

Abstract
Natural hybridization accompanied by a shift in niche preference by hybrid genotypes can lead to hybrid speciation. Natural selection may cause the fixation of advantageous alleles in the ecologically diverged hybrids, and the loci experiencing selection should exhibit a reduction in allelic diversity relative to neutral loci. Here, we analyzed patterns of genetic diversity at 59 microsatellite loci associated with expressed sequence tags (ESTs) in a homoploid hybrid sunflower species, Helianthus anomalus. We used two indices, ln RV and ln RH, to compare variation and heterozygosity (respectively) at each locus between the hybrid species and its two parental species, H. annuus and H. petiolaris. Mean values of ln RV and ln RH were significantly lower than zero, which implies that H. anomalus experienced a population bottleneck during its recent evolutionary history. After correcting for the apparent bottleneck, we found six loci with a significant reduction in variation or with heterozygosity in the hybrid species, compared to one or both of the parental species. These loci should be viewed as a ranked list of candidate loci, pending further sequencing and functional analyses. Sequence data were generated for two of the candidate loci, but population genetics tests failed to detect deviations from neutral evolution at either locus. Nonetheless, a greater than eight-fold excess of nonsynonymous substitutions was found near a putative N-myristoylation motif at the second locus (HT998), and likelihood-based models indicated that the protein has been under selection in H. anomalus in the past and, perhaps, in one or both parental species. Finally, our data suggest that selective sweeps may have united populations of H. anomalus isolated by a mountain range, indicating that even low gene-flow species may be held together by the spread of advantageous alleles.
Keywords: ecological divergence, EST-based microsatellite markers, homoploid hybrid speciation, natural selection
Introduction
Hybridization has been documented between many plant and animal species (Arnold 2006). While most hybrid genotypes are less fit than the parental genotypes (Coyne & Orr 2004), hybrids may contribute to the adaptation of the parental species by serving as a bridge for adaptive introgression (Anderson 1949; Arnold 2006). In addition, fit hybrid combinations that are rare may become established if they can escape from hybrid zones before being broken up by recombination (Barton 2001). This may occur through polyploidy, asexual reproduction, selfing, ecogeographical isolation and/or homoploid hybrid speciation. The latter mechanism, also referred to as recombinational speciation (Grant 1981), describes the formation of a new species by hybridization without change in ploidy level. Reproductive isolation of the new hybrid genotype from the parental species may emerge almost immediately via karyotypic changes if the parental chromosomes are rearranged (Grant 1981; Rieseberg 1997). However, ecological divergence may also act as a strong reproductive barrier, provided that the parental species are less fit than the hybrid in its habitat (McCarthy et al. 1995; Buerkle et al. 2000). A combination of these two factors (chromosomal rearrangement and ecological divergence) dramatically increases the probability of homoploid hybrid speciation.
Hybrids have considerable genetic flexibility that may facilitate adaptation to a new habitat (Barton 2001). Hybrids can exhibit intermediate trait values, combine traits from both parents and/or exhibit extreme trait values compared to the parental species (Gross & Rieseberg 2005). Unlike the slow accumulation of novel mutations, homoploid hybrid species can benefit immediately from transgressive trait values generated by complementary gene action (Rieseberg et al. 2003). Natural selection can cause fixation of advantageous alleles (or chromosomal segments) in the ecologically diverged hybrid, and the footprint of selection at these loci can be detected using various methods (Schlötterer 2002; Lexer et al. 2003; Beaumont 2005; Edelist et al. 2006).
Regions of the genome that are under selection are likely to exhibit a strong reduction in allelic variation relative to neutral loci (Harr et al. 2002; Schlötterer 2002; Sáez et al. 2003). Variation is expected to be lowest near the target of selection and to increase with genetic distance from the selected site, a phenomenon known as ‘hitch-hiking’ (Maynard-Smith & Haigh 1974). The predicted size of the affected region is unknown in homoploid hybrids, but it is reasonable to speculate that regions that are swept to fixation during the origin of a hybrid species are large because of the speed of hybrid speciation and interspecific linkage disequilibrium (Ungerer et al. 1998; Buerkle et al. 2000; Edelist et al. 2006). However, sweeps that occur after speciation are probably no different than in taxa that have more conventional origins. Because hybrid speciation is often accompanied by a population bottleneck, it is important to use highly variable markers to study selective sweeps. Microsatellites are ideal because they are highly variable, frequently associated with genes and amenable to high-throughput genotyping (e.g. Vigouroux et al. 2002; Dieringer et al. 2005; Muir & Schlötterer 2005; Song et al. 2006).
Schlötterer and coworkers (see Harr et al. 2002; Schlötterer 2002; Schlötterer & Dieringer 2005; Wiehe et al. 2007) have developed two quantitative model-free statistics to identify loci that exhibit the largest reduction in microsatellite diversity, ln RV and ln RH. Ln RV is the natural logarithm of the ratio between variances of two populations:
and ln RH compares the expected heterozygosity of the populations, based on a stepwise mutation model (Ohta & Kimura 1973):
These indices account for heterogeneity among loci in mutation rates by using the ratio of the observed variance or heterozygosity at each locus. In computer simulations, both ln RV and ln RH were robust to neutral drift, mutation rates and sample sizes (Schlötterer 2002; Schlötterer & Dieringer 2005). Because heterozygosity is expected to return to its expected equilibrium value more rapidly than variance in allele size, ln RH should be more powerful in detecting recent selective sweeps (Storz 2005). The joint use of ln RV and ln RH reduces the false positive rate about three-fold (Schlötterer & Dieringer 2005). Under neutrality, the distributions of ln RV and ln RH can be approximated by a Gaussian distribution. If one of the statistics for a locus falls beyond the predetermined confidence bounds (i.e. 95% of a standard normal distribution), it signals a significant reduction in genetic diversity (Harr et al. 2002; Dieringer et al. 2005).
In this study, we tested for locus-specific reductions in genetic diversity at 59 microsatellite loci in the homoploid hybrid sunflower species, Helianthus anomalus Blake, compared to its widely distributed parental species, H. annuus L. and H. petiolaris Nutt. (Rieseberg 1991; Schwarzbach & Rieseberg 2002). H. anomalus is a sand-dune adapted species that appears to have arisen multiple times between 116 000 and 144 000 years ago (Schwarzbach & Rieseberg 2002). Putatively adaptive differences between H. anomalus and its parental species, such as large seeds, smaller leaves and higher leaf nitrogen concentration, have been documented (Schwarzbach et al. 2001; Rosenthal et al. 2002; Brouillette et al. 2006), and directional selection on some of the divergent traits has been detected in early generation hybrids grown in the dune habitat of H. anomalus (Ludwig et al. 2004). H. anomalus is therefore a logical species in which to look for candidate genes for ecological speciation.
The microsatellites tested mostly derive from the flanking untranslated regions of complementary DNA (cDNA) developed via expressed sequence tag (EST) sequencing. A few of the microsatellites tested occur within coding sequences. Thus, locus-specific reductions in microsatellite diversity are assumed to result from selection on very tightly linked genes (Maynard-Smith & Haigh 1974; Kim & Stephan 2002; Schlötterer 2002). We also generated sequence data for two genes that may be targets of selection based on the microsatellite screens. We ask the following specific questions: (i) can divergently selected genes and/or sites be identified through this comparative hitch-hiking approach? (ii) Do the candidate genes identified relate functionally to sand-dune adaptation? And (iii) what is the geographical extent of the putative selective sweeps?
Materials and methods
Study species and collection sites
Helianthus anomalus is a narrowly distributed endemic of Utah and Arizona (USA), adapted to moving and semistable sand dunes in a relatively arid climate (Schwarzbach et al. 2001; Ludwig et al. 2004). Many of the extreme phenotypic traits that are adaptive in H. anomalus appear to have arisen via hybridization (Rieseberg et al. 2003; Ludwig et al. 2004; Rosenthal et al. 2005). H. anomalus also is strongly reproductively isolated from H. annuus and H. petiolaris by a chromosomal sterility barrier (Lai et al. 2005).
