

Abstract
The nature of the “toxic gain of function” that results from amyotrophic lateral sclerosis (ALS)-, Parkinson-, and Alzheimer-related mutations is a matter of debate. As a result no adequate model of any neurodegenerative disease etiology exists. We demonstrate that two synergistic properties, namely, increased protein aggregation propensity (increased likelihood that an unfolded protein will aggregate) and decreased protein stability (increased likelihood that a protein will unfold), are central to ALS etiology. Taken together these properties account for 69% of the variability in mutant Cu/Zn-superoxide-dismutase-linked familial ALS patient survival times. Aggregation is a concentration-dependent process, and spinal cord motor neurons have higher concentrations of Cu/Zn-superoxide dismutase than the surrounding cells. Protein aggregation therefore is expected to contribute to the selective vulnerability of motor neurons in familial ALS.
Author Summary
Amyotrophic lateral sclerosis (ALS), also known in America as Lou Gehrig's disease, is a fatal neurodegenerative disease with no effective treatment. Paralysis occurs as the result of the death of cells that connect the brain to various muscles, namely, the motor neurons of the brain and spinal cord. Ninety percent of ALS is sporadic and of unknown cause. A landmark discovery in ALS research was that mutations in the gene coding for Cu/Zn-superoxide dismutase cause at least 2% of ALS, and researchers have since discovered at least 119 such mutations. Neurologists also discovered that different mutations have remarkably different prognoses. For example, patients with the A4V mutation survive an average of 1 year after diagnosis, whereas patients with the H46R mutation survive an average of 18 years. Biochemists discovered that different mutations result in remarkably different physical properties, for example, stability of Cu/Zn-superoxide dismutase. In this article we apply an algorithm that predicts how fast a given Cu/Zn-superoxide dismutase will aggregate (stick to other proteins) and demonstrate that faster aggregation relates to faster death of ALS patients. We also demonstrate that loss of Cu/Zn-superoxide dismutase stability relates to faster ALS patient death. Our findings imply that aggregation of unfolded SOD1 is toxic for ALS patients, and in fact accounts for 69% of the variability in mutant Cu/Zn-superoxide-dismutase-linked familial ALS patient survival times.
Increased protein aggregation and decreased protein stability are shown to be central to familial amyotrophic lateral sclerosis etiology.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease with roughly 10% of the cases being inherited or familial [1]. The cause of sporadic ALS (sALS) is unknown while familial ALS (fALS) is known to be caused by mutations in six different genes and six different chromosomal loci [2–4]. One of these genes encoding Cu/Zn-superoxide dismutase (SOD1) was found to associate with 20% of fALS, and at least 119 fALS-associated SOD1 mutations have been characterized in humans [1,5].
Fifteen years after the discovery that SOD1 mutations can cause ALS [1], the mechanisms of toxicity are still not well understood. The dominant inheritance of most SOD1 mutations and the literature as a whole indicate that SOD1 mutations result in a toxic gain of function rather than a loss of function [6–9]. Numerous hypotheses have been proposed, reviewed in [10,11], and can be broken down conceptually into the (nonexclusive) toxic mechanisms that converge to SOD1 protein structure–function and those that converge elsewhere (downstream effects). Popular hypotheses for SOD1 variant structure and function changes include decreased stability of apo or metallated SOD1 [12–15], increased hydrophobicity [16] and aggregation propensity [17,18], susceptibility to posttranslational modification [19–25], loss of metals [22,26–32], and aberrant chemistry [33–37]. Popular hypotheses for downstream effects [38–40] include impairment of axonal transport [41–43], impairment of proteasome [39,44,45] or chaperone activity [46,47], and mitochondrial [9,48–53] or endoplasmic reticulum–Golgi dysfunction [54,55]. Notably, the only potentially toxic property thus far shared by all fALS SOD1 variants is an increased propensity to form proteinaceous aggregates [56–62]. Due to the clinical similarities between fALS and sALS, research into SOD1 mutation-related fALS may provide insight into sporadic cases. Here we demonstrate that two properties, namely, increased aggregation propensity and instability (loss of stability), are major contributors to SOD1 toxicity in ALS patients. On the basis of these results we rationalize the determinants of aggregation, the selective vulnerability of neurons, and patient survival times.
Results
SOD1 Mutations Have Inherently Different Toxicities
The goal of this study is to discover the mechanisms of toxicity of fALS SOD1 mutations. Neurologists often publish the age at onset and the time from disease onset to death (also termed survival or disease duration) for their ALS patients, thereby enabling epidemiological studies that assess the risk of a given variable [63–66], which for this study include given mutations' relative toxicity and physical characteristics (physicochemical parameters). Previous studies revealed that different SOD1 mutations have inherently different toxicities (encode different mean disease durations) [67]. We expanded upon these studies with a larger set of fALS-causing SOD1 mutations as well as larger patient cohorts. Hazard ratios (relative risk of dying at a given time) of fALS SOD1 mutations and non-SOD1-related fALS compared to that of sALS were obtained from the Cox proportional hazard model (Table 1). From this result, fALS SOD1 mutations with data from at least five individual patients (full criteria for inclusion are defined in the Materials and Methods section) were significantly related to different hazards. Kaplan–Meier survival curves from patients with fALS-causing SOD1 mutations, non-SOD1-related fALS, and sALS were generated. Figure 1 illustrates that fALS SOD1 mutations encode different prognoses, ranging from considerably better (e.g., H46R, hazard rate = 0.075 × sALS) to considerably worse (e.g., A4V, hazard rate = 5.7 × sALS) than sALS. Moreover, the Log rank, Breslow, and Tarone–Ware tests, which also compare patient survival rates (i.e., each mutation versus every other fALS-causing SOD1 mutation, non-SOD1-related fALS, and sALS) using different mathematical functions, confirm that different SOD1 mutations have inherently different prognoses (Table 2).
Table 1.
SOD1 Mutations Encode Inherently Different Prognoses
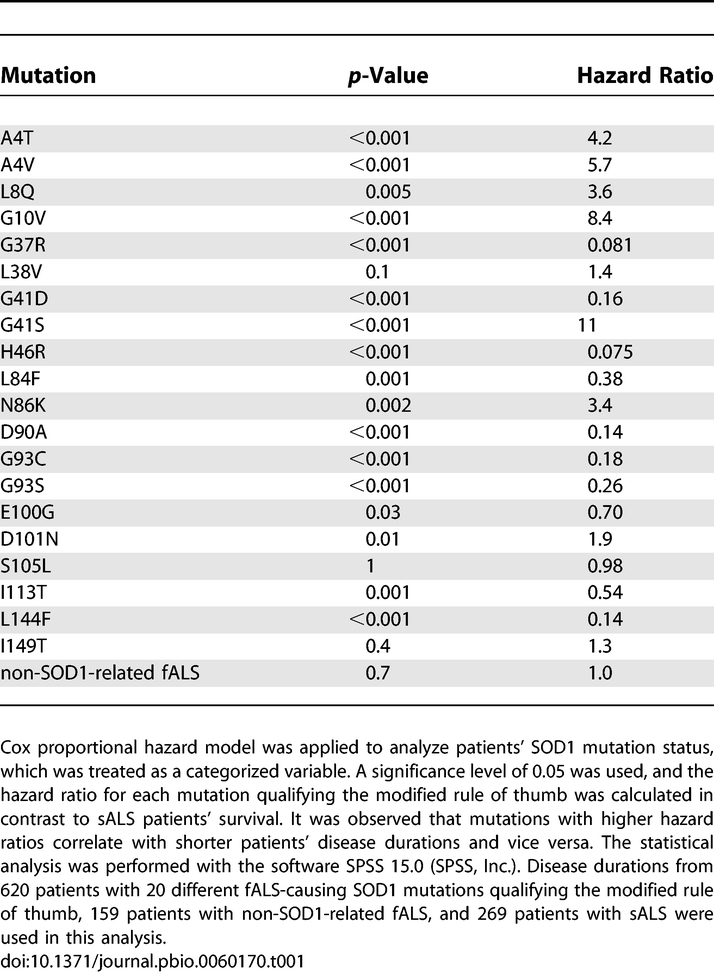
Figure 1. fALS Patients' Kaplan–Meier Survival Curves Illustrating Low and High Risk SOD1 Mutations.

Kaplan–Meier survival curves from patients with A4V (red) and H46R (blue) SOD1 mutations and sALS (green) are as shown. Disease durations from 205 patients with A4V SOD1 mutation, 63 patients with H46R SOD1 mutation, and 269 patients with sALS were used to generate these Kaplan–Meier survival curves.
Table 2.
SOD1 Mutations Are Inherently Related to ALS Patients' Disease Duration

SOD1 Variants' Gain of Hydrophobicity, Loss of α-Helix, and Gain of β-Sheet Propensity Are fALS Risk Factors, While Loss of Net Charge Is Protective
To test the hypothesis that changes to the physicochemical properties of SOD1 variants are toxic, specifically those properties known to influence protein aggregation, physicochemical properties for each protein variant (hydrophobicity, propensity to lose α-helices, form β-sheets, protein net charge, etc.) were evaluated in a Cox proportional hazard model (Table 3). The hazard ratios were significantly higher than 1.0 for mutations that either increase hydrophobicity, lose α-helices, or form β-sheets. In contrast, mutations that decrease the magnitude of the protein net charge correlate with hazard ratios significantly smaller than 1.0. These results indicate that changes in the SOD1 variants' properties, specifically increases in hydrophobicity and propensity to lose α-helices and to form β-sheets, correlate with decreased fALS patient survival, while decreases of the magnitude of net charge correlate with increased fALS patient survival, in contradiction with previous reports [15,68].
Table 3.
SOD1 Variants' Gain of Hydrophobicity, Loss of α-Helix, and Gain of β-Sheet Propensity Are ALS Risk Factors, While Loss of Net Charge Is Protective

Dobson and co-workers [69] introduced an equation (termed the Chiti–Dobson equation herein) to predict the changes of aggregation rates of unfolded peptides or proteins upon point mutations by their physicochemical properties. This equation was derived empirically by modeling how three physicochemical properties, hydrophobicity, secondary structure (including loss of α-helix and gain of β-sheet), and protein net charge, change upon mutations (the hazard analyses for each of these properties were reported in the previous paragraph). These physicochemical property changes then were related to changes in protein aggregation rate, yielding an equation that predicts how any mutation will change the rate of protein aggregation (the predicted change of aggregation rate is referred to as the aggregation propensity). Although this equation is empirical, it is based upon first physical/chemical principles and approximates how a given mutation will change the energy (and thus the equilibrium) between a solvated and an aggregated protein. The Chiti–Dobson equation is ln(νmut/νwt) = 0.633ΔHydr + 0.198(ΔΔG coil-α + ΔΔG β-coil) – 0.491Δcharge, in which ln(νmut/νwt) represents change of aggregation rate upon mutation, and ΔHydr, ΔΔG coil-α, ΔΔG β-coil, and Δcharge represent the changes of hydrophobicity, free energy change for the process from α-helix to random coil, free energy change for the process from random coil to β-sheet, and protein net charge from the mutation, respectively. In their landmark study, it was demonstrated that increases in hydrophobicity, losses of α-helices, gains of β-sheets, and decreases in the magnitude of protein net charge increase the rate of protein aggregation.
Protein Aggregation Propensity Is a Risk Factor of fALS
The Chiti–Dobson equation and the many equations it inspired are robust and versatile, having successfully predicted aggregation rates of diverse disease-associated proteins [70], including amyloid β-peptide [69], tau [69], α-synuclein [69], amylin [69], lysozyme [71], etc. Moreover, increases in the predicted rates of aggregation of various mutations in amyloid β-peptide were shown to relate to increased neuronal dysfunction and degeneration in a Drosophila model of Alzheimer's disease [72]. To test the hypothesis that protein aggregation propensity is related to fALS patient survival, the Chiti–Dobson equation was used to predict the aggregation propensities of fALS-causing SOD1 mutations. We started this study by validating the Chiti–Dobson equation, taking all experimental protein aggregation rate data available at the inception of our study (data reported as of 2005, listed in Table 4) and recalibrating the equation. The detailed results of the validation are reported in Figure 2. In summary, the Chiti–Dobson equation was verified for use in fALS, and the statistical correlation between the physicochemical parameters (hydrophobicity, net charge, and secondary structure) and the aggregation propensity remained and changed only marginally. Since the time we validated the Chiti–Dobson equation, a number of papers also validated their general approach [73–75]. Even so, we have included our own analysis since it provides exposure to the physical basis of aggregation propensity. Furthermore, inclusion of this data makes this study self-contained so that all of the data necessary to support or disprove our model are contained herein. Notably, this paper's conclusions were the same using both the original and the recalibrated Chiti–Dobson equation.
