondary structure, in proteins, from lattice models. (c) Experiments supporting panel b, showing that compactness correlates with secondary structure content in nonnative states of many different proteins (218). Reprinted from Reference 218 with permission.
Designing New Proteins and Nonbiological Foldamers
Although our knowledge of the forces of folding remains incomplete, it has not hindered the emergence of successful practical protein design. Novel proteins are now designed as variants of existing proteins (43, 94, 99, 145, 173, 243), or from broadened alphabets of nonnatural amino acids (226), or de novo (129) (Figure 2). Moreover, folding codes are used to design new polymeric materials called foldamers (76, 86, 120). Folded helix bundles have now been designed using nonbiological backbones (134). Foldamers are finding applications in biomedicine as antimicrobials (179, 185), lung surfactant replacements (235), cytomegalovirus inhibitors (62), and siRNA delivery agents (217). Hence, questions of deep principle are no longer bottlenecks to designing foldable polymers for practical applications and new materials.
Figure 2.
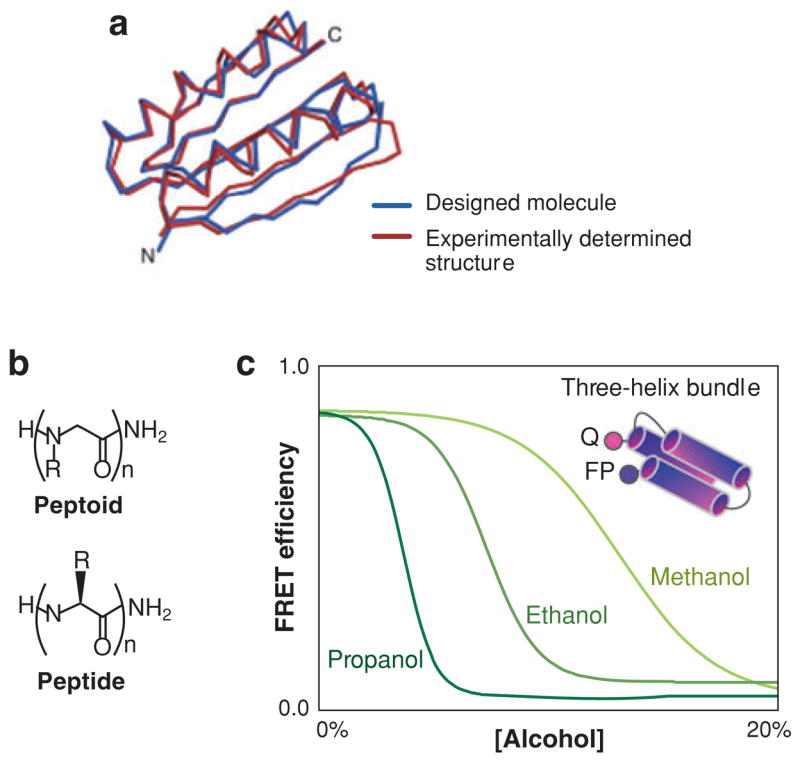
(a) A novel protein fold, called Top7, designed by Kuhlman et al. (129). Designed molecule (blue) and the experimental structure determined subsequently (red). From Reference 129; reprinted with permission from AAAS. (b) Three-helix bundle foldamers have been made using nonbiological backbones (peptoids, i.e., N-substituted glycines).
(c) Their denaturation by alcohols indicates they have hydrophobic cores characteristic of a folded molecule (134).
COMPUTATIONAL PROTEIN STRUCTURE PREDICTION IS INCREASINGLY SUCCESSFUL
A major goal of computational biology has been to predict a protein’s three-dimensional native structure from its amino acid sequence. This could help to (a) accelerate drug discovery by replacing slow, expensive structural biology experiments with faster, cheaper computer simulations, and (b) annotate protein function from genome sequences (9). With the rapid growth of experimentally determined structures available in the Protein Databank (PDB), protein structure prediction has become as much a problem of inference and machine learning as it is of protein physics.
Among the earliest uses of protein databases to infer protein structures were secondary structure prediction algorithms (33, 34, 190). In the mid-1980s, several groups began using the methods of computational physics—atomic force fields plus Monte Carlo sampling—to compute the structures of the Metenkephalin, a five-residue peptide (95, 141). The early 1990s saw significant strides in using databases and homology detection algorithms to assemble structures from homologous sequences (192) and to recognize folds by threading unknown sequences onto three-dimensional structures from a database (111). A key advance was the exploitation of evolutionary relationships among sequences through the development of robust sequence alignment methods (32, 64, 224).
CASP: A Community-Wide Experiment
In 1994, John Moult invented CASP (Critical Assessment of Techniques for Protein Structure Prediction) (165), a biennial, community-wide blind test to predict the unknown structures of proteins. Organizers identify proteins likely to be solved or whose structures have not yet been released, and predictors have roughly 3–5 weeks to predict their native structures. CASP has grown from 35 predictor groups and 24 target sequences in CASP1 in 1994 to over 200 groups and 100+ targets in CASP7 in 2006.
Over the seven CASPs, two trends are clear (164, 219). First, although much remains to be done, there has been substantial improvement in protein structure prediction. Web servers and software packages often predict the native structure of small, single-domain proteins to within about 2–6 Å of their experimental structures (8, 17, 242). In addition, fast-homology methods are computing approximate folds for whole genomes (182, 214). Figure 3 shows a quantitative assessment of performance at the first five CASP meetings. The most significant gains have occurred in the alignments of targets to homologs, the detection of evolutionarily distant homologs, and the generation of reasonable models for difficult targets that do not have templates (new folds). Since CASP5, predictions have also benefited from the use of metaservers, which solicit and establish consensus among predictions from multiple algorithms. Second, while most methods rely on both physics and bioinformatics, the most successful methods currently draw heavily from knowledge contained in native structural databases. Bioinformatics methods have benefited from the growth in size of the PDB (9, 219).
Figure 3.
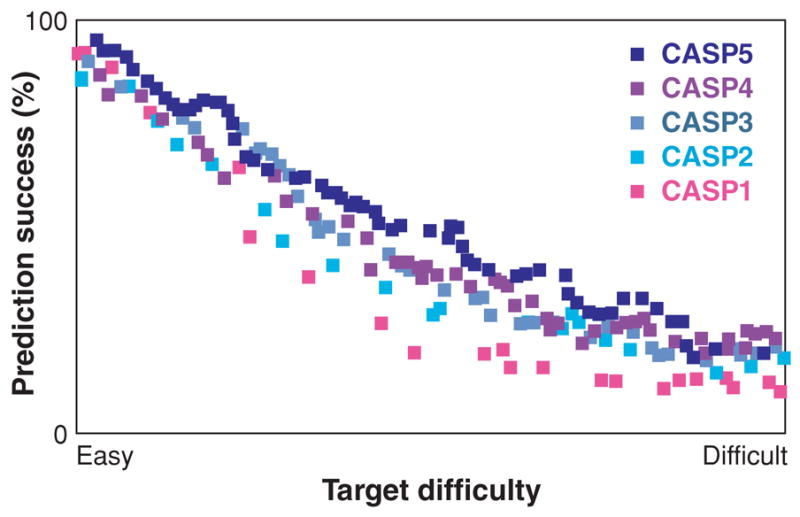
Progress in protein structure prediction in CASP1–5 (219). The y-axis contains the GDT TS score, the percentage of model residues that can be superimposed on the true native structure, averaged over four resolutions from 1 to 8 Å (100% is perfect). The x-axis is the ranked target difficulty, measured by sequence and structural similarities to proteins in the PDB at the time of the respective CASP. This shows that protein structure prediction on easy targets is quite good and is improving for targets of intermediate difficulty. Reprinted from Reference 219 with permission.
The following challenges remain (8, 164, 219): (a) to refine homology models beyond those of the best template structures; (b) to reduce errors to routinely better than 3 Å, particularly for proteins that are large, have significant β content, are new folds, or have low homology; (c) to handle large multidomain or domain-swapped proteins; (d) to address membrane proteins; and (e) to predict protein-protein interactions. Structural genomics is likely to help here (87, 222). In any case, the current successes in computer-based predictions of native protein structures are far beyond what was expected thirty years ago, when structure prediction seemed impossible.
ARE THERE MECHANISMS OF PROTEIN FOLDING?
In 1968, Cyrus Levinthal first noted the puzzle that even though they have vast conformational spaces, proteins can search and converge quickly to native states, sometimes in microseconds. How do proteins find their native states so quickly? It was postulated that if we understood the physical mechanism of protein folding, it could lead to fast computer algorithms to predict native structures from their amino acid sequences. In its description of the 125 most important unsolved problems in science, Science magazine framed the problem this way: “Can we predict how proteins will fold? Out of a near infinitude of possible ways to fold, a protein picks one in just tens of microseconds. The same task takes 30 years of computer time” (1).
The following questions of principle have driven the field: How can all the denatured molecules in a beaker find the same native structure, starting from different conformations? What conformations are not searched? Is folding hierarchical (10, 119)? Which comes first: secondary or tertiary structure (80, 239)? Does the protein collapse to compact structures before structure formation, or before the rate-limiting step (RLS), or are they concurrent (7, 89, 101, 195, 205, 213)? Are there folding nuclei (58, 152)?
Several models have emerged. In the diffusion-collision model, microdomain structures form first and then diffuse and collide to form larger structures (115, 116). The nucleation-condensation mechanism (70) proposes that a diffuse transition state ensemble (TSE) with some secondary structure nucleates tertiary contacts. Some proteins, such as helical bundles, appear to follow a hierarchical diffusion-collision model (155, 169) in which secondary structure forms and assembles in a hierarchical order. In hierarchic condensation (139), the chain searches for compact, contiguous structured units, which are then assembled into the folded state. Or, proteins may fold via the stepwise assembly of structural subunits called foldons (22, 126), or they may search for topomers, which are largely unfolded states that have native-like topologies (45, 150). These models are not mutually exclusive.
There Have Been Big Advances in Experimental and Theoretical Methods
The search for folding mechanisms has driven major advances in experimental protein science. These include fast laser temperature-jump methods (22); mutational methods that give quantities called φ-values (71, 84, 106) [now also used for ion-channel kinetics and other rate processes (42)] or ψ-values (204), which can identify those residues most important for folding speed; hydrogen exchange methods that give monomer-level information about folding events (125, 149); and the extensive exploration of protein model systems, including cytochrome c, CI2, barnase, apomyoglobin, the src, α-spectrin, and fyn SH3 domains, proteins L and G, WW domains, trpzip, and trp cage (154). In addition, peptide model experimental test systems provide insights into the fast early-folding events (14, 109, 124). Furthermore, single-molecule methods are beginning to explore the conformational heterogeneity of folding (23, 133, 166, 194).
There have been corresponding advances in theory and computation. Computer-based molecular minimization methods were first applied to protein structures in the 1960s (18, 79, 171), followed by molecular dynamics (140, 155), improved force fields (40) (reviewed in Reference 114), weighted sampling and multi-temperature methods (130, 210), highly parallelized codes for supercomputers (2, 57), and distributed grid computing methods such as Folding@home (198, 241). Models having less atomic detail also emerged to address questions more global and less detailed in nature about protein conformational spaces: (a) The Go model (82), which was intended to see if a computer could find the native structure if native guiding constraints were imposed, is now widely used to study folding kinetics of proteins having known native structures (37, 38, 113, 197, 225). (b) More physical models, typically based on polymer-like lattices, are used to study the static and dynamic properties of conformational spaces (19, 50). (c) Master-equation approaches can explore dynamics in heterogeneous systems (26, 36, 77, 138, 161, 176, 231, 232). Below, we describe some of what has been learned from these studies.
The PSB Plot: Folding Speed Correlates with the Localness of Contacts in the Native Structure
One of the few universal features of protein folding kinetics was first observed by Plaxco, Simons, and Baker (PSB), namely, that the folding speed of a protein is correlated with a topological property of its native structure (88, 184). As shown in Figure 4, Plaxco et al. found that the folding rates of two-state proteins—now known to vary more than 8 orders of magnitude—correlate with the average degree to which native contacts are local within the chain sequence: Fast-folders usually have mostly local structure, such as helices and tight turns. Slow-folders usually have more nonlocal structure, such as β-sheets (184), although there are exceptions (237). Folding rates have been subsequently found to correlate well with other native topological parameters such as the protein’s effective chain length (chain length minus the number of amino acids in helices) (107), secondary structure length (104), sequence-distant contacts per residue (90), the fraction of contacts that are sequence distant (163), the total contact distance (245), and intrinsic propensities, for example, of α-helices (131). And, there are now also methods that predict the folding rate from the sequence (91, 186). It follows that a protein typically forms smaller loops and turns faster than it forms larger loops and turns, consistent with the so-called zipping and assembly (ZA) mechanism, described below, which postulates that search speed is governed by the effective loop sizes [the effective contact order (ECO) (53, 73)] that the chain must search at any step.
