

Abstract
We report fluorescence in situ hybridization (FISH) mapping of 152, mostly de novo, apparently balanced chromosomal rearrangement (ABCR) breakpoints in 76 individuals, 30 of whom had no obvious phenotypic abnormality (control group) and 46 of whom had an associated disease (case group). The aim of this study was to identify breakpoint characteristics that could discriminate between these groups and which might be of predictive value in de novo ABCR (DN-ABCR) cases detected antenatally. We found no difference in the proportion of breakpoints that interrupted a gene, although in three cases, direct interruption or deletion of known autosomal-dominant or X-linked recessive Mendelian disease genes was diagnostic. The only significant predictor of phenotypic abnormality in the group as a whole was the localization of one or both breakpoints to an R-positive (G-negative) band with estimated predictive values of 0.69 (95% CL 0.54–0.81) and 0.90 (95% CL 0.60–0.98), respectively. R-positive bands are known to contain more genes and have a higher guanine-cytosine (GC) content than do G-positive (R-negative) bands; however, whether a gene was interrupted by the breakpoint or the GC content in the 200kB around the breakpoint had no discriminant ability. Our results suggest that the large-scale genomic context of the breakpoint has prognostic utility and that the pathological mechanism of mapping to an R-band cannot be accounted for by direct gene inactivation.
Introduction
With conventional microscopic analysis of banded chromosomes, structural chromosome rearrangements without apparent gain or loss of chromosome material are observed to have an estimated combined live-birth prevalence of 0.52%.1 The observed frequencies of different classes of apparently balanced chromosome rearrangements (ABCR) in unselected-newborn studies via banded chromosome analysis are as follows: reciprocal translocations (0.17%), robertsonian translocations (0.1%), pericentric inversions (0.03%), and paracentric inversions (0.01). Fourteen percent of ABCRs occur de novo, and the combined mutation rate has been estimated as 3.7 × 10−4. Familial ABCR cases are inherited maternally and paternally at almost equal rates.
Most individuals carrying an ABCR will have no medical problems as a result of the chromosomal anomaly. However, ABCRs can cause disease in a minority of cases and families. The most direct evidence for this comes from the study of families in which an ABCR segregates with a Mendelian disorder. Here, molecular characterization of chromosomal breakpoint in such families has been of great utility in the identification of many human-disease genes, including dystrophin,2 Menkes disease,3 FOXC1,4 and NSD1.5 Less direct evidence for the pathogenic nature of a minority of ABCRs is provided by the consistent observation of an increased frequency in cases with undiagnosed learning disabilities.6–13 Clinical follow-up studies of de novo ABCRs (DN-ABCRs) identified through prenatal diagnostic cytogenetics show a 3%–10% risk of serious malformations and learning disabilities.14,15
In cases in which a fetus has no detectable structural malformation, counseling prospective parents regarding the clinical significance of a de novo ABCR detected prenatally is difficult because there is no method of discriminating the minority of breakpoints associated with major medical effects. We hypothesized that rapid molecular characterization of ABCR breakpoints could aid such discrimination, and to test this we have used a strategy analogous to a case-control study, mapping the DN-ABCR breakpoints in a series of cases with adverse phenotypes and in a similar number of phenotypically normal cases. We show that only one breakpoint characteristic, localization to an R-positive band, was “predictive” of abnormal outcome in the group as a whole. However, in individual cases, the presence of a submicroscopic deletion or duplication or the direct interruption of a known autosomal-dominant or X-linked disease gene provides clinically useful information.