Table 4.
Data Used To Rederive the Chiti–Dobson Equation

Figure 2. Rederivation and Validation of the Chiti–Dobson Equation.

The original Chiti–Dobson equation was rederived by correlating all known empirically measured mutation-related changes in protein aggregation rates as of 2005 with the corresponding changes in the physicochemical properties of charge, hydrophobicity, and secondary structure. Importantly, aggregation rate data were not taken directly from the publication that presented the Chiti–Dobson equation; instead these values were calculated from their respective original publication if applicable (Table 4). The dependence of observed ln(νmut/νwt) on hydrophobicity, secondary structure, and charge changes were still observed after the addition of extra protein aggregation data.
(A) The relationship between observed ln(νmut/νwt) and Δhydrophobicity. To insure that the effect of hydrophobicity change was considered independent of other physiochemical properties, only mutations that had a Δcharge of 0 and a |ΔΔG coil-α + ΔΔG β-coil| of less than 2.5 kJ/mol were considered.
(B) The relationship between observed ln(νmut/νwt) and ΔΔG coil-α + ΔΔG β-coil. To insure that the effect of secondary structure change was considered independent of other physiochemical properties, only mutations which had a Δcharge of 0 and a |Δhydrophobicity| of less than 3 kcal/mol were considered.
(C) The relationship between the observed ln(νmut/νwt) and Δcharge. To ensure that the effect of charge change was considered independent of other physiochemical properties, only mutations that had a |Δhydrophobicity| of less than 3 kcal/mol and a |ΔΔG coil-α + ΔΔG β-coil| of less than 2.5 kJ/mol were considered. Wild-type protein was used as a data point at (0,0) in all of the three graphs. The rederived slopes from this figure for the three factors, 0.95 for hydrophobicity, 0.18 for secondary structure, and −0.78 for charge, were applied to calculate aggregation propensities of fALS-causing SOD1 variants presented in Figure 4. Patient survival times were plotted against these aggregation propensities; the corresponding slope and R values differ less than 5% compared to the results in Figure 4 (unpublished data), validating the Chiti–Dobson equation.
The average patient survival times for different SOD1 variants with measured thermodynamic stabilities were plotted against corresponding predicted aggregation propensities, and linear regression analysis weighted by the number of patients for each mutation yielded R (multiple correlation coefficient, with a larger value indicating a stronger relationship) and P (value less than 0.05 implies a significant result) values of 0.58 and <0.001, respectively (Figure 3A). The severity of fALS thus is related to mutation-induced increases in SOD1 aggregation propensity. The same plot was performed with the linear regression analysis not weighted by the number of patients (Figure 4A), yielding R and P values of 0.23 and 0.2, respectively. Unfortunately, the published epidemiology data do not provide the information necessary to stratify for known ALS covariates, including lifestyle (diet and smoking) [76–79], palliative care [80], bulbar onset, etc., and weighted data are more likely to account for differences in these factors. The Chiti–Dobson equation results for all fALS-causing SOD1 mutations with patients' survival data also were evaluated in a univariate Cox proportional hazard model (Table 3). The hazard ratio for the Chiti–Dobson equation result was significantly higher than 1.0, which also indicates that aggregation propensity is a risk factor for fALS. Previous studies of Huntington's disease revealed an inverse relationship between the length of glutamine repeat of huntingtin and age of disease onset. The authors of this previous study concluded that disease onset correlates with rate of nucleation of aggregation [81]. We demonstrate here an inverse relationship between the rate of aggregation elongation after nucleation and the disease duration after onset.
Figure 3. Synergistic Increases in SOD1 Aggregation Propensity and Increases of Instability Are Associated with Decreases in Survival for fALS Patients (Data Weighted by the Number of Patients).

(A) An increase in aggregation propensity is associated with a decrease in fALS patient survival. Single point mutations of SOD1 found in fALS patients with corresponding reported stability values in (B) were considered. Aggregation propensities for each fALS mutation were calculated using the Chiti–Dobson equation, normalized such that 0 represents the least and 1 represents the most aggregation prone proteins, and the corresponding disease duration (survival) was plotted versus this value.
(B) An increase in SOD1 instability is associated with severe disease. All instability values of apo SOD1 reported in the literature were normalized such that 0 represents the most and 1 represents the least stable proteins, and corresponding disease durations were plotted versus these values.
(C) Increase in SOD1 aggregation propensity and gain of instability synergistically decrease patient survival. Normalized aggregation propensity in (A) and instability in (B) were summed and normalized to the range from 0 to 1, and patient survival was plotted versus this value. The data used in these three graphs (disease durations from 580 patients with 28 different fALS-causing SOD1 mutations with reported stability values) were weighted based on the number of patients for each mutation using SPSS version 15.0.
Note that the correlation between the size of each data point and the number of patients for (A–C) is shown as an inset in (C).
Figure 4. Synergistic Increases in SOD1 Aggregation Propensity and Losses of Thermodynamic Stability Are Associated with Decreased Survival of fALS Patients (Using Unweighted Data).

(A) An increase in aggregation propensity is associated with decreased fALS patient survival. Single point mutations found in fALS patients with corresponding reported thermodynamic stabilities in (B) were considered.
(B) A loss of SOD1 thermodynamic stability is associated with severe disease.
(C) Increase in SOD1 aggregation propensity and gain of instability synergistically decrease patient survival. The data used in these three graphs (disease durations from 580 patients with 28 different fALS-causing SOD1 mutations with reported stability values) were treated equally regardless of the number of patients using the software SigmaPlot 9.0 (Systat Software, Inc.). SPSS 15.0 also was used on this analysis, and identical results were obtained.
Protein Instability Is a Risk Factor for fALS
On the basis of our observation that predicted increased protein aggregation correlates with increased disease severity and previous data indicating that protein unfolding or misfolding promote aggregation [82–85], we tested the hypothesis that a loss of protein stability also could be a risk factor for ALS. For the sake of simplicity, we use the term instability throughout this article, with instability defined as the inverse of either the normalized ΔΔG (unfolding free energy change difference between mutant and wild-type SOD1) or normalized ΔT m (melting point difference between mutant and wild-type SOD1). Instability was considered for two reasons: (1) the Chiti–Dobson equation predicts the aggregation rates of unfolded proteins (it was derived from the aggregation rates of proteins in high trifluoroethanol concentrations that contained secondary but no tertiary structure), and therefore, formally, unfolding must occur prior to aggregation, and (2) unfolding is known to speed protein aggregation in vitro to the extent that without chemically induced unfolding induction periods extend from months to years, as demonstrated for SOD1 [32]. Aggregation in vivo therefore may require protein unfolding. Before using stability data published by different laboratories using different methods (melting point, which yields ΔT m, or chaotroph-induced unfolding, which yields ΔΔG), we sought to determine the reliability of the data. If different laboratories reported similar values of stability for the same mutants, then the data could be deemed reliable. Therefore, all published measurements of apo SOD1 stability (metallated SOD1 calorimetry data often bear the characteristics of irreversible denaturation, probably via Cu-catalyzed disulfide bond formation, and is therefore less reliable) [15,31,60,86–88] were compiled, and the experimental values of ΔΔG and ΔT m were normalized to the range from 0 to 1 (described in the Materials and Methods section), with 0 representing the least stable, and 1 representing the most stable (highest stability) variant. Through the use of all of the data from mutants where ΔΔG and ΔT m were measured by different laboratories, a plot of normalized ΔΔG versus normalized ΔT m was created. Good interlaboratory correlation of measured stability values was observed (slope = 0.94, R = 0.90, P = 0.002; Figure 5), and we therefore deemed the stability data reliable for use.
Figure 5. Correlation between Measures of Stability, Specifically Normalized ΔT m and ΔΔG, Reported from Different Groups for Apo SOD1 Variants.

The variants with both measured ΔT m and ΔΔG were plotted, and a good correlation between normalized ΔT m and normalized ΔΔG was observed. This correlation provides the rationale for averaging ΔT m and ΔΔG and taking the average value as an indicator for stability of fALS-related variants. The normalized ΔT m and ΔΔG were obtained as described in the Materials and Methods section.
Next, patient survival data for fALS-causing SOD1 variants were plotted against corresponding instability values, and linear regression analysis weighted by the number of patients for each mutation yielded R and P values of 0.71 and <0.001, respectively (Figure 3B). The same plot was performed with the linear regression analysis not weighted by the number of patients (Figure 4B), yielding R = 0.34 and P = 0.07. A gain of SOD1 instability (loss of stability) upon mutation therefore is related to decreased fALS patient survival. Increased in vitro instability is consistent with previous findings that the in vivo half-lives of SOD1 variants are decreased [89].
Protein Aggregation Propensity and Protein Instability Are Synergistic Risk Factors for fALS
Previous results from computer simulations indicate a multistep process for aggregation via destabilization [90], encouraging us to understand the combined effect of aggregation propensity and protein instability upon ALS patient survival. On the basis of their respective multiple correlation coefficients and slopes, aggregation propensity and instability are equal contributors to fALS patients' survival. Moreover, no obvious correlation between protein instability and aggregation propensity was observed for the SOD1 variants used in Figure 3 (Figure S1), indicating that increased instability is not responsible for the increased predicted protein aggregation propensity. The combination of instability and aggregation propensity represents the relative energy in proceeding from folded to unfolded apo SOD1 and then from unfolded to aggregated states. Patient survival was plotted against corresponding summed instability and aggregation propensity values. A linear regression analysis weighted by the number of patients for each mutation yielded R and P values of 0.83 and <0.001, respectively (Figure 3C). The same plot was performed with the linear regression analysis not weighted by the number of patients (Figure 4C), yielding R = 0.47 and P = 0.01. The improved statistical result of predicting patient survival after combining instability and aggregation propensity indicates that aggregation occurs from unfolded or partially unfolded SOD1. The stability data used herein were for apo SOD1, and therefore the absence of metals is implicit. The R 2 value was 0.69 from the weighted data, indicating that 69% of the intrinsic variability these fALS patients' survival resulted from the combination of increased aggregation propensity and instability. Additionally, aggregation propensity and instability were evaluated in a Cox proportional hazard model (Table 5). The hazard ratios were significantly higher than 1.0 for both factors. The sum of aggregation propensity and instability also was evaluated in a univariate Cox proportional hazard model (Table 5). The hazard ratio for this sum was also significantly higher than 1.0, further indicating that aggregation propensity and instability are synergistic risk factors for fALS. Note that the aggregation propensity and instability tested in Table 5 were normalized to the range from 0 to 1 (as in Figures 3 and 4), while the values tested in Table 3 were not normalized. As a result of normalization, which decreased the value range of tested factors, the hazard ratios of Table 5 are much larger than Table 3, and therefore the large values of hazard ratios reported in Table 5 should not be overinterpreted. Significantly, a fALS patient with an SOD1 mutation of relatively low aggregation propensity and high stability is expected to survive longer after disease onset. It has not escaped our attention that the rate of protein aggregation has implications in both sporadic diseases and aging; for example, the toxicity of a given posttranslational modification is a function of its effect on protein stability and aggregation propensity.
Table 5.
SOD1 Variants' Aggregation Propensity and Instability Are ALS Risk Factors

Discussion
We describe here synergistic gains of toxic functions of SOD1 in ALS. These are the first results in any neurodegenerative disease demonstrating that protein instability and aggregation propensity are synergistic risk factors. The fact that there are two synergistic risk factors rather than a single toxic gain of function probably has delayed the discovery of the mechanisms of fALS mutant SOD1 toxicity. The SOD1 stability data used in this paper were measured from apo SOD1, and the aggregation rate data used to create the Chiti–Dobson model were from in vitro unfolded proteins. Therefore, formally, the combination of instability and aggregation propensity represents the relative energy in proceeding from apo folded to unfolded SOD1 and then from unfolded to aggregated states. It has been demonstrated experimentally that apo SOD1 has a faster rate of aggregation than that of holo forms [32]. Partial unfolding/misfolding also can lead to aggregation [28,91–96], and our results cannot rule out a role for the aggregation of partially folded, including metallated, SOD1. Previous studies revealed a correlation [15] and conversely a lack of correlation [86] between SOD1 variant stability and patient disease duration. Correlation between SOD1 variant stability and patient disease duration, however, required that the authors omit stability data of 4 of the 15 variants from their regression analysis (on the basis that these variants change the net charge of SOD1).