Figure 4.
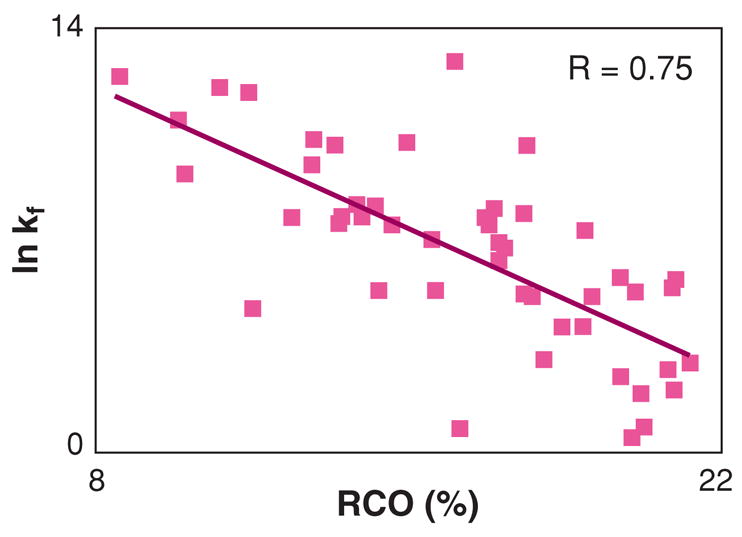
Folding rate versus relative contact order (a measure of localness of contacts in the native structure) for the 48 two-state proteins given in Reference 91, showing that proteins with the most local contacts fold faster than proteins with more nonlocal contacts.
Proteins Fold on Funnel-Shaped Energy Landscapes
Why has folding been regarded as so challenging? The issue is the astronomical number of conformations a protein must search to find its native state. Models arose in the 1980s to study the nature of the conformational space (19, 47), i.e., the shape of the energy landscape, which is the mathematical function F(φ, ϕ, X) that describes the intramolecular-plus-solvation free energy of a given protein as a function of the microscopic degrees of freedom. A central goal has been to quantify the statistical mechanical partition function, a key component of which is the density of states (DOS), i.e., the number of conformations at each energy level. In simple cases, the logarithm of the DOS is the conformational entropy. Such entropies have not been determinable through all-atom modeling, because that would require astronomical amounts of computational sampling (although replica-exchange methods can now do this for very small peptides). Hence understanding a protein’s DOS and its entropies has required simplified models, such as mean-field polymer and lattice treatments (51), spin-glass theories (19, 47), or exact enumerations in minimalist models (132).
A key conclusion is that proteins have funnel-shaped energy landscapes, i.e., many high-energy states and few low-energy states (19, 49, 50, 52, 138). What do we learn from the funnel idea? Funnels have both qualitative and quantitative uses. First, cartoonizations of funnels (Figure 5) provide a useful shorthand language for communicating the statistical mechanical properties and folding kinetics of proteins. Figure 5 illustrates fast folding (simple funnel), kinetic trapping (moats or wells), and slow random searching (golf course). These pictures show a key distinction between protein folding and simple classical chemical reactions. A simple chemical reaction proceeds from its reactant A to its product B, through a pathway, i.e., a sequence of individual structures. A protein cannot do this because its reactant, the denatured state, is not a single microscopic structure. Folding is a transition from disorder to order, not from one structure to another. Simple one-dimensional reaction path diagrams do not capture this tremendous reduction in conformational degeneracy.
Figure 5.
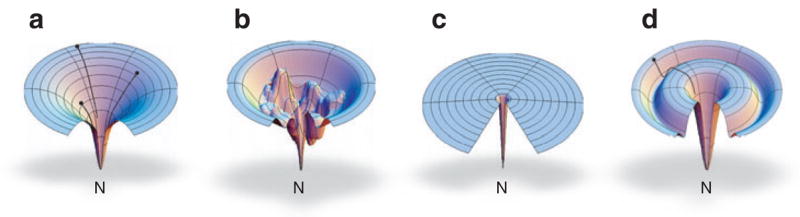
One type of energy landscape cartoon: the free energy F(φ, ϕ, κ) of the bond degrees of freedom. These pictures give a sort of simplified schematic diagram, useful for illustrating a protein’s partition function and density of states. (a) A smooth energy landscape for a fast folder, (b) a rugged energy landscape with kinetic traps, (c) a golf course energy landscape in which folding is dominated by diffusional conformational search, and (d) a moat landscape, where folding must pass through an obligatory intermediate.
A funnel describes a protein’s conformational heterogeneity
Conformational heterogeneity has been found in the few experiments that have been designed to look for it (16, 143, 157, 206, 209, 244). For example, using time-resolved FRET with four different intramolecular distances, Sridevi et al. (206) found in Barstar that (a) that the chain entropy increases as structures become less stable, (b) that there are multiple folding routes, and (c) that different routes dominate under different folding conditions. Moreover, changing the denaturant can change the dominant pathway, implying heterogeneous kinetics (143). Figure 6 shows the funnel landscape that has been determined by extensive mutational analysis of the seven ankyrin sequence repeats of the Notch ankyrin repeat domain (16, 157, 209).
Figure 6.
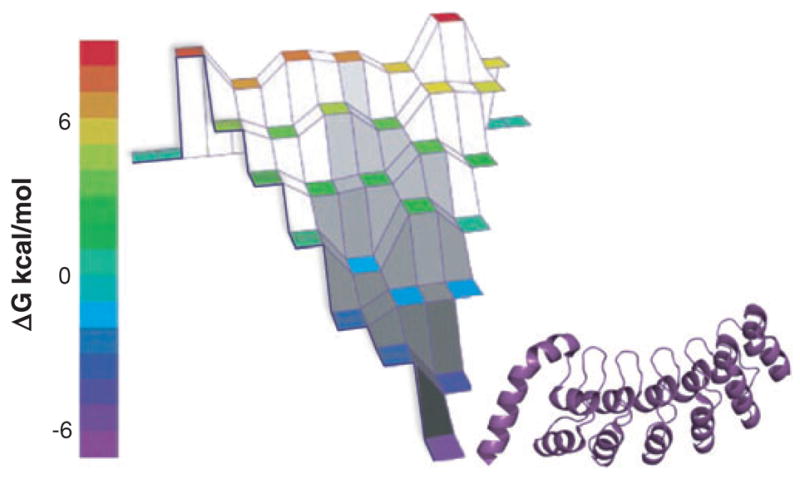
The experimentally determined energy landscape of the seven ankyrin repeats of the Notch receptor (16, 157, 209). The energy landscape is constructed by measuring the stabilities of folded fragments for a series of overlapping modular repeats. Each horizontal tier presents the partially folded fragments with the same number of repeats. Reprinted from Reference 157 with permission.
A funnel describes a protein’s chain entropy
The funnel idea first arose to explain denaturation, the balance between the chain entropy and the forces of folding (48). Proteins denature at high temperatures because there are many states of high energy and fewer states of low energy, that is, the landscape is funneled. For cold unfolding, the shape of the funnel changes with temperature because of free-energy changes of the aqueous solvent. When you can accurately compute a protein’s DOS, you can predict the protein’s free energy of folding and its denaturation and cooperativity properties. Figure 7 shows an example in which the DOS (set onto its side to illustrate the funnel) was found by extensive lattice enumeration for F13W*, a three-helix bundle, with predictions compared to experiments (146).
Figure 7.
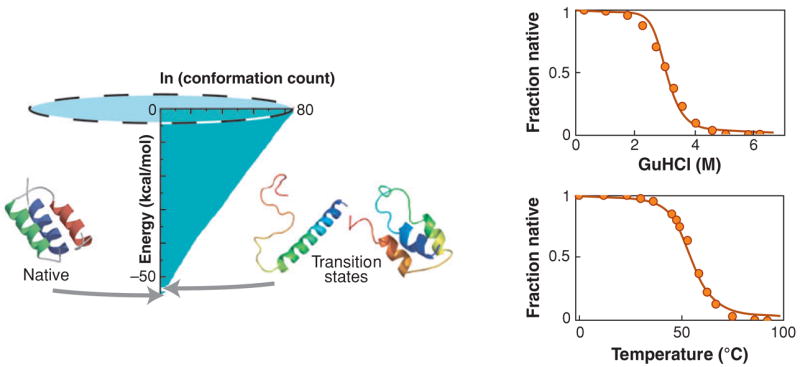
(Left) The density of states (DOS) cartoonized as an energy landscape for the three-helix bundle protein F13W*: DOS (x-axis) versus the energy (y-axis). (Right) Denaturation predictions versus experiments (146). The peak free energy (here, where the DOS is minimum), typically taken to be the transition state, is energetically very close to native.
Funnels provide a microscopic framework for folding kinetics
Folding kinetics is traditionally described by simple mass action models, such as D →I →N (on-path intermediate I between the denatured state D and native state N) or X →D →N (off-path intermediate X), where the symbol I or X represents macrostates that are invoked for the purpose of curve-fitting experimental kinetics data. In contrast, funnel models or master-equation models aim to explain the kinetics in terms of underlying physical forces. They aim to predict the microstate composition of those macrostates, for example. The states in master-equation models differ from those in mass-action models insofar as the former are more numerous, more microscopic, are defined by structural or energetic criteria, and are arranged kinetically to reflect the underlying funnel-like organization of the dynamical flows.
For example, Figure 8b shows a master-equation model for the folding of SH3, illustrating the apparently paradoxical result that folding can be serial and parallel at the same time. The protein has multiple routes available. However, one of the dominant series pathways is U →B →BD →BDE →BCDE →N. BD is the TSE because it is the dynamically least populated state. B precedes BD diagrammatically in series in this pathway. Yet, the probability bucket labeled B does not first fill up and then empty into BD; rather the levels in both buckets, B and BD, rise and fall together and hence are dynamically in parallel. Such series and parallel steps are also seen in computer simulations of CI2 (189), for example. Sometimes a chain contact A forms before another contact B in nearly all the simulation trajectories (series-like). But another contact C may form before B in some trajectories and after B in others (parallel-like). Some folding is sequential, as in Fyn SH3 (123), cytochrome (66), T4 lysozyme (24), and Im7 (74), and some folding is parallel, as in cytochrome C (83) and HEW lyzosyme (151).
Figure 8.
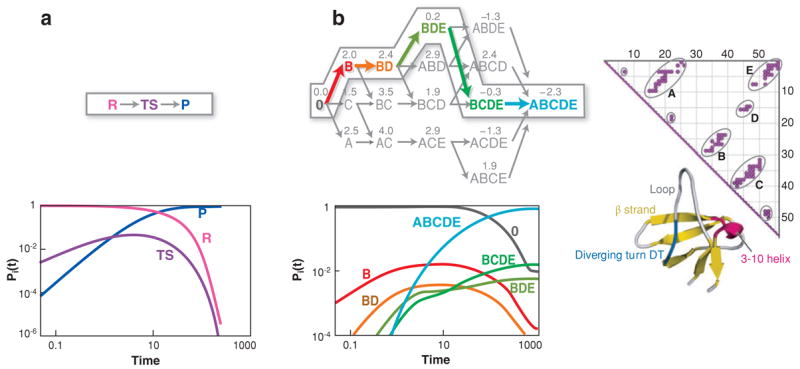
(a) A simple single-pathway system. R, reactant; TS, transition state; P, product. (b) Pathway diagram of SH3 folding from a master-equation model. The native protein has five contact clusters: A = RT loop; B = β2β3; C = β3β4; D = RT loop-β4; and E = β1β5. Combined letters, such as BD, mean that multiple contact clusters have formed. Funneling occurs toward the right, because the symbols on the left indicate large ensembles, whereas the symbols on the right are smaller ensembles. The numbers indicate free energies relative to the denatured state. The arrows between the states are colored to indicate transition times between states. The slowest steps are in red; the fastest steps in green. BD is the transition state ensemble because it is the highest free energy along the dominant route. While B and BD would seem to be obligatorily in series, the time evolutions of these states show that they actually rise and fall in parallel (232).