Subjects and Methods
Patient Data
Apparently balanced chromosome rearrangements (ABCRs) for both the case and control groups were ascertained via the clinical and cytogenetic records of regional genetics services. Written consent was obtained from individuals or parents/guardians for the mapping of the translocation breakpoints with a mechanism approved by the UK Multicenter Regions Ethics Committee. The main inclusion criterion was the presence of a chromosomal rearrangement that was considered balanced on conventional banding analysis. De novo cases were chosen for analysis where possible. ABCRs were assigned to the case group if a phenotypic abnormality was present, was plausibly genetically determined, and could not be explained by another cause. Cases were assigned to the control group only if they were > 6 years old and considered to have normal neurocognitive development—regarding developmental milestones and educational performance—and no other obvious phenotypic abnormality on the basis of clinical reassessment by an experienced clinical geneticist.
General Aspects of FISH Analysis
Metaphase chromosomes were prepared from fixed cell suspensions from peripheral blood, cultured amniocytes, or lymphoblastoid cultures via standard clinical methods. One case, T86-0404, was mapped on isolated nuclei from paraffin sections with a combination of chromosome paints and locus-specific BAC (Bacterial Artificial Chromosome) probes, as previously described.16 BAC and PAC (P1-derived Artificial Chromosome) clones were obtained from the BACPAC Resource Center (Children's Hospital Oakland Research Institute, Oakland, CA, USA) or the Wellcome Trust Sanger Institute (Cambridge, UK), and DNA was prepared in 96-well plates by use of a Biomek 2000K robot (Beckman) and the Wizard MagneSil Plasmid Purification kit (Promega) or the Montage BAC96 miniprep kit (Millipore). Individual BAC- or PAC-clone DNA was prepared via a standard mini-prep method recommended by the BACPAC Resource Center. Probes were labeled with digoxigenin-11-dUTP or biotin-16-dUTP (Roche) by nick translation. Probe labeling, DNA hybridization, and antibody detection were carried out via methods described previously.43 At least five metaphases were analyzed for each hybridization with a Zeiss Axioskop 2 microscope with the appropriate filters (#83000 for DAPI, FITC, and rhodamine; Chroma Technology). Images were collected and merged via a Coolsnap HQ CCD camera (Photometrics) and SmartCapture 2 software (Digital Scientific).
Mapping Strategy
A strategy for rapid fluorescence in situ hybridization (FISH) mapping was developed with a staged approach determined by the genomic context of each breakpoint. A set of large-insert BAC and PAC clones (a kind gift from Dr. Nigel Carter of Wellcome Trust Sanger Institute), spaced at intervals of approximately 1Mb throughout the genome, were utilized during the first stage.17 FISH probes were chosen to cover 10–20Mb around the cytogenetic breakpoint band to both flank the breakpoint and screen for large deletions, duplications, or rearrangements at or close to the breakpoint.
The second stage began once breakpoint-flanking probes were identified from the 1Mb clones. A contig of BAC, PAC, or fosmid clones that span the breakpoint interval of ∼1Mb was identified with UCSC or Ensembl genome browsers and the clones. FISH analysis of all of the probes in the contig was done to identify breakpoint-spanning clones in most cases. This approach allowed us to “walk through” the breakpoint with a minimum of 600 Kb (three clones) and a maximum of 2 Mb (ten clones) of sequence checked for BAC-sized deletions around the breakpoint. If no genes were present in the breakpoint clone or if a single gene was interrupted, then no further mapping was performed, because our goal was to determine the gross genetic pathology rather that the precise breakpoint.
A third stage of mapping was performed if the breakpoint clone contained multiple genes, in order to determine which gene, if any, was disrupted. The breakpoints were then further delineated with FISH probes generated by PCR. Primers were designed, via the Primer3 program (see Web Resources), from genomic sequence to generate 10 Kb probes. PCR reactions were performed with the Expand Long Template PCR kit (Roche) according to the manufacturer's conditions. Products were purified with a Qiaquick PCR purification kit (QIAGEN) and labeled by nick translation with digoxigenin-11-dUTP. Details of all primers are available on request.