As presented in Table 3, SOD1 variants' loss of net charge correlates with increased patient survival, while gain of hydrophobicity, loss of α-helix, and gain of β-sheet propensity are ALS risk factors. On the basis of Dobson and co-workers' related work [69,73,97], a loss of net charge is predicted to increase the aggregation propensity of unfolded proteins. If aggregation is toxic, then one would expect loss of net charge to be toxic. In contrast to the synergistic effects for aggregation propensity and instability presented in Table 5, the correlation of loss of net charge with increased survival has an effect of decreasing the hazard ratio presented in the univariate model presented in Table 3. We demonstrate that mutations causing the entire protein to approach neutrality are protective in the context of fALS (Table 3) rather than deleterious as proposed by Oliveberg and co-workers [15,68]. These results should be cautiously interpreted since in contrast to our Cox proportional hazard model result that loss of net charge is protective, the mean patient survival for loss of net charge and gain of net charge mutations, unweighted by the number of patients, are 7.1 and 6.9 years, respectively. Further study clearly is required to understand the role of charge in ALS etiology.
In contrast with the strong familiality shown for disease duration after onset (Table 1), SOD1-mediated ALS showed modest familiality with respect to onset, accounting for only 42% of the variability in A4V and D90A fALS patients [98], and with only G37R and L38V mutations of SOD1 being significant covariates of age of onset [67]. The same analysis shown in Figures 3 and 4 was performed using age at disease onset rather than disease duration as the dependant variable (Figure S2), and little or no relationship between disease onset and aggregation propensity or instability was observed. The Chiti–Dobson equation predicts the rate of aggregation after nucleation (rate of elongation). It is tempting therefore to speculate that the rate of nucleation is a determinant of age at onset. Testing this hypothesis would require the development of a model that can predict nucleation rates based upon physicochemical parameters, a task that is hampered by the stochastic nature of in vitro nucleation times [99,100] but that should now be possible given our recent development of methods for modeling in vitro nucleation kinetics [101].
Although our model accounts for 69% of the variability in fALS patient survival after onset, there are clearly genetic components of fALS that our model cannot account for. For example, while D90A is normally a dominantly inherited mutation in North America, 2.5% of people in Sweden and Finland are heterozygous asymptomatic carriers of the D90A SOD1 mutation [102,103] and require two mutant alleles before presenting ALS symptoms. Notably, our results and conclusions were unaffected by including or excluding D90A survival times during data analysis.
It is postulated that diseases for which protein aggregation contributes to patient death will (1) develop in cells with the highest concentration of the aggregation-prone protein in accordance with the concentration dependence of aggregation rates [101,104] and (2) have a prognosis influenced by the aggregation propensity of the aggregating protein, in accordance with the results reported herein. Motor neurons are the cells in the ventral horn of the spinal cord with the highest SOD1 concentration [39,105], perhaps explaining an aspect of the selective vulnerability of these cells.
Protein aggregation is a hallmark of many neurodegenerative diseases, including ALS. The toxicity of aggregation is fiercely debated (reviewed in [106,107]), fueled by reproducible evidence that aggregates can be toxic [108,109], have no effect [110], or be protective [111]. We propose that aggregates on either extreme of size, i.e., small protofibrillar aggregates [108,112] or aggregates large enough to clog axons, are more toxic, while midsize microscopically visible aggregates are less toxic [106]. Our data indicate that the increased aggregation propensity of SOD1 is related to decreased survival of ALS patients. Notably aggregation of SOD1 has been demonstrated in fALS [113,114] and a subset of sALS patients [114,115], 18 fALS rodent models of 13 different SOD1 mutations [7–9,23,59,116–128], and at least 13 SOD1 mutants in cell models [56,58,59,116,118,119,129–131].
Materials and Methods
Familial ALS patients' disease duration and age of onset.
Familial ALS patients' data were taken from all of the available literature. Disease duration was initiated with onset of the first symptoms until the patient's death or when respiratory assistance was required for patients' survival. The average duration and onset for each mutation were calculated as the weighted average based on the number of patients (Table 6). If the patients were still reported to be alive without respiratory assistance at the end of a study, then their disease durations were not used to calculate the average unless the known duration value was larger than the average calculated with only durations from patients deceased or with respiratory assistance. For studies reporting average disease duration and Kaplan–Meier curves, the reported average durations were used to calculate the weighted averages. The current unavailability of http://www.alsod.org/ made it impossible to review the references provided by the website (from which we had taken survival times before it became unavailable), which created the risk of counting a patient's disease onset or survival twice, and made reproducing our study impossible for other groups. We therefore opted not to report data from this website in this study, thereby eliminating no more than 67 (there were 67 http://www.alsod.org/ patients' data without accompanying literature references that may, or may not, have been represented by our literature search) of 1319 patients' data. However, we did perform a complete, alternative set of analyses that did include http://www.alsod.org/ data (unpublished data), and the statistical correlations in the figures and tables shown herein persisted. Mean values of disease durations also were obtained from Kaplan–Meier curves and tested on SOD1 mutations with known experimental thermodynamic stabilities, and the results were comparable to those in Figure 3. Since the weighted average method can provide disease duration regardless of the number of patients, we opted for its use.
Table 6.
Disease Durations and Age of Onsets for SOD1 Mutation Related fALS, Non-SOD1-Related fALS, and sALS Patients

Kaplan–Meier survival curves, the log rank tests, and the Cox proportional hazard model.
Kaplan–Meier curves of survival for different fALS-causing SOD1 mutations, non-SOD1-related fALS, and sALS were generated. The hazard ratios of different fALS-causing SOD1 mutations and non-SOD1-related fALS compared to sALS were tested as a category variable by Cox proportional hazard model analysis. For studies reporting Kaplan–Meier curves but without individual patients' data, the Engauge Digitizer 4.1 software was used to obtain coordinates for cumulative survival at each time point. This information was used to calculate the number of patients not surviving at each time point under the assumption that there is no censored patient (with unknown exact survival time because of being alive at the end of study, lost to follow-up, or withdrawal from the study) within the course of survival curves. For cumulative survival not reaching 0 at the end of study, those fractions of patients were treated as censored. The error of the estimated number of patients is less than 5% of the number reported. To eliminate the chance that one or two patients' survival data bias the analysis result, a rule of thumb [132] requiring that each tested fALS-related SOD1 mutation includes at least five noncensored patients was applied. Since patients' survival was reported only as an average from a group of patients and individual patient's survival information was not described in some publications, one or two publications' survival data might bias the analysis result. To eliminate this chance of bias, the rule of thumb was modified as requiring at least five independent descriptions of noncensored patients' survival data (a reported average without individual patient's survival information was treated as one description). The statistical analysis was performed with the software SPSS 15.0 (SPSS, Inc.).
Aggregation propensity calculated from the Chiti–Dobson equation.
The hydrophobicity, β-sheet propensity, and charge values for the amino acid residues were obtained from the Supplementary Information of [69]. While applying the AGADIR algorithm at http://www.embl-heidelberg.de/Services/serrano/agadir/agadir-start.html to obtain α-helical propensities for wild-type (wt) and mutant (mut) P α wt and P α mut values for ΔΔG coil-α calculation for human SOD1, the parameters of pH 7, 310 K, and ionic strength of 0.100 were used. For the protein human SOD1, the N terminus is acetylated, and the C terminus is free in vivo. After the prediction at the residue level was output, the value in the column “Hel” at a specific residue was taken as P α. If a value of 0 for P α was obtained, then 0.1 was added to both P α wt and P α mut values for the correct mathematical meaning of ln(P α wt/P α mut) (F. Chiti, personal communication). The Chiti–Dobson equation terms, Δhydrophobicity, Δcharge, ΔΔG coil-α, and ΔΔG β-coil, were calculated based on equations illustrated in the legend of Table 1 of [69]. The ln(νmut/νwt) values were calculated based on Equation 1 from [69] and normalized from 0 to 1 using the equation normalized aggregation propensity = (aggregation propensity before normalization – MINap)/(MAXap – MINap), with MINap and MAXap as the minimum and maximum aggregation propensities of fALS-causing mutations with known thermodynamic stabilities, respectively, so that the larger normalized values correlate to larger aggregation propensities.
Normalized ΔΔG.
The free energy change difference (ΔΔG) and melting temperature difference (ΔT m) of unfolding a pathogenic variant and wild-type protein are parameters used to characterize the thermodynamic stability of a protein. ΔΔG values were taken from Table 2 of [15]. To graph with other protein stability data, the ΔΔG values were normalized by applying the equation normalized ΔΔG = (ΔΔG values before normalization – MINΔΔG)/(MAXΔΔG – MINΔΔG), with MINΔΔG and MAXΔΔG as the minimum and maximum values of ΔΔG in this dataset, respectively.
Normalized ΔT m.
ΔT m values were taken from Table 1 of [86], Table 1 of [60], Table 2 of [87], Table 3 of [88], and Table II of [31]. ΔT m values from [60,87,88] were averaged for each mutation. Those results then were averaged with the ΔT m values from [31,86] to determine the ΔT m values for given mutations. The normalized ΔT m values were obtained by applying the equation normalized ΔT m = (average ΔT m values before normalization – MINΔTm)/(MAXΔTm – MINΔTm), with MINΔTm and MAXΔTm as the minimum and maximum values of averaged ΔT m values in this dataset, respectively.
Thermodynamic instability of SOD1 variants.
The instability values for SOD1 variants were obtained from the equation normalized instability = 1 – average of normalized ΔΔG and normalized ΔT m for each mutation, so instability values are simply (1 – normalized ΔΔG or ΔT m), and larger values correlate to less stable variants.
The normalized aggregation propensity and instability for each variant were summed and normalized to the range from 0 to 1 to consider the two factors together.
Supporting Information
(62 KB PDF)
The same dataset for fALS-associated SOD1 mutations shown in Figures 3 and 4 were considered. The predicted aggregation propensities and instabilities from 28 different fALS-causing SOD1 mutations were plotted using the software SigmaPlot 9.0 (Systat Software, Inc.).
(213 KB AI).
The relationship between SOD1 aggregation propensity (A, D), instability (B, E), or sum of aggregation propensity and instability (C, F) with fALS patients' age of onset are presented. The linear regressions presented in (A–C) were weighted by the number of patients for each mutation using SPSS version 15.0 (SPSS, Inc.). The correlation between the size of each data point and the number of patients for (A-C) is shown as an inset in (C). The linear regressions presented in (D–F) were treated equally regardless of the number of patients for each mutation (unweighted) using the software SigmaPlot 9.0 (Systat Software, Inc.). The age of onset data presented in these six graphs are from 649 patients with 29 different fALS-causing SOD1 mutations with reported stability values. Aggregation propensity, instability, and sum of aggregation propensity and instability were obtained as described in the Materials and Methods section. Aggregation propensity, instability, or sum of aggregation propensity and instability has little or no correlation with patients' age of onset.
(261 KB AI).
Acknowledgments
We thank Professor F. Chiti for help with understanding the application of the Chiti–Dobson equation. We thank Drs. G. A. Petsko, D. Ringe, M. Rosbash, E. Marder, R. Finke, N. Bonini, and I. Epstein, the entire Agar laboratory, and A. Morris for comments on the manuscript. We thank J. Lani from Statistics Solutions for advice on statistical analysis. We especially thank the neurologists and patients worldwide for the patient outcome data used in this study, while noting that improving upon this study and extending this study to sporadic disease requires that physicians begin to include epidemiological information, including patient lifestyle and palliative care for all neurodegenerative disease patients.