Or, consider the traditional idea that a reaction’s RLS coincides with the point of the highest free energy, ΔG‡, along the reaction coordinate. As a matter of principle, the RLS need not necessarily coincide with the free-energy barrier. There is evidence that they may not coincide for some protein folding processes (13, 15, 27). What’s the distinction? To find the RLS, you need a dynamical model. You would find the eigenvector corresponding to the slowest eigenvalue (in two-state kinetics). Microscopic master-equation modeling typically finds that this eigenvector only identifies the process U →N, with no finer pinpointing of any special structures along the way. In contrast, ΔG‡ is a thermodynamic quantity—the maximum free energy along the fastest route, which usually does correspond to some specific ensemble of structures. Below, we describe why these matters of principle are important.
How do we convert folding experiments into insights about molecular behavior?
To interpret data, we must use models. φ-value experiments aim to identify RLSs in folding. But how we understand the molecular events causing a given φ-value depends on whether we interpret it by funnels or pathways. A φ-value measures how a folding rate changes when a protein is mutated (42, 67, 71, 105, 144, 153, 154, 176, 178, 193) (see Reference 231 and references 1–24 therein). φ equals the change in the logarithm of the folding rate caused by the mutation, divided by the change in the logarithm of the folding equilibrium constant. If we then seek a structural interpretation of φ, we need a model. Using the Bronsted-Hammond pathway model of chemical reactions, φ is often assumed to indicate the position of the TSE along the folding reaction coordinate: φ = 0 means the mutation site is denatured in the TSE; φ = 1 means the mutation site is native in the TSE. In this pathway view, φ can never lie outside the range from 0 to 1; in the funnel view, φ is not physically restricted to this range. For example, φ < 0 or φ > 1 has been predicted for mutations that stabilize a helix but that destabilize the bundle’s tertiary structure (231). Unfortunately, experiments are not yet definitive. While some φ-values are indeed observed to be negative or greater than 1 (44, 85, 176, 193), those values might be experimental artifacts (193). Other challenges in interpreting φ have also been noted (65, 188). To resolve the ambiguities in interpreting φ, we need to deepen our understanding beyond the single-reaction-coordinate idea.
How do we convert computer simulations into insights about molecular behavior?
Similarly, insights about folding events are often sought from computer modeling. It is much easier to calculate structural or energetic quantities than kinetic quantities. For example, some modeling efforts compute φ-values by assuming some particular structure for the TSE (78, 233) or some particular reaction coordinate, such as the RMSD to native structure, radius of gyration, number of hydrogen bonds, or number of native contacts (196). Alternatively, a quantity called pfold (56), which defines a separatrix (a sort of continental divide between folded and unfolded states), is sometimes computed. Although pfold predicts well the RLSs for simple landscapes (147), it can give less insight into protein landscapes having multiple barriers or other complexity (30). To go beyond classical assumptions, there has been an extensive and growing effort to use master-equation approaches (13, 26, 31, 36, 60, 61, 77, 110, 161, 172, 176, 201, 202, 208, 211, 212, 231, 232) to explore underlying assumptions about reaction coordinates, pathways, transition states, and RLSs.
Funnel models can explain some non-canonical behaviors in ultrafast folding
More than a dozen proteins fold in microseconds (128). Some fold in hundreds of nanoseconds (127, 237). Is there a state of protein folding that is so fast that there is no free-energy barrier at all (156)? This has sometimes been called downhill folding (93, 122, 128, 187). There is currently an intensive search for downhill folders—and much controversy about whether or not such folding has yet been observed, mainly in BBL, a 40-residue helical protein. That controversy hinges on questions of experimental analysis (68, 69, 75, 103, 168, 170, 191): establishing proper baselines and ionization states to find denaturation temperatures and to determine whether the equilibrium is two-state, for example, which would imply a barrier between D and N.
Remarkably, all known ultrafast-folders have anti-Arrhenius thermal kinetics. That is, heating those proteins at high temperatures slows down folding, the opposite of what is expected from traditional activation barriers. Here too, any molecular interpretation requires a model, and the common expectation is based on the classical Arrhenius/Eyring pathway model. Is the Arrhenius model sufficient for funnels involving many fast processes? Ultrafast folding kinetics has recently been explored in various models (59, 77, 122, 148, 167). One funnel model (77) explains that the reason why increasing the temperature leads to slower folding is because of thermal unfolding of the denatured chain, leading to a larger conformational space that must be searched for the chain to find route to native downhill. It predicts that the ultimate speed limit to protein folding, at temperatures that will disappear all other barriers, is the conformational search through the denatured basin. Near the speed limit of protein folding, the heterogeneity and searching that are intrinsic to funnels can be an important component of the folding physics. That model also explains that helical proteins fold faster than β-sheets, on average, because helices have more parallel microscopic folding routes (because a helix can nucleate at many different points along the chain).
The Zipping and Assembly Hypothesis for the Folding Routes
Protein folding is a stochastic process: One protein molecule in a beaker follows a different microscopic trajectory than another molecule because of thermal fluctuations. Hence, protein folding is often studied using Monte Carlo or molecular dynamics sampling. However, computations seeking the native state using purely physical models are prohibitively expensive, because this is a challenging needle-in-a-haystack global optimization problem (96, 132, 216). Since the beginnings of experimental folding kinetics, there has been the view that the Levinthal paradox—of how a protein searches its conformational space so quickly—might be explained by a folding mechanism, i.e., by some higher-level description (beyond the statement that it is stochastic) that clarifies how the protein decides which structures to form and avoids searching vast stretches of the conformational space in the first place.
Zipping and assembly (ZA) is a hypothesis for a general folding mechanism. On fast timescales, small fragments of the chain can search their conformations more completely than larger fragments can (53, 73). There are certain problems of global optimization—including the ZA mechanism of protein folding and the Cocke-Kasami-Younger method for parsing sentences (54, 102)—in which the globally optimal solution (native structure, in this case) can almost always be found (although not guaranteed) by a divide-and-conquer strategy, a fast process of cobbling together smaller locally optimal decisions. Accordingly, in the earliest time steps after folding is initiated (picoseconds to nanoseconds), each of the different peptide fragments of the chain searches for small local metastable structures, such as helical turns, β-turns, or small loops. Each peptide segment searches its own conformations, at the same time that other segments are searching. Not stable on their own, a few of those local structures are sufficiently metastable to survive to the next longer timescale, where they grow (or zip) into increasingly larger and more stable structures. On still longer timescales, pairs or groups of these substructures can assemble into structures that are still larger and more native-like, and metastability gives way to stability (97, 102, 103, 199, 215, 223, 228–230, 232).
The ZA mechanism shares much in common with other mechanisms, such as diffusion-collision, hierarchical, and foldon models. The last two mechanisms, however, are descriptors of experiments. They do not prescribe how to compute a protein’s folding route from its amino acid sequence. In contrast, the ZA mechanism is such a microscopic recipe, starting from the amino acid sequence and specifying a time series of ensembles of conformations the chain searches at each stage of folding. ZA is a funnel process: There are many parallel microscopic routes at the beginning, and fewer and more sequential routes at the end. The ZA mechanism provides a plausible answer to Levinthal’s paradox of what vast stretches of conformational space the protein never bothers to search. For any compact native polymer structure, there are always routes to the native state that take only small-conformational-entropy-loss steps. ZA otherwise explores very little of conformational space. These few routes constitute the dominant folding processes in the ZA mechanism. One test of this mechanism is the prediction of the change of folding routes (229), measured by the change in φ-value distributions (143), upon circular permutation of the chain. Proteins can be circularly permuted if the chain termini are adjacent to each other in the wild-type native structure. In such cases, the ends are covalently linked and the chain is broken elsewhere. This alters the native topology (contact map) dramatically, and sometimes the folding routes, but does not appear to substantially change the native structure (20, 142, 143, 160, 221).
PHYSICS-BASED MODELING OF FOLDING AND STRUCTURE PREDICTION
Computer simulations of purely physics-based models are becoming useful for structure prediction and for studying folding routes. Here the metric of success is not purely performance in native structure prediction; it is to gain a deeper understanding of the forces and dynamics that govern protein properties. When purely physical methods are successful, it will allow us to go beyond bioinformatics to (a) predict conformational changes, such as induced fit, important for computational drug discovery; (b) understand protein mechanisms of action, motions, folding processes, enzymatic catalysis, and other situations that require more than just the static native structure; (c) understand how proteins respond to solvents, pH, salts, denaturants, and other factors; and (d) design synthetic proteins having noncanonical amino acids or foldameric polymers with nonbiological backbones.
A key issue has been whether semiempirical atomic physical force fields are good enough to fold up a protein in a computer. Physics-based methods are currently limited by large computational requirements owing to the formidable conformational search problem and, to a lesser extent, by weaknesses in force fields. Nevertheless, there have been notable successes in the past decade enabled by the development of large supercomputer resources and distributed computing systems. The first milestone was a supercomputer simulation by Duan and Kollman in 1998 of the folding of the 36-residue villin headpiece in explicit solvent, for nearly a microsecond of computed time, reaching a collapsed state 4.5 Å from the NMR structure (57). Another milestone was the development by Pande and colleagues of Folding@home, a distributed grid computing system running on the screensavers of volunteer computers worldwide. Pande and colleagues (241) have studied the folding kinetics of villin. High-resolution structures of villin have recently been reached by Pande and colleagues (110) and Duan and colleagues (136, 137). In addition, three groups have folded the 20-residue Trp-cage peptide to ~1 Å: Simmerling et al. (200), the IBM Blue Gene group of Pitera and Swope (183), and Duan and colleagues (35). Recently, Lei & Duan (135) folded the albumin-binding domain, a 47-residue, three-helix bundle, to 2.0 Å. Physics-based approaches are also folding small helices and β-hairpin peptides of up to ~20 residues that have stable secondary structures (63, 81, 108, 240, 246; M.S. Shell, R. Ritterson & K.A. Dill, unpublished data). Physical potential models have also been sampled using non-Boltzmann stochastic and deterministic optimization strategies (121, 174, 207, 220).
Here are some of the key conclusions. First, a powerful way to sample conformations and obtain proper Boltzmann averages is replica exchange molecular dynamics (REMD) (210). Second, although force fields are good, they need improvements in backbone torsional energies to address the balance between helical and extended conformations (81, 108, 240), and in implicit solvation, which dramatically reduces the expense relative to explicit water simulations but which frequently overstabilizes ion-pairing interactions, in turn destabilizing native structures (63, 246).
Can modern force fields with Boltzmann sampling predict larger native structures? Recent work indicates that, when combined with a conformational search technique based on the ZA folding mechanism, purely physics-based methods can arrive at structures close to the native state for chains up to ~100 monomers (177; M.S. Shell, S.B. Ozkan, V.A. Voelz, G.H.A. Wu & K.A. Dill, unpublished data). The approach, called ZAM (zipping and assembly method), uses replica exchange and the AMBER96 force field and works by (a) breaking the full protein chain into small fragments (initially 8-mers), which are simulated separately using REMD; (b) then growing or zipping the fragments having metastable structures by adding a few new residues or assembling two such fragments together, with further REMD and iterations; and (c) locking in place any stable residue-residue contacts with a harmonic spring, enforcing emerging putative physical folding routes, without the need to sample huge numbers of degrees of freedom at a time.
ZAM was tested through the folding of eight of nine small proteins from the PDB to within 2.5 Å, using a 70-processor cluster over 6 months (177), giving good agreement with the φ-values known for four of them. Figure 9 shows the ZAM folding process for one of these proteins, and Figure 10 shows the predicted versus experimental structures for all nine. In a more stringent test, ZAM was applied in CASP7 to the folding of six small proteins from 76 to 112 residues (M.S. Shell, S.B. Ozkan, V.A. Voelz, G.H.A. Wu & K.A. Dill, unpublished data). Of the four proteins attempted in CASP7 that were not domain-swapped, ZAM predicted roughly correct tertiary structures, segments of more than 40 residues with an average RMSD of 5.9 angstroms, and secondary structures with 73% accuracy. From these studies it has been concluded that ZA routes can identify limited-sampling routes to the native state from unfolded states, directed by all-atom force fields, and that the AMBER96 plus a generalized Born implicit solvent model is a reasonable scoring function. Fragments that adopt incorrect secondary structures early in the simulations are frequently corrected in later-stage folding because the emerging tertiary structure of the protein often will not tolerate them.
