

Abstract
RNA folding is a remarkably complex problem that involves ion-mediated electrostatic interaction, conformational entropy, base pairing and stacking, and noncanonical interactions. During the past decade, results from a variety of experimental and theoretical studies pointed to (a) the potential ion correlation effect in Mg2+ -RNA interactions, (b) the rugged energy landscapes and multistate RNA folding kinetics even for small RNA systems such as hairpins and pseudoknots, (c) the intraloop interactions and sequence-dependent loop free energy, and (d) the strong nonadditivity of chain entropy in RNA pseudoknot and other tertiary folds. Several related issues, which have not been thoroughly resolved, require combined approaches with thermodynamic and kinetic experiments, statistical mechanical modeling, and all-atom computer simulations.
Keywords: chain entropy, stability, conformational change, nonadditivity, ion correlation effect
INTRODUCTION
Over the past three decades, many high-resolution nuclear magnetic resonance (NMR) and X-ray crystallographic structures have been solved for RNAs ranging from tRNAs to the complete ribosome (60, 65). On the basis of these structures, RNA biologists gained great insight into RNA mechanisms in gene regulation and biochemical catalysis. However, we are still far from predicting RNA functions from the sequence. Why? Because a static structure does not tell us how RNA undergoes structural changes in the biochemical process. In order to understand RNA functions, we would like to predict not only the native structure from the nucleotide sequence (23, 56, 82), but also the statistical distribution of the conformational ensemble, from which we can predict the folding stability and the rates and pathways for the structural changes. This review is focused primarily on the thermodynamics and kinetics of RNA folding.
Knowledge-based modeling is highly successful in predicting structures if there exist multiple homologous sequences with known structures (42, 56, 82). The method can become convoluted when applied to deciphering the alternative structures and structural switches. In contrast, first-principles statistical mechanical modeling predicts the structures and structural changes without relying on homologous structures. In addition, with the continuous improvement of the force field and the conformational sampling, computer simulations (51, 79) have provided significant insight into RNA dynamics and stability for hairpins (37, 83, 104), pseudoknots (110), and large RNA machinery such as ribosome (77). In this review, we address the recent advances in RNA folding thermodynamics, kinetics, and ion electrostatic effect with emphasis on the theoretical developments.
In simple terms, RNA folding is a process of navigating the huge conformational space of the polynucleotide chain. The process is driven by several interrelated factors: thermally driven chain fluctuation, ion-mediated electrostatics, base pairing and stacking, and other noncanonical interactions. Therefore, a predictive understanding of RNA folding requires a synthesis of several key components of the problem, including the characterization of RNA conformational space, the equilibrium distribution of the chain conformations, and the kinetics of folding and conformational changes. Of particular importance to RNA folding is the role of metal ions. In recent years, we have witnessed important advances in both theoretical and experimental fronts on RNA folding. Here I focus on several recent results on the above issues. These new advances, especially the results on the nonadditive, long-range, and correlated interactions in tertiary folds and ion effects, challenge the widespread speculation that the RNA folding problem is less difficult than the protein folding problem.
RNA CONFORMATIONAL STATISTICS AND ENTROPY
Each RNA nucleotide has eight degrees of freedom: six backbone torsions, one torsion between the ribose ring and the base, and two types of configurations (C3′—endo and C2′—endo) for the pucker of the ribose ring (Figure 1c). It would be a formidable task to model RNA folding using all eight parameters because of the large number of possible conformations. In search for a Ramachandran plot for RNA, Murthy et al. (63) performed ab initio computation for a small system with two nucleotide-5′, 3′-diphosphates and one truncated dinucleotide. Their study demonstrated that steric interactions alone can account for well-separated regions in the conformational space. The result suggests that a denatured RNA has a much smaller conformational space (and entropy) than a completely flexible chain. The findings for the conformational statistics of the denatured state have significant implications for RNA folding stability (i.e., the free energy difference between the folded and the unfolded states), as well as for the folding kinetics, which involves searching for the folded structure from the unfolded conformational ensemble. For a folded RNA, the torsions are further restricted by the long-range (distant along the sequence but proximate in space) contacts and volume exclusion between the nucleotides.
Figure 1.
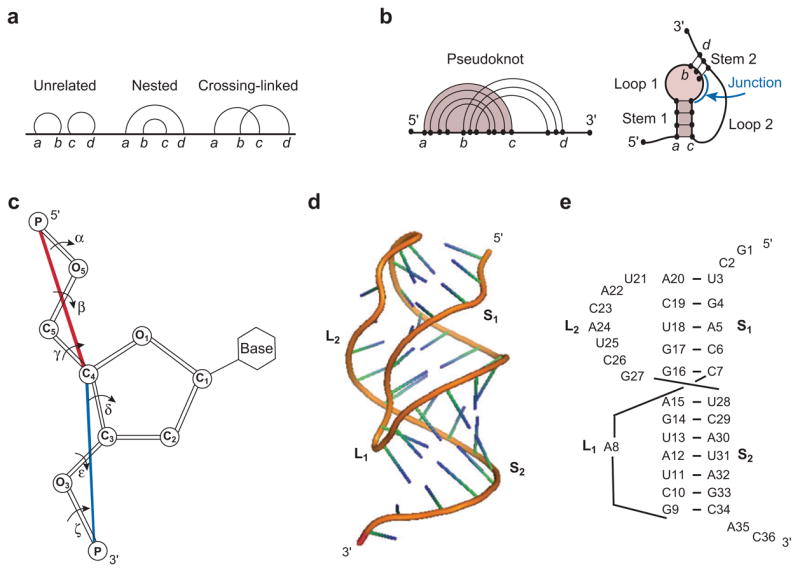
(a) Graphical representation for an RNA structure, where vertices (i.e., nucleotide monomers) are connected by straight lines (i.e., backbone covalent bonds) and base pairs are denoted by curved links. (b) Graphical representation for a pseudoknot. (c) The virtual bonds (shown as the red and blue lines) for RNA backbone. (d) The three-dimensional structure and (e) base-pairing structure for a typical pseudoknot (gene 32 mRNA pseudoknot of bacteriophage T2; PDB ID: 2TPK). Loop 1 (L 1) and loop 2 (L2) span across the major (narrow, deep) groove of helix 2 (S 2) and the minor (wide, shallow) groove of helix 1 (S1), respectively. The structure in (d) is adopted from the PDB (PDB ID: 2TPK) (39).
An alternative approach to the investigation of the Ramachandran plot for RNA is based on the survey of the existing known RNA structures. The studies from several groups on this approach have led to two observations. (a) The RNA backbone is rotameric (61). The finding represents a significant development beyond the earlier studies by Olson (66) that led to the virtual bond model for nucleic acids. The model reduces the original six backbone torsions into two (virtual) bonds because the backbone C-O torsions (β and ε in Figure 1c) have a preferred trans (t) rotational isomeric state. (b) RNA virtual bonds manifest separated, preferred conformational regions (31). This finding of the discrete distributions of the virtual bonds is remarkable because it significantly reduces the the conformational space for any virtual bond-based RNA models.
Motivated by the observation by Duarte & Pyle (31) on the discrete distributions of the virtual bonds, Cao & Chen (12–14) developed a virtual bond-based RNA folding model (Vfold). The model is based on the experimentally measured atomic coordinates for the helix conformation and the self-avoiding random walks of the virtual bonds in a diamond lattice for the loop conformations. Because the model has the ability to treat structural details such as intraloop base pairing, loop-helix structural interferences (volume exclusion) in multibranched loops (102), and tertiary contacts, through conformational count it can predict the chain entropy for a given RNA structure such as a loop.
LOOP STABILITY AND RNA SECONDARY STRUCTURE FOLDING THERMODYNAMICS
Chastain & Tinoco (18) defined RNA secondary (tertiary) structures as non-cross-linked (cross-linked) helices and loops (Figure 1a,b). Such a definition has the advantage to directly connect the structure to the additivity principle of folding energetics. For a secondary structure, which is relatively extended, different base stacks and loops are weakly correlated and the total free energy of the structure is equal to the sum of the free energies of the base stacks and loops (additivity). For a tertiary structure, however, different helices and loops are brought into close contact and become strongly correlated. As a result, the free energy additivity that applies to secondary structure is doomed to fail (26).