Abbreviations
- ALS
amyotrophic lateral sclerosis
- fALS
familial ALS
- mut
mutant
- sALS
sporadic ALS
- SOD1
Cu/Zn-superoxide dismutase
- wt
wild-type
Footnotes
Author contributions. JNA and NYRA conceived the hypotheses. QW and JNA designed the analysis. QW, JLJ, and JNA performed the analysis. QW and JNA wrote the paper.
Funding. This work was made possible by award W81XWH-04–0158 from the Department of Defense and grant 1392 from the ALS Association. NYRA was supported by a research award from the American Brain Tumor Association and a research grant from the Brain Science Foundation.
Competing interests. The authors have declared that no competing interests exist.
References
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Mitchell JD, Borasio GD. Amyotrophic lateral sclerosis. Lancet. 2007;369:2031–2041. doi: 10.1016/S0140-6736(07)60944-1. [DOI] [PubMed] [Google Scholar]
- Valdmanis PN, Rouleau GA. Genetics of familial amyotrophic lateral sclerosis. Neurology. 2008;70:144–152. doi: 10.1212/01.wnl.0000296811.19811.db. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Andersen PM. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr Neurol Neurosci Rep. 2006;6:37–46. doi: 10.1007/s11910-996-0008-9. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, et al. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Ripps ME, Huntley GW, Hof PR, Morrison JH, Gordon JW. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 1995;92:689–693. doi: 10.1073/pnas.92.3.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, et al. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Hayward LJ. Mutant SOD1 instability: implications for toxicity in amyotrophic lateral sclerosis. Neurodegener Dis. 2005;2:115–127. doi: 10.1159/000089616. [DOI] [PubMed] [Google Scholar]
- Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Valentine JS, Eggers DK, Roe JA, Tiwari A, et al. Familial amyotrophic lateral sclerosis-associated mutations decrease the thermal stability of distinctly metallated species of human copper/zinc superoxide dismutase. J Biol Chem. 2002;277:15932–15937. doi: 10.1074/jbc.M112088200. [DOI] [PubMed] [Google Scholar]
- Hough MA, Grossmann JG, Antonyuk SV, Strange RW, Doucette PA, et al. Dimer destabilization in superoxide dismutase may result in disease-causing properties: structures of motor neuron disease mutants. Proc Natl Acad Sci U S A. 2004;101:5976–5981. doi: 10.1073/pnas.0305143101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg MJ, Tibell L, Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci U S A. 2002;99:16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg MJ, Bystrom R, Boknas N, Andersen PM, Oliveberg M. Systematically perturbed folding patterns of amyotrophic lateral sclerosis (ALS)-associated SOD1 mutants. Proc Natl Acad Sci U S A. 2005;102:9754–9759. doi: 10.1073/pnas.0501957102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari A, Xu Z, Hayward LJ. Aberrantly increased hydrophobicity shared by mutants of Cu,Zn-superoxide dismutase in familial amyotrophic lateral sclerosis. J Biol Chem. 2005;280:29771–29779. doi: 10.1074/jbc.M504039200. [DOI] [PubMed] [Google Scholar]
- Brown RH., Jr. SOD1 aggregates in ALS: cause, correlate or consequence. Nat Med. 1998;4:1362–1364. doi: 10.1038/3945. [DOI] [PubMed] [Google Scholar]
- Shaw BF, Valentine JS. How do ALS-associated mutations in superoxide dismutase 1 promote aggregation of the protein. Trends Biochem Sci. 2007;32:78–85. doi: 10.1016/j.tibs.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Ellerby LM, Hart PJ, Wiedau-Pazos M, Valentine JS. Do posttranslational modifications of CuZnSOD lead to sporadic amyotrophic lateral sclerosis. Ann Neurol. 1997;42:135–137. doi: 10.1002/ana.410420202. [DOI] [PubMed] [Google Scholar]
- Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, et al. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- Tiwari A, Hayward LJ. Familial amyotrophic lateral sclerosis mutants of copper/zinc superoxide dismutase are susceptible to disulfide reduction. J Biol Chem. 2003;278:5984–5992. doi: 10.1074/jbc.M210419200. [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Rodriguez JA, Kim JW, Tiwari A, Goto JJ, et al. Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2002;277:15923–15931. doi: 10.1074/jbc.M112087200. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa Y, Fu R, Deng HX, Siddique T, O'Halloran TV. Disulfide cross-linked protein represents a significant fraction of ALS-associated Cu, Zn-superoxide dismutase aggregates in spinal cords of model mice. Proc Natl Acad Sci U S A. 2006;103:7148–7153. doi: 10.1073/pnas.0602048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agar J, Durham H. Relevance of oxidative injury in the pathogenesis of motor neuron diseases. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:232–242. doi: 10.1080/14660820310011278. [DOI] [PubMed] [Google Scholar]
- Crow JP, Sampson JB, Zhuang Y, Thompson JA, Beckman JS. Decreased zinc affinity of amyotrophic lateral sclerosis-associated superoxide dismutase mutants leads to enhanced catalysis of tyrosine nitration by peroxynitrite. J Neurochem. 1997;69:1936–1944. doi: 10.1046/j.1471-4159.1997.69051936.x. [DOI] [PubMed] [Google Scholar]
- Roberts BR, Tainer JA, Getzoff ED, Malencik DA, Anderson SR, et al. Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS. J Mol Biol. 2007;373:877–890. doi: 10.1016/j.jmb.2007.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elam JS, Taylor AB, Strange R, Antonyuk S, Doucette PA, et al. Amyloid-like filaments and water-filled nanotubes formed by SOD1 mutant proteins linked to familial ALS. Nat Struct Biol. 2003;10:461–467. doi: 10.1038/nsb935. [DOI] [PubMed] [Google Scholar]
- Strange RW, Antonyuk S, Hough MA, Doucette PA, Rodriguez JA, et al. The structure of holo and metal-deficient wild-type human Cu, Zn superoxide dismutase and its relevance to familial amyotrophic lateral sclerosis. J Mol Biol. 2003;328:877–891. doi: 10.1016/s0022-2836(03)00355-3. [DOI] [PubMed] [Google Scholar]
- Arnesano F, Banci L, Bertini I, Martinelli M, Furukawa Y, et al. The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status. J Biol Chem. 2004;279:47998–48003. doi: 10.1074/jbc.M406021200. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, O'Halloran TV. Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation. J Biol Chem. 2005;280:17266–17274. doi: 10.1074/jbc.M500482200. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Durazo A, Girotto S, Gralla EB, et al. Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS. Proc Natl Acad Sci U S A. 2007;104:11263–11267. doi: 10.1073/pnas.0704307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Beal MF, Becher MW, Schulz JB, Wong PC, et al. Elevated free nitrotyrosine levels, but not protein-bound nitrotyrosine or hydroxyl radicals, throughout amyotrophic lateral sclerosis (ALS)-like disease implicate tyrosine nitration as an aberrant in vivo property of one familial ALS-linked superoxide dismutase 1 mutant. Proc Natl Acad Sci U S A. 1997;94:7606–7611. doi: 10.1073/pnas.94.14.7606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckman JS, Carson M, Smith CD, Koppenol WH. ALS, SOD and peroxynitrite. Nature. 1993;364:584. doi: 10.1038/364584a0. [DOI] [PubMed] [Google Scholar]
- Crow JP, Ye YZ, Strong M, Kirk M, Barnes S, et al. Superoxide dismutase catalyzes nitration of tyrosines by peroxynitrite in the rod and head domains of neurofilament-L. J Neurochem. 1997;69:1945–1953. doi: 10.1046/j.1471-4159.1997.69051945.x. [DOI] [PubMed] [Google Scholar]
- Wiedau-Pazos M, Goto JJ, Rabizadeh S, Gralla EB, Roe JA, et al. Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis. Science. 1996;271:515–518. doi: 10.1126/science.271.5248.515. [DOI] [PubMed] [Google Scholar]
- Yim HS, Kang JH, Chock PB, Stadtman ER, Yim MB. A familial amyotrophic lateral sclerosis-associated A4V Cu, Zn-superoxide dismutase mutant has a lower Km for hydrogen peroxide. Correlation between clinical severity and the Km value. J Biol Chem. 1997;272:8861–8863. doi: 10.1074/jbc.272.14.8861. [DOI] [PubMed] [Google Scholar]
- Durham HD, Kabashi E, Taylor DM, Agar JN. The proteasome in neurodegeneration. In: Stefanis L, Keller JN, editors. Motor Neuron Disease. New York: Springer; 2007. pp. 247–264. [Google Scholar]
- Kabashi E, Agar JN, Taylor DM, Minotti S, Durham HD. Focal dysfunction of the proteasome: a pathogenic factor in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2004;89:1325–1335. doi: 10.1111/j.1471-4159.2004.02453.x. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Durham HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1038–1050. doi: 10.1016/j.bbadis.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Leigh PN, Dodson A, Swash M, Brion JP, Anderton BH. Cytoskeletal abnormalities in motor neuron disease. An immunocytochemical study. Brain. 1989;112:521–535. doi: 10.1093/brain/112.2.521. [DOI] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Collard JF, Cote F, Julien JP. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature. 1995;375:61–64. doi: 10.1038/375061a0. [DOI] [PubMed] [Google Scholar]
- Allen S, Heath PR, Kirby J, Wharton SB, Cookson MR, et al. Analysis of the cytosolic proteome in a cell culture model of familial amyotrophic lateral sclerosis reveals alterations to the proteasome, antioxidant defenses, and nitric oxide synthetic pathways. J Biol Chem. 2003;278:6371–6383. doi: 10.1074/jbc.M209915200. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- Bruening W, Roy J, Giasson B, Figlewicz DA, Mushynski WE, et al. Up-regulation of protein chaperones preserves viability of cells expressing toxic Cu/Zn-superoxide dismutase mutants associated with amyotrophic lateral sclerosis. J Neurochem. 1999;72:693–699. doi: 10.1046/j.1471-4159.1999.0720693.x. [DOI] [PubMed] [Google Scholar]
- Tummala H, Jung C, Tiwari A, Higgins CM, Hayward LJ, et al. Inhibition of chaperone activity is a shared property of several Cu,Zn-superoxide dismutase mutants that cause amyotrophic lateral sclerosis. J Biol Chem. 2005;280:17725–17731. doi: 10.1074/jbc.M501705200. [DOI] [PubMed] [Google Scholar]
- Borthwick GM, Johnson MA, Ince PG, Shaw PJ, Turnbull DM. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann Neurol. 1999;46:787–790. doi: 10.1002/1531-8249(199911)46:5<787::aid-ana17>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Haasdijk ED, Grashorn JA, Hawkins R, van Duijn W, et al. Human Cu/Zn superoxide dismutase (SOD1) overexpression in mice causes mitochondrial vacuolization, axonal degeneration, and premature motoneuron death and accelerates motoneuron disease in mice expressing a familial amyotrophic lateral sclerosis mutant SOD1. Neurobiol Dis. 2000;7:623–643. doi: 10.1006/nbdi.2000.0299. [DOI] [PubMed] [Google Scholar]
- Jaarsma D, Rognoni F, van Duijn W, Verspaget HW, Haasdijk ED, et al. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001;102:293–305. doi: 10.1007/s004010100399. [DOI] [PubMed] [Google Scholar]