Figure 9.
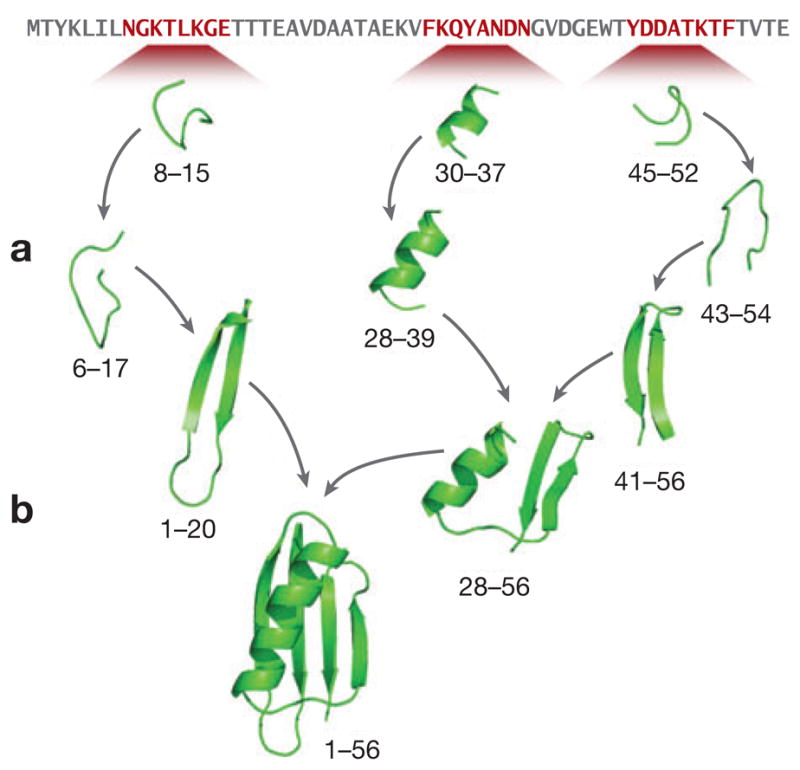
The folding routes found in the ZAM conformational search process for protein G, from the work described in Reference 177. The chain is first parsed into many short, overlapping fragments. After sampling by replica exchange molecular dynamics, stable hydrophobic contacts are identified and restrained. Fragments are then either (a) grown or zipped though iterations of adding new residues, sampling, and contact detection, or (b) assembled together pairwise using rigid body alignment followed by further sampling until a completed structure is reached.
Figure 10.
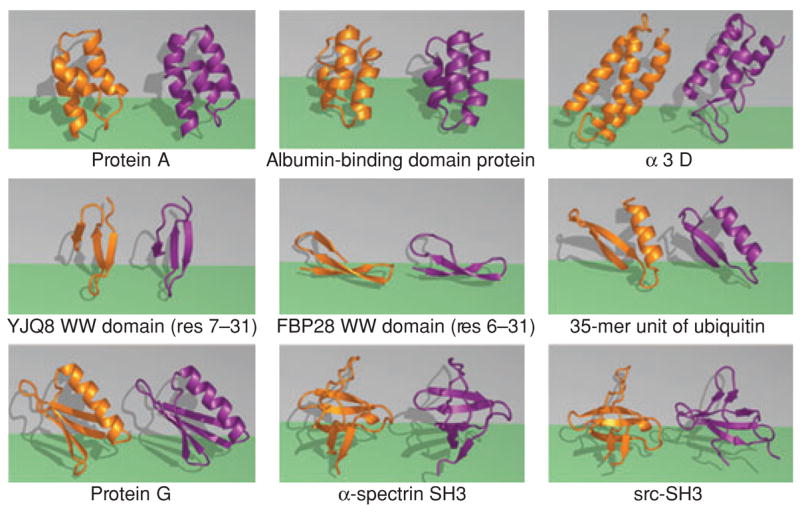
Ribbon diagrams of the predicted protein structures using the ZAM search algorithm (purple) versus experimental PDB structures (orange). The backbone C-RMSDs with respect to PDB structures are protein A (1.9 Å), albumin domain binding protein (2.4 Å), alpha-3D [2.85 Å (excluding loop residues) or 4.6 Å], FBP26 WW domain (2.2 Å), YJQ8 WW domain (2.0 Å), 1–35 residue fragment of Ubiquitin (2.0 Å), protein G (1.6 Å), and -spectrin SH3 (2.2 Å). ZAM fails to find the src-SH3 structure: Shown is a conformation that is 6 Å from the experimental structure. The problem in this case appears to be overstabilization of nonnative ion pairs in the GB/SA implicit solvation model.
SUMMARY
The protein folding problem has seen enormous advances over the last fifty years. New experimental techniques have arisen, including hydrogen exchange, φ-value methods that probe mutational effects on folding rates, single-molecule methods that can explore heterogeneity of folding and energy landscapes, and fast temperature-jump methods. New theoretical and computational approaches have emerged, including methods of bioinformatics, multiple-sequence alignments, structure-prediction Web servers, physics-based force fields of good accuracy, physical models of energy landscapes, fast methods of conformational sampling and searching, master-equation methods to explore the physical mechanisms of folding, parallel and distributed grid-based computing, and the CASP community-wide event for protein structure prediction.
Protein folding no longer appears to be the insurmountable grand challenge that it once appeared to be. Current knowledge of folding codes is sufficient to guide the successful designs of new proteins and foldameric materials. For the once seemingly intractable Levinthal puzzle, there is now a viable hypothesis: A protein can fold quickly and solve its big global optimization puzzle by piecewise solutions of smaller component puzzles. Other matters of principle are now yielding to theory and physics-based modeling. And current computer algorithms are now predicting native structures of small proteins remarkably accurately, promising growing value in drug discovery and proteomics.
SUMMARY POINTS
The protein folding code is mainly embodied in side chain solvation interactions. Novel protein folds and nonbiological foldamers are now being successfully designed and are moving toward practical applications.
Thanks to CASP, the growing PDB, and fast-homology and sequence alignment methods, computer methods now can often predict correct native structures of small proteins.
The protein folding problem has both driven—and benefited from—big advances in experimental and theoretical/computational methods.
Proteins fold on funnel-shaped energy landscapes, which describe the conformational heterogeneity among the nonnative states. This heterogeneity is key to the entropy that opposes folding and thus to folding equilibria. This heterogeneity is also important for understanding folding kinetics at the level of the individual chain processes.
A protein can fold quickly to its native structure by ZA, making independent local decisions first and then combining those substructures. In this way, a protein can avoid searching most of its conformational space. ZA appears to be a useful search method for computational modeling.
Acknowledgments
For very helpful comments and insights, both on this review and through ongoing discussions over the years, we are deeply grateful to D. Wayne Bolen, Hue Sun Chan, John Chodera, Yong Duan, Walter Englander, Frank Noe, José Onuchic, Vijay Pande, Jed Pitera, Kevin Plaxco, Adrian Roitberg, George Rose, Tobin Sosnick, Bill Swope, Dave Thirumalai, Vince Voelz, Peter G Wolynes, and Huan-Xiang Zhou. We owe particular thanks and appreciation to Buzz Baldwin, to whom this volume of the Annual Review of Biophysics is dedicated, not only for his interest and engagement with us on matters of protein folding over the many years, but also for his pioneering and founding leadership of the whole field. We appreciate the support from NIH grant GM 34993, the Air Force, and the Sandler Foundation.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any biases that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.So much more to know. . . Science. 2005;309:78–102. doi: 10.1126/science.309.5731.78b. [DOI] [PubMed] [Google Scholar]
- 2.Allen F, Coteus P, Crumley P, Curioni A, Denneau M, et al. Blue gene: a vision for protein science using a petaflop supercomputer. IBM Syst J. 2001;40:310–27. [Google Scholar]
- 3.Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–30. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 4.Anfinsen CB, Scheraga HA. Experimental and theoretical aspects of protein folding. Adv Protein Chem. 1975;29:205–300. doi: 10.1016/s0065-3233(08)60413-1. [DOI] [PubMed] [Google Scholar]
- 5.Auton M, Holthauzen LM, Bolen DW. Anatomy of energetic changes accompanying urea-induced protein denaturation. Proc Natl Acad Sci USA. 2007;104:15317–22. doi: 10.1073/pnas.0706251104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Avbelj F, Baldwin RL. Role of backbone solvation in determining thermodynamic β propensities of the amino acids. Proc Natl Acad Sci USA. 2002;99:1309–13. doi: 10.1073/pnas.032665499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bachmann A, Kiefhaber T. Apparent two-state tendamistat folding is a sequential process along a defined route. J Mol Biol. 2001;306:375–86. doi: 10.1006/jmbi.2000.4399. [DOI] [PubMed] [Google Scholar]
- 8.Baker D. Prediction and design of macromolecular structures and interactions. Philos Trans R Soc B Biol Sci. 2006;361:459–63. doi: 10.1098/rstb.2005.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker D, Sali A. Protein structure prediction and structural genomics. Science. 2001;294:93–96. doi: 10.1126/science.1065659. [DOI] [PubMed] [Google Scholar]
- 10.Baldwin RL, Rose GD. Is protein folding hierarchic? I. Local structure and peptide folding. Trends Biochem Sci. 1999;24:26–33. doi: 10.1016/s0968-0004(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 11.Banavar JR, Maritan A. Physics of proteins. Annu Rev Biophys Biomol Struct. 2007;36:261–80. doi: 10.1146/annurev.biophys.36.040306.132808. [DOI] [PubMed] [Google Scholar]
- 12.Banavar JR, Maritan A, Micheletti C, Trovato A. Geometry and physics of proteins. Proteins. 2002;47:315–22. doi: 10.1002/prot.10091. [DOI] [PubMed] [Google Scholar]
- 13.Best RB, Hummer G. Reaction coordinates and rates from transition paths. Proc Natl Acad Sci USA. 2005;102:6732–37. doi: 10.1073/pnas.0408098102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bieri O, Wirz J, Hellrung B, Schutkowski M, Drewello M, Kiefhaber T. The speed limit for protein folding measured by triplet-triplet energy transfer. Proc Natl Acad Sci USA. 1999;96:9597–601. doi: 10.1073/pnas.96.17.9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolhuis PG. Kinetic pathways of β-hairpin (un)folding in explicit solvent. Biophys J. 2005;88:50–61. doi: 10.1529/biophysj.104.048744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bradley CM, Barrick D. The Notch ankyrin domain folds via a discrete, centralized pathway. Structure. 2006;14:1303–12. doi: 10.1016/j.str.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 17.Bradley P, Misura KMS, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–71. doi: 10.1126/science.1113801. [DOI] [PubMed] [Google Scholar]
- 18.Brant DA, Flory PJ. The role of dipole interactions in determining polypeptide configurations. J Am Chem Soc. 1965;87:663–64. [Google Scholar]
- 19.Bryngelson JD, Wolynes PG. Spin glasses and the statistical mechanics of protein folding. Proc Natl Acad Sci USA. 1987;84:7524–28. doi: 10.1073/pnas.84.21.7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bulaj G, Koehn RE, Goldenberg DP. Alteration of the disulfide-coupled folding pathway of BPTI by circular permutation. Protein Sci. 2004;13:1182–96. doi: 10.1110/ps.03563704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrne MP, Manuel RL, Lowe LG, Stites WE. Energetic contribution of side chain hydrogen bonding to the stability of staphylococcal nuclease. Biochemistry. 1995;34:13949–60. doi: 10.1021/bi00042a029. [DOI] [PubMed] [Google Scholar]
- 22.Callender RH, Dyer RB, Gilmanshin R, Woodruff WH. Fast events in protein folding: the time evolution of primary processes. Annu Rev Phys Chem. 1998;49:173–202. doi: 10.1146/annurev.physchem.49.1.173. [DOI] [PubMed] [Google Scholar]
- 23.Cecconi C, Shank EA, Bustamante C, Marqusee S. Direct observation of the three-state folding of a single protein molecule. Science. 2005;309:2057–60. doi: 10.1126/science.1116702. [DOI] [PubMed] [Google Scholar]
- 24.Cellitti J, Bernstein R, Marqusee S. Exploring subdomain cooperativity in T4 lysozyme. II. Uncovering the C-terminal subdomain as a hidden intermediate in the kinetic folding pathway. Protein Sci. 2007;16:852–62. doi: 10.1110/ps.062632807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan HS, Dill KA. Origins of structure in globular proteins. Proc Natl Acad Sci USA. 1990;87:6388–92. doi: 10.1073/pnas.87.16.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chan HS, Dill KA. Transition states and folding dynamics of proteins and heteropolymers. J Chem Phys. 1994;100:9238–57. [Google Scholar]
- 27.Chekmarev SF, Krivov SV, Karplus M. Folding time distributions as an approach to protein folding kinetics. J Phys Chem B. 2005;109:5312–30. doi: 10.1021/jp047012h. [DOI] [PubMed] [Google Scholar]
- 28.Chen J, Stites WE. Packing is a key selection factor in the evolution of protein hydrophobic cores. Biochemistry. 2001;40:15280–89. doi: 10.1021/bi011776v. [DOI] [PubMed] [Google Scholar]
- 29.Chikenji G, Fujitsuka Y, Takada S. Shaping up the protein folding funnel by local interaction: lesson from a structure prediction study. Proc Natl Acad Sci USA. 2006;103:3141–46. doi: 10.1073/pnas.0508195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho SS, Levy Y, Wolynes PG. P versus Q: structural reaction coordinates capture protein folding on smooth landscapes. Proc Natl Acad Sci USA. 2006;103:586–91. doi: 10.1073/pnas.0509768103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chodera JD, Singhal N, Pande VS, Dill KA, Swope WC. Automatic discovery of metastable states for the construction of Markov models of macromolecular conformational dynamics. J Chem Phys. 2007;126:155101. doi: 10.1063/1.2714538. [DOI] [PubMed] [Google Scholar]