A structure defined by the base pairs and flexible loops can assume many possible conformations. The statistical average over all the possible conformations gives the partition function Z: Z = Σconformations e−Δ/kBT, where T is the temperature, kB (3.3 × 10−24 cal/mol·K) is the Boltzmann constant, and ΔE is the energy of the conformation. Z is central to the folding thermodynamics because it gives all the thermodynamic properties (free energy ΔG, enthalpy ΔH, and entropy ΔS).
The favorable stacking energy and the adverse conformational entropy are the two largest factors that determine the RNA folding stability (80). Different secondary structure models use the same set of empirical thermodynamic parameters (Turner rules) (56) for the helix. What distinguishes the different models is how to treat the loop free energy ΔGloop. Most models (38, 57, 115) assume a purely entropic (i.e., ΔGloop = −TΔSloop) and sequence-independent loop free energy. The complication of loop stability stems from the intraloop interactions such as non-canonical base pairing, mismatched (non-Waston-Crick) base stacks, single-strand base stacking, and other ion- and solvent-mediated interactions. The sequence-dependent energies of the intraloop interactions cause (a) the sequence dependence and (b) the enthalpic component of the loop free energy.
A statistical approach to the loop free energy requires the enumeration of different intraloop contacts. A key issue is how to evaluate the entropy for given intraloop contacts. No experiment can measure such a conditional entropy. The entropy can only come from an entropy theory such as Vfold. Cao & Chen applied their Vfold model to all the possible intraloop mismatched base stacks and achieved improved predictions for the folding thermodynamics and the structure for RNA secondary structures (12) and RNA/RNA complexes (3, 13, 27). Further improvements of the model should include the detailed information for the base configurations and other important intraloop interactions (such as noncanonical base pairing).
RNA PSEUDOKNOT: STRUCTURE AND FOLDING STABILITY
Pseudoknots (Figure 1d,e) play a critical role in many biochemical processes from regulation of viral gene expression to the catalysis of mRNA splicing and human telomerase RNA (hTR) (85). Central to the pseudoknot stability is the loop entropy (Figure 1d,e). Earlier pseudoknot algorithms were severely limited by the nearly complete lack of loop entropy parameters (28, 29, 74). Accurate calculation of loop entropy is challenging because of the loop-stem correlation through the volume exclusion effect. Abrahams et al. (1) introduced a uniform single value of TΔSloop = 4.2 kcal/mol for all pseudoknot loops. Later, Gultyaev et al. (36) proposed a set of much-improved loop entropy parameters by introducing several fitting parameters to account for the loop-stem interference. The model with fitted parameters correctly predicted that the known pseudoknots have lower free energies than the hairpins. More recently, with the Vfold model, which employs no fitting parameter, Cao & Chen (14) systematically computed the loop entropy parameters (Table 1) for different stem and loop size using rigorous polymer statistical mechanics. These parameters give reliable predictions for the folding thermodynamics (Figure 2) as well as the native structures (14).
Table 1.
Pseudoknot loop entropy parametersa
| Stem size | Loop size (nt) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (bp) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 |
| (S2) | L1 (across the deep groove of S2) | |||||||||||
| 2 | – | – | 6.2 | 6.4 | 6.4 | 6.6 | 6.8 | 6.9 | 7.1 | 7.2 | ||
| 3 | – | 6.4* | 6.4* | 6.4 | 6.6 | 6.6 | 6.8 | 6.9 | 7.1 | 7.3 | 7.5 | |
| 4 | 4.4* | 4.4* | 4.5 | 5.4 | 5.6 | 6.0 | 6.3 | 6.6 | 6.9 | 7.1 | 7.3 | |
| 5 | 2.3 | 4.4 | 4.6 | 5.7 | 6.0 | 6.5 | 6.9 | 7.2 | 7.5 | 7.8 | 8.0 | |
| 6 | 2.3 | 4.4 | 4.8 | 5.8 | 6.0 | 6.5 | 6.8 | 7.1 | 7.4 | 7.6 | 7.8 | |
| 7 | 2.3 | 4.4 | 5.0 | 5.9 | 6.2 | 6.8 | 7.0 | 7.3 | 7.6 | 7.8 | 8.0 | |
| 8 | – | 4.4 | 5.2 | 5.7 | 6.4 | 6.7 | 7.1 | 7.3 | 7.5 | 7.7 | 7.9 | |
| 9 | – | 5.5* | 5.5 | 6.4 | 6.7 | 7.2 | 7.5 | 7.9 | 8.1 | 8.3 | 8.5 | |
| 10 | – | 6.9* | 6.9* | 6.9 | 7.5 | 7.7 | 8.1 | 8.3 | 8.6 | 8.8 | 8.9 | |
| 11 | – | – | – | – | 8.7 | 8.8 | 8.9 | 9.1 | 9.2 | 9.3 | 9.3 | |
| 12 | – | – | – | – | 9.8 | 9.2 | 9.5 | 9.6 | 9.7 | 9.8 | 9.8 | |
| (S1) | L2 (across the shallow groove of S1) | |||||||||||
| 2 | – | – | – | 7.6 | 7.0 | 7.0 | 7.1 | 7.2 | 7.3 | 7.4 | 7.5 | 7.7 |
| 3 | – | 6.5* | 6.5* | 6.5 | 6.6 | 6.7 | 6.9 | 7.1 | 7.2 | 7.4 | 7.6 | 7.7 |
| 4 | – | – | 9.2* | 9.2* | 9.2 | 8.9 | 8.9 | 8.9 | 9.0 | 9.0 | 9.1 | 9.2 |
| 5 | – | – | – | 9.8* | 9.8* | 9.8 | 9.1 | 8.9 | 8.8 | 8.8 | 8.8 | 8.8 |
| 6 | – | – | – | 11.9* | 11.9* | 11.9* | 11.9 | 11.0 | 10.4 | 10.1 | 9.9 | 9.8 |
| 7 | – | – | – | – | 12.4* | 12.4* | 12.4* | 12.4 | 11.4 | 11.0 | 10.7 | 10.5 |
| 8 | – | – | – | – | 12.1* | 12.1* | 12.1* | 12.1 | 11.6 | 11.4 | 11.2 | 11.1 |
| 9 | – | – | – | – | – | 13.7* | 13.7* | 13.7* | 13.7 | 12.6 | 12.0 | 11.5 |
| 10 | – | – | – | – | – | 13.7* | 13.7* | 13.7 | 12.7 | 12.2 | 11.8 | 11.5 |
| 11 | – | – | – | – | – | – | – | – | 15.9 | 14.1 | 13.0 | 12.4 |
| 12 | – | – | – | – | – | – | – | – | 18.7 | 15.8 | 14.2 | 13.2 |
The upper and lower tables give (− ΔSloop/kB) for loop 1 and loop 2, respectively; see Figure 1. The entries with an asterisk (*) denote the long stem and short loop structures that cannot be realized in the diamond lattice but may be viable for a realistic pseudoknot. For these restricted loops, we use the entropies of the minimal loop lengths for the same helix length. This table is adapted from Reference 14.
Figure 2.

Predicting pseudoknot stability using the loop entropy parameters in Table 1 (14). For loop 1, length of stem 2 is 7 bp, length of loop 1 is 1 nt, so the entropy is read from Table 1 as ΔS1(7, 1). Similarly, for loop 2, stem 1 is 5 bp, loop 2 is 4 nt, so the loop entropy is ΔS2(5, 4). The total free energy of the pseudoknot is ΔG = ΔG1 + ΔG2 − T ΔS1(7, 1) − T ΔS2(5, 4) + ΔGassemble = (−6.6) kcal/mol + (−11.2) kcal/mol + (kB T)(2.3) + (kB T)(9.8) + 1.3 kcal/mol = −9.0 kcal/mol, where ΔGassemble = 1.3 kcal/mol accounts for the multiple ways to connect the two loops (14) and kBT = 0.62 kcal/mol at T = 37°C. See Reference 14 for more examples.