- Kong J, Xu Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J Neurosci. 1998;18:3241–3250. doi: 10.1523/JNEUROSCI.18-09-03241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Vielhaber S, Kunz D, Winkler K, Wiedemann FR, Kirches E, et al. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain. 2000;123:1339–1348. doi: 10.1093/brain/123.7.1339. [DOI] [PubMed] [Google Scholar]
- Dal Canto MC, Gurney ME. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am J Pathol. 1994;145:1271–1279. [PMC free article] [PubMed] [Google Scholar]
- Mourelatos Z, Gonatas NK, Stieber A, Gurney ME, Dal Canto MC. The Golgi apparatus of spinal cord motor neurons in transgenic mice expressing mutant Cu,Zn superoxide dismutase becomes fragmented in early, preclinical stages of the disease. Proc Natl Acad Sci U S A. 1996;93:5472–5477. doi: 10.1073/pnas.93.11.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR. High molecular weight complexes of mutant superoxide dismutase 1: age-dependent and tissue-specific accumulation. Neurobiol Dis. 2002;9:139–148. doi: 10.1006/nbdi.2001.0471. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Borchelt DR. Mapping superoxide dismutase 1 domains of non-native interaction: roles of intra- and intermolecular disulfide bonding in aggregation. J Neurochem. 2006;96:1277–1288. doi: 10.1111/j.1471-4159.2005.03642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- Wang J, Slunt H, Gonzales V, Fromholt D, Coonfield M, et al. Copper-binding-site-null SOD1 causes ALS in transgenic mice: aggregates of non-native SOD1 delineate a common feature. Hum Mol Genet. 2003;12:2753–2764. doi: 10.1093/hmg/ddg312. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Rumfeldt JA, Scholz GA, Irani RA, Frey HE, et al. Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis show enhanced formation of aggregates in vitro. Proc Natl Acad Sci U S A. 2003;100:7021–7026. doi: 10.1073/pnas.1237797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw BF, Lelie HL, Durazo A, Nersissian AM, Xu G, et al. Detergent-insoluble Aggregates Associated with Amyotrophic Lateral Sclerosis in Transgenic Mice Contain Primarily Full-length, Unmodified Superoxide Dismutase-1. J Biol Chem. 2008;283:8340–8350. doi: 10.1074/jbc.M707751200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Boca M, Girotto S, Martinelli M, et al. SOD1 and Amyotrophic Lateral Sclerosis: Mutations and Oligomerization. PLoS ONE. 2008;3:e1677. doi: 10.1371/journal.pone.0001677. doi: 10.1371/journal.pone.0001677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armon C, Moses D. Linear estimates of rates of disease progression as predictors of survival in patients with ALS entering clinical trials. J Neurol Sci. 1998;160:S37–S41. doi: 10.1016/s0022-510x(98)00196-8. [DOI] [PubMed] [Google Scholar]
- Czaplinski A, Yen AA, Appel SH. Amyotrophic lateral sclerosis: early predictors of prolonged survival. J Neurol. 2006;253:1428–1436. doi: 10.1007/s00415-006-0226-8. [DOI] [PubMed] [Google Scholar]
- Czaplinski A, Yen AA, Simpson EP, Appel SH. Predictability of disease progression in amyotrophic lateral sclerosis. Muscle Nerve. 2006;34:702–708. doi: 10.1002/mus.20658. [DOI] [PubMed] [Google Scholar]
- Czaplinski A, Yen AA, Simpson EP, Appel SH. Slower disease progression and prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the natural history of amyotrophic lateral sclerosis changing. Arch Neurol. 2006;63:1139–1143. doi: 10.1001/archneur.63.8.1139. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41:210–221. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- Sandelin E, Nordlund A, Andersen PM, Marklund SS, Oliveberg M. Amyotrophic lateral sclerosis-associated copper/zinc superoxide dismutase mutations preferentially reduce the repulsive charge of the proteins. J Biol Chem. 2007;282:21230–21236. doi: 10.1074/jbc.M700765200. [DOI] [PubMed] [Google Scholar]
- Chiti F, Stefani M, Taddei N, Ramponi G, Dobson CM. Rationalization of the effects of mutations on peptide and protein aggregation rates. Nature. 2003;424:805–808. doi: 10.1038/nature01891. [DOI] [PubMed] [Google Scholar]
- Caflisch A. Computational models for the prediction of polypeptide aggregation propensity. Curr Opin Chem Biol. 2006;10:437–444. doi: 10.1016/j.cbpa.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]
- Luheshi LM, Tartaglia GG, Brorsson AC, Pawar AP, Watson IE, et al. Systematic in vivo analysis of the intrinsic determinants of amyloid β pathogenicity. PLoS Biol. 2007;5:e290. doi: 10.1371/journal.pbio.0050290. doi: 10.1371/journal.pbio.0050290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBay KF, Pawar AP, Chiti F, Zurdo J, Dobson CM, et al. Prediction of the absolute aggregation rates of amyloidogenic polypeptide chains. J Mol Biol. 2004;341:1317–1326. doi: 10.1016/j.jmb.2004.06.043. [DOI] [PubMed] [Google Scholar]
- Tartaglia GG, Cavalli A, Pellarin R, Caflisch A. Prediction of aggregation rate and aggregation-prone segments in polypeptide sequences. Protein Sci. 2005;14:2723–2734. doi: 10.1110/ps.051471205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez de Groot N, Pallares I, Aviles FX, Vendrell J, Ventura S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct Biol. 2005;5:18. doi: 10.1186/1472-6807-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutedja NA, Veldink JH, Fischer K, Kromhout H, Wokke JH, et al. Lifetime occupation, education, smoking, and risk of ALS. Neurology. 2007;69:1508–1514. doi: 10.1212/01.wnl.0000277463.87361.8c. [DOI] [PubMed] [Google Scholar]
- Nelson LM, McGuire V, Longstreth WT, Jr., Matkin C. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. I. Cigarette smoking and alcohol consumption. Am J Epidemiol. 2000;151:156–163. doi: 10.1093/oxfordjournals.aje.a010183. [DOI] [PubMed] [Google Scholar]
- Nelson LM, Matkin C, Longstreth WT, Jr., McGuire V. Population-based case-control study of amyotrophic lateral sclerosis in western Washington State. II. Diet. Am J Epidemiol. 2000;151:164–173. doi: 10.1093/oxfordjournals.aje.a010184. [DOI] [PubMed] [Google Scholar]
- Desport JC, Preux PM, Truong TC, Vallat JM, Sautereau D, et al. Nutritional status is a prognostic factor for survival in ALS patients. Neurology. 1999;53:1059–1063. doi: 10.1212/wnl.53.5.1059. [DOI] [PubMed] [Google Scholar]
- Kamimoto K, Murakami N, Muroga T, Matsubara M, Yamamoto M. [A comparative study between amyotrophic lateral sclerosis patients with and without mechanical ventilation] Rinsho Shinkeigaku. 1989;29:989–993. [PubMed] [Google Scholar]
- Perutz MF, Windle AH. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature. 2001;412:143–144. doi: 10.1038/35084141. [DOI] [PubMed] [Google Scholar]
- Booth DR, Sunde M, Bellotti V, Robinson CV, Hutchinson WL, et al. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature. 1997;385:787–793. doi: 10.1038/385787a0. [DOI] [PubMed] [Google Scholar]
- Fandrich M, Forge V, Buder K, Kittler M, Dobson CM, et al. Myoglobin forms amyloid fibrils by association of unfolded polypeptide segments. Proc Natl Acad Sci U S A. 2003;100:15463–15468. doi: 10.1073/pnas.0303758100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MA, Lazo ND, Lomakin A, Condron MM, Arai H, et al. Familial Alzheimer's disease mutations alter the stability of the amyloid beta-protein monomer folding nucleus. Proc Natl Acad Sci U S A. 2007;104:16522–16527. doi: 10.1073/pnas.0705197104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokvist M, Grobner G. Misfolding of amyloidogenic proteins at membrane surfaces: the impact of macromolecular crowding. J Am Chem Soc. 2007;129:14848–14849. doi: 10.1021/ja076059o. [DOI] [PubMed] [Google Scholar]
- Rodriguez JA, Shaw BF, Durazo A, Sohn SH, Doucette PA, et al. Destabilization of apoprotein is insufficient to explain Cu,Zn-superoxide dismutase-linked ALS pathogenesis. Proc Natl Acad Sci U S A. 2005;102:10516–10521. doi: 10.1073/pnas.0502515102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Rumfeldt JA, Karbassi F, Siddall CA, Lepock JR, et al. Calorimetric analysis of thermodynamic stability and aggregation for apo and holo amyotrophic lateral sclerosis-associated Gly-93 mutants of superoxide dismutase. J Biol Chem. 2006;281:6184–6193. doi: 10.1074/jbc.M509496200. [DOI] [PubMed] [Google Scholar]
- Vassall KA, Stathopulos PB, Rumfeldt JA, Lepock JR, Meiering EM. Equilibrium thermodynamic analysis of amyotrophic lateral sclerosis-associated mutant apo Cu,Zn superoxide dismutases. Biochemistry. 2006;45:7366–7379. doi: 10.1021/bi0600953. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Lee MK, Slunt HS, Guarnieri M, Xu ZS, et al. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc Natl Acad Sci U S A. 1994;91:8292–8296. doi: 10.1073/pnas.91.17.8292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellarin R, Caflisch A. Interpreting the aggregation kinetics of amyloid peptides. J Mol Biol. 2006;360:882–892. doi: 10.1016/j.jmb.2006.05.033. [DOI] [PubMed] [Google Scholar]
- Kelly JW. Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struct Biol. 1996;6:11–17. doi: 10.1016/s0959-440x(96)80089-3. [DOI] [PubMed] [Google Scholar]
- Khurana R, Gillespie JR, Talapatra A, Minert LJ, Ionescu-Zanetti C, et al. Partially folded intermediates as critical precursors of light chain amyloid fibrils and amorphous aggregates. Biochemistry. 2001;40:3525–3535. doi: 10.1021/bi001782b. [DOI] [PubMed] [Google Scholar]
- DiDonato M, Craig L, Huff ME, Thayer MM, Cardoso RM, et al. ALS mutants of human superoxide dismutase form fibrous aggregates via framework destabilization. J Mol Biol. 2003;332:601–615. doi: 10.1016/s0022-2836(03)00889-1. [DOI] [PubMed] [Google Scholar]
- Plakoutsi G, Bemporad F, Calamai M, Taddei N, Dobson CM, et al. Evidence for a mechanism of amyloid formation involving molecular reorganisation within native-like precursor aggregates. J Mol Biol. 2005;351:910–922. doi: 10.1016/j.jmb.2005.06.043. [DOI] [PubMed] [Google Scholar]
- Rumfeldt JA, Stathopulos PB, Chakrabarrty A, Lepock JR, Meiering EM. Mechanism and thermodynamics of guanidinium chloride-induced denaturation of ALS-associated mutant Cu,Zn superoxide dismutases. J Mol Biol. 2006;355:106–123. doi: 10.1016/j.jmb.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Shaw BF, Durazo A, Nersissian AM, Whitelegge JP, Faull KF, et al. Local unfolding in a destabilized, pathogenic variant of superoxide dismutase 1 observed with H/D exchange and mass spectrometry. J Biol Chem. 2006;281:18167–18176. doi: 10.1074/jbc.M600623200. [DOI] [PubMed] [Google Scholar]
- Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, et al. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J Mol Biol. 2005;350:379–392. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Fogh I, Rijsdijk F, Andersen PM, Sham PC, Knight J, et al. Age at onset in sod1-mediated amyotrophic lateral sclerosis shows familiality. Neurogenetics. 2007;8:235–236. doi: 10.1007/s10048-007-0092-2. [DOI] [PubMed] [Google Scholar]
- Ivanova M, Jasuja R, Kwong S, Briehl RW, Ferrone FA. Nonideality and the nucleation of sickle hemoglobin. Biophys J. 2000;79:1016–1022. doi: 10.1016/S0006-3495(00)76355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopeit T, Hortschansky P, Schroeckh V, Guhrs K, Zandomeneghi G, et al. Mutagenic analysis of the nucleation propensity of oxidized Alzheimer's beta-amyloid peptide. Protein Sci. 2005;14:2125–2131. doi: 10.1110/ps.051470405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AM, Watzky MA, Agar JN, Finke RG. Fitting neurological protein aggregation kinetic data via a 2-step, minimal/“Ockham's razor” model: the Finke–Watzky mechanism of nucleation followed by autocatalytic surface growth. Biochemistry. 2008;47:2413–2427. doi: 10.1021/bi701899y. [DOI] [PubMed] [Google Scholar]