- 32.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–26. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry. 1974;13:222–45. doi: 10.1021/bi00699a002. [DOI] [PubMed] [Google Scholar]
- 34.Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem. 1978;47:251–76. doi: 10.1146/annurev.bi.47.070178.001343. [DOI] [PubMed] [Google Scholar]
- 35.Chowdhury S, Lee MC, Xiong G, Duan Y. Ab initio folding simulation of the Trp-cage mini-protein approaches NMR resolution. J Mol Biol. 2003;327:711–17. doi: 10.1016/s0022-2836(03)00177-3. [DOI] [PubMed] [Google Scholar]
- 36.Cieplak M, Henkel M, Karbowski J, Banavar JR. Master equation approach to protein folding and kinetic traps. Phys Rev Lett. 1998;80:3654–57. [Google Scholar]
- 37.Cieplak M, Xuan Hoang T. Scaling of folding properties in Go models of proteins. J Biol Phys. 2000;26:273–94. doi: 10.1023/A:1010359024559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: What determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J Mol Biol. 2000;298:937–53. doi: 10.1006/jmbi.2000.3693. [DOI] [PubMed] [Google Scholar]
- 39.Cordes MHJ, Davidsont AR, Sauer RT. Sequence space, folding and protein design. Curr Opin Struct Biol. 1996;6:3–10. doi: 10.1016/s0959-440x(96)80088-1. [DOI] [PubMed] [Google Scholar]
- 40.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, et al. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J Am Chem Soc. 1995;117:5179–97. [Google Scholar]
- 41.Creamer TP, Rose GD. A-helix-forming propensities in peptides and proteins. Proteins. 1994;19:85–97. doi: 10.1002/prot.340190202. [DOI] [PubMed] [Google Scholar]
- 42.Cymes GD, Grosman C, Auerbach A. Structure of the transition state of gating in the acetylcholine receptor channel pore: a φ-value analysis. Biochemistry. 2002;41:5548–55. doi: 10.1021/bi011864f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 44.de Los Rios MA, Daneshi M, Plaxco KW. Experimental investigation of the frequency and substitution dependence of negative φ-values in two-state proteins. Biochemistry. 2005;44:12160–67. doi: 10.1021/bi0505621. [DOI] [PubMed] [Google Scholar]
- 45.Debe DA, Carlson MJ, Goddard WA. The topomer-sampling model of protein folding. Proc Natl Acad Sci USA. 1999;96:2596–601. doi: 10.1073/pnas.96.6.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deechongkit S, Dawson PE, Kelly JW. Toward assessing the position-dependent contributions of backbone hydrogen bonding to β-sheet folding thermodynamics employing amide-to-ester perturbations. J Am Chem Soc. 2004;126:16762–71. doi: 10.1021/ja045934s. [DOI] [PubMed] [Google Scholar]
- 47.Dill KA. Theory for the folding and stability of globular proteins. Biochemistry. 1985;24:1501–9. doi: 10.1021/bi00327a032. [DOI] [PubMed] [Google Scholar]
- 48.Dill KA. Dominant forces in protein folding. Biochemistry. 1990;29:7133–55. doi: 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- 49.Dill KA. Polymer principles and protein folding. Protein Sci. 1999;8:1166–80. doi: 10.1110/ps.8.6.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dill KA, Alonso DOV, Hutchinson K. Thermal stabilities of globular proteins. Biochemistry. 1989;28:5439–49. doi: 10.1021/bi00439a019. [DOI] [PubMed] [Google Scholar]
- 51.Dill KA, Bromberg S, Yue KZ, Fiebig KM, Yee DP, et al. Principles of protein folding: a perspective from simple exact models. Protein Sci. 1995;4:561–602. doi: 10.1002/pro.5560040401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dill KA, Chan HS. From Levinthal to pathways to funnels. Nat Struct Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 53.Dill KA, Fiebig KM, Chan HS. Cooperativity in protein-folding kinetics. Proc Natl Acad Sci USA. 1993;90:1942–46. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dill KA, Lucas A, Hockenmaier J, Huang L, Chiang D, Joshi AK. Computational linguistics: a new tool for exploring biopolymer structures and statistical mechanics. Polymer. 2007;48:4289–300. [Google Scholar]
- 55.Drozdov AN, Grossfield A, Pappu RV. Role of solvent in determining conformational preferences of alanine dipeptide in water. J Am Chem Soc. 2004;126:2574–81. doi: 10.1021/ja039051x. [DOI] [PubMed] [Google Scholar]
- 56.Du R, Pande VS, Grosberg AY, Tanaka T, Shakhnovich ES. On the transition coordinate for protein folding. J Chem Phys. 1998;108:334–50. [Google Scholar]
- 57.Duan Y, Kollman PA. Pathways to a protein folding intermediate observed in a 1-microsecond simulation in aqueous solution. Science. 1998;282:740–44. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 58.Dyson HJ, Wright PE, Scheraga HA. The role of hydrophobic interactions in initiation and propagation of protein folding. Proc Natl Acad Sci USA. 2006;103:13057–61. doi: 10.1073/pnas.0605504103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ellison PA, Cavagnero S. Role of unfolded state heterogeneity and en-route ruggedness in protein folding kinetics. Protein Sci. 2006;15:564–82. doi: 10.1110/ps.051758206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elmer SP, Park S, Pande VS. Foldamer dynamics expressed via Markov state models. I. Explicit solvent molecular-dynamics simulations in acetonitrile, chloroform, methanol, and water. J Chem Phys. 2005;123:114902. doi: 10.1063/1.2001648. [DOI] [PubMed] [Google Scholar]
- 61.Elmer SP, Park S, Pande VS. Foldamer dynamics expressed via Markov state models. II. State space decomposition. J Chem Phys. 2005;123:114903. doi: 10.1063/1.2008230. [DOI] [PubMed] [Google Scholar]
- 62.English EP, Chumanov RS, Gellman SH, Compton T. Rational development of β-peptide inhibitors of human cytomegalovirus entry. J Biol Chem. 2006;281:2661–67. doi: 10.1074/jbc.M508485200. [DOI] [PubMed] [Google Scholar]
- 63.Felts AK, Harano Y, Gallicchio E, Levy RM. Free energy surfaces of β-hairpin and α-helical peptides generated by replica exchange molecular dynamics with the AGBNP implicit solvent model. Proteins. 2004;56:310–21. doi: 10.1002/prot.20104. [DOI] [PubMed] [Google Scholar]
- 64.Feng DF, Doolittle RF. Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol Evol. 1987;25:351–60. doi: 10.1007/BF02603120. [DOI] [PubMed] [Google Scholar]
- 65.Feng H, Vu ND, Zhou Z, Bai Y. Structural examination of φ-value analysis in protein folding. Biochemistry. 2004;43:14325–31. doi: 10.1021/bi048126m. [DOI] [PubMed] [Google Scholar]
- 66.Feng H, Zhou Z, Bai Y. A protein folding pathway with multiple folding intermediates at atomic resolution. Proc Natl Acad Sci USA. 2005;102:5026–31. doi: 10.1073/pnas.0501372102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferguson N, Sharpe TD, Johnson CM, Fersht AR. The transition state for folding of a peripheral subunit-binding domain contains robust and ionic-strength dependent characteristics. J Mol Biol. 2006;356:1237–47. doi: 10.1016/j.jmb.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 68.Ferguson N, Sharpe TD, Johnson CM, Schartau PJ, Fersht AR. Structural biology: analysis of “downhill” protein folding. Nature. 2007;445:14–15. doi: 10.1038/nature05643. [DOI] [PubMed] [Google Scholar]
- 69.Ferguson N, Sharpe TD, Schartau PJ, Sato S, Allen MD, et al. Ultra-fast barrier-limited folding in the peripheral subunit-binding domain family. J Mol Biol. 2005;353:427–46. doi: 10.1016/j.jmb.2005.08.031. [DOI] [PubMed] [Google Scholar]
- 70.Fersht AR. Nucleation mechanisms in protein folding. Curr Opin Struct Biol. 1997;7:3–9. doi: 10.1016/s0959-440x(97)80002-4. [DOI] [PubMed] [Google Scholar]
- 71.Fersht AR, Sato S. Φ-value analysis and the nature of protein-folding transition states. Proc Natl Acad Sci USA. 2004;101:7976–81. doi: 10.1073/pnas.0402684101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fersht AR, Shi JP, Knill-Jones J, Lowe DM, Wilkinson AJ, et al. Hydrogen bonding and biological specificity analysed by protein engineering. Nature. 1985;314:235–38. doi: 10.1038/314235a0. [DOI] [PubMed] [Google Scholar]
- 73.Fiebig KM, Dill KA. Protein core assembly processes. J Chem Phys. 1993;98:3475–87. [Google Scholar]
- 74.Friel CT, Beddard GS, Radford SE. Switching two-state to three-state kinetics in the helical protein Im9 via the optimisation of stabilising non-native interactions by design. J Mol Biol. 2004;342:261–73. doi: 10.1016/j.jmb.2004.06.076. [DOI] [PubMed] [Google Scholar]
- 75.Garcia-Mira MM, Sadqi M, Fischer N, Sanchez-Ruiz JM, Munoz V. Experimental identification of downhill protein folding. Science. 2002;298:2191–95. doi: 10.1126/science.1077809. [DOI] [PubMed] [Google Scholar]
- 76.Gellman SH. Foldamers: a manifesto. Acc Chem Res. 1998;31:173–80. [Google Scholar]
- 77.Ghosh K, Ozkan SB, Dill K. The ultimate speed limit to protein folding is conformational searching. J Am Chem Soc. 2007;129:11920–27. doi: 10.1021/ja066785b. [DOI] [PubMed] [Google Scholar]
- 78.Gianni S, Guydosh NR, Khan F, Caldas TD, Mayor U, et al. Unifying features in protein-folding mechanisms. Proc Natl Acad Sci USA. 2003;100:13286–91. doi: 10.1073/pnas.1835776100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gibson KD, Scheraga HA. Minimization of polypeptide energy I. Preliminary structures of bovine pancreatic ribonuclease s-peptide. Proc Natl Acad Sci USA. 1967;58:420–27. doi: 10.1073/pnas.58.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gilmanshin R, Williams S, Callender RH, Woodruff WH, Dyer RB. Fast events in protein folding: relaxation dynamics of secondary and tertiary structure in native apomyoglobin. Proc Natl Acad Sci USA. 1997;94:3709–13. doi: 10.1073/pnas.94.8.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gnanakaran S, Garcia AE. Validation of an all-atom protein force field: from dipeptides to larger peptides. J Phys Chem B. 2003;107:12555–57. [Google Scholar]
- 82.Go N, Taketomi H. Respective roles of short- and long-range interactions in protein folding. Proc Natl Acad Sci USA. 1978;75:559–63. doi: 10.1073/pnas.75.2.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goldbeck RA, Thomas YG, Chen E, Esquerra RM, Kliger DS. Multiple pathways on a protein-folding energy landscape: kinetic evidence. Proc Natl Acad Sci USA. 1999;96:2782–87. doi: 10.1073/pnas.96.6.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goldenberg DP. Genetic studies of protein stability and mechanisms of folding. Annu Rev Biophys Biomol Struct. 1988;17:481–507. doi: 10.1146/annurev.bb.17.060188.002405. [DOI] [PubMed] [Google Scholar]
- 85.Goldenberg DP. Finding the right fold. Nat Struct Biol. 1999;6:987–90. doi: 10.1038/14866. [DOI] [PubMed] [Google Scholar]