Despite the promising progress, current pseudoknot theories are unable to treat many biologically significant pseudoknotted structures, such as pseudoknots with (a) a junction between the helix stems (Figure 1b), (b) internal or bulge loops in the helix stems, or (c) loop-stem or loop-loop base pair and base triple interactions (105). These structures are critical to the function of anti-HIV RNA aptamers (11), telomerase RNAs (96), and frameshifting RNAs (22). Three key issuesfocus on how to compute the chain entropy for these complex structures (47, 48), how to treat loop-stem and loop-loop tertiary interaction energies, and how to treat the possible coaxial stacking between the helices. The current theories can treat only H(airpin)-type pseudoknots with at most one nucleotide that connects the two helix stems, in which case the coaxially stack between the helices contributes a sequence-dependent free energy of ΔGstack ~ 1 – 5 kcal/mol at 37°C (106).
HAIRPIN AND PSEUDOKNOT FOLDING KINETICS
Hairpin is an elementary building block of RNA structure. The system is simple enough for exhaustive detailed studies yet sufficiently sophisticated to serve as a paradigm for more complex RNA folding kinetics. There have been extensive studies on DNA hairpin folding kinetics using laser temperature-jump (4, 55) and fluorescence correlation spectroscopy (8, 43, 44, 52) techniques. In contrast, there are only very limited experiments on RNA hairpin (and pseudoknot) folding kinetics (54, 64, 69, 111, 113, 114). The majority of RNA folding kinetics experiments address the folding of a large tertiary structure (76, 88, 99, 101, 109) after the formation of the secondary structure in the initial fast phase (μsec) of the measurements. Although RNA and DNA share remarkably similar biophysical properties in many aspects, without detailed measurements, assuming DNA and RNA hairpins to have the same folding kinetics remains unwarranted. In fact, DNA and RNA have different enthalpy and entropy parameters (56, 78), which imply that they can have different folding thermodynamics and kinetics.
Recent experimental (54) and theoretical studies (111, 113, 114) revealed complex multistate, nonexponential folding kinetics even for small RNA hairpins, suggesting a complex bumpy energy landscape. These new results, especially the loop and stem sequence dependence of the hairpin folding kinetics, should dispel the common misconception about hairpin folding kinetics, namely, that folding is always rate-limited by the loop closure and the rate is determined by the loop size (68).
Tetraloop Hairpin Folding Kinetics
A tetraloop is stabilized by excess base pairing and stacking in the loop (103). Experimental measurements for tetraloop hairpin folding kinetics indicated that (a) changing a regular non-GNRA tetraloop to a GNRA tetraloop did not alter the folding rate (64), and (b) the stable (YNMG) UUCG tetraloop and the excessively less stable tetraloop UUUU have nearly the same folding rate (69) and hence the difference in stability between the two loops comes from the different unfolding rates. These experimental findings suggest that the rate-limiting step is not the (sequence-dependent) intraloop stabilization.
All-atom molecular dynamics simulation by Sorin et al. (83) revealed multiple pathways for the folding of a tetraloop RNA hairpin. The simulation showed two types of folding pathways. The first type is zipping (dominant pathway), in which the loop is closed by the marginally stable base pair (with partial formation of hydrogen bonding), followed by subsequent zipping of the full helix stem. The rate-limiting step is the loop closure instead of intraloop stabilization, which is consistent with the experiments. The second folding pathway type is compaction, in which the chain collapses through nonspecific base pairing to form a heterogeneous structural ensemble, followed by (native-like or misfolded) helix growth from the nucleation centers. In their simulations, the initial collapsed state remained stable over the simulation timescale (μsec). Further simulation is required in order to show how the structure converges to the final, completely folded state.
RNA Hairpin Folding Kinetics: Theoretical Studies
A fundamental ingredient embodied in theoretical approaches to the folding kinetics is the rate constant for an elementary transition from state i to state j, where is the activation barrier and k0 is the fundamental rate constant, which is solvent dependent. Currently there are three types of rate models:
Base stack model (111, 113, 114): An elementary kinetic move is the addition and breaking of a base stack, with ΔGi → j equal to the change in the entropic free energy TΔS and the enthalpy ΔH, respectively.
Base pair model (20, 32): An elementary kinetic step is the formation or disruption of a base pair, with ΔGi → j equal to ΔG/2 (ΔG is the free energy change from state i to state) or equal j to ΔG for ΔG > 0 and 0 otherwise.
Helix stem model (41): A kinetic move is the creation or deletion of a helix stem, with rate constant determined by the helix free energy change (see the Base pair model, above).
The different rate models can lead to different folding pathways. The key factor that distinguishes the different rate models is whether the barrier is determined by (ΔH, ΔS) or by ΔG. The (ΔH, ΔS) values for different RNA base stacks show well-separated discrete hierarchies, whereas the ΔG values show no such large separation. For two typical base stacks, 5′AU-AU3′ and 5′UC-GA3′, the difference Δ (ΔHstack, ΔSstack) = (7.4 kcal/mol, 20 cal/mol/K) is much larger than the difference Δ (Δ Gstack) = 1.4 kcal/mol (81). Because of this fact, different models can give different folding kinetics. For instance, in the base stack model, a kinetic trap can be formed by an ensemble of conformations that all contain a nonnative base stack with an outstanding ΔHstack, whereas in the other two models, a trap can only be formed by a stable misfolded helix stem (with multiple stabilizing base stacks). The competition between the loop formation, stem elongation, and detrapping determines the complex sequence dependence of the folding kinetics (54).
For a nontetraloop hairpin, Zhang & Chen (111) predicted a non-Arrhenius kinetics characterized by the folding-transition temperature Tf and the glass-transition temperature Tg (Figure 3). There are three caveats for the predictions. First, the kinetic behavior varies between different sequences. Second, other mechanisms, such as kinetic traps, heat capacity changes (ΔCp) in loop and base pair/stack formation, and the change of the single-strand base stacking, can also cause the non-Arrhenius behavior. Without further experimental studies, none of the possibilities can be excluded. Third, the single exponential kinetics at T > Tg does not exclude the formation of kinetic intermediates. An ensemble of states, including the kinetic intermediates, can transiently form an effective single macrostate if they can quickly reach mutual local equilibrium. A single exponential kinetics only means the transition between the macrostates is a two-state process.
Figure 3.
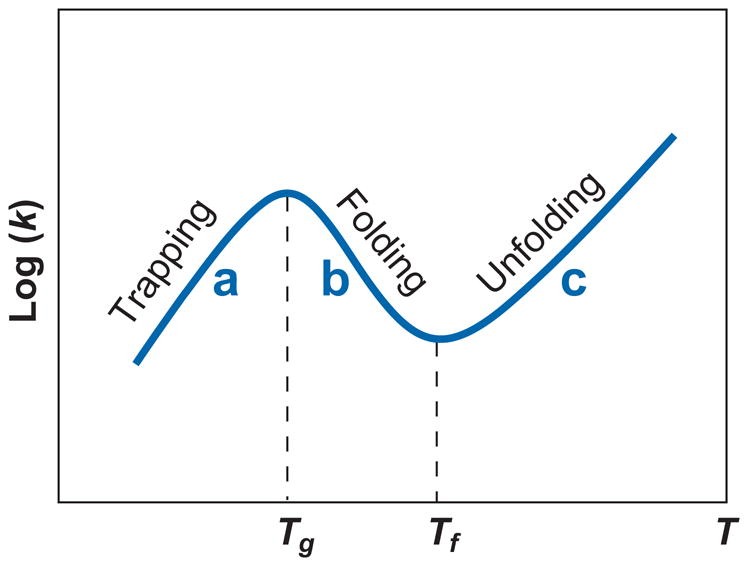
Temperature dependence of the reaction rate k (111). (a) T > Tf > Tg: The relaxation process is predominantly unfolding, with single exponential kinetics along an unzipping pathway. The apparent activation barrier [= − d ln(k)/d (1/T)] is positive, equal to the enthalpic cost of breaking the rate-limiting base stack(s). (b) Tg > T > Tf: The relaxation process is predominantly folding, with single exponential kinetics along a zipping pathway. The activation barrier is negative, equal to the stabilizing enthalpy of the rate-limiting base stack(s). (c) T < Tg: The relaxation process is predominantly folding, with multistate multiple exponential kinetics involving multiple pathways, including the zipping pathway and detrapping-refolding pathways. The activation barrier is positive, equal to the average enthalpic cost to detrap the misfolded states.