- Sjalander A, Beckman G, Deng HX, Iqbal Z, Tainer JA, et al. The D90A mutation results in a polymorphism of Cu,Zn superoxide dismutase that is prevalent in northern Sweden and Finland. Hum Mol Genet. 1995;4:1105–1108. doi: 10.1093/hmg/4.6.1105. [DOI] [PubMed] [Google Scholar]
- Robberecht W, Aguirre T, Van den Bosch L, Tilkin P, Cassiman JJ, et al. D90A heterozygosity in the SOD1 gene is associated with familial and apparently sporadic amyotrophic lateral sclerosis. Neurology. 1996;47:1336–1339. doi: 10.1212/wnl.47.5.1336. [DOI] [PubMed] [Google Scholar]
- O'Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen S, et al. Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay. Methods Enzymol. 2006;413:34–74. doi: 10.1016/S0076-6879(06)13003-7. [DOI] [PubMed] [Google Scholar]
- Sau D, De Biasi S, Vitellaro-Zuccarello L, Riso P, Guarnieri S, et al. Mutation of SOD1 in ALS: a gain of a loss of function. Hum Mol Genet. 2007;16:1604–1618. doi: 10.1093/hmg/ddm110. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Opinion: what is the role of protein aggregation in neurodegeneration. Nat Rev Mol Cell Biol. 2005;6:891–898. doi: 10.1038/nrm1742. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–511. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- Taylor DM, Gibbs BF, Kabashi E, Minotti S, Durham HD, et al. Tryptophan 32 potentiates aggregation and cytotoxicity of a copper/zinc superoxide dismutase mutant associated with familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282:16329–16335. doi: 10.1074/jbc.M610119200. [DOI] [PubMed] [Google Scholar]
- Witan H, Kern A, Koziollek-Drechsler I, Wade R, Behl C, et al. Heterodimer formation of wild-type and amyotrophic lateral sclerosis-causing mutant Cu/Zn-Superoxide dismutase induces toxicity independent of protein aggregation. Hum Mol Genet. 2008;17:1373–1385. doi: 10.1093/hmg/ddn025. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Shibata N, Hirano A, Kobayashi M, Siddique T, Deng HX, et al. Intense superoxide dismutase-1 immunoreactivity in intracytoplasmic hyaline inclusions of familial amyotrophic lateral sclerosis with posterior column involvement. J Neuropathol Exp Neurol. 1996;55:481–490. doi: 10.1097/00005072-199604000-00011. [DOI] [PubMed] [Google Scholar]
- Shibata N, Asayama K, Hirano A, Kobayashi M. Immunohistochemical study on superoxide dismutases in spinal cords from autopsied patients with amyotrophic lateral sclerosis. Dev Neurosci. 1996;18:492–498. doi: 10.1159/000111445. [DOI] [PubMed] [Google Scholar]
- Wood JD, Beaujeux TP, Shaw PJ. Protein aggregation in motor neurone disorders. Neuropathol Appl Neurobiol. 2003;29:529–545. doi: 10.1046/j.0305-1846.2003.00518.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, et al. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, et al. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Yasui K, Nakano T, Doi K, Fukada Y, et al. Mouse motor neuron disease caused by truncated SOD1 with or without C-terminal modification. Brain Res Mol Brain Res. 2005;135:12–20. doi: 10.1016/j.molbrainres.2004.11.019. [DOI] [PubMed] [Google Scholar]
- Jonsson PA, Ernhill K, Andersen PM, Bergemalm D, Brannstrom T, et al. Minute quantities of misfolded mutant superoxide dismutase-1 cause amyotrophic lateral sclerosis. Brain. 2004;127:73–88. doi: 10.1093/brain/awh005. [DOI] [PubMed] [Google Scholar]
- Chang-Hong R, Wada M, Koyama S, Kimura H, Arawaka S, et al. Neuroprotective effect of oxidized galectin-1 in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2005;194:203–211. doi: 10.1016/j.expneurol.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Tobisawa S, Hozumi Y, Arawaka S, Koyama S, Wada M, et al. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun. 2003;303:496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- Jonsson PA, Bergemalm D, Andersen PM, Gredal O, Brannstrom T, et al. Inclusions of amyotrophic lateral sclerosis-linked superoxide dismutase in ventral horns, liver, and kidney. Ann Neurol. 2008;63:671–675. doi: 10.1002/ana.21356. [DOI] [PubMed] [Google Scholar]
- Zetterstrom P, Stewart HG, Bergemalm D, Jonsson PA, Graffmo KS, et al. Soluble misfolded subfractions of mutant superoxide dismutase-1s are enriched in spinal cords throughout life in murine ALS models. Proc Natl Acad Sci U S A. 2007;104:14157–14162. doi: 10.1073/pnas.0700477104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson PA, Graffmo KS, Brannstrom T, Nilsson P, Andersen PM, et al. Motor neuron disease in mice expressing the wild type-like D90A mutant superoxide dismutase-1. J Neuropathol Exp Neurol. 2006;65:1126–1136. doi: 10.1097/01.jnen.0000248545.36046.3c. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Li H, Gonzales V, Fromholt D, et al. Somatodendritic accumulation of misfolded SOD1-L126Z in motor neurons mediates degeneration: αB-crystallin modulates aggregation. Hum Mol Genet. 2005;14:2335–2347. doi: 10.1093/hmg/ddi236. [DOI] [PubMed] [Google Scholar]
- Gurney ME. The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci. 1997;152:S67–S73. doi: 10.1016/s0022-510x(97)00247-5. [DOI] [PubMed] [Google Scholar]
- Nagai M, Aoki M, Miyoshi I, Kato M, Pasinelli P, et al. Rats expressing human cytosolic copper–zinc superoxide dismutase transgenes with amyotrophic lateral sclerosis: associated mutations develop motor neuron disease. J Neurosci. 2001;21:9246–9254. doi: 10.1523/JNEUROSCI.21-23-09246.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Slunt HH, Gonzales V, Coonfield M, et al. Coincident thresholds of mutant protein for paralytic disease and protein aggregation caused by restrictively expressed superoxide dismutase cDNA. Neurobiol Dis. 2005;20:943–952. doi: 10.1016/j.nbd.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Allgulander C, Fisher LD. Survival analysis (or time to an event analysis), and the Cox regression model—methods for longitudinal psychiatric research. Acta Psychiatr Scand. 1986;74:529–535. doi: 10.1111/j.1600-0447.1986.tb06279.x. [DOI] [PubMed] [Google Scholar]
- Chiti F, Calamai M, Taddei N, Stefani M, Ramponi G, et al. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc Natl Acad Sci U S A. 2002;99:16419–16426. doi: 10.1073/pnas.212527999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azriel R, Gazit E. Analysis of the minimal amyloid-forming fragment of the islet amyloid polypeptide. An experimental support for the key role of the phenylalanine residue in amyloid formation. J Biol Chem. 2001;276:34156–34161. doi: 10.1074/jbc.M102883200. [DOI] [PubMed] [Google Scholar]
- Sakagashira S, Hiddinga HJ, Tateishi K, Sanke T, Hanabusa T, et al. S20G mutant amylin exhibits increased in vitro amyloidogenicity and increased intracellular cytotoxicity compared to wild-type amylin. Am J Pathol. 2000;157:2101–2109. doi: 10.1016/S0002-9440(10)64848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmona M, Malesani P, De Gioia L, Gorla S, Bruschi M, et al. Molecular determinants of the physicochemical properties of a critical prion protein region comprising residues 106–126. Biochem J. 1999;342:207–214. [PMC free article] [PubMed] [Google Scholar]
- Thompson AJ, Barnham KJ, Norton RS, Barrow CJ. The Val-210-Ile pathogenic Creutzfeldt-Jakob disease mutation increases both the helical and aggregation propensities of a sequence corresponding to helix-3 of PrPC . Biochim Biophys Acta. 2001;1544:242–254. doi: 10.1016/s0167-4838(00)00225-9. [DOI] [PubMed] [Google Scholar]
- Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, et al. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Murray IV, Trojanowski JQ, Lee VM. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem. 2001;276:2380–2386. doi: 10.1074/jbc.M008919200. [DOI] [PubMed] [Google Scholar]
- Van Nostrand WE, Melchor JP, Cho HS, Greenberg SM, Rebeck GW. Pathogenic effects of D23N Iowa mutant amyloid β -protein. J Biol Chem. 2001;276:32860–32866. doi: 10.1074/jbc.M104135200. [DOI] [PubMed] [Google Scholar]
- Miravalle L, Tokuda T, Chiarle R, Giaccone G, Bugiani O, et al. Substitutions at codon 22 of Alzheimer's Aβ peptide induce diverse conformational changes and apoptotic effects in human cerebral endothelial cells. J Biol Chem. 2000;275:27110–27116. doi: 10.1074/jbc.M003154200. [DOI] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, et al. The ‘Arctic' APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- Esler WP, Stimson ER, Ghilardi JR, Lu YA, Felix AM, et al. Point substitution in the central hydrophobic cluster of a human β-amyloid congener disrupts peptide folding and abolishes plaque competence. Biochemistry. 1996;35:13914–13921. doi: 10.1021/bi961302+. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, Berry RW, Binder LI. Tau polymerization: role of the amino terminus. Biochemistry. 2003;42:2252–2257. doi: 10.1021/bi0272510. [DOI] [PubMed] [Google Scholar]
- Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, et al. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry. 2000;39:11714–11721. doi: 10.1021/bi000850r. [DOI] [PubMed] [Google Scholar]
- Gamblin TC, King ME, Dawson H, Vitek MP, Kuret J, et al. In vitro polymerization of tau protein monitored by laser light scattering: method and application to the study of FTDP-17 mutants. Biochemistry. 2000;39:6136–6144. doi: 10.1021/bi000201f. [DOI] [PubMed] [Google Scholar]
- Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, et al. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- Li L, von Bergen M, Mandelkow EM, Mandelkow E. Structure, stability, and aggregation of paired helical filaments from tau protein and FTDP-17 mutants probed by tryptophan scanning mutagenesis. J Biol Chem. 2002;277:41390–41400. doi: 10.1074/jbc.M206334200. [DOI] [PubMed] [Google Scholar]
- Symmons MF, Buchanan SG, Clarke DT, Jones G, Gay NJ. X-ray diffraction and far-UV CD studies of filaments formed by a leucine-rich repeat peptide: structural similarity to the amyloid fibrils of prions and Alzheimer's disease β-protein. FEBS Lett. 1997;412:397–403. doi: 10.1016/s0014-5793(97)00809-0. [DOI] [PubMed] [Google Scholar]
- Orpiszewski J, Benson MD. Induction of β-sheet structure in amyloidogenic peptides by neutralization of aspartate: a model for amyloid nucleation. J Mol Biol. 1999;289:413–428. doi: 10.1006/jmbi.1999.2768. [DOI] [PubMed] [Google Scholar]
- Wirehn J, Carlsson K, Herland A, Persson E, Carlsson U, et al. Activity, folding, misfolding, and aggregation in vitro of the naturally occurring human tissue factor mutant R200W. Biochemistry. 2005;44:6755–6763. doi: 10.1021/bi047388l. [DOI] [PubMed] [Google Scholar]
- Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, et al. D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis. Biochemistry. 2003;42:6656–6663. doi: 10.1021/bi027319b. [DOI] [PubMed] [Google Scholar]