- 86.Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as versatile frameworks for the design and evolution of function. Nat Chem Biol. 2007;3:252–62. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Grabowski M, Joachimiak A, Otwinowski Z, Wladek M. Structural genomics: keeping up with expanding knowledge of the protein universe. Nucleic Acids Seq Topol. 2007;17:347–53. doi: 10.1016/j.sbi.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grantcharova V, Alm EJ, Baker D, Horwich AL. Mechanisms of protein folding. Curr Opin Struct Biol. 2001;11:70–82. doi: 10.1016/s0959-440x(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 89.Grater F, Grubmuller H. Fluctuations of primary ubiquitin folding intermediates in a force clamp. J Struct Biol. 2007;157:557–69. doi: 10.1016/j.jsb.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 90.Gromiha MM, Selvaraj S. Comparison between long-range interactions and contact order in determining the folding rate of two-state proteins: application of long-range order to folding rate prediction. J Mol Biol. 2001;310:27–32. doi: 10.1006/jmbi.2001.4775. [DOI] [PubMed] [Google Scholar]
- 91.Gromiha MM, Thangakani AM, Selvaraj S. FOLD-RATE: prediction of protein folding rates from amino acid sequence. Nucleic Acids Res. 2006;34:W70–74. doi: 10.1093/nar/gkl043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Haber E, Anfinsen CB. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J Biol Chem. 1962;237:1839–44. [PubMed] [Google Scholar]
- 93.Hagen SJ. Probe-dependent and nonexponential relaxation kinetics: unreliable signatures of downhill protein folding. Proteins. 2007;68:205–17. doi: 10.1002/prot.21342. [DOI] [PubMed] [Google Scholar]
- 94.Handel T, Degrado WF. Denovo design of a Zn2+-binding protein. J Am Chem Soc. 1990;112:6710–11. [Google Scholar]
- 95.Hansmann UHE, Okamoto Y. Prediction of peptide conformation by multicanonical algorithm: new approach to the multiple-minima problem. J Comput Chem. 1993;14:1333–38. [Google Scholar]
- 96.Hart WE, Istrail S. Robust proofs of NP-hardness for protein folding: general lattices and energy potentials. J Comput Biol. 1997;4:1–22. doi: 10.1089/cmb.1997.4.1. [DOI] [PubMed] [Google Scholar]
- 97.Haspel N, Tsai CJ, Wolfson H, Nussinov R. Reducing the computational complexity of protein folding via fragment folding and assembly. Protein Sci. 2003;12:1177–87. doi: 10.1110/ps.0232903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hecht MH, Das A, Go A, Bradley LH, Wei Y. De novo proteins from designed combinatorial libraries. Protein Sci. 2004;13:1711–23. doi: 10.1110/ps.04690804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hecht MH, Richardson JS, Richardson DC, Ogden RC. De novo design, expression, and characterization of felix: a four-helix bundle protein of native-like sequence. Science. 1990;249:884–91. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 100.Ho BK, Dill KA. Folding very short peptides using molecular dynamics. PLoS Comput Biol. 2006;2:e27. doi: 10.1371/journal.pcbi.0020027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoang L, Bedard S, Krishna MMG, Lin Y, Englander SW. Cytochrome c folding pathway: kinetic native-state hydrogen exchange. Proc Natl Acad Sci USA. 2002;99:12173–78. doi: 10.1073/pnas.152439199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hockenmaier J, Joshi AK, Dill KA. Routes are trees: the parsing perspective on protein folding. Proteins. 2006;66:1–15. doi: 10.1002/prot.21195. [DOI] [PubMed] [Google Scholar]
- 103.Huang F, Sato S, Sharpe TD, Ying L, Fersht AR. Distinguishing between cooperative and unimodal downhill protein folding. Proc Natl Acad Sci USA. 2007;104:123–27. doi: 10.1073/pnas.0609717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huang JT, Cheng JP, Chen H. Secondary structure length as a determinant of folding rate of proteins with two- and three-state kinetics. Proteins. 2007;67:12–17. doi: 10.1002/prot.21282. [DOI] [PubMed] [Google Scholar]
- 105.Hubner IA, Shimada J, Shakhnovich EI. Φ values and the folding transition state of protein G: utilization and interpretation of experimental data through simulation. Abstr Pap Am Chem Soc. 2003;226:U450. [Google Scholar]
- 106.Itzhaki LS, Otzen DE, Fersht AR. The structure of the transition state for folding of chymotrypsin inhibitor 2 analysed by protein engineering methods: evidence for a nucleation-condensation mechanism for protein folding. J Mol Biol. 1995;254:260–88. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 107.Ivankov DN, Finkelstein AV. Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc Natl Acad Sci USA. 2004;101:8942–44. doi: 10.1073/pnas.0402659101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jang S, Kim E, Pak Y. Direct folding simulation of α-helices and β-hairpins based on a single all-atom force field with an implicit solvation model. Proteins. 2007;66:53–60. doi: 10.1002/prot.21173. [DOI] [PubMed] [Google Scholar]
- 109.Jas GS, Eaton WA, Hofrichter J. Effect of viscosity on the kinetics of α-helix and β-hairpin formation. J Phys Chem B. 2001;105:261–72. [Google Scholar]
- 110.Jayachandran G, Vishal V, Pande VS. Using massively parallel simulation and Markovian models to study protein folding: examining the dynamics of the villin headpiece. J Chem Phys. 2006;124:164902. doi: 10.1063/1.2186317. [DOI] [PubMed] [Google Scholar]
- 111.Jones DT, Taylor WR, Thornton JM. A new approach to protein fold recognition. Nature. 1992;358:86–89. doi: 10.1038/358086a0. [DOI] [PubMed] [Google Scholar]
- 112.Kamtekar S, Schiffer JM, Xiong H, Babik JM, Hecht MH. Protein design by binary patterning of polar and nonpolar amino acids. Science. 1993;262:1680–85. doi: 10.1126/science.8259512. [DOI] [PubMed] [Google Scholar]
- 113.Karanicolas J, Brooks CL. The origins of asymmetry in the folding transition states of protein L and protein G. Protein Sci. 2002;11:2351–61. doi: 10.1110/ps.0205402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci USA. 2005;102:6679–85. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Karplus M, Weaver DL. Diffusion-collision model for protein folding. Biopolymers. 1979;18:1421–37. doi: 10.1002/bip.360260404. [DOI] [PubMed] [Google Scholar]
- 116.Karplus M, Weaver DL. Protein folding dynamics: the diffusion-collision model and experimental data. Protein Sci. 1994;3:650–68. doi: 10.1002/pro.5560030413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kendrew JC. The three-dimensional structure of a protein molecule. Sci Am. 1961;205:96–110. doi: 10.1038/scientificamerican1261-96. [DOI] [PubMed] [Google Scholar]
- 118.Kim DE, Gu H, Baker D. The sequences of small proteins are not extensively optimized for rapid folding by natural selection. Proc Natl Acad Sci USA. 1998;95:4982–86. doi: 10.1073/pnas.95.9.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim PS, Baldwin RL. Specific intermediates in the folding reactions of small proteins and the mechanism of protein folding. Annu Rev Biochem. 1982;51:459–89. doi: 10.1146/annurev.bi.51.070182.002331. [DOI] [PubMed] [Google Scholar]
- 120.Kirshenbaum K, Zuckermann RN, Dill KA. Designing polymers that mimic biomolecules. Curr Opin Struct Biol. 1999;9:530–35. doi: 10.1016/S0959-440X(99)80075-X. [DOI] [PubMed] [Google Scholar]
- 121.Klepeis JL, Floudas CA. ASTRO-FOLD: a combinatorial and global optimization framework for ab initio prediction of three-dimensional structures of proteins from the amino acid sequence. Biophys J. 2003;85:2119–46. doi: 10.1016/s0006-3495(03)74640-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Knott M, Chan HS. Criteria for downhill protein folding: calorimetry, chevron plot, kinetic relaxation, and single-molecule radius of gyration in chain models with subdued degrees of cooperativity. Proteins. 2006;65:373–91. doi: 10.1002/prot.21066. [DOI] [PubMed] [Google Scholar]
- 123.Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, et al. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature. 2004;430:586–90. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- 124.Krieger F, Fierz B, Bieri O, Drewello M, Kiefhaber T. Dynamics of unfolded polypeptide chains as model for the earliest steps in protein folding. J Mol Biol. 2003;332:265–74. doi: 10.1016/s0022-2836(03)00892-1. [DOI] [PubMed] [Google Scholar]
- 125.Krishna MM, Hoang L, Lin Y, Englander SW. Hydrogen exchange methods to study protein folding. Methods. 2004;34:51–64. doi: 10.1016/j.ymeth.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 126.Krishna MMG, Maity H, Rumbley JN, Lin Y, Englander SW. Order of steps in the cytochrome c folding pathway: evidence for a sequential stabilization mechanism. J Mol Biol. 2006;359:1411–20. doi: 10.1016/j.jmb.2006.04.035. [DOI] [PubMed] [Google Scholar]
- 127.Kubelka J, Chiu TK, Davies DR, Eaton WA, Hofrichter J. Sub-microsecond protein folding. J Mol Biol. 2006;359:546–53. doi: 10.1016/j.jmb.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 128.Kubelka J, Hofrichter J, Eaton WA. The protein folding ‘speed limit’. Curr Opin Struct Biol. 2004;14:76–88. doi: 10.1016/j.sbi.2004.01.013. [DOI] [PubMed] [Google Scholar]
- 129.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Design of a novel globular protein fold with atomic-level accuracy. Science. 2003;302:1364–68. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 130.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. The weighted histogram analysis method for free-energy calculations on biomolecules. 1. The method. J Comput Chem. 1992;13:1011–21. [Google Scholar]
- 131.Kuznetsov IB, Rackovsky S. Class-specific correlations between protein folding rate, structure-derived, and sequence-derived descriptors. Proteins. 2004;54:333–41. doi: 10.1002/prot.10518. [DOI] [PubMed] [Google Scholar]
- 132.Lau KF, Dill KA. A lattice statistical mechanics model of the conformational and sequence spaces of proteins. Macromolecules. 1989;22:3986–97. [Google Scholar]
- 133.Laurence TA, Kong X, Jäger M, Weiss S. Probing structural heterogeneities and fluctuations of nucleic acids and denatured proteins. Proc Natl Acad Sci USA. 2005;102:17348–53. doi: 10.1073/pnas.0508584102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lee BC, Zuckermann RN, Dill KA. Folding a nonbiological polymer into a compact multihelical structure. J Am Chem Soc. 2005;127:10999–1009. doi: 10.1021/ja0514904. [DOI] [PubMed] [Google Scholar]
- 135.Lei H, Duan Y. Ab initio folding of albumin binding domain from all-atom molecular dynamics simulation. J Phys Chem B. 2007;111:5458–63. doi: 10.1021/jp0704867. [DOI] [PubMed] [Google Scholar]
- 136.Lei H, Duan Y. Two-stage folding of Hp-35 from ab initio simulations. J Mol Biol. 2007;370:196–206. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lei H, Wu C, Liu H, Duan Y. Folding free-energy landscape of villin headpiece subdomain from molecular dynamics simulations. Proc Natl Acad Sci USA. 2007;104:4925–30. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Leopold PE, Montal M, Onuchic JN. Protein folding funnels: a kinetic approach to the sequence-structure relationship. Proc Natl Acad Sci USA. 1992;89:8721–25. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lesk AM, Rose GD. Folding units in globular proteins. Proc Natl Acad Sci USA. 1981;78:4304–8. doi: 10.1073/pnas.78.7.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Levitt M. Molecular dynamics of native protein. I. Computer simulation of trajectories. J Mol Biol. 1983;168:595–617. doi: 10.1016/s0022-2836(83)80304-0. [DOI] [PubMed] [Google Scholar]