Microarray Analysis
1Mb ‘CytoChip’ microarrays were used for array-based comparative genomic hybridization (array-CGH) analysis (BlueGnome Ltd, UK). Genomic DNA from the standard DNA and from each case was labeled by random priming (Invitrogen, UK) with Cy5-dCTP and Cy3-dCTP to allow for dye-swap analysis. Hybridization and washes were performed on a HS 400 Pro hybridization station (Tecan, UK). Each subarray was prehybridized for 45 min at 37°C with 1.5 μg of herring sperm DNA (Sigma-Aldrich, UK) in 75 μl of hybridization buffer (50% formamide, 7% dextran sulfate, X2 saline sodium citrate [SSC], 10 mM Tris-HCl pH 7.5, and 0.1% Tween 20). Test and reference samples were mixed, coprecipitated, and resuspended in a 75 μl hybridization solution that also contained 2.5 μg/μl Cot-1 (Invitrogen), denatured at 75°C for 15 min, incubated for two hours at 37°C to block repetitive sequences, and hybridized for 21 hr. Post-hybridization washes were performed via three wash cycles in each of PBS/0.05%Tween at 37°C, X 0.1 SSC at 54°C, and X1 PBS at 37°C, and a final wash was performed in PBS/0.05%Tween at 23°C. Slides were dried with high-purity nitrogen and then stored in darkness. Arrays were scanned with a GenePix Pro 5.0 array scanner (Axon Instruments, UK) and analyzed with BlueFuse for Microarrays analysis software version 3.4 (BlueGnome, UK). Exclusion criteria were set for clones of which (1) confidence values were less than 0.3, (2) replicates had a standard deviation > 0.1, (3) dye-swap replicates had a standard deviation > 0.2, or (4) a quality flag < 1 existed. For all array hybridizations, < 95% of clones were expected to be included for analysis. 99.5% of the clones were used in the final analysis and interpretation. Data lying beyond three standard deviations were considered to be within the region of copy-number change.
Bioinformatic Analysis and Statistical Methods
The genomic coordinates of each breakpoint-spanning clone were determined by searching of the publicly available human genome with UCSC or Ensembl Genome browsers. In order to compare breakpoints mapped to different resolutions, windows of various sizes (200, 500, 1000, and 5000Kb) were defined around each breakpoint. The Ensembl genes track was used to identify genes disrupted by breakpoints that had been mapped. The per-base conservation scores for each ∼200 Kb region around the breakpoint using the scores from the phastCons table, which is the primary table underlying the Conservation track in UCSC browser. The Gene-Ontology (GO) terms associated with each of the directly disrupted genes were retrieved with the Ensembl genome browser.
Comparisons between cases and controls in the numbers of observations in two different categories were made with Fishers Exact Test. A two-tailed test was used for all comparisons except that involving the comparison of the numbers of cases and controls with deletions in cis with one or both breakpoints where a one tailed test was used.
Binary Logistic Regression was used to identify those variables most important in distinguishing the breakpoints found in the normal and abnormal groups of individuals. The positive predictive value was estimated as the proportion of abnormal individuals to total individuals with a particular feature. A significance level of 0.05 was used throughout. Confidence limits were calculated via the method of Newcombe and Altman (2000).18 All other calculations were made with SPSS version 14.
Results
General Data
A total of 76 unrelated cases (152 cytogenetically visible breakpoints) were analyzed. FISH mapping was performed on 46 individuals in whom the ABCR was associated with an abnormal phenotype (termed cases) and on 30 individuals who were apparently normal at the time of clinical assessment (termed controls). Table 1 summarizes the clinical and cytogenetic features of these cases and controls. Forty-two of the 46 cases and 25 of the 30 controls were de novo translocations. Table 2 documents the numbers of cases in each of the general clinical categories.
Table 1.