Samples of the three species (Table 1) were collected in Utah and Arizona in the Summer of 2003 (H. annuus and H. petiolaris) and in the Summer of 2004 (H. anomalus). Samples were collected from at least 50 individuals along a transect across the population or from all individuals in small populations.
Table 1.
Location of populations sampled for this study. UT, Utah; AZ, Arizona
| Species | Population code | Location | Coordinates |
|---|---|---|---|
| Helianthus anomalus | ANO1244 | Little Sahara; UT | 39°44′N, 112°19′W |
| H. anomalus | ANO1273 | Hanksville South; UT | 38°01′N, 110°33′W |
| H. anomalus | ANO1260 | Hanksville North; UT | 38°35′N, 110°35′W |
| H. anomalus | ANO1260b | Hanksville Airport; UT | 38°27′N, 110°39′W |
| H. annuus | ANN1307 | West of Kayenta; AZ | 36°66′N, 110°41′W |
| H. annuus | ANN1310 | Toquerville; UT | 37°26′N, 113°28′W |
| H. annuus | ANN1312 | East of Little Sahara; UT | 39°72′N, 112°23′W |
| H. petiolaris | PET1322 | San Juan County; UT | 37°55′N, 109°79′W |
| H. petiolaris | PET1323 | Mexican Hat; UT | 37°03′N, 110°13′W |
| H. petiolaris | PET1325 | Iron county; UT | 38°08′N, 112°68′W |
Leaf tissue was collected in the field and placed in 15 mL Falcon tubes with desiccant for transport and storage. Tissue was ground, and DNA was extracted using a QIAGEN DNeasy kit. DNA quality and concentration were determined using a spectrophotometer, and the concentration of each sample was diluted to 2 ng/μL with purified water.
Polymerase chain reaction (PCR) and microsatellite genotyping
The microsatellite loci and primer sequences used in this study were derived from a database of ESTs developed for sunflowers by the Compositae Genome Project database (CGP; http://cgpdb.ucdavis.edu/). Only those loci that amplified well across all species and had been genetically mapped (Burke et al. 2002; Tang et al. 2002; Lai et al. 2005) were employed for population surveys. The 5′ end of each forward primer had an extension, complementary to M13 universal primer, labelled with one of four dyes FAM, VIC, NED and PET (Schuelke 2000).
Amplification was done using a touchdown polymerase chain reaction (PCR) protocol, designed to reduce nonspecific primer associations and subsequent arbitrary fragment amplification (Don et al. 1991). An initial denaturing step of 3 min at 95° was followed by 10 touchdown cycles of 30 s at 94 °C, 30 s at the annealing temperature (58° with a decrease of 1° each cycle), and 45 s at 72°. The touchdown cycles were followed by 29 cycles of 30 s at 94°, 30 s at 48° and 45 s at 72°. The final step was a 20 min elongation phase at 72°.
Allele identification was performed using an ABI3730 capillary sequencer (Applied Biosystems, Foster City, CA). One μL of amplification product from each of four loci, each labelled with a different dye, were pooled and diluted in 54 μL ddH2O product (1: 60). One μL of the diluted PCR was then added to a 9 μL mixture of 0.1 μL of GenScan-500 Liz Size Standard (Applied Biosystems) and 8.9 μL ddH2O. Samples were denatured for 5 min at 95° and cooled immediately on ice before they were loaded on the sequencer. Chromatographs of genotyping data were generated and fragment lengths were scored using GENEMAPPER 3.0 (Applied Biosystems).
Analyses of microsatellite data
Variance in repeat number, genetic diversity and global FST (Weir & Cockerham 1984) were computed for each locus using MSAnalyser (Dieringer & Schlötterer 2003). For three loci, only one allele was detected for H. anomalus (HT715 and HT941) or H. petiolaris (HT716). In these cases, the calculated variances are zero. To avoid dividing by zero in calculations of ln RV, we followed Edelist et al. (2006) by adding a single allele that differed by one repeat unit from the most abundant allele. This results in a slight underestimation of ln RV or ln RH, respectively, for the three loci.
Both ln RV and ln RH were computed for all loci for the following three comparisons: H. anomalus vs. H. annuus (ln RVano/ann and ln RHano/ann), H. anomalus vs. H. petiolaris (ln RVano/pet) and ln RHano/pet and H. annuus vs. H. petiolaris (ln RVann/pet and ln RHann/pet). To account for multiple comparisons, we used the sequential Bonferroni correction (Rice 1989) as follows. For each index we ranked all the values from the three interspecific comparisons. Because the indices did not show significant deviations from a normal distribution, significance levels could be computed as the mean of the normal probability distribution for each category (ano/ann, ano/pet or ann/pet) minus 1 − (alpha/k), where alpha is 0.05 (one-tailed test) and k is the rank of the ln RV or ln RH value (one is the highest value and 177 the lowest). Given that our goal is to identify candidate loci for further investigation, it might be argued that the use of a sequential Bonferroni correction is overly conservative and some positively selected genes may be missed (Moran 2003). However, as described later, there is no shortage of candidates, so it seemed best to err on the side of caution.
Because selection leaves reduced variability in the genomic region flanking the selected site (Maynard-Smith & Haigh 1974), analyses of multiple, linked markers can reduce false positives (Wiehe et al. 2007). The width of the sweep can be inferred from the same pattern, if there is sufficient density of markers.
Here, we employed a modification of the multilocus test of Wiehe et al. (2007) to ask whether the pattern of heterozygosity at loci linked to outlier ln RH values is more correlated than expected by chance. We first standardized all ln RH values:
For each outlier locus i we summed the values for adjacent loci from both sides along the linkage group to create the test statistic
where n is the number of loci included in the test and the ln RH value of the outlier locus is not included in the summation. Because ln RH does not show significant deviation from a normal distribution (see Results), the distribution of T(n) is approximately normal with a mean of ca. 0 and variance n (Wiehe et al. 2007). For each outlier locus we estimated the effect of the putative sweep by testing the significance of T(n) at increasing recombinational distances from the target locus.
To estimate the proportion of genetic variance distributed within and among populations and species, we performed hierarchical analysis of molecular variance (AMOVA) using ARLEQUIN 3.0 (Excoffier et al. 2005). Relationships among hybrid and parental species genotypes were revealed by Principle Coordinate Analysis (PCoA), using GENALEX6 (Peakall & Smouse 2006). We analyzed the dataset twice: first with all the loci, and then only with loci that showed significant reductions in variation or heterozygosity. The goal of the separate analyses was to investigate the possible role of selection in differentiating the species, as opposed to reduced gene flow resulting from reproductive isolation. Lastly, we computed FST for each locus as a measure of genetic differentiation among species, using F-STAT (version 2.9.3; http://www.unil.ch/izea/softwares/fstat.html).
Gene-product attributes
cDNA sequences of outlier loci (see Results) were obtained from the CGP database and compared to the TAIR7 Arabidopsis thaliana genome release (www.arabidopsis.org). The best A. thaliana hit for each transcript was determined using BLASTX (Altschul et al. 1997) with a minimum e-value of 10−10. Genes with A. thaliana annotations were categorized according to cellular component, molecular function and biological process using Gene Ontology (GO) functional groups, based on the Arabidopsis genome annotation (The Gene Ontology Consortium 2000). Attributes of the putative products of the outlier loci were further explored using InterProScan (http://www.ebi.ac.uk/InterProScan/index.html; The InterPro Consortium 2001; Zdobnov & Apweiler 2001). This web-based program searches for protein families, domains and sites by comparing an input sequence with all six reading frames against various protein databases. Minimum open-reading-frame size was set to 90 bases.