Experimental verification of the rate model is a challenge because the microscopic elementary processes are buried in the ensemble averages of the measured kinetics. Single-molecule experiments, with careful extrapolation to the force-free case, may provide a discerning measure. All-atom simulations with reliable force field and sampling method are highly valuable for providing detailed atomistic configurations for the transition state. Alternatively, systematic theory-experiment tests for designed sequences can also provide critical assessment for the different rate models.
There is an important component missed in all the above theoretical models, namely, the sequence-dependent conformational statistics of the single-stranded coil and loop states. The importance of the unfolded state to hairpin folding kinetics has been suggested by several experimental studies. Experiments by Bonnet et al. (8) and Nagel et al. (64) indicated that a pyrimidine-rich loop (such U-loop) folds faster than a purine-rich loop (A-loop) because an A-rich single-stranded coil is prone to form single-stranded stacking, which must be disrupted prior to loop formation. To further examine how the loop composition affects folding kinetics, Proctor et al. (69) substituted a syn-G at position 4 in the UUCG tetraloop by a more bulky nucleotide analogy, 8-bromoguanosine, and found the substitution caused a fourfold increase in the folding rate. Thermodynamic measurements (69) suggested that the stronger steric constraint due to the bulky nucleotide analogy reduces the conformational space (and entropy) of the unfolded state, reducing the time for searching the loop conformation from the ensemble of the unfolded conformations.
Pseudoknot Folding Kinetics
Pseudoknot, which forms stable helix stems, is prone to form intermediate states (with a timescale of tens of microseconds). Depending on the sequence, the intermediate state often contains nonnative base pairs and thus must be disrupted (in milliseconds to minutes) before refolding (15, 41, 70). Several theoretical studies (15, 41) indicated that pseudoknot folding involves parallel pathways: a fraction of population folds without being trapped to the misfolded state while the rest of population is first trapped and then refolds through slow detrapping. A recent study by Cao & Chen (15) suggested the dominant pathway shows biphasic kinetics through a misfolded hairpin intermediate. These results have sig-nificant implications for the cotranscriptional RNA folding (67) when the RNA chain elongation effect is considered (35, 41) (but protein binding and ion effects are ignored). Simulations for several systems by Isambert & Siggia (41) and Gultyaev at el. (35) suggested that a fast RNA chain elongation would more likely cause kinetic trapping. For instance, kinetic Monte Carlo simulation for hepatitis delta virus ribozyme (HDV pseudoknot) predicted that an increase of RNA synthesis rate from 50 to 1000 nt/s causes the trapped population to increase from less than 10% to about 30% (41).
hTR pseudoknot is a catalytically essential region of human telomerase RNA (85, 96). The hTR pseudoknot structure has been solved by Theimer et al. (95). Several studies for the structure-activity relationship pointed to the essential role of the pseudoknot structure in hTR activity (19, 21, 96, 97).
Comolli et al. (21) proposed a molecular switch between hairpin and pseudoknot as an essential step for the telomerase activity (97). In contrast, Chen & Greider (19) proposed that only the static pseudoknot structure is required for the function, because their experiment showed that (a) a hairpin-destabilizing mutant retained telomerase activity and (b) destabilizing the pseudoknot caused reduction in telomerase activity. Recently, motivated by the controversy, Cao & Chen (15) applied the kinetic cluster approach (112) to examine the pseudoknot folding kinetics. Their results indicated that the folding of the mutant in the experiment of Chen & Greider (a) has a long-lived hairpin intermediate and (b) undergoes a hairpin-to-pseudoknot transition. The theoretical study suggested a possible kinetic control of the telomerase activity. Discerning the validity of these different models requires more detailed kinetic, biochemical, and functional experiments.
ELECTROSTATIC INTERACTIONS IN RNA FOLDING
RNA backbone is highly (negatively) charged. The formation of the compact tertiary structure requires the polyanionic chain to overcome the massive charge repulsion from the backbone. The divalent Mg2+ ions are unusually efficient to promote RNA tertiary structure folding. Physically, the distribution of the cations surrounding an RNA molecule is continuous and dynamic. However, it is useful to classify discrete ion types (Figure 4). A small number of the ions (16, 17, 25, 58, 59), which bind to specific sites of RNA, are classified as specific bound ions, while the majority of ions, which move diffusively without binding to any specific sites, are classified as (nonspecific) diffusive ions.
Figure 4.

Two types of cations surrounding the P4-P6 RNA structure (17): specific binding and diffusive binding. A recent Mg2+ ion titration study suggested that two metal ions induce cooperative folding of the P4-P6 metal ion core (25).
Central to the ion electrostatics of RNA folding is are the Mg2+ ions. Several excellent articles (9, 30, 101, 109) have extensively reviewed this important problem. In this review, we focus on the recent progress in several specific issues related to Mg2+-mediated RNA folding (5, 10, 24, 45, 99).
First, RNA structure is an important determinant for ion-RNA interaction. A rigorous treatment for the ion electrostatics should be based on the complete conformational ensemble of RNA structure. To examine the effect of RNA conformational ensemble, Grilley et al. (34) performed a series of rigorous experiments to measured the Mg2+-dependent stability of an rRNA segment and the BWYV pseudoknot. The experimental data pointed to the importance of considering the (ensemble of) (84) the intermediate states of RNA for the effect of Mg2+ on RNA stability. Despite the possible uncertainties associated with the validity of the Poisson-Boltzmann equation for Mg2+-RNA interactions in the analysis, the results suggest that one must be cautious when using the traditional two-state approximation for ion-dependent RNA stability.
Second, ion charge and size (in addition to RNA structure) are also important determinants for ion-mediated RNA folding (2, 46). Ion size is crucial not only because it determines the closest distance between an ion and the RNA but also because it affects ion-ion distance and hence the strength of Coulombic and excluded volume correlation. By measuring the folding stability of the Tetrahymena ribozyme in the solution of four different types of divalent ions (Mg2+, Ca2+, Sr2+, Ba2+), Koculi et al. (45) found a remarkable linear relationship between the increase of the folding stability of the Tetrahymena ribozyme and the increase of the (divalent) ion charge density (i.e., charge/volume of the ion).
Subsequent Brownian dynamics simulation by Koculi et al. (45) for the ion effect on Tetrahymena ribozyme folding led to two additional results. First, the distribution of the condensed ions around the RNA showed the liquid-like ion correlation. Second, for the Tetrahymena ribozyme, nonspecific electrostatic interaction alone (without specific metal ion coordination effect) can account for the collapsed state as well as the dependence (at least qualitatively) of the folding stability on ion charge density. Koculi et al. hypothesized that for a large RNA such as Tetrahymena ribozyme, the large number of nonspecifically bound ions outcompetes the specifically bound ions (16, 25) for global stability. However, other experiments suggested that the contribution from the specific site-bound ions may vary significantly between different RNAs (34, 84). These contrasting findings suggest the necessity to account for the full ensemble of not only the RNA structures but also the ion distributions (including the presence and absence of the possible buried ions) in the theory.
In a different line of approach, several theoretical (90–94) and experimental (49, 76, 89, 108) groups focused on the mechanism of the unusually efficient Mg2+-mediated driving force in RNA compaction (formation of compact structures lacking stable native tertiary contacts). The remarkable difference between the required ion concentrations (mM for Mg2+ and M for Na+) cannot be simply explained by the effect of the ionic strength (30). Motivated by the desire to quantify the Mg2+-mediated force, two experimental groups (5, 71) performed a series of remarkable experiments for a simple paradigm system: two short DNA helices immersed in a salt solution. The system is interesting for two reasons. First, helix assembly is the elementary step in the initial collapse transition. Second, it helps to quantitatively understand how the electrostatic force stabilizes the helix-helix packing in the observed RNA structural database (62).
For the system with two helices (each with 12 base pairs) tethered by a electrically neutral junction, Bai et al. (5) performed small-angle X-ray scattering (SAXS) experiments and observed (a) no attractive force that is strong enough to induce a collapsed state even in the presence of a 0.6 M (high) Mg2+ concentration, and (b) that the Mg2+-induced compact state is an electrostatically relaxed (neutralized) random state.