Clinical Features of Cases and Cytogenetic Localization of Breakpoints in Cases and Controls
| Group | Number | ABa | Code | Cytogenic BPb | Clinical Features |
|---|---|---|---|---|---|
| Cases | 1 | XATc | GILLE | t(X;11)(p22.3;p12) | Superior atypical coloboma, foveal hypoplasia, inferior vermis cerebellum |
| 2 | Recipd | MARTA | t(8;9)(q21;q21) | Peters anomaly | |
| 3 | Recip | SG-3301 | t(2;17)(q32;q24)pat | Micrognathia,glossoptosis, cleft palate | |
| 4 | Recip | ROOJA | 45,X,t(5;7)(q21;q33) | Hemolytic anemia, Turner syndrome, prenatal onset short stature Developmental delay? | |
| 5 | Recip | MAGAN | t(3;4)(q23;q31) | Severe short stature, hemolytic anemia, recurrent hypoglycemia | |
| 6 | Inve | CRENA | inv(4)(q21q35) | Bilateral coloboma, hypotonia | |
| 7 | Recip | F92-2253 | t(5;11)(q15;p15.5) | Global developmental delay | |
| 8 | Recip | T86-0593 | t(5;12)(q11.2;q12) | Testicular atrophy | |
| 9 | Inv | GILL | inv(12)(p11.2q24.3) | Cleft palate, severe learning disabilities | |
| 10 | Recip | B97-1182 | t(2;10)(q11.2;q22.3) | Mild learning disability | |
| 11 | Recip | B01-2958 | t(4;14)(q31.22;q11.2) | Moderate learning difficulties, particular problems with numeracy | |
| 12 | Recip | B96-0581 | t(2;3)(q21.3;q21.3) | Attention deficit hyperactivity disorder | |
| 13 | XAT | F03-0432 | t(X;8)(q26.?1;p23.?1) | Abnormal ultrasound | |
| 14 | Recip | B01-2957 | t(14;18)(q24.1;p11.32) | Learning difficulties: spelling dificulties, on learning support. Height and weight normal | |
| 15 | Recip | B99-1983 | t(13;18)(q31.1/q22.3;q22.1) | Global developmental delay. dysmorphic features. IUGR, brachydactyly | |
| 16 | Recip | B04-0611 | t(1;9)(p32.3?;q33.2) | Precocious puberty | |
| 17 | Inv | B90748 | inv(1)(p36.1q25.3) | Familial MDSf, ALLg | |
| 18 | Recip | CMS5859 | t(3;7)(q28;p21.3) by FISH | Anophthalmia with AEGh | |
| 19 | Recip | B04-0880 | t(2;3)(/q32.2/q33;q26.3/q27) | Autistic spectrum disorder, severe learning disability, Piere Robin sequence | |
| 20 | Recip | T86-0404 | t(2;6)(p23;q14) | Sirenomelia | |
| 21 | Recip | CV1012 | t(5;17)(q15;q23)pat | Pierre Robin sequence | |
| 22 | Recip | NCL-B04-2316 | t(3;6)(q23;p21.1) | Complex congenital heart disease, asplenia, malrotation | |
| 23 | Recip | COLJA | t(11;13)(p15.3;q22) | Cleft palate, prominent ears, small chin, tapering fingers | |
| 24 | Recip | B00213 | t(2;7)(q33;p21) | Cleft palate, mild learning disability | |
| 25 | Recip | BB45 | t(2;11)(q32;p14) | Cleft palate, mild learning disability | |
| 26 | XAT | BL02-1299 | t(X;10)(q22.3;q24.3) | AVSDi, hypopituitary, posterior embryotoxin, myopia, dislocated hips | |
| 27 | XAT | BL02-0828 | t(X;11)(q21.2;q13.5) | Premature menopause | |
| 28 | Recip | F0124/00 | t(2;16)(p15;q22) | Low birth weight, hypotonia, learning difficulties, pulmonary hypertension, delayed visual maturation | |