- 141.Li Z, Scheraga HA. Monte Carlo–minimization approach to the multiple-minima problem in protein folding. Proc Natl Acad Sci USA. 1987;84:6611–15. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lindberg MO, Haglund E, Hubner IA, Shakhnovich EI, Oliveberg M. Identification of the minimal protein-folding nucleus through loop-entropy perturbations. Proc Natl Acad Sci USA. 2006;103:4083–88. doi: 10.1073/pnas.0508863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lindberg MO, Oliveberg M. Malleability of protein folding pathways: a simple reason for complex behaviour. Curr Opin Struct Biol. 2007;17:21–29. doi: 10.1016/j.sbi.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 144.Lindorff-Larsen K, Paci E, Serrano L, Dobson CM, Vendruscolo M. Calculation of mutational free energy changes in transition states for protein folding. Biophys J. 2003;85:1207–14. doi: 10.1016/S0006-3495(03)74556-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Looger LL, Dwyer MA, Smith JJ, Hellinga HW. Computational design of receptor and sensor proteins with novel functions. Nature. 2003;423:185–90. doi: 10.1038/nature01556. [DOI] [PubMed] [Google Scholar]
- 146.Lucas A, Huang L, Joshi A, Dill KA. Statistical mechanics of helix bundles using a dynamic programming approach. J Am Chem Soc. 2007;129:4272–81. doi: 10.1021/ja067153s. [DOI] [PubMed] [Google Scholar]
- 147.Lucent D, Vishal V, Pande VS. Protein folding under confinement: a role for solvent. Proc Natl Acad Sci USA. 2007;104:10430–34. doi: 10.1073/pnas.0608256104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ma H, Gruebele M. Low barrier kinetics: dependence on observables and free energy surface. J Comput Chem. 2006;27:125–34. doi: 10.1002/jcc.20311. [DOI] [PubMed] [Google Scholar]
- 149.Maity H, Maity M, Krishna MM, Mayne L, Englander SW. Protein folding: the stepwise assembly of foldon units. Proc Natl Acad Sci USA. 2005;102:4741–46. doi: 10.1073/pnas.0501043102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Makarov DE, Keller CA, Plaxco KW, Metiu H. How the folding rate constant of simple, single-domain proteins depends on the number of native contacts. Proc Natl Acad Sci USA. 2002;99:3535–39. doi: 10.1073/pnas.052713599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Matagne A, Radford SE, Dobson CM. Fast and slow tracks in lysozyme folding: insight into the role of domains in the folding process. J Mol Biol. 1997;267:1068–74. doi: 10.1006/jmbi.1997.0963. [DOI] [PubMed] [Google Scholar]
- 152.Matheson RR, Jr, Scheraga HA. A method for predicting nucleation sites for protein folding based on hydrophobic contacts. Macromolecules. 1978;11:819–29. [Google Scholar]
- 153.Matouschek A, Kellis JT, Jr, Serrano L, Fersht AR. Mapping the transition state and pathway of protein folding by protein engineering. Nature. 1989;340:122–26. doi: 10.1038/340122a0. [DOI] [PubMed] [Google Scholar]
- 154.Maxwell KL, Wildes D, Zarrine-Afsar A, De Los Rios MA, Brown AG, et al. Protein folding: defining a “standard” set of experimental conditions and a preliminary kinetic data set of two-state proteins. Protein Sci. 2005;14:602–16. doi: 10.1110/ps.041205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McCammon JA, Gelin BR, Karplus M. Dynamics of folded proteins. Nature. 1977;267:585–90. doi: 10.1038/267585a0. [DOI] [PubMed] [Google Scholar]
- 156.Meisner WK, Sosnick TR. Barrier-limited, microsecond folding of a stable protein measured with hydrogen exchange: implications for downhill folding. Proc Natl Acad Sci USA. 2004;101:15639–44. doi: 10.1073/pnas.0404895101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mello CC, Barrick D. An experimentally determined protein folding energy landscape. Proc Natl Acad Sci USA. 2004;101:14102–7. doi: 10.1073/pnas.0403386101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Mezei M. Chameleon sequences in the PDB. Protein Eng. 1998;11:411–14. doi: 10.1093/protein/11.6.411. [DOI] [PubMed] [Google Scholar]
- 159.Micheletti C, Banavar JR, Maritan A, Seno F. Protein structures and optimal folding from a geometrical variational principle. Phys Rev Lett. 1999;82:3372–75. [Google Scholar]
- 160.Miller EJ, Fischer KF, Marqusee S. Experimental evaluation of topological parameters determining protein-folding rates. Proc Natl Acad Sci USA. 2002;99:10359–63. doi: 10.1073/pnas.162219099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Miller R, Danko CA, Fasolka MJ, Balazs AC, Chan HS, Dill KA. Folding kinetics of proteins and copolymers. J Chem Phys. 1992;96:768–80. [Google Scholar]
- 162.Minor DL, Kim PS. Context-dependent secondary structure formation of a designed protein sequence. Nature. 1996;380:730–34. doi: 10.1038/380730a0. [DOI] [PubMed] [Google Scholar]
- 163.Mirny L, Shakhnovich E. Protein folding theory: from lattice to all-atom models. Annu Rev Biophys Biomol Struct. 2001;30:361–96. doi: 10.1146/annurev.biophys.30.1.361. [DOI] [PubMed] [Google Scholar]
- 164.Moult J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr Opin Struct Biol. 2005;15:285–89. doi: 10.1016/j.sbi.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 165.Moult J, Pedersen JT, Judson R, Fidelis K. A large-scale experiment to assess protein structure prediction methods. Proteins. 1995;23:ii–iv. doi: 10.1002/prot.340230303. [DOI] [PubMed] [Google Scholar]
- 166.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. A natively unfolded yeast prion monomer adopts an ensemble of collapsed and rapidly fluctuating structures. Proc Natl Acad Sci USA. 2007;104:2649–54. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Munoz V. Thermodynamics and kinetics of downhill protein folding investigated with a simple statistical mechanical model. Int J Quant Chem. 2002;90:1522–28. [Google Scholar]
- 168.Munoz V, Sanchez-Ruiz JM. Exploring protein-folding ensembles: a variable-barrier model for the analysis of equilibrium unfolding experiments. Proc Natl Acad Sci USA. 2004;101:17646–51. doi: 10.1073/pnas.0405829101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Myers JK, Oas TG. Preorganized secondary structure as an important determinant of fast protein folding. Nat Struct Biol. 2001;8:552–58. doi: 10.1038/88626. [DOI] [PubMed] [Google Scholar]
- 170.Naganathan AN, Perez-Jimenez R, Sanchez-Ruiz JM, Munoz V. Robustness of downhill folding: guidelines for the analysis of equilibrium folding experiments on small proteins. Biochemistry. 2005;44:7435–49. doi: 10.1021/bi050118y. [DOI] [PubMed] [Google Scholar]
- 171.Nemethy G, Scheraga HA. Theoretical determination of sterically allowed conformations of a polypeptide chain by a computer method. Biopolymers. 1965;3:155–84. [Google Scholar]
- 172.Noé F, Horenko I, Schütte C, Smith JC. Hierarchical analysis of conformational dynamics in biomolecules: transition networks of metastable states. J Chem Phys. 2007;126:155102. doi: 10.1063/1.2714539. [DOI] [PubMed] [Google Scholar]
- 173.O’Neil KT, Hoess RH, DeGrado WF. Design of DNA-binding peptides based on the leucine zipper motif. Science. 1990;249:774–78. doi: 10.1126/science.2389143. [DOI] [PubMed] [Google Scholar]
- 174.Oldziej S, Czaplewski C, Liwo A, Chinchio M, Nanias M, et al. Physics-based protein-structure prediction using a hierarchical protocol based on the UNRES force field: assessment in two blind tests. Proc Natl Acad Sci USA. 2005;102:7547–52. doi: 10.1073/pnas.0502655102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Onuchic JN, Wolynes PG. Energy landscapes, glass transitions, and chemical-reaction dynamics in biomolecular or solvent environment. J Chem Phys. 1993;98:2218–24. [Google Scholar]
- 176.Ozkan SB, Bahar I, Dill KA. Transition states and the meaning of φ-values in protein folding kinetics. Nat Struct Biol. 2001;8:765–69. doi: 10.1038/nsb0901-765. [DOI] [PubMed] [Google Scholar]
- 177.Ozkan SB, Wu GHA, Chodera JD, Dill KA. Protein folding by zipping and assembly. Proc Natl Acad Sci USA. 2007;104:11987–92. doi: 10.1073/pnas.0703700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Paci E, Vendruscolo M, Dobson CM, Karplus M. Determination of a transition state at atomic resolution from protein engineering data. J Mol Biol. 2002;324:151–63. doi: 10.1016/s0022-2836(02)00944-0. [DOI] [PubMed] [Google Scholar]
- 179.Patch JA, Barron AE. Helical peptoid mimics of magainin-2 amide. J Am Chem Soc. 2003;125:12092–93. doi: 10.1021/ja037320d. [DOI] [PubMed] [Google Scholar]
- 180.Pauling L, Corey RB. Atomic coordinates and structure factors for two helical configurations of polypeptide chains. Proc Natl Acad Sci USA. 1951;37:235–40. doi: 10.1073/pnas.37.5.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Pauling L, Corey RB, Branson HR. The structure of proteins: two hydrogen-bonded helical configurations of the polypeptide chain. Proc Natl Acad Sci USA. 1951;37:205–11. doi: 10.1073/pnas.37.4.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pieper U, Eswar N, Braberg H, Madhusudhan MS, Davis FP, et al. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004;32:D217–22. doi: 10.1093/nar/gkh095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pitera JW, Swope W. Understanding folding and design: replica-exchange simulations of “Trp-cage” miniproteins. Proc Natl Acad Sci USA. 2003;100:7587–92. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Plaxco KW, Simons KT, Baker D. Contact order, transition state placement and the refolding rates of single domain proteins. J Mol Biol. 1998;277:985–94. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 185.Porter EA, Wang X, Lee HS, Weisblum B, Gellman SH. Non-haemolytic β-aminoacid oligomers. Nature. 2000;404:565. doi: 10.1038/35007145. [DOI] [PubMed] [Google Scholar]
- 186.Punta M, Rost B. Protein folding rates estimated from contact predictions. J Mol Biol. 2005;348:507–12. doi: 10.1016/j.jmb.2005.02.068. [DOI] [PubMed] [Google Scholar]
- 187.Qiu L, Hagen SJ. A limiting speed for protein folding at low solvent viscosity. J Am Chem Soc. 2004;126:3398–99. doi: 10.1021/ja049966r. [DOI] [PubMed] [Google Scholar]
- 188.Raleigh DP, Plaxco KW. The protein folding transition state: What are φ-values really telling us? Protein Pept Lett. 2005;12:117–22. doi: 10.2174/0929866053005809. [DOI] [PubMed] [Google Scholar]
- 189.Reich L, Weikl TR. Substructural cooperativity and parallel versus sequential events during protein unfolding. Proteins. 2006;63:1052–58. doi: 10.1002/prot.20966. [DOI] [PubMed] [Google Scholar]
- 190.Rost B, Eyrich VA. EVA: large-scale analysis of secondary structure prediction. Proteins. 2001;45:192–99. doi: 10.1002/prot.10051. [DOI] [PubMed] [Google Scholar]
- 191.Sadqi M, Fushman D, Munoz V. Atom-by-atom analysis of global downhill protein folding. Nature. 2006;442:317–21. doi: 10.1038/nature04859. [DOI] [PubMed] [Google Scholar]
- 192.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 193.Sanchez IE, Kiefhaber T. Origin of unusual φ-values in protein folding: evidence against specific nucleation sites. J Mol Biol. 2003;334:1077–85. doi: 10.1016/j.jmb.2003.10.016. [DOI] [PubMed] [Google Scholar]
- 194.Schuler B, Lipman EA, Eaton WA. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature. 2002;419:743–47. doi: 10.1038/nature01060. [DOI] [PubMed] [Google Scholar]