In a separate study for DNA helices of 25 base pairs dispersed in the solution, Qiu et al. (71) observed an enhanced local clustering of DNA helices with increasing Mg2+ concentration. Quantitative analysis for the SAXS profile pointed to a short-range attractive force induced by a small amount of Mg2+ ions (~16.7 mM). Unlike the helices tethered by short loops (5), which are optimal for possible side-by-side helix attraction, the dispersed short helices (71) tend to form end-to-end helix stacking (attraction), and the attraction increases with decreasing helix length.
A central issue here concerns the strength of the Mg2+-induced force, specifically, whether the force is strong enough to hold the two helices together against the random motion. Further experimental studies on the origin of the Mg2+-induced attraction (71) and the effect of the different tethers, helix length, and the ion different concentration (5) would be highly valuable.
Ion-mediated attraction between polyelectrolytes has been an extensively studied problem (many excellent reviews have been published; see Reference 6). Several models, such as ion entropic driving force (73), ion-ion correlation effect (75), and solvent effects (72, 87), have been proposed to explain the attraction. In the ion correlation mechanism, the correlation causes the ions to efficiently reach low-lying states whose energies are much lower than the mean-field energy. As the helices approach each other, the strong correlation (for multivalent ions) makes ions quickly reach low-energy (correlated) states and causes a net attraction.
In a more recent measurement for the Mg2+ -induced compaction in a tethered duplex system, researchers (Y. Bai, V. Chu, J. Lipfert, V. Pande, D. Herschlag & S. Doniach, manuscript submitted) found that the mean-field Poisson-Boltzmann equation, which ignores the ion correlation effect, would overestimate the required [Mg2+] for compaction more than 10 times higher than the experimentally measured value! This experimental result suggested that the traditional Poisson-Boltzmann equation, which ignores the ion correlation and the finite size of the ions, may not be appropriate for Mg2+ ions.
Motivated by the importance of the ion correlation effect and of the ensemble of ion distributions, Tan & Chen (90–94) developed the tightly bound ion (TBI) model. The essence of the theory is to provide a rigorous treatment for the strongly correlated ions (tightly bound ions). Later experiments by Stellwagen et al. (86) suggested that the experimentally observed bound cations around a 26-bp DNA helix may correspond to the tightly bound ions proposed in the TBI theory. The TBI model predicted an Mg2+-induced compaction transition at [Mg2+] = 1 ~ 5 mM, which is consistent with the experimental findings (49, 76, 108). In addition, the model made several new predictions. (a) Polynucleotide tether instead of neutral tether (5), smaller counterions, and multiple helices would make the compaction transition more cooperative. (b) The most compact state is somewhere between the random state and the fully collapsed state and occurs at [Mg2+] = 0.1 M. A higher [Mg2+] (>0.1 M) makes the system less compact. The validation of these predictions requires further experimental tests.
SUMMARY AND CONCLUSIONS
RNA folding is remarkably complex. It is a result of the delicate balance between multiple factors: the chain entropy, ion-mediated electrostatic interactions and solvation effect, base pairing and stacking, and other non-canonical interactions. Recent experimental and theoretical studies on large ribozyme as well as small RNA systems (hairpin, pseudoknots, and two-helix assembly) are beginning to reveal the detailed folding mechanism that will lead us to a quantitative and predictive model for RNA folding. Currently, the greatest challenge lies in resolving several key issues such as the magnitude of the electrostatic (including ion correlation) and the nonelectrostatic (such as entropic) forces in RNA, the contribution of the specific versus nonspecific electrostatic effects, and the validity of the different rate models for the elementary steps. On the theoretical side, the challenge lies in the accurate calculation for the conformational entropy for RNA tertiary folds, for which the additivity principle fails, proper treatment of Mg2+-induced correlation and solvation effect, and evaluation of the energetics for various noncanonical tertiary interactions. Finally, I would like to emphasize that this review is by no means complete. I refer readers to other review articles (98) on the issues that are not covered here, such as single-molecule experiments for force-induced RNA folding (7, 33, 40, 50, 53, 100, 107), knowledge-based approaches for structural prediction, and computer simulations on RNA folding.
SUMMARY POINTS
The rotameric nature of RNA backbone leads to reduced low-resolution models that enable reliable predictions for the chain entropy and folding thermodynamics.
Intraloop interactions can cause sequence-dependent loop free energy. Theory considering intraloop interactions can lead to improved predictions for RNA folding thermodynamics.
The folding stability of pseudoknot and other RNA tertiary structures are nonadditive. Accurate modeling for the loop-stem interference is the key for pseudoknot prediction.
The different rate constant models for the elementary steps in RNA folding can cause different folding pathways. It is critical to identify the correct rate model through well-designed discerning experiments.
The ion correlation effect may contribute to the unusually efficient role of Mg2+ in RNA tertiary structure folding.
FUTURE ISSUES
Is RNA hairpin and pseudoknot folding two-state or multistate? What is the transition state of folding? To resolve the issue requires accurate all-atom simulation, statistical mechanical modeling, and temperature-jump experiments.
There exist different rate models for the elementary steps in RNA folding, yet which rate model is valid? Single-molecule experiments would be able to draw a decisive conclusion for the validity of the different models.
How should the stability contribution from the site-bound ions (metal ion core) in RNA tertiary structures be properly estimated? This requires a predictive model for the electrostatic and solvation effects, as well as the ion correlation effect.
How can we predict the chain entropy for large complex RNA tertiary folds with multiple junctions, and loop-stem and loop-loop tertiary interactions?
How can we extract the (Turner rules–like) thermodynamic parameters for tertiary interactions from the melting experiments? This requires systematic thermodynamic experiments for a series of designed RNA tertiary motifs.
From the ion electrostatic model, the chain entropy model, and the secondary and tertiary energy parameters, how do we establish a unified predictive model for RNA folding?
Acknowledgments
The author wishes to express his deep appreciation to Dr. Song Cao and Dr. Zhi-Jie Tan, who contributed to this review through useful discussions and preparation of the figures, as well as to Dr. David Draper, Dr. Daniel Herschlag, and Dr. Lois Pollack for critical reading of the manuscript. This project is supported by NIH grant (R01-GM063732) and in part by the Institute for Mathematics and its Applications with funds provided by the National Science Foundation.