Gene sequencing
We sequenced two of the seven loci that showed reduced allelic diversity (HT953 and HT998) both to confirm the results of the microsatellite analyses and to identify sites that might be the target of selection. Only two loci were sequenced. The other five were not sequenced because of difficulties obtaining sequences from the cDNA library of the ESTs or designing primers that would amplify the coding region. Twelve individuals each from H. petiolaris, H. annuus and H. anomalus, providing broad coverage of the geographical distribution in Utah, were sampled for sequencing. Loci were PCR-amplified from genomic DNA using PCR primers designed utilizing sequence data from the sunflower EST database (953F-GTAATGAAAGACGATGACG; 953R-GTTTAGTTTCAACGACATTACTG; 998F-GGAGTGCAGAACCTTAGACATC; 998R-CATGAAATATATGCCATGCCAAGATC). Ten μL PCR reactions included 2 ng/μL DNA, 1 U taq DNA polymerase, 10 ng/μL each primer, 2.4 μL taq buffer (200 mM tris-HCl, 100 mM KCl, 100 mM (NH4)2SO4, 20 mM MgCl2, 1%Triton, 1 mg/mL BSA) and H2O to volume. Fragments were amplified using a touchdown protocol: an initial denaturing cycle of 3 min at 95 °C was followed by 10 touchdown cycles (temperature drops 1 °C each cycle) of 30 s at 94 °C, 30 s starting at 62 °C and 45 s at 72 °C. These 10 cycles were followed by 29 cycles of 30 s at 94 °C, 30 s at 52 °C and 45 s at 72 °C followed by an elongation period of 10 min at 72 °C. Final product was cleaned using ExoSap-It (USB, Cleveland, OH). PCR products were diluted to 10–20 ng/μL for sequencing reactions. Sequencing reactions included 1 μL PCR product, 7.5 μL ddH2O, 0.6 μL of 25 mM MgCl, 0.4 μL primer (953F, 998F & R) and 0.5 μL BigDye (Applied Biosystems). Sequences obtained for locus 953 were relatively short (see Results), and the forward primer (953F) was sufficient to sequence it completely. Locus 998 was longer (see Results) and was sequenced from opposite directions using forward and reverse primers (998F & R). Sequencing was performed using an ABI3730 capillary sequencer (Applied Biosystems). Sequence chromatograms were visualized and edited using SEQUENCHER 4.1.2 (Gene Code Corp.). Alignments were performed using MACCLADE with amino-acid translations (Maddison & Maddison 2000). When heterozygotes were detected in HT998 in which frame shifts led to unreadable sequence, the PCR product from these accessions was cloned using the TOPO TA cloning kit (Invitrogen™). Given our specific interest in sequence variation, haplotypes from heterozygotes with simple polymorphic sites were estimated using homozygote sequences as template. These haplotypes may not be exact but represent the variability in sequence data present in the two heterozygote phases. Following alignment, coding regions were identified by translating the consensus DNA sequence to protein sequence, using both ORF Finder (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and ExPASy (http://www.expasy.ch/tools/dna.html). The UTR sequence was removed before further analyses. Sequences and alignments are available in GenBank (Accession nos EF165367–EF165469). We used DNASP version 4.10.4 (Rozas et al. 2003) for calculating variability statistics and performing neutrality tests.
To detect reductions in variation in the sequence data, we used an index ln RVi (analogous to ln RV), which is the natural logarithm of the ratio of variance at a locus between two populations:
where π is nucleotide diversity in a sequence (Nei & Li 1979; Nei & Kumar 2000) in populations i and j. In a random mating population, π is simply heterozygosity in the nucleotide level (Nei & Kumar 2000) and hence has some similarity to ln RH. However, this condition is violated in our study by the pooling of populations within a species. Hence, we treat ln RVi as a measure of reduction in sequence diversity, equivalent to ln RV. Ln RVi was computed by calculating π for each of the three species at each locus by using a ‘sliding window’ (sizes 100 and 50) with step sizes of 10 and 25 under each window size along the sequences in DNASP and then calculated ln RVi for each window across the two loci. The sliding window analysis was intended to determine whether putative sweeps affect variation across the entire locus (as assumed by the general hitch-hiking mapping approach employed here) or whether the affects of the sweeps are more local.
Two neutrality tests were performed: Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997). Both tests detect local skews in the frequency spectrum of polymorphisms due to population expansion or selection, but Fu’s Fs is more sensitive to the former (Fu 1997). Tajima’s D was calculated using DNASP (Rozas et al. 2003), whereas Fs determined using ARLEQUIN 3.0 (Excoffier et al. 2005). In addition, we performed a sliding window analysis (window sizes 50 and 100, step size 10 and 25) of the ratio between nonsynonymous (Ka) and synonymous substitutions (Ks) using each species individually as the reference population in comparison to the other two species. Ka and Ks values were normalized as the number of nonsynonymous or synonymous substitutions per nonsynonymous or synonymous site, respectively. The analysis of Ka/Ks is based on the expectation that within coding regions, synonymous substitutions occur more frequently than nonsynonymous (KA) changes. In general, Ka/Ks ratios of > 1 are considered evidence of positive selection. Ka/Ks ratios were estimated following Nei & Gojobori (1986), and the sliding window analysis was implemented in DNASP (Rozas et al. 2003).
Lastly, maximum likelihood (ML) methods were employed to test for positive selection and positively selected sites (Suzuki & Gojobori 1999) in HT998, using codeml as implemented in PAML (Yang 2000). Additional Helianthus taxa recovered from GenBank and the Helianthus EST database were added to the data set (H. ciliaris Cl313, H. exilis CHES7914, H. paradoxus CL488 & CHUS6029, H. petiolaris CL143.1 and 2, H. tuberosus CHTM1878.b1). These taxa provided outgroups and increased power under likelihood methods (Yang et al. 2000). Models M0 (one-ratio), M1 (neutral), M2 (selection), M7 (beta) and M8 (beta & ω) were implemented (Nielsen & Yang 1998; Yang et al. 2000) using an underlying neighbour-joining tree estimated using PAUP* (Swofford 2000). Models M2 and M8 are designed to test for positive selection (Yang et al. 2000) and have been shown to perform well under both real and simulated data (Wong et al. 2004). We tested for positive selection using −2[ln(H0) − ln(H1)] as a test statistic, where M1 and M7 are H0 and M2 and M8 are H1, respectively. Significance was obtained from a chi-square distribution using a likelihood-ratio test (LRT), with one degree of freedom (Yang et al. 2000).
Results
Pattern of variation across the genome
A total of 59 loci were genotyped across all three species. The values for all comparisons with significant values for either of the two indices are given in Table 2. For all loci and all species comparisons, the distribution of values of ln RV and ln RH did not differ significantly from normality (Kolmogorov–Smirnov test; P > 0.1 for all analyses), as expected from computer simulations (Schlötterer 2002; Schlötterer & Dieringer 2005). However, the mean values of ln RV and ln RH were negative in comparisons of Helianthus anomalus to either H. annuus (mean ln RVano/ann = −0.47, mean ln RHano/ann = −0.65) or H. petiolaris(mean ln RVano/pet = −0.89, mean ln RHano/pet = −0.79) and deviated significantly from zero (P < 0.05 for all analyses). These negative values may be due to a population bottleneck during the origin of H. anomalus (Ungerer et al. 1998). To account for the apparent bottleneck effect, we tested for significant deviations from the mean, rather than from zero (Wiehe et al. 2007). Note that this correction makes the tests for reduction in variation/heterozygosity more conservative, relative to the significance level proposed by Schlötterer (2002) and Schlötterer & Dieringer (2005). Values for all loci are provided in Table S1 (Supplementary materials).
Table 2.