- Kenig M, Berbic S, Krijestorac A, Kroon-Zitko L, Tusek M, et al. Differences in aggregation properties of three site-specific mutants of recombinant human stefin B. Protein Sci. 2004;13:63–70. doi: 10.1110/ps.03270904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du HN, Tang L, Luo XY, Li HT, Hu J, et al. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human α-synuclein. Biochemistry. 2003;42:8870–8878. doi: 10.1021/bi034028+. [DOI] [PubMed] [Google Scholar]
- Parrini C, Taddei N, Ramazzotti M, Degl'Innocenti D, Ramponi G, et al. Glycine residues appear to be evolutionarily conserved for their ability to inhibit aggregation. Structure. 2005;13:1143–1151. doi: 10.1016/j.str.2005.04.022. [DOI] [PubMed] [Google Scholar]
- Sato T, Nakanishi T, Yamamoto Y, Andersen PM, Ogawa Y, et al. Rapid disease progression correlates with instability of mutant SOD1 in familial ALS. Neurology. 2005;65:1954–1957. doi: 10.1212/01.wnl.0000188760.53922.05. [DOI] [PubMed] [Google Scholar]
- Aksoy H, Dean G, Elian M, Deng HX, Deng G, et al. A4T mutation in the SOD1 gene causing familial amyotrophic lateral sclerosis. Neuroepidemiology. 2003;22:235–238. doi: 10.1159/000070564. [DOI] [PubMed] [Google Scholar]
- Juneja T, Pericak-Vance MA, Laing NG, Dave S, Siddique T. Prognosis in familial amyotrophic lateral sclerosis: progression and survival in patients with Glu100Gly and Ala4Val mutations in Cu,Zn superoxide dismutase. Neurology. 1997;48:55–57. doi: 10.1212/wnl.48.1.55. [DOI] [PubMed] [Google Scholar]
- Nakano R, Sato S, Inuzuka T, Sakimura K, Mishina M, et al. A novel mutation in Cu/Zn superoxide dismutase gene in Japanese familial amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 1994;200:695–703. doi: 10.1006/bbrc.1994.1506. [DOI] [PubMed] [Google Scholar]
- Cudkowicz ME, McKenna-Yasek D, Chen C, Hedley-Whyte ET, Brown RH., Jr. Limited corticospinal tract involvement in amyotrophic lateral sclerosis subjects with the A4V mutation in the copper/zinc superoxide dismutase gene. Ann Neurol. 1998;43:703–710. doi: 10.1002/ana.410430604. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Bowling AC, Patterson D, Usdin TB, Sapp P, et al. A frequent Ala 4 to Val superoxide dismutase-1 mutation is associated with a rapidly progressive familial amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:981–987. doi: 10.1093/hmg/3.6.981. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Nilsson P, Keranen ML, Forsgren L, Hagglund J, et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain. 1997;120:1723–1737. doi: 10.1093/brain/120.10.1723. [DOI] [PubMed] [Google Scholar]
- Gellera C, Castellotti B, Riggio MC, Silani V, Morandi L, et al. Superoxide dismutase gene mutations in Italian patients with familial and sporadic amyotrophic lateral sclerosis: identification of three novel missense mutations. Neuromuscul Disord. 2001;11:404–410. doi: 10.1016/s0960-8966(00)00215-7. [DOI] [PubMed] [Google Scholar]
- Andersen PM, Sims KB, Xin WW, Kiely R, O'Neill G, et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: a decade of discoveries, defects and disputes. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:62–73. doi: 10.1080/14660820310011700. [DOI] [PubMed] [Google Scholar]
- Stewart HG, Andersen PM, Eisen A, Weber M. Corticomotoneuronal dysfunction in ALS patients with different SOD1 mutations. Clin Neurophysiol. 2006;117:1850–1861. doi: 10.1016/j.clinph.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Kohno S, Takahashi Y, Miyajima H, Serizawa M, Mizoguchi K. A novel mutation (Cys6Gly) in the Cu/Zn superoxide dismutase gene associated with rapidly progressive familial amyotrophic lateral sclerosis. Neurosci Lett. 1999;276:135–137. doi: 10.1016/s0304-3940(99)00803-4. [DOI] [PubMed] [Google Scholar]
- Morita M, Aoki M, Abe K, Hasegawa T, Sakuma R, et al. A novel two-base mutation in the Cu/Zn superoxide dismutase gene associated with familial amyotrophic lateral sclerosis in Japan. Neurosci Lett. 1996;205:79–82. doi: 10.1016/0304-3940(96)12378-8. [DOI] [PubMed] [Google Scholar]
- Hirano M, Fujii J, Nagai Y, Sonobe M, Okamoto K, et al. A new variant Cu/Zn superoxide dismutase (Val7→Glu) deduced from lymphocyte mRNA sequences from Japanese patients with familial amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 1994;204:572–577. doi: 10.1006/bbrc.1994.2497. [DOI] [PubMed] [Google Scholar]
- Bereznai B, Winkler A, Borasio GD, Gasser T. A novel SOD1 mutation in an Austrian family with amyotrophic lateral sclerosis. Neuromuscul Disord. 1997;7:113–116. doi: 10.1016/s0960-8966(96)00419-1. [DOI] [PubMed] [Google Scholar]
- Kim NH, Kim HJ, Kim M, Lee KW. A novel SOD1 gene mutation in a Korean family with amyotrophic lateral sclerosis. J Neurol Sci. 2003;206:65–69. doi: 10.1016/s0022-510x(02)00338-6. [DOI] [PubMed] [Google Scholar]
- Penco S, Schenone A, Bordo D, Bolognesi M, Abbruzzese M, et al. A SOD1 gene mutation in a patient with slowly progressing familial ALS. Neurology. 1999;53:404–406. doi: 10.1212/wnl.53.2.404. [DOI] [PubMed] [Google Scholar]
- Battistini S, Giannini F, Greco G, Bibbo G, Ferrera L, et al. SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J Neurol. 2005;252:782–788. doi: 10.1007/s00415-005-0742-y. [DOI] [PubMed] [Google Scholar]
- Gellera C. Genetics of ALS in Italian families. Amyotroph Lateral Scler Other Motor Neuron Disord. 2001;2:S43–S46. doi: 10.1080/14660820152415735. [DOI] [PubMed] [Google Scholar]
- Kim HY, Ki CS, Koh SH, Park KH, Sunwoo IN, et al. Clinical characteristics of familial amyotrophic lateral sclerosis with a Phe20Cys mutation in the SOD1 gene in a Korean family. Amyotroph Lateral Scler. 2007;8:73–78. doi: 10.1080/17482960701223154. [DOI] [PubMed] [Google Scholar]
- Jafari-Schluep HF, Khoris J, Mayeux-Portas V, Hand C, Rouleau G, et al. [Superoxyde dismutase 1 gene abnormalities in familial amyotrophic lateral sclerosis: phenotype/genotype correlations. The French experience and review of the literature] Rev Neurol (Paris) 2004;160:44–50. doi: 10.1016/s0035-3787(04)70846-2. [DOI] [PubMed] [Google Scholar]
- Camu W, Khoris J, Moulard B, Salachas F, Briolotti V, et al. Genetics of familial ALS and consequences for diagnosis. French ALS Research Group. J Neurol Sci. 1999;165:S21–S26. doi: 10.1016/s0022-510x(99)00022-2. [DOI] [PubMed] [Google Scholar]
- Inoue K, Fujimura H, Ogawa Y, Satoh T, Shimada K, et al. Familial amyotrophic lateral sclerosis with a point mutation (G37R) of the superoxide dismutase 1 gene: a clinicopathological study. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:244–247. doi: 10.1080/146608202760839012. [DOI] [PubMed] [Google Scholar]
- Gamez J, Corbera-Bellalta M, Nogales G, Raguer N, Garcia-Arumi E, et al. Mutational analysis of the Cu/Zn superoxide dismutase gene in a Catalan ALS population: should all sporadic ALS cases also be screened for SOD1. J Neurol Sci. 2006;247:21–28. doi: 10.1016/j.jns.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Garcia-Redondo A, Bustos F, Juan YSB, Del Hoyo P, Jimenez S, et al. Molecular analysis of the superoxide dismutase 1 gene in Spanish patients with sporadic or familial amyotrophic lateral sclerosis. Muscle Nerve. 2002;26:274–278. doi: 10.1002/mus.10193. [DOI] [PubMed] [Google Scholar]
- Regal L, Vanopdenbosch L, Tilkin P, Van den Bosch L, Thijs V, et al. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol. 2006;63:262–267. doi: 10.1001/archneur.63.2.262. [DOI] [PubMed] [Google Scholar]
- Rainero I, Pinessi L, Tsuda T, Vignocchi MG, Vaula G, et al. SOD1 missense mutation in an Italian family with ALS. Neurology. 1994;44:347–349. doi: 10.1212/wnl.44.2.347. [DOI] [PubMed] [Google Scholar]
- Arisato T, Okubo R, Arata H, Abe K, Fukada K, et al. Clinical and pathological studies of familial amyotrophic lateral sclerosis (FALS) with SOD1 H46R mutation in large Japanese families. Acta Neuropathol. 2003;106:561–568. doi: 10.1007/s00401-003-0763-5. [DOI] [PubMed] [Google Scholar]
- Ohi T, Nabeshima K, Kato S, Yazawa S, Takechi S. Familial amyotrophic lateral sclerosis with His46Arg mutation in Cu/Zn superoxide dismutase presenting characteristic clinical features and Lewy body-like hyaline inclusions. J Neurol Sci. 2004;225:19–25. doi: 10.1016/j.jns.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Ohi T, Saita K, Takechi S, Nabesima K, Tashiro H, et al. Clinical features and neuropathological findings of familial amyotrophic lateral sclerosis with a His46Arg mutation in Cu/Zn superoxide dismutase. J Neurol Sci. 2002;197:73–78. doi: 10.1016/s0022-510x(02)00054-0. [DOI] [PubMed] [Google Scholar]
- Aoki M, Abe K, Itoyama Y. Molecular analyses of the Cu/Zn superoxide dismutase gene in patients with familial amyotrophic lateral sclerosis (ALS) in Japan. Cell Mol Neurobiol. 1998;18:639–647. doi: 10.1023/A:1020681802277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmoy T, Bjorgo K, Roos PM. Slowly progressing amyotrophic lateral sclerosis caused by H46R SOD1 mutation. Eur Neurol. 2007;58:57–58. doi: 10.1159/000102170. [DOI] [PubMed] [Google Scholar]
- Shaw CE, Enayat ZE, Powell JF, Anderson VE, Radunovic A, et al. Familial amyotrophic lateral sclerosis. Molecular pathology of a patient with a SOD1 mutation. Neurology. 1997;49:1612–1616. doi: 10.1212/wnl.49.6.1612. [DOI] [PubMed] [Google Scholar]
- Orrell RW, Habgood JJ, Gardiner I, King AW, Bowe FA, et al. Clinical and functional investigation of 10 missense mutations and a novel frameshift insertion mutation of the gene for copper-zinc superoxide dismutase in UK families with amyotrophic lateral sclerosis. Neurology. 1997;48:746–751. doi: 10.1212/wnl.48.3.746. [DOI] [PubMed] [Google Scholar]
- Stewart HG, Mackenzie IR, Eisen A, Brannstrom T, Marklund SL, et al. Clinicopathological phenotype of ALS with a novel G72C SOD1 gene mutation mimicking a myopathy. Muscle Nerve. 2006;33:701–706. doi: 10.1002/mus.20495. [DOI] [PubMed] [Google Scholar]
- Orrell RW, Marklund SL, deBelleroche JS. Familial ALS is associated with mutations in all exons of SOD1: a novel mutation in exon 3 (Gly72Ser) J Neurol Sci. 1997;153:46–49. doi: 10.1016/s0022-510x(97)00181-0. [DOI] [PubMed] [Google Scholar]
- Segovia-Silvestre T, Andreu AL, Vives-Bauza C, Garcia-Arumi E, Cervera C, et al. A novel exon 3 mutation (D76V) in the SOD1 gene associated with slowly progressive ALS. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:69–74. doi: 10.1080/146608202760196039. [DOI] [PubMed] [Google Scholar]
- Ceroni M, Malaspina A, Poloni TE, Alimonti D, Rognoni F, et al. Clustering of ALS patients in central Italy due to the occurrence of the L84F SOD1 gene mutation. Neurology. 1999;53:1064–1071. doi: 10.1212/wnl.53.5.1064. [DOI] [PubMed] [Google Scholar]
- Shaw CE, Enayat ZE, Chioza BA, Al-Chalabi A, Radunovic A, et al. Mutations in all five exons of SOD-1 may cause ALS. Ann Neurol. 1998;43:390–394. doi: 10.1002/ana.410430319. [DOI] [PubMed] [Google Scholar]
- Curti D, Rognoni F, Alimonti D, Malaspina A, Feletti F, et al. SOD1 activity and protective factors in familial ALS patients with L84F SOD1 mutation. Amyotroph Lateral Scler Other Motor Neuron Disord. 2002;3:115–122. doi: 10.1080/146608202760834111. [DOI] [PubMed] [Google Scholar]