- Vfold
virtual bond folding model
- hTR
human telomerase RNA
- SAXS
small angle X-ray scattering
- TBI
tightly bound ion model
Footnotes
The Annual Review of Biophysics is online at biophys.annualreviews.org
DISCLOSURE STATEMENT
The author is not aware of any biases that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Abrahams JP, van den Berg M, van Batenburg E, Pleij C. Prediction of RNA secondary structure, including pseudoknot, by computer simulation. Nucleic Acids Res. 1990;18:3035–44. doi: 10.1093/nar/18.10.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andresen K, Das R, Park HY, Smith H, Kwok LW, et al. Spatial distribution of competing ions around DNA in solution. Phys Rev Lett. 2004;93:248103. doi: 10.1103/PhysRevLett.93.248103. [DOI] [PubMed] [Google Scholar]
- 3.Andronescu M, Zhang ZC, Condon A. Secondary structure prediction of interacting RNA molecules. J Mol Biol. 2005;345:987–1001. doi: 10.1016/j.jmb.2004.10.082. [DOI] [PubMed] [Google Scholar]
- 4.Ansari A, Kuznetsov SV, Shen Y. Configurational diffusion down a folding funnel describes the dynamics of DNA hairpins. Proc Natl Acad Sci USA. 2001;98:7771–76. doi: 10.1073/pnas.131477798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai Y, Das R, Millett IS, Herschlag D, Doniach S. Probing counterion modulated repulsion and attraction between nucleic acid duplexes in solution. Proc Natl Acad Sci USA. 2005;102:1035–40. doi: 10.1073/pnas.0404448102. Experiment demonstrates that the Mg2+-induced force is not sufficient to cause a collapsed state for two short DNA helices tethered by a neutral short loop. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bloomfield VA. DNA condensation by multivalent cations. Biopolymers. 1997;44:269–82. doi: 10.1002/(SICI)1097-0282(1997)44:3<269::AID-BIP6>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 7.Bokinsky G, Zhuang XW. Single-molecule RNA folding. Acc Chem Res. 2005;38:566–73. doi: 10.1021/ar040142o. [DOI] [PubMed] [Google Scholar]
- 8.Bonnet G, Krichevsky O, Libchaber A. Kinetics of conformational fluctuations in DNA hairpin-loops. Proc Natl Acad Sci USA. 1998;95:8602–6. doi: 10.1073/pnas.95.15.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brenowitz M, Chance MR, Dhavan G, Takamoto K. Probing the structural dynamics of nucleic acids by quantitative time-resolved and equilibrium hydroxyl radical ‘footprinting’. Curr Opin Struct Biol. 2002;12:648–53. doi: 10.1016/s0959-440x(02)00366-4. [DOI] [PubMed] [Google Scholar]
- 10.Buchmueller KL, Weeks KM. Near native structure in an RNA collapsed state. Biochemistry. 2003;42:13869–78. doi: 10.1021/bi035476k. [DOI] [PubMed] [Google Scholar]
- 11.Burke DH, Scates L, Andrews K, Gold L. Bent pseudoknots and novel RNA inhibitors of type 1 human immunodeficiency virus (HIV-1) reverse transcriptase. J Mol Biol. 1996;264:650–66. doi: 10.1006/jmbi.1996.0667. [DOI] [PubMed] [Google Scholar]
- 12.Cao S, Chen SJ. Predicting RNA folding thermodynamics with a reduced chain representation model. RNA. 2005;11:1884–97. doi: 10.1261/rna.2109105. The first paper to introduce the virtual-bond-based RNA folding model (Vfold) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cao S, Chen SJ. Free energy landscapes of RNA/RNA complexes: with applications to snRNA complexes in spliceosomes. J Mol Biol. 2006;357:292–312. doi: 10.1016/j.jmb.2005.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao S, Chen SJ. Predicting RNA pseudoknot folding thermodynamics. Nucleic Acids Res. 2006;34:2634–52. doi: 10.1093/nar/gkl346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao S, Chen SJ. Biphasic folding kinetics of RNA pseudoknots and telomerase RNA activity. J Mol Biol. 2007;367:909–24. doi: 10.1016/j.jmb.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cate JH, Gooding AR, Podell E, Zhou KH, Golden BL, et al. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996;273:1678–85. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 17.Cate JH, Hanna RL, Doudna JA. A magnesium ion core at the heart of a ribozyme domain. Nat Struct Biol. 1997;4:553–58. doi: 10.1038/nsb0797-553. [DOI] [PubMed] [Google Scholar]
- 18.Chastain M, Tinoco I., Jr Structural elements in RNA. Prog Nucleic Acids Res Mol Biol. 1991;41:131–77. doi: 10.1016/S0079-6603(08)60008-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen JL, Greider CW. Functional analysis of the pseudoknot structure in human telomerase RNA. Proc Natl Acad Sci USA. 2005;102:8080–85. doi: 10.1073/pnas.0502259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cocco S, Marko JF, Monasson R. Slow nucleic acid unzipping kinetics from sequence-defined barriers. Eur Phys J E. 2003;10:153–61. doi: 10.1140/epje/e2003-00019-8. [DOI] [PubMed] [Google Scholar]
- 21.Comolli LR, Smirnov I, Xu L, Blackburn EH, James TL. A molecular switch underlies a human telomerase disease. Proc Natl Acad Sci USA. 2002;99:16998–7003. doi: 10.1073/pnas.262663599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cornish PV, Hennig M, Giedroc DP. A loop 2 cytidine-stem 1 minor groove interaction as a positive determinant for pseudoknot-stimulated-1 ribosomal frameshifting. Proc Natl Acad Sci USA. 2005;102:12694–99. doi: 10.1073/pnas.0506166102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Das R, Baker D. Automated de novo prediction of native-like RNA tertiary structures. Proc Natl Acad Sci USA. 2007;104:14664–69. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Das R, Kwok LW, Millett IS, Bai Y, Mills TT, et al. The fastest global events in RNA folding: electrostatic relaxation and tertiary collapse of the Tetrahymena ribozyme. J Mol Biol. 2003;332:311–19. doi: 10.1016/s0022-2836(03)00854-4. [DOI] [PubMed] [Google Scholar]
- 25.Das R, Travers KJ, Bai Y, Herschlag D. Determining the Mg2+ stoichiometry for folding an RNA metal ion core. J Am Chem Soc. 2005;127:8272–73. doi: 10.1021/ja051422h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dill KA. Additivity principles in biochemistry. J Biol Chem. 1997;272:701–4. doi: 10.1074/jbc.272.2.701. [DOI] [PubMed] [Google Scholar]
- 27.Dimitrov RA, Zuker M. Prediction of hybridization and melting for double-stranded nucleic acids. Biophys J. 2004;87:215–26. doi: 10.1529/biophysj.103.020743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ding Y. Statistical and Bayesian approaches to RNA secondary structure prediction. RNA. 2006;12:323–31. doi: 10.1261/rna.2274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dirks RM, Pierce NA. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J Comput Chem. 2004;25:1295–304. doi: 10.1002/jcc.20057. [DOI] [PubMed] [Google Scholar]
- 30.Draper DE, Grilley D, Soto AM. Ions and RNA folding. Annu Rev Biophys Biomol Struct. 2005;34:221–43. doi: 10.1146/annurev.biophys.34.040204.144511. [DOI] [PubMed] [Google Scholar]
- 31.Duarte CM, Pyle AM. Stepping through an RNA structure: a novel approach to conformational analysis. J Mol Biol. 1998;284:1465–78. doi: 10.1006/jmbi.1998.2233. [DOI] [PubMed] [Google Scholar]
- 32.Flamm C, Fontana W, Hofacker IL, Schuster P. RNA folding at elementary step resolution. RNA. 2000;6:325–38. doi: 10.1017/s1355838200992161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerland U, Bundschuh R, Hwa T. Mechanically probing the folding pathway of single RNA molecules. Biophys J. 2003;84:2831–40. doi: 10.1016/S0006-3495(03)70012-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grilley D, Soto AM, Draper DE. Mg2+ -RNA interaction free energies and their relationship to the folding of RNA tertiary structures. Proc Natl Acad Sci USA. 2006;103:14003–8. doi: 10.1073/pnas.0606409103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gultyaev AP, van Batenburg FHD, Pleij CWA. The computer-simulation of RNA folding pathways using a genetic algorithm. J Mol Biol. 1995;250:37–51. doi: 10.1006/jmbi.1995.0356. [DOI] [PubMed] [Google Scholar]