Outlier loci for comparisons between Helianthus anomalus and its two parental species (H. annuus and H. petiolaris): ano, H. anomalus; ann, H. annuus; pet, H. petiolaris; LG, linkage group; cM, map location on the LG. Asterisks denote significant reduction of variation or heterozygosity following the sequential Bonferroni correction for multiple comparisons
| ln RV |
ln RH |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Locus | LG | cM | FST | ano-ann | ano-pet | ann-pet | ano-ann | ano-pet | ann-pet |
| HT715 | 17 | 29.1 | 0.566 | −7.20* | −5.53* | 1.67 | −7.97* | −7.65* | 0.32 |
| HT719 | 17 | 76.5 | 0.405 | −3.44 | −4.11 | −0.66 | −4.93* | −4.21 | 0.72 |
| HT813 | 15 | 104.8 | 0.355 | −4.42* | −4.68* | −0.26 | −1.06 | −1.98 | −0.92 |
| HT941 | 17 | 23.6 | 0.142 | −3.93* | −3.41 | 0.52 | −3.11 | −4.26* | −1.15 |
| HT945 | 17 | 83 | 0.236 | 7.54 | 0.03 | −7.52* | 0.30 | −0.53 | −0.83 |
| HT953 | 15 | 109.1 | 0.429 | −4.91* | −5.26* | −0.34 | −6.40* | −7.17* | −0.77 |
| HT998 | 17 | 68.1 | 0.048 | −0.32 | −5.73* | −5.41* | −0.41 | −0.28 | 0.12 |
Six loci exhibited a significant reduction in variation or heterozygosity in H. anomalus, compared to one or both of the parental species (Table 2). Two of these (HT715 and HT953) represent strong candidates because both test statistics were significant for comparisons of H. anomalus to both parents. The remaining four loci (HT719, HT813, HT941 and HT998) are weak candidates because they were significant in only a subset of the comparisons made (Table 2). Interestingly, HT998 exhibited reduced variability in comparisons of H. anomalus to H. annuus and H. petiolaris to H. annuus, but not in comparisons of H. anomalus to H. petiolaris, implying a loss of variation in multiple species, a hypothesis confirmed by sequence data (see below). An additional locus (HT945) differed in variability levels between the parental species.
For three linkage groups (LG13, LG15 and LG17), there were sufficient loci to examine the pattern of selection along a chromosome (Fig. 1). Map distances are from the framework map for the common sunflower (Tang et al. 2002; S. K. Knapp, personal communication). Markers linked to outlier values of ln RH showed no correlation in variability levels (based on significance of T(k); see Methods), indicating that these are false positives or that swept segments are smaller than the distance to the nearest locus genotyped. Given the low density of markers sampled, the latter hypothesis seems likely. Note that LG13 contains no significant reduction in any of the loci.
Fig. 1.

Pattern of interspecific variation along three linkage groups. ln RV and ln RH (vertical axis) of Helianthus anomalus compared to genotypes of parental species H. annuus (ano-ann) and H. petiolaris (ano-pet) and plotted against the recombinational distance (in centi-Morgans) along the linkage group. Asterisks denote loci with significant outlier values of ln RV or ln RH, indicating strong reductions in variation or heterozygosity in H. anomalus compared to the correspondent parental species.
Gene-product attributes
The predicted proteins for three of the seven outlier loci had no recognizable domains (Table 3), possibly indicating that they are sunflower-specific genes or rapidly evolving genes lacking recognizable homologies with proteins from other organisms. InterPro Scan detected a protein domain match for the other four outlier loci, and GO annotations could be assigned to three of them (Table 3). HT719 contains a domain that is possibly related to light regulation (InterPro annotation: IPR009856) but was not significantly assigned to any GO biological process or molecular function. HT813 showed homology to Carotene hydroxylase (IPR005596; GO:0042411). HT953 appears to be a Zinc finger, PHD-type protein (IPR001965), which are thought to be involved in chromatin-mediated transcriptional regulation. Lastly, HT998 contains a Ca2+-dependent membrane-targeting module (IPR000008) that may target proteins to the vacuole (GO:0000326). However, these data are too few and too general to make useful conclusions about function or to relate them to ecological adaptation in H. anomalus.
Table 3.
Attributes of putative gene products of seven outlier loci
| Locus | GO biological process | GO cellular component | GO molecular function | InterPro domain annotation (Acc. no.) |
|---|---|---|---|---|
| HT715 | Unknown | Unknown | Unknown | Unknown |
| HT719 | Unknown | Chloroplast (GO:0009507) | Unknown | Light regulated Lir1 protein (IPR009856) |
| HT813 | Carotene metabolic process (GO:0016119); Xanthophyll biosynthetic process (GO:0016123) | Chloroplast (GO:0009507) | β-Carotene hydroxylase activity (GO:0042411) | Carotene hydroxylase (IPR005596) |
| HT941 | Unknown | Unknown | Unknown | Unknown |
| HT945 | Unknown | Unknown | Unknown | Unknown |
| HT953 | regulation of transcription, DNA-dependent (GO:0006355) | Unknown | DNA binding (GO:0003677) | Zinc finger, PHD-type (IPR001965) |
| HT998 | Protein targeting to vacuole (GO:0006623) | Protein storage vacuole (GO:0000326); Endoplasmic reticulum (GO:0005783) | Protein binding (GO:0005515) | C2 Calcium-dependent membrane targeting (IPR000008) |
Differentiation among populations and species
Hierarchical analysis of molecular variation (AMOVA; Table 4) revealed that approximately half of the molecular variance is partitioned among species, with the remainder approximately equally distributed within and among populations within species. Surprisingly, a larger proportion of the among-species variation is due to comparisons between H. anomalus and its parents (22.2% H. annuus and 16. 8% H. petiolaris) rather than comparisons between the parental species (13.4%).
Table 4.
Analysis of molecular variance (AMOVA) within and among populations and species of Helianthus
| Source of variation | d.f. | Sum of squares | Variance components | % of variation | Fixation index | P | |
|---|---|---|---|---|---|---|---|
| H. anomalus vs. H. annuus | Among species | 1 | 130.345 | 0.46 | 22.20 | FCT = 0.222 | < 0.001 |
| Among populations within species | 5 | 68.841 | 0.15 | 7.05 | FSC = 0.091 | < 0.001 | |
| Within populations | 281 | 452.943 | 0.14 | 7.01 | FIT = 0.363 | < 0.001 | |
| Total | 575 | 1032.628 | 2.07 | 100 | |||
| H. anomalus vs. H. petiolaris | Among species | 1 | 101.056 | 0.33 | 16.78 | FCT = 0.167 | < 0.001 |
| Among populations within species | 5 | 89.477 | 0.19 | 9.93 | FSC = 0.119 | < 0.001 | |
| Within populations | 281 | 444.365 | 0.13 | 6.81 | FIT = 0.335 | < 0.001 | |
| Total | 575 | 1012.898 | 1.97 | 100 | |||
| H. annuus vs. H. petiolaris | Among species | 1 | 65.091 | 0.25 | 13.41 | FCT = 0.134 | < 0.001 |
| Among populations within species | 4 | 67.375 | 0.24 | 12.62 | FSC = 0.146 | < 0.001 | |
| Within populations | 186 | 316.703 | 0.32 | 16.87 | FIT = 0.429 | < 0.001 | |
| Total | 383 | 1407.948 | 1.87 | 100 |
PCoAs of individual plant genotypes clearly separated H. anomalus and the two parental species along the first and third axes, whereas in the second axis, H. anomalus occurs intermediate between the two parental species (Fig. 2A,B). Interestingly, H. anomalus individuals segregated along the third axis according to geography, with individuals from eastern (Hanksville) and western (Little Sahara) Utah occurring in discrete clusters (Fig. 2B). No geographical separation was found after re-analysis using only the seven outlier loci (Fig. 2C, D).
Fig. 2.

Principal Coordinate Analysis of individual plant genotypes. Axes titles include the percentage variance explained by that axis in parentheses. anoB, Helianthus anomalus, Hanksville area populations; anoC, H. anomalus, Little Sahara populations; ann, H. annuus; pet, H. petiolaris. A and B are plots for all loci, C and D are plots for only the six loci with significant reductions in variation.