| 29 | Recip | BL02-3104 | t(9;22)(q34.2;q11.21) | Learning difficulties, mild bilateral hearing loss | |
| 30 | Recip | AC114 | t(10;11)(q24;p13) | Ventricular septal defect, cleft palate, XY sex reversal, hydronephrosis | |
| 31 | Recip | BL03-1791 | t(7;8)(q32.2;q22) | Marfanoid habitus, learning difficulty | |
| 32 | Recip | CV345 | t(6;9)(p23;q22.3) | Cleft lip and palate, other dysmorphism | |
| 33 | Inv | BL02-2567 | inv(1)(q32.1q44) | Agenesis of the corpus callosum, Dandy Walker, dysmorphisms | |
| 34 | Recip | BL03-0967 | t(9;20)(p13.1;p13) | Cataracts, microcephaly | |
| 35 | Recip | BL03-1789 | t(5;9)(p13.1;q22.1) | Spastic paraparesis | |
| 36 | Recip | BL03-2425 | t(11;13)(p15;q32) | Infantile seizures, myopia, ptosis | |
| 37 | Recip | CV1222 | t(1;7)(q21.3;q34) | Learning disability, celiac disease | |
| 38 | Recip | BL04-0107 | t(5;6)(q13;q23) | Non specific dysmorphism, small hands,bilateral hernias, tremor, delayed puberty | |
| 39 | Recip | BL03-3077 | t(1;20)(q32.1;q13.3) | Non-specific dysmorphism, slim hands and feet, high arched palate, clinodactyly | |
| 40 | Recip | CV1456 | t(1;2)(q24.2;q31.3) | Mowat-Wilson syndrome | |
| 41 | Recip | BL04-2026 | t(5;6)(q15;q25.1) | Severe mental handicap | |
| 42 | Recip | BL04-2240 | t(9;15)(q21.2;q26) | Abnormal baby, mild dysmorphism, developmental delay | |
| 43 | XAT | BL04-3772 | t(X;4)(p11.4;p16) | Primary ammenorhea | |
| 44 | Recip | BL04-1899 | t(6;16)(q15;q13) | Renal cancer, brother also has translocation and rare cancer | |
| 45 | Recip | BL05-0692 | t(1;13)(q44;q32) | Agenesis of the corpus callosum | |
| 46 | Recip | BL03-2183 | t(5;14)(q15;q24) | Primary amenorrhoea | |
| Controls | 1 | Recip | B98-0026 | t(1;15)(q24.3;q22.3) | N/A |
| 2 | Recip | F00-1558 | t(15;16)(q22.3;q22.1) | N/A | |
| 3 | Recip | B97-0349 | t(3;11)(p13;q12.2) | N/A | |
| 4 | Recip | B04-0088 | t(3;9)(p26;p22) | N/A | |
| 5 | Recip | F96-1781 | t(5;6)(q31.3;p21.3) | N/A | |
| 6 | Inv | SISCO | inv(1)(p31q43) | N/A | |
| 7 | Recip | PETEN | t(7;12)(q32;p13) | N/A | |
| 8 | Recip | EDINN | t(1;14)(q42;q11) | N/A | |
| 9 | Recip | JOGAR | t(X;14)(p11.21;q32.2/32.3) | N/A | |
| 10 | Recip | B01-2804 | t(4;12)(p16.1;q24.31) | N/A | |
| 11 | Recip | BL03-0187 | t(2;8)(p15;q13) | N/A | |
| 12 | Recip | BL03-0186 | t(1;7)(p32;q34) | N/A | |
| 13 | Recip | BL03-0185 | t(2;4)(q37.1;p15.32) | N/A | |
| 14 | Recip | BL03-0362 | t(2;5)(q31;q33.3) | N/A | |
| 15 | Recip | BL03-0356 | t(3;12)(q27;p13) | N/A | |
| 16 | Recip | BL03-0426 | t(4;11)(q31;p13) | N/A | |
| 17 | Recip | BL03-1970 | t(6;15)(q23.1;q21.2) | N/A | |
| 18 | Recip | BL03-1979 | t(6;14)(q32;q32.1) | N/A | |
| 19 | Recip | F0440 | t(8;15)(q22;q22.1) | N/A | |
| 20 | Recip | BL03-0876 | t(1;11)(q42.1;p15.5) | N/A | |
| 21 | Recip | BL03-3032 | t(5;18)(p15.1;p11.32) | N/A | |
| 22 | Recip | BL04-0339 | t(5;6)(q11.2;q23.1) | N/A | |
| 23 | Inv | BL03-2725 | inv(14)(q22.1q32.1) | N/A | |