- 195.Segel DJ, Bachmann A, Hofrichter J, Hodgson KO, Doniach S, Kiefhaber T. Characterization of transient intermediates in lysozyme folding with time-resolved small-angle X-ray scattering. J Mol Biol. 1999;288:489–99. doi: 10.1006/jmbi.1999.2703. [DOI] [PubMed] [Google Scholar]
- 196.Shea JE, Brooks CL., III From folding theories to folding proteins: a review and assessment of simulation studies of protein folding and unfolding. Annu Rev Phys Chem. 2001;52:499–535. doi: 10.1146/annurev.physchem.52.1.499. [DOI] [PubMed] [Google Scholar]
- 197.Shea JE, Onuchic JN, Brooks CL. Exploring the origins of topological frustration: design of a minimally frustrated model of fragment B of protein A. Proc Natl Acad Sci USA. 1999;96:12512–17. doi: 10.1073/pnas.96.22.12512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Shirts M, Pande VS. Computing: Screen savers of the world unite! Science. 2000;290:1903–4. doi: 10.1126/science.290.5498.1903. [DOI] [PubMed] [Google Scholar]
- 199.Shmygelska A. Search for folding nuclei in native protein structures. Bioinformatics. 2005;21:394–402. doi: 10.1093/bioinformatics/bti1050. [DOI] [PubMed] [Google Scholar]
- 200.Simmerling C, Strockbine B, Roitberg AE. All-atom structure prediction and folding simulations of a stable protein. J Am Chem Soc. 2002;124:11258–59. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 201.Singhal N, Snow CD, Pande VS. Using path sampling to build better Markovian state models: predicting the folding rate and mechanism of a tryptophan zipper β hairpin. J Chem Phys. 2004;121:415–25. doi: 10.1063/1.1738647. [DOI] [PubMed] [Google Scholar]
- 202.Snow CD, Rhee YM, Pande VS. Kinetic definition of protein folding transition state ensembles and reaction coordinates. Biophys J. 2006;91:14–24. doi: 10.1529/biophysj.105.075689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sohl JL, Jaswal SS, Agard DA. Unfolded conformations of α-lytic protease are more stable than its native state. Nature. 1998;395:817–19. doi: 10.1038/27470. [DOI] [PubMed] [Google Scholar]
- 204.Sosnick TR, Dothager RS, Krantz BA. Differences in the folding transition state of ubiquitin indicated by φ and ψ analyses. Proc Natl Acad Sci USA. 2004;101:17377–82. doi: 10.1073/pnas.0407683101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sridevi K, Juneja J, Bhuyan AK, Krishnamoorthy G, Udgaonkar JB. The slow folding reaction of barstar: The core tryptophan region attains tight packing before substantial secondary and tertiary structure formation and final compaction of the polypeptide chain. J Mol Biol. 2000;302:479–95. doi: 10.1006/jmbi.2000.4060. [DOI] [PubMed] [Google Scholar]
- 206.Sridevi K, Lakshmikanth GS, Krishnamoorthy G, Udgaonkar JB. Increasing stability reduces conformational heterogeneity in a protein folding intermediate ensemble. J Mol Biol. 2004;337:699–711. doi: 10.1016/j.jmb.2003.12.083. [DOI] [PubMed] [Google Scholar]
- 207.Srinivasan R, Rose GD. Ab initio prediction of protein structure using LINUS. Proteins. 2002;47:489–95. doi: 10.1002/prot.10103. [DOI] [PubMed] [Google Scholar]
- 208.Sriraman S, Kevrekidis IG, Hummer G. Coarse master equation from Bayesian analysis of replica molecular dynamics simulations. J Phys Chem B. 2005;109:6479–84. doi: 10.1021/jp046448u. [DOI] [PubMed] [Google Scholar]
- 209.Street TO, Bradley CM, Barrick D. Predicting coupling limits from an experimentally determined energy landscape. Proc Natl Acad Sci USA. 2007;104:4907–12. doi: 10.1073/pnas.0608756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–51. [Google Scholar]
- 211.Swope WC, Pitera JW, Suits F. Describing protein folding kinetics by molecular dynamics simulations. 1. Theory. J Phys Chem B. 2004;108:6571–81. [Google Scholar]
- 212.Swope WC, Pitera JW, Suits F, Pitman M, Eleftheriou M, et al. Describing protein folding kinetics by molecular dynamics simulations. 2. Example applications to alanine dipeptide and a B-hairpin peptide. J Phys Chem B. 2004;108:6582–94. [Google Scholar]
- 213.Travaglini-Allocatelli C, Cutruzzolà F, Bigotti MG, Staniforth RA, Brunori M. Folding mechanism of Pseudomonas aeruginosa cytochrome c. J Mol Biol. 1999;289:1459–67. doi: 10.1006/jmbi.1999.2852. [DOI] [PubMed] [Google Scholar]
- 214.Tress M, Ezkurdia I, Graña O, López G, Valencia A. Assessment of predictions submitted for the CASP6 comparative modelling category. Proteins. 2005;61:27–45. doi: 10.1002/prot.20720. [DOI] [PubMed] [Google Scholar]
- 215.Tsai CJ, Maizel JV, Jr, Nussinov R. Anatomy of protein structures: visualizing how a one-dimensional protein chain folds into a three-dimensional shape. Proc Natl Acad Sci USA. 2000;97:12038–43. doi: 10.1073/pnas.97.22.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Unger R, Moult J. Finding the lowest free energy conformation of a protein is an NP-hard problem: proof and implications. Bull Math Biol. 1993;55:1183–98. doi: 10.1007/BF02460703. [DOI] [PubMed] [Google Scholar]
- 217.Utku Y, Dehan E, Ouerfelli O, Piano F, Zuckermann RN, et al. A peptidomimetic siRNA transfection reagent for highly effective gene silencing. Mol Biosyst. 2006;2:312–17. doi: 10.1039/b603229j. [DOI] [PubMed] [Google Scholar]
- 218.Uversky VN, Fink AL. The chicken-egg scenario of protein folding revisited. FEBS Lett. 2002;515:79–83. doi: 10.1016/s0014-5793(02)02441-9. [DOI] [PubMed] [Google Scholar]
- 219.Venclovas C, Zemla A, Fidelis K, Moult J. Assessment of progress over the CASP experiments. Proteins. 2003;53:585–95. doi: 10.1002/prot.10530. [DOI] [PubMed] [Google Scholar]
- 220.Verma A, Schug A, Lee KH, Wenzel W. Basin hopping simulations for all-atom protein folding. J Chem Phys. 2006;124:044515. doi: 10.1063/1.2138030. [DOI] [PubMed] [Google Scholar]
- 221.Viguera AR, Serrano L, Wilmanns M. Different folding transition states may result in the same native structure. Nat Struct Biol. 1996;3:874–80. doi: 10.1038/nsb1096-874. [DOI] [PubMed] [Google Scholar]
- 222.Vitkup D, Melamud E, Moult J, Sander C. Completeness in structural genomics. Nat Struct Biol. 2001;8:559–66. doi: 10.1038/88640. [DOI] [PubMed] [Google Scholar]
- 223.Voelz VA, Dill KA. Exploring zipping and assembly as a protein folding principle. Proteins. 2006;66:877–88. doi: 10.1002/prot.21234. [DOI] [PubMed] [Google Scholar]
- 224.Wallace IM, Blackshields G, Higgins DG. Multiple sequence alignments. Curr Opin Struct Biol. 2005;15:261–66. doi: 10.1016/j.sbi.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 225.Wallin S, Chan HS. Conformational entropic barriers in topology-dependent protein folding: perspectives from a simple native-centric polymer model. J Phys Condens Matter. 2006;18:S307–28. [Google Scholar]
- 226.Wang L, Xie J, Schultz PG. Expanding the genetic code. Annu Rev Biophys Biomol Struct. 2006;35:225–49. doi: 10.1146/annurev.biophys.35.101105.121507. [DOI] [PubMed] [Google Scholar]
- 227.Wang Z, Mottonen J, Goldsmith EJ. Kinetically controlled folding of the serpin plasminogen activator inhibitor 1. Biochemistry. 1996;35:16443–48. doi: 10.1021/bi961214p. [DOI] [PubMed] [Google Scholar]
- 228.Weikl TR. Loop-closure events during protein folding: rationalizing the shape of φ-value distributions. Proteins. 2005;60:701–11. doi: 10.1002/prot.20504. [DOI] [PubMed] [Google Scholar]
- 229.Weikl TR, Dill KA. Folding kinetics of two-state proteins: effect of circularization, permutation, and crosslinks. J Mol Biol. 2003;332:953–63. doi: 10.1016/s0022-2836(03)00884-2. [DOI] [PubMed] [Google Scholar]
- 230.Weikl TR, Dill KA. Folding rates and low-entropy-loss routes of two-state proteins. J Mol Biol. 2003;329:585–98. doi: 10.1016/s0022-2836(03)00436-4. [DOI] [PubMed] [Google Scholar]
- 231.Weikl TR, Dill KA. Transition-states in protein folding kinetics: the structural interpretation of φ values. J Mol Biol. 2007;365:1578–86. doi: 10.1016/j.jmb.2006.10.082. [DOI] [PubMed] [Google Scholar]
- 232.Weikl TR, Palassini M, Dill KA. Cooperativity in two-state protein folding kinetics. Protein Sci. 2004;13:822–29. doi: 10.1110/ps.03403604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.White GWN, Gianni S, Grossmann JG, Jemth P, Fersht AR, Daggett V. Simulation and experiment conspire to reveal cryptic intermediates and a slide from the nucleation-condensation to framework mechanism of folding. J Mol Biol. 2005;350:757–75. doi: 10.1016/j.jmb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 234.Wolfenden R. Experimental measures of amino acid hydrophobicity and the topology of transmembrane and globular proteins. J Gen Physiol. 2007;129:357–62. doi: 10.1085/jgp.200709743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wu CW, Seurynck SL, Lee KY, Barron AE. Helical peptoid mimics of lung surfactant protein C. Chem Biol. 2003;10:1057–63. doi: 10.1016/j.chembiol.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 236.Wurth C, Kim W, Hecht MH. Combinatorial approaches to probe the sequence determinants of protein aggregation and amyloidogenicity. Protein Pept Lett. 2006;13:279–86. doi: 10.2174/092986606775338506. [DOI] [PubMed] [Google Scholar]
- 237.Xu Y, Purkayastha P, Gai F. Nanosecond folding dynamics of a three-stranded β-sheet. J Am Chem Soc. 2006;128:15836–42. doi: 10.1021/ja064865+. [DOI] [PubMed] [Google Scholar]
- 238.Yang JS, Chen WW, Skolnick J, Shakhnovich EI. All-atom ab initio folding of a diverse set of proteins. Structure. 2006;15:53–63. doi: 10.1016/j.str.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 239.Yeh SR, Rousseau DL. Hierarchical folding of cytochrome c. Nat Struct Biol. 2000;7:443–45. doi: 10.1038/75831. [DOI] [PubMed] [Google Scholar]
- 240.Yoda T, Sugita Y, Okamoto Y. Secondary structure preferences of force fields for proteins evaluated by generalized-ensemble simulations. Chem Phys. 2004;307:269–83. [Google Scholar]
- 241.Zagrovic B, Snow CD, Shirts MR, Pande VS. Simulation of folding of a small α-helical protein in atomistic detail using worldwide-distributed computing. J Mol Biol. 2002;323:927–37. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 242.Zhang Y, Arakaki AK, Skolnick J. TASSER: an automated method for the prediction of protein tertiary structures in CASP6. Proteins. 2005;61:91–98. doi: 10.1002/prot.20724. [DOI] [PubMed] [Google Scholar]
- 243.Zhao H, Giver L, Shao Z, Affholter JA, Arnold FH. Molecular evolution by staggered extension process (StEP) in vitro recombination. Nat Biotechnol. 1998;16:258–61. doi: 10.1038/nbt0398-258. [DOI] [PubMed] [Google Scholar]
- 244.Zhong S, Rousseau DL, Yeh SR. Modulation of the folding energy landscape of cytochrome c with salt. J Am Chem Soc. 2004;126:13934–35. doi: 10.1021/ja0459431. [DOI] [PubMed] [Google Scholar]
- 245.Zhou HY, Zhou YQ. Folding rate prediction using total contact distance. Biophys J. 2002;82:458–63. doi: 10.1016/S0006-3495(02)75410-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Zhou R. Free energy landscape of protein folding in water: explicit vs. implicit solvent. Proteins. 2003;53:148–61. doi: 10.1002/prot.10483. [DOI] [PubMed] [Google Scholar]