Candidate genes — sequence analyses
One hundred and sixty five bases of coding sequence were obtained from 29 individuals for locus HT953. Among the three species there were six polymorphic sites in HT953, all substitutions were synonymous and none were fixed in any of the three species. For locus HT998, 34 individuals were sequenced for 699 bases. There were 75 polymorphic sites with a total of 54 synonymous and 21 nonsynonymous changes. One nonsynonymous change is found solely in all H. anomalus, although three heterozygote individuals retain an allele lacking the change (sequence alignments provided in Tables S2 and S3, Supplementary materials). General measurements of variability within and among taxa are presented in Table 5. H. anomalus sequences from locus HT998 are more similar to H. petiolaris than to H. annuus, implying that the hybrid inherited this locus from H. petiolaris. Unfortunately, there was too little sequence variation in HT953 to infer parentage at this locus.
Table 5.
Sequence variability within and among species for locus HT998. N, # individuals; H, # heterozygotes; Pi, nucleotide diversity; Nps, # polymorphic sites within species; Ka/Ks, # nonsynonymous/synonymous polymorphisms among species or within species (last three columns)
| N | H | Pi | Nps | Ka/Ks | Ka/Ks ann | Ka/Ks pet | Ka/Ks ano | |
|---|---|---|---|---|---|---|---|---|
| Helianthus annuus | 12 | 9 | 0.022 | 48 | 8/36 | — | 19/44 | 16/47 |
| H. petiolaris | 11 | 8 | 0.014 | 40 | 8/18 | 19/44 | — | 21/44 |
| H. anomalus | 11 | 8 | 0.020 | 45 | 6/22 | 16/47 | 21/44 | — |
Tajima’s D (Tajima 1989) and Fu’s Fs were not significant for either locus in any of the three species, although for HT953, Fs was close to the significance threshold in H. annuus [Fs = −2.8, P = 0.029; significance cut-off = 0.02 (Fu 1997)]. The sliding window analysis of HT998 identified a high Ka/Ks ratio (> 1) between DNA sequence positions 350–450 (Fig. 3) regardless of which species is treated as the source species and under all analysis parameters (i.e. window sizes 100 or 50 bases, step size 10 or 25 bases). ln RVi does show a decline for H. anomalus relative to H. annuus or H. petiolaris in this sequence region under all analysis parameters (i.e. there is lower sequence variability for H. anomalus), but this is not unique to this region of the sequence with other regions where ln RVi is much lower (Fig. 4). Interestingly, the H. anomalus sequence variability is roughly similar when compared to either of the parental species from the start of the sequence roughly to position 450, while the pattern of sequence variability between H. petiolaris vs. H. annuus is unique. Between positions 535 and 590, H. annuus shows low variability compared to H. petiolaris and H. anomalus (Fig. 4).
Fig. 3.
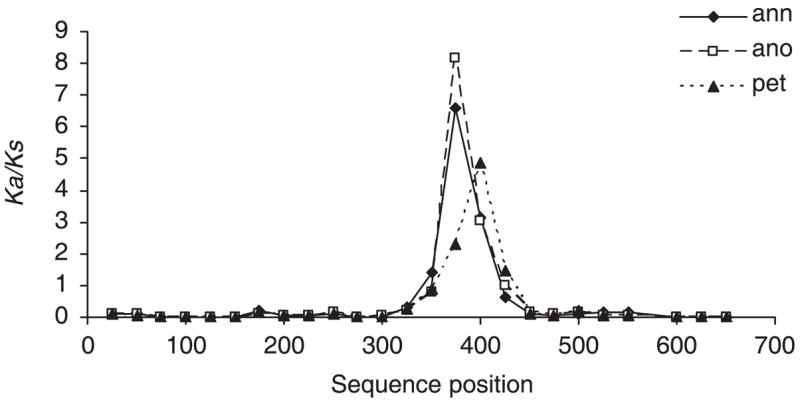
Ka/Ks for HT998 treating each species’ sequence as the source data for comparison. ano, Helianthus anomalus; ann, H. annuus; pet, H. petiolaris. Considered significant at Ka/Ks > 1. Values are for sliding window of 50 bases with step size of 25 bases.
Fig. 4.
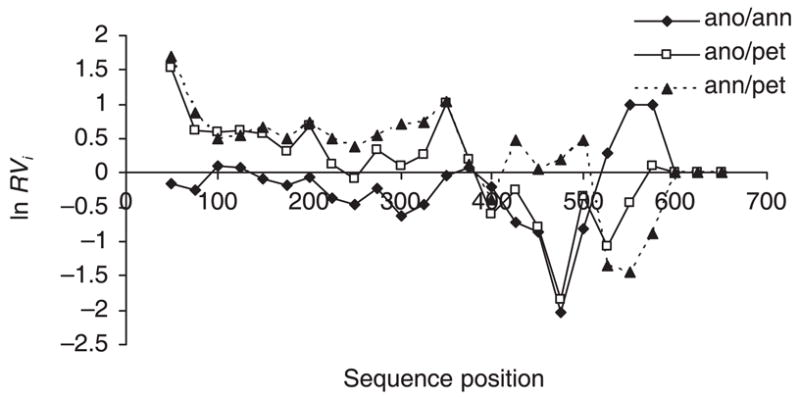
The ratio of nucleotide diversity between Helianthus anomalus and parental species as well as H. annuus vs. H. petiolaris for HT998. ln RVi is the natural logarithm of the ratio between π (H. anomalus) and π (parental species). Values are for a sliding window of 50 bases with step size of 25 bases.
In addition to point mutations, length variation was observed at multiple positions. Notably, at positions 373–375 we found a gap of three bases (one codon; Ala) in sequences of H. annuus, which was not found in H. anomalus and H. petiolaris. The amino acid immediately adjacent to this domain is conserved among H. annuus and H. petiolaris (Ala), while there is a unique variant (Thr) among most H. anomalus individuals at this position in the sequence, although two haplotypes retain (Ala). We used PredictProtein (Rost et al. 2003) to identify putative functional domains in the peptide coded by HT998. Results from the PROSITE motif search (Hulo et al. 2006) imply that the site with a codon deletion in H. annuus (positions 373–375 of the DNA sequence) is located adjacent to the start of an N-myristoylation motif (PROSITE Accession no.: PS00008). Myristate is a fatty acid added to the N-terminal of a protein and alters the physical and functional properties of the modified protein (Towler et al. 1988; Farazi et al. 2001). In addition to the N-myristoylation motif identified, a C2-domain was also recognized by InterProScan. This suggests that HT998 codes for a signalling protein that interacts with cellular membrane proteins and could potentially be involved in a broad array of biological processes (Nalefski & Falke 1996).
Multiple indels were detected between positions 502 and 612. This length variation is not consistent across taxa and varied among haplotypes in heterozygous H. anomalus and H. petiolaris. The microsatellite sequences appear to be positioned within the coding region and not in the UTR, as is the microsatellite in HT953.
Under ML tests for selection of HT998, the models that specify parameters for selection (M2, M8) outperformed those that do not [M1, M7; see Yang et al. (2000) for model details] as measured by likelihood estimates. Under the likelihood ratio test, M2 outperformed M1 (P < 0.01), as did M8 compared to M7 (P < 0.001). Both M2 and M8 identified positively selected sites (P < 0.05; Table 6). These sites did not correspond to the sequence region (350–450) displaying a high Ka/Ks ratio. M2 and M8 identified 1.4% and 2.7% of sites, respectively, under strong positive selection.
Table 6.