| 24 | Recip | BL04-2024 | t(5;7)(p15.1;q31.2) | N/A | |
| 25 | Recip | BL04-2025 | t(3;6)(q25;q15) | N/A | |
| 26 | Recip | CV1510 | t(6;14)(p23;q13) | N/A | |
| 27 | Recip | BL04-3994 | t(3;9)(p25;q13) | N/A | |
| 28 | Recip | BL04-4270 | t(6;8)(q25;q12) | N/A | |
| 29 | Recip | BL05-0379 | t(5;9)(p14;p13.1) | N/A | |
| 30 | Recip | BL03-2435 | t(6;12)(q21;q24.1) | N/A |
The breakpoints shown in this table are those reported by the referring clinical cytogenics laboratories. For consistency, these breakpoints are used throughout the text. The FISH-mapped breakpoints and the flanking or breakpoint-spanning clone identifiers are available online in Tables S1 and S2, respectively.
“AB” denotes “aberration category.”
“BP” denotes “breakpoint.”
“XAT” denotes “X-autosome translocation.”
“Recip” denotes “reciprocal translocation.”
“Inv” denotes “inversion.”
“MDS” denotes “myelodysplastic syndrome.”
“ALL” denotes “acute lymphoblastoid leukemia.”
“AEG” denotes “anophthalmia.”
“AVSD” denotes “atrioventricular septal defect.”
Table 2.
Clinical Categories within Case Group
| Phenotype Category | Number |
|---|---|
| Learning Disability ± Dysmorphism | 15 |
| Cleft Lip or Cleft Palate | 8 |
| Eye Malformation | 5 |
| Reproductive Abnormality | 5 |
| Multiple Congenital Anomalies | 3 |
| Brain Malformation | 3 |
| Abnormal Prenatal Ultrasound Scan | 2 |
| Cancer Syndrome | 2 |
| Unknown Metabolic Disease | 1 |
| Neurological disease | 1 |
| Known Monogenic Syndrome | 1 |
Of the 152 ABCR breakpoints, 138 (90.8%) were mapped at a resolution equivalent to a single BAC clone (∼200 Kb). Fourteen breakpoints were mapped at a lower resolution, due to repetitive genomic regions (e.g., pericentromic repeats) or exhaustion of clinical material.
Microdeletions or Microduplications
The staged FISH strategy detected deletions in cis with one or both breakpoints in 6/46 (0.13, 95% CI 0.06–0.26) of the cases and in 0/30 (0.0, 95% CI 0.0–0.11) of the control ABCRs (p < 0.05). Three cases had deletions contiguous with or very close to an ABCR breakpoint: Case BL02-0828, with premature menopause, had a single BAC deletion at the 11 breakpoint of t (X;11)(q21.2;q13.5), no genes were identified in either the deleted or 200Kb of the flanking genomic sequence. Case BL02-2567, with agenesis of the corpus callosum, Dandy Walker malformation, and craniofacial dysmorphisms with inv(1)(q32.1q44), had a deletion ≤ 3 Mb in size encompassing at least nine genes, at or close to a telomeric breakpoint as part of a complex cryptic rearrangement.19 Case BL03-2183, with primary amenorrhoea associated with t(5;14)(q15;q24), was shown to have a cryptic complex rearrangement at the 5q breakpoint with a 1.8 Mb deletion encompassing three genes, FBXL2, FER, and PJA2, flanked by a region of chromosome 5q that was duplicated through an insertional translocation to 6q.