- 36.Gultyaev AP, van Batenburg FHD, Pleij CWA. An approximation of loop free energy values of RNA H-pseudoknots. RNA. 1999;5:609–17. doi: 10.1017/s135583829998189x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hall KB, Williams DJ. Dynamics of the IRE RNA hairpin loop probed by 2-aminopurine fluorescence and stochastic dynamics simulations. RNA. 2004;10:34–47. doi: 10.1261/rna.5133404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003;31:3429–31. doi: 10.1093/nar/gkg599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holland JA, Hansen MR, Du Z, Hoffman DW. An examination of coaxial stacking of helical stems in a pseudoknot motif: the gene 32 messenger RNA pseudoknot of bacteriophage T2. RNA. 1999;5:257–71. doi: 10.1017/s1355838299981360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hyeon C, Thirumalai D. Mechanical unfolding of RNA: from hairpins to structures with internal multiloops. Biophys J. 2007;92:731–43. doi: 10.1529/biophysj.106.093062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isambert H, Siggia ED. Modeling RNA folding paths with pseudoknots: Application to hepatitis D-virus ribozyme. Proc Natl Acad Sci USA. 2000;97:6515–20. doi: 10.1073/pnas.110533697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jossinet F, Westhof E. Sequence to structure (S2S): display, manipulate and interconnect RNA data from sequence to structure. Bioinformatics. 2005;21:3320–21. doi: 10.1093/bioinformatics/bti504. [DOI] [PubMed] [Google Scholar]
- 43.Jung J, Van Orden A. A three-state mechanism for DNA hairpin folding characterized by multiparameter fluorescence fluctuation spectroscopy. J Am Chem Soc. 2006;128:1240–49. doi: 10.1021/ja0560736. [DOI] [PubMed] [Google Scholar]
- 44.Kim J, Doose S, Neuweiler H, Sauer M. The initial step of DNA hairpin folding: a kinetic analysis using fluorescence correlation spectroscopy. Nucleic Acids Res. 2006;34:2516–27. doi: 10.1093/nar/gkl221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koculi E, Hyeon C, Thirumalai D, Woodson SA. Charge density of divalent metal cations determines RNA stability. J Am Chem Soc. 2007;129:2676–82. doi: 10.1021/ja068027r. A combined experimental and theoretical study shows the correlated ion distribution for the condensed ions and a linear relationship between the ion charge density and RNA stability. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koculi E, Lee NK, Thirumalai D, Woodson SA. Folding of the Tetrahymena ribozyme by polyamines: importance of counterion valence and size. J Mol Biol. 2004;341:27–36. doi: 10.1016/j.jmb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 47.Kopeikin Z, Chen SJ. Statistical thermodynamics for chain molecules with simple RNA tertiary contacts. J Chem Phys. 2005;122:094909. doi: 10.1063/1.1857831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kopeikin Z, Chen SJ. Folding thermodynamics of pseudoknotted chain conformations. J Chem Phys. 2006;124:154903. doi: 10.1063/1.2188940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwok LW, Shcherbakova I, Lamb JS, Park HY, Andresen K, et al. Concordant exploration of the kinetics of RNA folding from global and local perspectives. J Mol Biol. 2006;355:282–93. doi: 10.1016/j.jmb.2005.10.070. [DOI] [PubMed] [Google Scholar]
- 50.Lee TH, Lapidus LJ, Zhao W, Travers KJ, Herschlag D, Chu S. Measuring the folding transition time of single RNA molecules. Biophys J. 2007;92:3275–83. doi: 10.1529/biophysj.106.094623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levitt M. Detailed molecular model for transfer ribonucleic acid. Nature. 1969;224:759–63. doi: 10.1038/224759a0. [DOI] [PubMed] [Google Scholar]
- 52.Li H, Ren X, Ying L, Balasubramanian S, Klenerman D. Measuring single-molecule nucleic acid dynamics in solution by two-color filtered ratiometric fluorescence correlation spectroscopy. Proc Natl Acad Sci USA. 2004;101:14425–30. doi: 10.1073/pnas.0404295101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liphardt J, Onoa B, Smith SB, Tinoco I, Bustamante C. Reversible unfolding of single RNA molecules by mechanical force. Science. 2001;292:733–37. doi: 10.1126/science.1058498. [DOI] [PubMed] [Google Scholar]
- 54.Ma H, Proctor DJ, Kierzek E, Kierzek R, Bevilacqua PC, Gruebele M. Exploring the energy landscape of a small RNA hairpin. J Am Chem Soc. 2006;128:1523–30. doi: 10.1021/ja0553856. Demonstrates the rugged energy landscape even for a system as small as a short RNA tetraloop hairpin. [DOI] [PubMed] [Google Scholar]
- 55.Ma H, Wan C, Wu A, Zewail AH. DNA folding and melting observed in real time redefine the energy landscape. Proc Natl Acad Sci USA. 2007;104:712–16. doi: 10.1073/pnas.0610028104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mathews DH, Schroeder SJ, Turner DH, Zuker M. Predicting RNA secondary structure. In: Gesteland RF, Cech RT, Atkins JF, editors. The RNA World. New York: Cold Spring Harbor Press; 2006. pp. 631–57. [Google Scholar]
- 57.McCaskill JS. The equilibrium partition function and base pair binding probabilities for RNA secondary structure. Biopolymers. 1990;29:1105–19. doi: 10.1002/bip.360290621. [DOI] [PubMed] [Google Scholar]
- 58.Misra VK, Draper DE. A thermodynamic framework for Mg2+ binding to RNA. Proc Natl Acad Sci USA. 2001;98:12456–61. doi: 10.1073/pnas.221234598. Study in which the overall free energy of placing an Mg2+ ion at a specific site in a crystal structure is computed (based on the Born model) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Misra VK, Draper DE. The linkage between magnesium binding and RNA folding. J Mol Biol. 2002;317:507–21. doi: 10.1006/jmbi.2002.5422. [DOI] [PubMed] [Google Scholar]
- 60.Moore PB, Steitz TA. The structural basis of large ribosomal subunit function. Annu Rev Biochem. 2003;72:813–50. doi: 10.1146/annurev.biochem.72.110601.135450. [DOI] [PubMed] [Google Scholar]
- 61.Murray LJ, Arendall WB, Richardson DC, Richardson JS. RNA backbone is rotameric. Proc Natl Acad Sci USA. 2003;100:13904–9. doi: 10.1073/pnas.1835769100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murthy VL, Rose GD. Is counterion delocalization responsible for collapse in RNA folding? Biochemistry. 2000;39:14365–70. doi: 10.1021/bi001820r. [DOI] [PubMed] [Google Scholar]
- 63.Murthy VL, Srinivasan R, Draper DE, Rose GD. A complete conformational map for RNA. J Mol Biol. 1999;291:313–27. doi: 10.1006/jmbi.1999.2958. [DOI] [PubMed] [Google Scholar]
- 64.Nagel JH, Flamm C, Hofacker IL, Franke K, de Smit MH, et al. Structural parameters affecting the kinetics of RNA hairpin formation. Nucleic Acids Res. 2006;34:3568–76. doi: 10.1093/nar/gkl445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Noller HF. RNA structure: reading the ribosome. Science. 2005;309:1508–14. doi: 10.1126/science.1111771. [DOI] [PubMed] [Google Scholar]
- 66.Olson WK. Configuration statistical of polynucleotide chains a single virtual bond treatment. Macromolecules. 1975;8:272–75. doi: 10.1021/ma60045a006. [DOI] [PubMed] [Google Scholar]
- 67.Pan T, Sosnick T. RNA folding during transcription. Annu Rev Biophys Biomol Struct. 2006;35:161–75. doi: 10.1146/annurev.biophys.35.040405.102053. [DOI] [PubMed] [Google Scholar]
- 68.Porschke D. Thermodynamic and kinetic parameters of an oligonucleotide hairpin helix. Biophys Chem. 1974;1:381–86. doi: 10.1016/0301-4622(74)85008-8. [DOI] [PubMed] [Google Scholar]
- 69.Proctor DJ, Ma H, Kierzek E, Kierzek R, Gruebele M, Bevilacqua PC. Folding thermodynamics and kinetics of YNMG RNA hairpins: Specific incorporation of 8-bromoguanosine leads to stabilization by enhancement of the folding rate. Biochemistry. 2004;43:14004–14. doi: 10.1021/bi048213e. [DOI] [PubMed] [Google Scholar]
- 70.Puglisi JD, Wyatt JR, Tinoco I., Jr Conformation of an RNA pseudoknot. J Mol Biol. 1990;214:437–53. doi: 10.1016/0022-2836(90)90192-O. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qiu X, Kwok LW, Park HY, Lamb JS, Andresen K, Pollack L. Measuring inter-DNA potentials in solution. Phys Rev Lett. 2006;96:138101. doi: 10.1103/PhysRevLett.96.138101. Demonstration of the Mg2+-induced attraction between DNA helices. [DOI] [PubMed] [Google Scholar]
- 72.Rau DC, Lee B, Parsegian VA. Measurement of the repulsive force between polyelectrolyte molecules in ionic solution: hydration forces between parallel DNA double helices. Proc Natl Acad Sci USA. 1984;81:2621–25. doi: 10.1073/pnas.81.9.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ray J, Manning GS. An attractive force between two rodlike polyions mediated by the sharing of condensed counterions. Langmiur. 1994;10:2450–61. [Google Scholar]
- 74.Rivas E, Eddy SR. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J Mol Biol. 1999;285:2053–68. doi: 10.1006/jmbi.1998.2436. [DOI] [PubMed] [Google Scholar]