Likelihood values (ln L) and parameters for locus HT998 using paml. For details of the parameters estimated in each model see Yang et al. (2007)
| Models | ln L | dn/ds | Estimated parameters | Selected sites |
|---|---|---|---|---|
| M0 (one-ratio) | −1786.072 | 0.189 | ||
| M1 (neutral) | −1736.020 | 0.158 | p0 = 0.862 p1 = 0.138 | |
| M2 (selection) | −1731.296 | 0.197 | p0 = 0.863 p1 = 0.123 p2 = 0.014 | 80 |
| M7 (beta) | −1737.501 | 0.143 | p = 0.043 q = 0.259 | |
| M8 (beta&ω) | −1731.206 | 0.195 | p0 = 0.973 p1 = 0.027 p = 0.080 | 68, 80 |
| q = 0.580 w = 2.902 |
Discussion
Patterns of reduced genetic diversity in Helianthus anomalus
In this study, we identified several genomic regions that had experienced significant reductions in allelic variation or genetic diversity in a homoploid hybrid species, Helianthus anomalus. Previous studies showed that H. anomalus is reproductively isolated from its parental species by chromosomal rearrangements (Lai et al. 2005), and that niche differentiation of H. anomalus created ecological isolation from the parental species (Ludwig et al. 2004). Here, we show that some genomic regions in the hybrid species are less diverse than the corresponding regions in the parental species’ genome. The abrupt changes in variability we found along linkage groups (Fig. 1) suggest that reduced variability in these outlier loci may be due to positive selection on advantageous alleles (although see below) rather than the fixation of a new homokaryotype; otherwise larger regions along the chromosome would show correlated reductions in variation. Also, the outlier loci do not map closely to chromosomal breakpoints.
While selection is one possible cause of reduced allelic variability at a given locus (e.g. Maynard-Smith & Haigh 1974; Harr et al. 2002; McDaniel & Shaw 2005; Edelist et al. 2006), reduced variance may also result from intermediately severe population bottlenecks or selection at a linked locus (Wiehe et al. 2007). Thus, while we provide a ranked list of candidate genes, the role of selection needs to be assessed by additional sequencing of the candidate genes, as well as by sequencing of neutral genes for comparative purposes.
Reduction in genetic variation in a species relative to its ancestors can be explored in various ways. The indices we used, ln RV and ln RH, control for variation in mutation rates among loci as well as genome-wide effects on variation due to population-size expansions or contractions, sampling design and population admixture (Schlötterer 2002; Schlötterer & Dieringer 2005). These ‘model-free indices’ are advantageous over model-based approaches when the historical demography of populations is poorly understood, when the assumptions of models are potentially violated and when mutation rates vary across loci, such as in the present study (Beaumont 2005; Hedrick 2005; Storz 2005).
Despite the supposed robustness of the ln RV and ln RH statistics to fluctuations in population size, the mean values of ln RV or ln RH, respectively, were significantly lower than zero. This implies that H. anomalus experienced a population bottleneck during its recent evolutionary history, which decreased overall genetic variation compared to the parental species. By standardizing to a zero mean, however, we were able to identify loci with more extreme reductions in genetic variation. Molecular analyses of numerous loci currently underway will provide better insight into the historical demography of the species (J. Strasburg and L. Rieseberg, unpublished).
Following correction for the apparent bottleneck, seven outlier loci passed our significance thresholds, of which six were due to reduction in variability in H. anomalus compared to one or both the parental species. While at least three false positives are expected by chance at a 95% confidence level (Storz 2005; Wiehe et al. 2007) and reduced variation may have other causes (above), the six loci can be viewed as candidate loci, with the two loci exhibiting low ln RV and ln RH indices when compared with both parental species, representing the strongest candidates for further study.
Identification of genes and specific molecular changes underlying adaptive divergence in wild populations is an important goal of evolutionary biology (Orr & Coyne 1992), but only recently has this become possible outside of model systems (e.g. Hoekstra et al. 2006). Unfortunately, the products of the outlier genes in the present study either could not be annotated or could only be annotated with attributes that are too general to evaluate a possible link with dune adaptation. We are currently developing transformation methods for sunflowers, which will make it possible to evaluate the function of these loci more directly.
We also generated sequence data for two of the candidate loci, but population genetic tests, i.e. Tajima’s D and Fu’s Fs, failed to detect deviations from neutral evolution at either locus. Nonetheless, ML analyses detected past selection in one of the two genes (HT998; Table 6) with strong support for two specific selected sites (68 and 80). Additionally, an increased ratio of nonsynonymous to synonymous site substitutions was found in all three species between positions 350 and 450 (Fig. 3) and was accompanied by changes in the physical length of the gene. This genic domain was associated with a putative N-myristoylation motif, but because protein myristoylation contributes to many pathways, such as salt tolerance, apoptosis and signal transduction activity (Ishitani et al. 2000; Farazi et al. 2001; de Vries et al. 2006), it is not possible to infer its role in ecological adaptation of these sunflower species at the present time. Even the recognition of the N-myristoylation motif should be taken with caution, because it has taxon-specific differences, and the motif recognized by the PROSITE program appears to have a high rate of false positives (Maurer-Stroh et al. 2002). The low specificity may explain why the N-myristolation mofit was not found by INTERPROSCAN.
Although ln RVi exhibited a decline in sequence variability in H. anomalus relative to its parental species at locus HT998, this reduction was not limited to a specific site, but rather to the entire sequence analyzed. The loss of diversity could result from a bottleneck during the speciation process or to a recent selective sweep as implied by the ln RH/ln RV data. Lastly, it is unclear why the sites identified by ML tests as being under selection were different from those identified by other analyses. Clearly, further investigation is needed to explore the role of selection in shaping the pattern of sequence and protein evolution at this locus.
Population and species differentiation
AMOVA results, as well as the PCoA, showed high differentiation among species relative to the differentiation among populations within species (Table 4 and Fig. 2). The hybrid species was separated from its parents along the first multivariate axis, whereas the parental species were separated on the second axis only. This seemingly anomalous observation appears to be due to continued gene flow between H. annuus and H. petiolaris subsequent to the origin of H. anomalus (Yatabe et al. 2007). This pattern is similar to that found in another homoploid hybrid sunflower species, H. paradoxus (Edelist et al. 2006) and may result from both the independent evolution of H. anomalus after hybrid speciation, as well as ongoing gene flow or introgression between the parental species.
Interestingly, there was genetic differentiation between the eastern and western populations of H. anomalus when all loci were included in the analysis, but not when the analysis was restricted to outlier loci (Fig. 2). This implies that (i) the putative sweeps detected in the present study are species-wide rather than restricted to local populations; and (ii) the genetic differences at neutral loci are a consequence of reduced gene flow rather than differential adaptation.
Implications for ecological speciation
So why is there such a high frequency (ca. 10%) of loci that showed extreme reductions in variability in the H. anomalus genome? One possibility is that a relatively large number of genetic changes have been targeted by selection during the origin and evolution of H. anomalus. Alternatively, the high proportion of outlier loci in the H. anomalus genome might be a byproduct of one or more population bottlenecks, which are known to increase the variance in diversity among loci (Aldrich et al. 1998).
The abrupt changes in variability across the genome, even between very closely linked genes (Fig. 1), implies that the sizes of the hitch-hiked fragments detected in this study were small; seemingly much smaller than in another sunflower hybrid species, H. paradoxus, where microsatellites several centi-Morgans from selected QTLs exhibited a significant reduction in variability (Edelist et al. 2006). The reason for this difference is not clear, although H. anomalus appears to have a more ancient origin (Schwarzbach & Rieseberg 2002) and the sweeps detected here probably occurred subsequently to its origin, and their hitch-hiking effect would therefore be smaller due to recombination.
Finally, our results suggest that selective sweeps may have united populations of H. anomalus that are isolated by a mountain range (eastern vs. western Utah; Fig. 2). This finding provides additional support for the view that even species with low gene flow and a highly subdivided population structure may be held together by the spread of advantageous alleles (Morjan & Rieseberg 2004).
Supplementary Material
The following supplementary material is available for this article:
Table S1 Loci genotyped in this study and values for relative reduction in variation (lnRV) and heterozygosity (lnRH) in the hybrid species (Helianthus anomalus) compared to each of its parental species (H. annuus, and H. petiolaris).
Table S2 Sequence alignment for locus HT953 sequenced in three Helianthus species.
Table S3 Sequence alignment for locus HT998 sequenced in three Helianthus species.