Three cases had deletions in cis that were a significant distance from an ABCR breakpoint: B99-1983 had an unusual brachydactyly associated with microcephaly and learning disability with t(13;18)(q31.1;q22.1). A 5 Mb deletion involving up to five genes was identified ∼6 Mb from the 13q31.1 ABCR breakpoint. CMS5859, with anophthalmia, esophageal atresia, genital malformations (AEG) syndrome, had a 2.1 Mb deletion, including SOX2, 9 Mb from the 3q translocation breakpoint. CV1456 has Mowat-Wilson syndrome caused by a 3 Mb deletion that includes SIP1. This deletion was 30 Mb from the 2q breakpoint in the associated t(1;2)(q24.2;q31.3).
In order to exclude the possibility that duplications or deletions elsewhere in the genome were contributing to the phenotypic effects in the case group, array CGH analysis was performed via CytoChip V1.0 on 12 (26.1%) ABCR cases. No deletion or duplication was identified that was not documented as a copy-number polymorphism.
Genes Directly Disrupted by Breakpoints
The genes directly disrupted by the mapped breakpoints are documented in Table 3. In 72/149 (48%) breakpoints, FISH mapping was able to exclude a direct gene disruption. A gene was considered disrupted if (1) the gene was larger in size than the breakpoint-spanning BAC (10/149 [6.7%] breakpoints) or (2) the breakpoint could be confidently inferred by FISH with overlapping BAC clone, Fosmids, or long-range PCR products (34/149 [23%] breakpoints). In 29/149 (19%), the available FISH mapping was not able to determine whether a gene was disrupted within the breakpoint-spanning BAC.
Table 3.
Genes Disrupted by Translocation Breakpoints in Cases and Controls
| Code | BP Band by FISH | Gene Disrupted | Description | ||
|---|---|---|---|---|---|
| Cases | 1 | GILLE | Xp22.2 | ARHGAP6 | Rho-GTPase-activating protein 6 |
| 2 | GILLE | 11p11.2 | PHF21A | PHD finger protein 21A | |
| 3 | MARTA | 9q21.13 | ANXA1 | annexin A1 | |
| 4 | ROOJA | 5q14.3 | RASA1 | Ras GTPase-activating protein 1 (GTPase-activating protein) (GAP) | |
| 5 | ROOJA | 7q22.1 | FBXL13 | F-box/LRR-repeat protein 13 (F-box and leucine-rich repeat protein 13) | |
| 6 | MAGAN | 3q23 | RASA2 | Ras GTPase-activating protein 2 (GAP1m) | |
| 7 | MAGAN | 4q31.22 | SLC10A7 | Sodium/bile acid cotransporter 7 | |
| 8 | F92-2253 | 5q15 | MCTP1 | multiple C2-domains with two transmembrane regions 1 isoform S | |
| 9 | GILL | 12q24.31 | PSL4∗ | Signal peptide peptidase-like 3 (SPP-like 3 protein) (Intramembrane protease 2) (IMP2) (Presenilin-like protein 4) | |
| 10 | F03-0432 | Xq26.2 | GPC3 | Glypican-3 precursor (Intestinal protein OCI-5) (GTR2-2) (MXR7) | |
| 11 | B04-0611 | 9q31.3 | EDG2 | endothelial differentiation gene, lysophosphatidic acid G-protein-coupled receptor, 2 | |
| 12 | B90748 | 1q25.1 | TNN | Tenascin-N precursor (TN-N) | |
| 13 | NCL-B04-2316 | 3q21.3 | EEFSEC | Selenocysteine-specific elongation factor | |
| 14 | NCL-B04-2316 | 6p21.1 | RUNX2 | Runt-related transcription factor 2 | |