- 75.Rouzina I, Bloomfield VA. Macroion attraction due to electrostatic correlation between screening counterions. 1. Mobile surface-adsorbed ions and diffuse ion cloud. J Phys Chem. 1996;100:9977–89. [Google Scholar]
- 76.Russell R, Millett IS, Tate MW, Kwok LW, Nakatani B, et al. Rapid compaction during RNA folding. Proc Natl Acad Sci USA. 2002;99:4266–71. doi: 10.1073/pnas.072589599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sanbonmatsu KY. High performance computing simulations of tRNA movement through the ribosome. J Biomol Struct Dyn. 2007;24:631. [Google Scholar]
- 78.SantaLucia J, Hicks D. The thermodynamics of DNA structural motifs. Annu Rev Biophys Biomol Struct. 2004;33:415–40. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 79.Scheraga HA, Khalili M, Liwo A. Protein-folding dynamics: overview of molecular simulation techniques. Annu Rev Phys Chem. 2007;58:57–83. doi: 10.1146/annurev.physchem.58.032806.104614. [DOI] [PubMed] [Google Scholar]
- 80.Searle MS, Williams DH. On the stability of nucleic acid structures in solution: enthalpy-entropy compensations, internal rotations and reversibility. Nucleic Acids Res. 1993;21:2051–56. doi: 10.1093/nar/21.9.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Serra MJ, Turner DH. Predicting thermodynamic properties of RNA. Methods Enzymol. 1995;259:242–61. doi: 10.1016/0076-6879(95)59047-1. [DOI] [PubMed] [Google Scholar]
- 82.Shapiro BA, Yingling YG, Kasprzak W, Bindewald E. Bridging the gap in RNA structure prediction. Curr Opin Struct Biol. 2007;17:157–65. doi: 10.1016/j.sbi.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 83.Sorin EJ, Rhee YM, Nakatani BJ, Pande VS. Insights into nucleic acid conformational dynamics from massively parallel stochastic simulations. Biophys J. 2003;85:790–803. doi: 10.1016/S0006-3495(03)74520-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Soto AM, Misra V, Draper DE. Tertiary structure of an RNA pseudoknot is stabilized by “diffuse” Mg2+ ions. Biochemistry. 2007;46:2973–83. doi: 10.1021/bi0616753. [DOI] [PubMed] [Google Scholar]
- 85.Staple DW, Butcher SE. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005;3:956–59. doi: 10.1371/journal.pbio.0030213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stellwagen E, Dong Q, Stellwagen NC. Quantitative analysis of monovalent counterion binding to random-sequence, double-stranded DNA using the replacement ion method. Biochemistry. 2007;46:2050–58. doi: 10.1021/bi062132w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takach JC, Mikulecky PJ, Feig AL. Salt-dependent heat capacity changes for RNA duplex formation. J Am Chem Soc. 2004;126:6530–31. doi: 10.1021/ja0316263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takamoto K, Das R, He Q, Doniach S, Brenowitz M, et al. Principles of RNA compaction: insights from the equilibrium folding pathway of the P4-P6 RNA domain in monovalent cations. J Mol Biol. 2004;343:1195–206. doi: 10.1016/j.jmb.2004.08.080. [DOI] [PubMed] [Google Scholar]
- 89.Takamoto K, He Q, Morris S, Chance MR, Brenowitz M. Monovalent cations mediate formation of native tertiary structure of the Tetrahymena thermophila ribozyme. Nat Struct Biol. 2002;9:928–33. doi: 10.1038/nsb871. [DOI] [PubMed] [Google Scholar]
- 90.Tan ZJ, Chen SJ. Electrostatic correlations and fluctuations for ion binding to a finite length polyelectrolyte. J Chem Phys. 2005;122:044903. doi: 10.1063/1.1842059. The first paper to introduce the TBI model to treat the effects of ion correlation and ensemble of ion distributions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tan ZJ, Chen SJ. Electrostatic free energy landscapes for nucleic acid helix assembly. Nucleic Acids Res. 2006;34:6629–39. doi: 10.1093/nar/gkl810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tan ZJ, Chen SJ. Ion-mediated nucleic acid helix-helix interactions. Biophys J. 2006;91:518–36. doi: 10.1529/biophysj.106.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys J. 2006;90:1175–90. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tan ZJ, Chen SJ. RNA helix stability in mixed Na2+/Mg2+ solution. Biophys J. 2007;92:3615–32. doi: 10.1529/biophysj.106.100388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Theimer CA, Blois CA, Feigon J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol Cell. 2005;17:671–82. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 96.Theimer CA, Feigon J. Structure and function of telomerase RNA. Curr Opin Struct Biol. 2006;16:307–18. doi: 10.1016/j.sbi.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 97.Theimer CA, Finger LD, Trantirek L, Feigon J. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc Natl Acad Sci USA. 2003;100:449–54. doi: 10.1073/pnas.242720799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thirumalai D, Hyeon C. RNA and protein folding: common themes and variations. Biochemistry. 2005;44:4957–70. doi: 10.1021/bi047314+. [DOI] [PubMed] [Google Scholar]
- 99.Thirumalai D, Lee N, Woodson SA, Klimov D. Early events in RNA folding. Annu Rev Phys Chem. 2001;52:751–62. doi: 10.1146/annurev.physchem.52.1.751. [DOI] [PubMed] [Google Scholar]
- 100.Tinoco I, Jr, Li PTX, Bustamante C. Determination of thermodynamics and kinetics of RNA reactions by force. Q Rev Biophys. 2006;39:325–60. doi: 10.1017/S0033583506004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Treiber DK, Williamson JR. Beyond kinetic traps in RNA folding. Curr Opin Struct Biol. 2001;11:309–14. doi: 10.1016/s0959-440x(00)00206-2. [DOI] [PubMed] [Google Scholar]
- 102.Tyagi R, Mathews DH. Predicting helical coaxial stacking in RNA multibranch loops. RNA. 2007;13:939–51. doi: 10.1261/rna.305307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Uhlenbeck OC. Tetraloops and RNA folding. Nature. 1990;346:613–14. doi: 10.1038/346613a0. [DOI] [PubMed] [Google Scholar]
- 104.Villa A, Stock G. What NMR relaxation can tell us about the internal motion of an RNA hairpin: a molecular dynamics simulation study. J Chem Theor Comput. 2006;2:1228–36. doi: 10.1021/ct600160z. [DOI] [PubMed] [Google Scholar]
- 105.Walberer BJ, Cheng AC, Frankel AD. Structural diversity and isomorphism of hydrogen-bonded base interactions in nucleic acids. J Mol Biol. 2003;327:767–80. doi: 10.1016/s0022-2836(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 106.Walter AE, Turner DH. Sequence dependence of stability for coaxial stacking of RNA helices with Watson-Crick base paired interfaces. Biochemistry. 1994;33:12715–19. doi: 10.1021/bi00208a024. [DOI] [PubMed] [Google Scholar]
- 107.Woodside MT, Anthony PC, Behnke-Parks WM, Larizadeh K, Herschlag D, Block SM. Direct measurement of the full, sequence-dependent folding landscape of a nucleic acid. Science. 2006;314:1001, 4. doi: 10.1126/science.1133601. Direct experimental measurements for DNA hairpin folding energy landscapes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Woodson SA. Compact but disordered states of RNA. Nat Struct Biol. 2000;7:349–52. doi: 10.1038/75106. [DOI] [PubMed] [Google Scholar]
- 109.Woodson SA. Metal ions and RNA folding: a highly charged topic with a dynamic future. Curr Opin Chem Biol. 2005;9:104–9. doi: 10.1016/j.cbpa.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 110.Yingling YG, Shapiro BA. The impact of dyskeratosis congenita mutations on the structure and dynamics of the human telomerase RNA pseudoknot domain. J Biomol Struct Dyn. 2007;24:303–19. doi: 10.1080/07391102.2007.10531238. [DOI] [PubMed] [Google Scholar]
- 111.Zhang WB, Chen SJ. RNA hairpin-folding kinetics. Proc Natl Acad Sci USA. 2002;99:1931–36. doi: 10.1073/pnas.032443099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang WB, Chen SJ. Analyzing the biopolymer folding rates and pathways using kinetic cluster method. J Chem Phys. 2003;119:8716–29. doi: 10.1063/1.1613255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang WB, Chen SJ. Exploring the complex folding kinetics of RNA hairpins. I. General folding kinetics analysis. Biophys J. 2006;90:765–77. doi: 10.1529/biophysj.105.062935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang WB, Chen SJ. Exploring the complex folding kinetics of RNA hairpins. II. Effect of sequence, length, and misfolded states. Biophys J. 2006;90:778–87. doi: 10.1529/biophysj.105.062950. A theoretical exploration for the sequence-dependent RNA hairpin folding energy landscapes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–15. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]