- Ohnishi A, Miyazaki S, Murai Y, Ueno S, Sakai H. [Familial amyotrophic lateral sclerosis showing variable clinical courses with (Leu84→Val) mutation of Cu/Zn superoxide dismutase] Rinsho Shinkeigaku. 1996;36:485–487. [PubMed] [Google Scholar]
- Beck M, Sendtner M, Toyka KV. Novel SOD1 N86K mutation is associated with a severe phenotype in familial ALS. Muscle Nerve. 2007;36:111–114. doi: 10.1002/mus.20756. [DOI] [PubMed] [Google Scholar]
- Maeda T, Kurahashi K, Matsunaga M, Inoue K, Inoue M. [On intra-familial clinical diversities of a familial amyotrophic lateral sclerosis with a point mutation of Cu/Zn superoxide dismutase (Asn 86-Ser)] No To Shinkei. 1997;49:847–851. [PubMed] [Google Scholar]
- Rezania K, Yan J, Dellefave L, Deng HX, Siddique N, et al. A rare Cu/Zn superoxide dismutase mutation causing familial amyotrophic lateral sclerosis with variable age of onset, incomplete penetrance and a sensory neuropathy. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:162–166. doi: 10.1080/aml.4.3.162.166. [DOI] [PubMed] [Google Scholar]
- Khoris J, Moulard B, Briolotti V, Hayer M, Durieux A, et al. Coexistence of dominant and recessive familial amyotrophic lateral sclerosis with the D90A Cu,Zn superoxide dismutase mutation within the same country. Eur J Neurol. 2000;7:207–211. doi: 10.1046/j.1468-1331.2000.00028.x. [DOI] [PubMed] [Google Scholar]
- Morita M, Abe K, Takahashi M, Onodera Y, Okumura H, et al. A novel mutation Asp90Val in the SOD1 gene associated with Japanese familial ALS. Eur J Neurol. 1998;5:389–392. [Google Scholar]
- Esteban J, Rosen DR, Bowling AC, Sapp P, McKenna-Yasek D, et al. Identification of two novel mutations and a new polymorphism in the gene for Cu/Zn superoxide dismutase in patients with amyotrophic lateral sclerosis. Hum Mol Genet. 1994;3:997–998. doi: 10.1093/hmg/3.6.997. [DOI] [PubMed] [Google Scholar]
- Kawata A, Kato S, Hayashi H, Hirai S. Prominent sensory and autonomic disturbances in familial amyotrophic lateral sclerosis with a Gly93Ser mutation in the SOD1 gene. J Neurol Sci. 1997;153:82–85. doi: 10.1016/s0022-510x(97)00176-7. [DOI] [PubMed] [Google Scholar]
- Iwai K, Yamamoto M, Yoshihara T, Sobue G. Anticipation in familial amyotrophic lateral sclerosis with SOD1-G93S mutation. J Neurol Neurosurg Psychiatry. 2002;72:819–820. doi: 10.1136/jnnp.72.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosler BA, Nicholson GA, Sapp PC, Chin W, Orrell RW, et al. Three novel mutations and two variants in the gene for Cu/Zn superoxide dismutase in familial amyotrophic lateral sclerosis. Neuromuscul Disord. 1996;6:361–366. doi: 10.1016/0960-8966(96)00353-7. [DOI] [PubMed] [Google Scholar]
- Ince PG, Shaw PJ, Slade JY, Jones C, Hudgson P. Familial amyotrophic lateral sclerosis with a mutation in exon 4 of the Cu/Zn superoxide dismutase gene: pathological and immunocytochemical changes. Acta Neuropathol. 1996;92:395–403. doi: 10.1007/s004010050535. [DOI] [PubMed] [Google Scholar]
- Cervenakova L, Protas II, Hirano A, Votiakov VI, Nedzved MK, et al. Progressive muscular atrophy variant of familial amyotrophic lateral sclerosis (PMA/ALS) J Neurol Sci. 2000;177:124–130. doi: 10.1016/s0022-510x(00)00350-6. [DOI] [PubMed] [Google Scholar]
- Orrell RW, Habgood J, Rudge P, Lane RJ, de Belleroche JS. Difficulties in distinguishing sporadic from familial amyotrophic lateral sclerosis. Ann Neurol. 1996;39:810–812. doi: 10.1002/ana.410390620. [DOI] [PubMed] [Google Scholar]
- Orrell RW, Habgood JJ, Malaspina A, Mitchell J, Greenwood J, et al. Clinical characteristics of SOD1 gene mutations in UK families with ALS. J Neurol Sci. 1999;169:56–60. doi: 10.1016/s0022-510x(99)00216-6. [DOI] [PubMed] [Google Scholar]
- Sato T, Yamamoto Y, Nakanishi T, Fukada K, Sugai F, et al. Identification of two novel mutations in the Cu/Zn superoxide dismutase gene with familial amyotrophic lateral sclerosis: mass spectrometric and genomic analyses. J Neurol Sci. 2004;218:79–83. doi: 10.1016/j.jns.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Tan CF, Piao YS, Hayashi S, Obata H, Umeda Y, et al. Familial amyotrophic lateral sclerosis with bulbar onset and a novel Asp101Tyr Cu/Zn superoxide dismutase gene mutation. Acta Neuropathol. 2004;108:332–336. doi: 10.1007/s00401-004-0893-4. [DOI] [PubMed] [Google Scholar]
- Abe K. Clinical and molecular analysis of neurodegenerative diseases. Tohoku J Exp Med. 1997;181:389–409. doi: 10.1620/tjem.181.389. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhao H, Lu M, Zhang Y, Wang L, et al. A rare Cu/Zn superoxide dismutase mutation causing familial amyotrophic lateral sclerosis with variable age of onset and incomplete penetrance in China. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005;6:234–238. doi: 10.1080/14660820510044478. [DOI] [PubMed] [Google Scholar]
- Orrell RW, Habgood JJ, Shepherd DI, Donnai D, deBelleroche JS. A novel mutation of SOD-1 (Gly 108 Val) in familial amyotrophic lateral sclerosis. Eur J Neurol. 1997;4:48–51. doi: 10.1111/j.1468-1331.1997.tb00298.x. [DOI] [PubMed] [Google Scholar]
- Rouleau GA, Clark AW, Rooke K, Pramatarova A, Krizus A, et al. SOD1 mutation is associated with accumulation of neurofilaments in amyotrophic lateral sclerosis. Ann Neurol. 1996;39:128–131. doi: 10.1002/ana.410390119. [DOI] [PubMed] [Google Scholar]
- Kokubo Y, Kuzuhara S, Narita Y, Kikugawa K, Nakano R, et al. Accumulation of neurofilaments and SOD1-immunoreactive products in a patient with familial amyotrophic lateral sclerosis with I113T SOD1 mutation. Arch Neurol. 1999;56:1506–1508. doi: 10.1001/archneur.56.12.1506. [DOI] [PubMed] [Google Scholar]
- Jones CT, Swingler RJ, Simpson SA, Brock DJ. Superoxide dismutase mutations in an unselected cohort of Scottish amyotrophic lateral sclerosis patients. J Med Genet. 1995;32:290–292. doi: 10.1136/jmg.32.4.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince PG, Tomkins J, Slade JY, Thatcher NM, Shaw PJ. Amyotrophic lateral sclerosis associated with genetic abnormalities in the gene encoding Cu/Zn superoxide dismutase: molecular pathology of five new cases, and comparison with previous reports and 73 sporadic cases of ALS. J Neuropathol Exp Neurol. 1998;57:895–904. doi: 10.1097/00005072-199810000-00002. [DOI] [PubMed] [Google Scholar]
- Suthers G, Laing N, Wilton S, Dorosz S, Waddy H. “Sporadic” motoneuron disease due to familial SOD1 mutation with low penetrance. Lancet. 1994;344:1773. doi: 10.1016/s0140-6736(94)92913-0. [DOI] [PubMed] [Google Scholar]
- Kostrzewa M, Burck-Lehmann U, Muller U. Autosomal dominant amyotrophic lateral sclerosis: a novel mutation in the Cu/Zn superoxide dismutase-1 gene. Hum Mol Genet. 1994;3:2261–2262. doi: 10.1093/hmg/3.12.2261. [DOI] [PubMed] [Google Scholar]
- Enayat ZE, Orrell RW, Claus A, Ludolph A, Bachus R, et al. Two novel mutations in the gene for copper zinc superoxide dismutase in UK families with amyotrophic lateral sclerosis. Hum Mol Genet. 1995;4:1239–1240. doi: 10.1093/hmg/4.7.1239. [DOI] [PubMed] [Google Scholar]
- Takehisa Y, Ujike H, Ishizu H, Terada S, Haraguchi T, et al. Familial amyotrophic lateral sclerosis with a novel Leu126Ser mutation in the copper/zinc superoxide dismutase gene showing mild clinical features and lewy body-like hyaline inclusions. Arch Neurol. 2001;58:736–740. doi: 10.1001/archneur.58.5.736. [DOI] [PubMed] [Google Scholar]
- Murakami T, Warita H, Hayashi T, Sato K, Manabe Y, et al. A novel SOD1 gene mutation in familial ALS with low penetrance in females. J Neurol Sci. 2001;189:45–47. doi: 10.1016/s0022-510x(01)00558-5. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Aoki M, Abe K, Shoji M, Iizuka T, et al. A novel missense point mutation (S134N) of the Cu/Zn superoxide dismutase gene in a patient with familial motor neuron disease. Hum Mutat. 1997;9:69–71. doi: 10.1002/(SICI)1098-1004(1997)9:1<69::AID-HUMU14>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Nogales-Gadea G, Garcia-Arumi E, Andreu AL, Cervera C, Gamez J. A novel exon 5 mutation (N139H) in the SOD1 gene in a Spanish family associated with incomplete penetrance. J Neurol Sci. 2004;219:1–6. doi: 10.1016/j.jns.2003.10.018. [DOI] [PubMed] [Google Scholar]
- Naini A, Musumeci O, Hayes L, Pallotti F, Del Bene M, et al. Identification of a novel mutation in Cu/Zn superoxide dismutase gene associated with familial amyotrophic lateral sclerosis. J Neurol Sci. 2002;198:17–19. doi: 10.1016/s0022-510x(02)00052-7. [DOI] [PubMed] [Google Scholar]
- Mase G, Ros S, Gemma A, Bonfigli L, Carraro N, et al. ALS with variable phenotypes in a six-generation family caused by Leu144Phe mutation in the SOD1 gene. J Neurol Sci. 2001;191:11–18. doi: 10.1016/s0022-510x(01)00625-6. [DOI] [PubMed] [Google Scholar]
- Ferrera L, Caponnetto C, Marini V, Rizzi D, Bordo D, et al. An Italian dominant FALS Leu144Phe SOD1 mutation: genotype-phenotype correlation. Amyotroph Lateral Scler Other Motor Neuron Disord. 2003;4:167–170. doi: 10.1080/aml.4.3.167.170. [DOI] [PubMed] [Google Scholar]
- Ito K, Uchiyama T, Fukutake T, Arai K, Kanesaka T, et al. [Different clinical phenotypes of siblings with familial amyotrophic lateral sclerosis showing Cys146Arg point mutation of superoxide dismutase 1 gene] Rinsho Shinkeigaku. 2002;42:175–177. [PubMed] [Google Scholar]
- Fong GC, Kwok KH, Song YQ, Cheng TS, Ho PW, et al. Clinical phenotypes of a large Chinese multigenerational kindred with autosomal dominant familial ALS due to Ile149Thr SOD1 gene mutation. Amyotroph Lateral Scler. 2006;7:142–149. doi: 10.1080/17482960600732412. [DOI] [PubMed] [Google Scholar]
- Kostrzewa M, Damian MS, Muller U. Superoxide dismutase 1: identification of a novel mutation in a case of familial amyotrophic lateral sclerosis. Hum Genet. 1996;98:48–50. doi: 10.1007/s004390050157. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(62 KB PDF)
The same dataset for fALS-associated SOD1 mutations shown in Figures 3 and 4 were considered. The predicted aggregation propensities and instabilities from 28 different fALS-causing SOD1 mutations were plotted using the software SigmaPlot 9.0 (Systat Software, Inc.).
(213 KB AI).
The relationship between SOD1 aggregation propensity (A, D), instability (B, E), or sum of aggregation propensity and instability (C, F) with fALS patients' age of onset are presented. The linear regressions presented in (A–C) were weighted by the number of patients for each mutation using SPSS version 15.0 (SPSS, Inc.). The correlation between the size of each data point and the number of patients for (A-C) is shown as an inset in (C). The linear regressions presented in (D–F) were treated equally regardless of the number of patients for each mutation (unweighted) using the software SigmaPlot 9.0 (Systat Software, Inc.). The age of onset data presented in these six graphs are from 649 patients with 29 different fALS-causing SOD1 mutations with reported stability values. Aggregation propensity, instability, and sum of aggregation propensity and instability were obtained as described in the Materials and Methods section. Aggregation propensity, instability, or sum of aggregation propensity and instability has little or no correlation with patients' age of onset.
(261 KB AI).