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1365-294X.2007.03557.x (This link will take you to the article abstract).
Acknowledgments
We thank Mike Barker, Melania Cristescu, Yoko Yatabe, Nolan Kane, Amanda Posto, Jared Strasburg and other members of the Rieseberg’s lab for help, suggestions and comments during the study. We thank Briana Gross for extracting DNA from populations of parental species, Steve Knapp for sharing with us unpublished mapping data and Roger Bivand, Alex Buerkle, Daniel Dieringer, Christian Schlötterer and Peter Smouse for advice on various aspects of the statistical analysis. A previous version of the manuscript was improved by comments of two anonymous reviewers. This study was supported by a grant from the US National Institute of Health (GM059065) to LHR. YS was supported by a postdoctoral award no. FI-353–2004 from BARD, The United-States — Israel Binational Agricultural Research and Development Fund.
Biography
Yuval Sapir is interested in reproductive biology and ecological speciation of plants. Michael Moody is interested in phylogenetic approaches to detect reticulated evolution in plants. Beau Brouillette is interested in plant responses to abiotic stress. Lisa Donovan studies plant evolutionary ecophysiology, with an emphasis on stress and resource use traits as they relate to plant performance. Loren Rieseberg studies the genetics of hybridization and speciation in plants.
Footnotes
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aldrich PR, Hamrick JL, Chavarriaga P, Kochert G. Micro-satellite analysis of demographic genetic structure in fragmented populations of the tropical tree Symphonia globulifera. Molecular Ecology. 1998;7:933–944. doi: 10.1046/j.1365-294x.1998.00396.x. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson E. Introgressive Hybridization. John Wiley; New York: 1949. [Google Scholar]
- Arnold ML. Evolution Through Genetic Exchange. Oxford University Press; Oxford: 2006. [Google Scholar]
- Barton NH. The role of hybridization in evolution. Molecular Ecology. 2001;10:551–568. doi: 10.1046/j.1365-294x.2001.01216.x. [DOI] [PubMed] [Google Scholar]
- Beaumont MA. Adaptation and speciation: what can FST tell us? Trends in Ecology and Evolution. 2005;20:435–440. doi: 10.1016/j.tree.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Brouillette LC, Gebremedhin DM, Donovan LA. Testing hypothesized evolutionary shifts toward stress tolerance in hybrid Helianthus species. West North American Naturalist. 2006;66:409–419. [Google Scholar]
- Buerkle CA, Morris RJ, Asmussen MA, Rieseberg LH. The likelihood of homoploid hybrid speciation. Heredity. 2000;84:441–451. doi: 10.1046/j.1365-2540.2000.00680.x. [DOI] [PubMed] [Google Scholar]
- Burke JM, Tang S, Knapp SJ, Rieseberg LH. Genetic analysis of sunflower domestication. Genetics. 2002;161:1257–1267. doi: 10.1093/genetics/161.3.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. Speciation. Sinauer Associates; Sunderland, MA: 2004. [Google Scholar]
- Dieringer D, Schlötterer C. MICROSATELLITE ANALYSER (MSA): a platform independent analysis tool for large microsatellite data sets. Molecular Ecology Notes. 2003;3:167–169. [Google Scholar]
- Dieringer D, Nolte V, Schlötterer C. Population structure in African Drosophila melanogaster revealed by microsatellite analysis. Molecular Ecology. 2005;14:563–573. doi: 10.1111/j.1365-294X.2004.02422.x. [DOI] [PubMed] [Google Scholar]
- Don R, Cox P, Wainwright B, Baker K, Mattick J. Touchdown PCR to circumvent spurious priming during gene amplification. Nucleic Acids Research. 1991;19:4008–4008. doi: 10.1093/nar/19.14.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelist C, Lexer C, Dillmann C, Sicard D, Rieseberg LH. Microsatellite signature of ecological selection for salt tolerance in a wild sunflower hybrid species, Helianthus paradoxus. Molecular Ecology. 2006;15:4623–4634. doi: 10.1111/j.1365-294X.2006.03112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005;1:47–50. [PMC free article] [PubMed] [Google Scholar]
- Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N-Myristoylation. Journal of Biological Chemistry. 2001;276:39501–39504. doi: 10.1074/jbc.R100042200. [DOI] [PubMed] [Google Scholar]
- Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–925. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant V. Plant Speciation. 2. Columbia University Press; New York: 1981. [Google Scholar]
- Gross BL, Rieseberg LH. The ecological genetics of homoploid hybrid speciation. Journal of Heredity. 2005;96:241–252. doi: 10.1093/jhered/esi026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr B, Kauer M, Schlötterer C. Hitchhiking mapping: a population-based fine-mapping strategy for adaptive mutations in Drosophila melanogaster. Proceedings of the National Academy of Sciences, USA. 2002;99:12949–12954. doi: 10.1073/pnas.202336899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. A standardized genetic differentiation measure. Evolution. 2005;59:1633–1638. [PubMed] [Google Scholar]
- Hoekstra HE, Hirschmann RJ, Bundey RA, Insel PA, Crossland JP. A single amino acid mutation contributes to adaptive beach mouse color pattern. Science. 2006;313:101–104. doi: 10.1126/science.1126121. [DOI] [PubMed] [Google Scholar]
- Hulo N, Bairoch A, Bulliard V, et al. The PROSITE database. Nucleic Acids Research. 2006;34(Database Issue):D227–D230. doi: 10.1093/nar/gkj063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitani M, Liu J, Halfter U, et al. SOS3 function in plant salt tolerance requires N-myristoylation and calcium binding. Plant Cell. 2000;12:1667–1677. doi: 10.1105/tpc.12.9.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002;160:765–777. doi: 10.1093/genetics/160.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Z, Nakazato T, Salmaso M, et al. Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics. 2005;171:291–303. doi: 10.1534/genetics.105.042242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lexer C, Welch ME, Raymond O, Rieseberg LH. The origin of ecological divergence in Helianthus paradoxus (Asteraceae): Selection on transgressive characters in a novel hybrid habitat. Evolution. 2003;57:1989–2000. doi: 10.1111/j.0014-3820.2003.tb00379.x. [DOI] [PubMed] [Google Scholar]
- Ludwig F, Rosenthal DM, Johnston JA, et al. Selection on leaf ecophysiological traits in a desert hybrid Helianthus species and early-generation hybrids. Evolution. 2004;58:2682–2692. doi: 10.1111/j.0014-3820.2004.tb01621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison D, Maddison W. Macclade 4: Analysis of Phylogeny and Character Evolution, Version 4. Sinauer Associates; Sunderland, MA: 2000. [Google Scholar]
- Maurer-Stroh S, Eisenhaber B, Eisenhaber F. N-terminal N-Myristoylation of proteins: refinement of the sequence motif and its taxon-specific differences. Journal of Molecular Biology. 2002;317:523–540. doi: 10.1006/jmbi.2002.5425. [DOI] [PubMed] [Google Scholar]
- Maynard-Smith J, Haigh J. The hitch-hiking effect of a favorable gene. Genetical Research. 1974;23:23–35. [PubMed] [Google Scholar]
- McCarthy EM, Asmussen MA, Anderson WW. A theoretical assessment of recombinational speciation. Heredity. 1995;74:502–509. [Google Scholar]
- McDaniel SF, Shaw AJ. Selective sweeps and intercontinental migration in the cosmopolitan moss Ceratodon purpureus (Hedw.) Brid. Molecular Ecology. 2005;14:1121–1132. doi: 10.1111/j.1365-294X.2005.02484.x. [DOI] [PubMed] [Google Scholar]
- Moran MD. Arguments for rejecting the sequential Bonferroni in ecological studies. Oikos. 2003;100:403–405. [Google Scholar]
- Morjan CL, Rieseberg LH. How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles. Molecular Ecology. 2004;13:1341–1356. doi: 10.1111/j.1365-294X.2004.02164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir G, Schlötterer C. Evidence for shared ancestral polymorphism rather than recurrent gene flow at microsatellite loci differentiating two hybridizing oaks (Quercus spp.) Molecular Ecology. 2005;14:549–561. doi: 10.1111/j.1365-294X.2004.02418.x. [DOI] [PubMed] [Google Scholar]
- Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: Structural and functional diversity. Protein Science. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Molecular Biology and Evolution. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular Evolution and Phylogenetics. Oxford University Press; Oxford: 2000. [Google Scholar]
- Nei M, Li W-H. Mathematical model for studying genetic variation in terms of restriction nucleases. Proceedings of the National Academy of Sciences, USA. 1979;76:5269–5273. doi: 10.1073/pnas.76.10.5269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Kimura M. A model of mutation appropriate to estimate the number of electrophoretically detectable alleles in a finite population. Genetical Research. 1973;22:201–204. doi: 10.1017/s0016672300012994. [DOI] [PubMed] [Google Scholar]
- Orr HA, Coyne JA. The genetics of adaptation: a reassessment. American Naturalist. 1992;140:725–742. doi: 10.1086/285437. [DOI] [PubMed] [Google Scholar]
- Peakall R, Smouse PE. GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes. 2006;6:288–295. doi: 10.1093/bioinformatics/bts460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR. Analyzing tables of statistical tests. Evolution. 1989;43:223–225. doi: 10.1111/j.1558-5646.1989.tb04220.x. [DOI] [PubMed] [Google Scholar]
- Rieseberg LH. Homoploid reticulate evolution in Helianthus (Asteraceae): evidence from ribosomal genes. American Journal of Botany. 1991;78:1218–1237. [Google Scholar]
- Rieseberg LH. Hybrid origins of plant species. Annual Review of Ecology and Systematics. 1997;28:359–389. [Google Scholar]
- Rieseberg LH, Raymond O, Rosenthal DM, et al. Major ecological transitions in wild sunflowers facilitated by hybridization. Science. 2003;301:1211–1216. doi: 10.1126/science.1086949. [DOI] [PubMed] [Google Scholar]
- Rosenthal DM, Schwarzbach AE, Donovan LA, Raymond O, Rieseberg LH. Phenotypic differentiation between three ancient hybrid taxa and their parental species. International Journal of Plant Sciences. 2002;163:387–398. [Google Scholar]
- Rosenthal DM, Rieseberg LH, Donovan LA. Re-creating ancient hybrid species’ complex phenotypes from early-generation synthetic hybrids: three examples using wild sunflowers. American Naturalist. 2005;166:26–41. doi: 10.1086/430527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost B, Yachdav G, Liu J. The PredictProtein Server. Nucleic Acids Research. 2003;32(Web Server Issue):W321–W326. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas JS, Ánchez-Delbarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19:2496–2497. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- Sáez AG, Tatarenkov A, Barrio E, Becerra NH, Ayala FJ. Patterns of DNA sequence polymorphism at Sod vicinities in Drosophila melanogaster: Unraveling the footprint of a recent selective sweep. Proceedings of the National Academy of Sciences, USA. 2003;100:1793–1798. doi: 10.1073/pnas.242746799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C. A microsatellite-based multilocus screen for the identification of local selective sweeps. Genetics. 2002;160:753–763. doi: 10.1093/genetics/160.2.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlötterer C, Dieringer D. A novel test statistic for the identification of local selective sweeps based on microsatellite gene diversity. In: Nurminsky D, editor. Selective Sweep. Landes Bioscience; Georgetown, TX: 2005. pp. 55–64. [Google Scholar]
- Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nature Biotechnology. 2000;18:233–234. doi: 10.1038/72708. [DOI] [PubMed] [Google Scholar]
- Schwarzbach AE, Rieseberg LH. Likely multiple origins of a diploid hybrid sunflower species. Molecular Ecology. 2002;11:1703–1715. doi: 10.1046/j.1365-294x.2002.01557.x. [DOI] [PubMed] [Google Scholar]
- Schwarzbach AE, Donovan LA, Rieseberg LH. Transgressive character expression in a hybrid sunflower species. American Journal of Botany. 2001;88:270–277. [PubMed] [Google Scholar]
- Song B-H, Clauss MJ, Pepper A, Mitchell-Olds T. Geographic patterns of microsatellite variation in Boechera stricta, a close relative of Arabidopsis. Molecular Ecology. 2006;15:357–369. doi: 10.1111/j.1365-294X.2005.02817.x. [DOI] [PubMed] [Google Scholar]
- Storz JF. Using genome scans of DNA polymorphism to infer adaptive population divergence. Molecular Ecology. 2005;14:671–688. doi: 10.1111/j.1365-294X.2005.02437.x. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Gojobori T. A method for detecting positive selection at single amino acid sites. Molecular Biology and Evolution. 1999;16:1315–1328. doi: 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- Swofford D. Phylogenetic Analysis Using Parsimony, Version 4.0. Sinauer Associates; Sunderland, MA: 2000. [Google Scholar]
- Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Yu JK, Slabaugh MB, Shintani DK, Knapp SJ. Simple sequence repeat map of the sunflower genome. Theoretical and Applied Genetics. 2002;105:1124–1136. doi: 10.1007/s00122-002-0989-y. [DOI] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Gene ontology: tool for the unification of biology. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The InterPro Consortium. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Research. 2001;29:37–40. doi: 10.1093/nar/29.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towler DA, Gordon JI, Adams SP, Glaser L. The biology and enzymology of eukaryotic protein acylation. Annual Review of Biochemistry. 1988;57:69–99. doi: 10.1146/annurev.bi.57.070188.000441. [DOI] [PubMed] [Google Scholar]
- Ungerer MC, Baird SJE, Pan J, Rieseberg LH. Rapid hybrid speciation in wild sunflowers. Proceedings of the National Academy of Sciences, USA. 1998;95:11757–11762. doi: 10.1073/pnas.95.20.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigouroux Y, Jaqueth JS, Matsuoka Y, et al. Rate and pattern of mutation at microsatellite loci in maize. Molecular Biology and Evolution. 2002;19:1251–1260. doi: 10.1093/oxfordjournals.molbev.a004186. [DOI] [PubMed] [Google Scholar]
- de Vries JS, Andriotis VM, Wu AJ, Rathjen JP. Tomato Pto encodes a functional N-myristoylation motif that is required for signal transduction in Nicotiana benthamiana. Plant Journal. 2006;45:31–45. doi: 10.1111/j.1365-313X.2005.02590.x. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- Wiehe T, Nolte V, Zivkovic D, Schlötterer C. Identification of Selective Sweeps Using a Dynamically Adjusted Number of Linked Microsatellites. Genetics. 2007;175:207–218. doi: 10.1534/genetics.106.063677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong W, Goldman N, Nielsen R. Accuracy and Power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics. 2004;168:1041–1051. doi: 10.1534/genetics.104.031153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. Phylogenetic Analysis by Maximum Likelihood (PAML), Version 3.14. University College; London: 2000. [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen A-M. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics. 2000;155:431–449. doi: 10.1093/genetics/155.1.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatabe Y, Kane NC, Scotti-Saintagne C, Rieseberg LH. Rampant gene exchange across a strong reproductive barrier between the annual sunflowers, Helianthus annuus and H. petiolaris. Genetics. 2007;175:1883–1893. doi: 10.1534/genetics.106.064469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Apweiler R. InterProScan — an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17:847–848. doi: 10.1093/bioinformatics/17.9.847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following supplementary material is available for this article:
Table S1 Loci genotyped in this study and values for relative reduction in variation (lnRV) and heterozygosity (lnRH) in the hybrid species (Helianthus anomalus) compared to each of its parental species (H. annuus, and H. petiolaris).
Table S2 Sequence alignment for locus HT953 sequenced in three Helianthus species.
Table S3 Sequence alignment for locus HT998 sequenced in three Helianthus species.
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1365-294X.2007.03557.x (This link will take you to the article abstract).