| 15 | B00213 | 2q33.1 | SATB2 | DNA-binding protein SATB2 (Special AT-rich sequence-binding protein 2) | |
| 16 | BL02-3104 | 9q34.2 | RXRA | Retinoic acid receptor RXR-alpha | |
| 17 | AC114 | 10q24.2 | LOXL4 | Lysyl oxidase homolog 4 precursor | |
| 18 | BL03-0967 | 20p13 | C20ORF116 | Protein C20orf116 precursor | |
| 19 | BL03-1791 | 7q22.1 | CUX1 | CCAAT displacement protein (CDP) (Cut-like 1 | |
| 20a | CV345 | 9q22.33 | C9orf156 | Uncharacterized conserved protein [Function unknown] | |
| 20b | CV345 | 9q22.33 | HEMGN | Hemogen | |
| 21 | BL03-2425 | 13q32.1 | HS6ST3 | heparan sulfate 6-O-sulfotransferase 3 | |
| 22 | CV1222 | 7q34 | DENND2A | DENN/MADD domain containing 2A | |
| 23 | BL04-0107 | 6q25.1 | MAP3K7IP2 | mitogen-activated protein kinase kinase kinase 7 interacting protein 2 isoform 1 | |
| 24 | BL04-2026 | 6q25.1 | MTHFD1L | methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1-like | |
| Controls | 1 | B98-0026 | 1q25.2 | CEP350 | centrosome-associated protein 350 |
| 2 | B97-0349 | 3p14.1 | FAM19A1 | family with sequence similarity 19 (chemokine (C-C motif)-like), member A1 | |
| 3 | SISCO | 1p13.3 | MYBPHL | myosin binding protein H-like | |
| 4 | PETEN | 12p13.32 | EFCAB4B | EF-hand calcium binding domain 4B | |
| 5 | JOGAR | Xp11.3 | EFHC2 | EF-hand domain (C-terminal) containing 2 | |
| 6 | JOGAR | 14q32.33 | ENST00000342537∗ | Ensembl novel pseudogene | |
| 7 | B01-2804 | 12q24.23 | KSR2 | Kinase suppressor of ras-2 (hKSR2) | |
| 8 | BL03-0186 | 1p32.1 | FLJ10986∗ | no description | |
| 9 | BL03-0186 | 7q35 | TPK1 | Thiamin pyrophosphokinase 1 | |
| 10 | BL03-0185 | 2q37.2 | CENTG2 | centaurin, gamma 2 | |
| 11 | BL03-0185 | 4p15.33 | C1QTNF7 | Complement C1q tumor necrosis factor-related protein 7 precursor | |
| 12 | BL03-0876 | 1q44 | KIF26B | kinesin family member 26B | |
| 13 | BL03-0362 | 2q32.1 | ZNF804A | zinc finger protein 804A | |
| 14 | BL03-1979 | 6q22.33 | PTPRK | Receptor-type tyrosine-protein phosphatase kappa precursor | |
| 15 | BL03-3032 | 18p11.31 | MRCL2∗ | myosin regulatory light chain MRCL2 | |
| 16 | BL03-2725 | 14q21.3 | GALC | Galactocerebrosidase precursor | |
| 17 | BL04-2024 | 7q31.2 | MET | Hepatocyte growth factor receptor precursor, (Met proto- oncogene tyrosine kinase) (c-met) | |
| 18 | CV1510 | 14q12 | NOVA1 | RNA-binding protein Nova-1 (Neuro-oncological ventral antigen 1) | |
| 19 | BL04-3994 | 3p26.3/26.2 | CNTN4 | Contactin 4 precursor (Brain-derived immunoglobulin superfamily protein 2 BIG-2) | |
| 20 | BL04-0339 | 5q12.3 | SDCCAG10 | serologically defined colon cancer antigen 10 |
indicates that no HGNC official gene name is available.
Disrupted genes in both cases and controls were large, mostly extending over 100 kb or more of the genome, with many large introns, characteristic of the genes found in AT-rich regions20 (Table 4). Disrupted genes ranged in size from ANXA1 (13 exons, covering 18 kb) to HS6ST3 (2 exons, covering 742 kb). Only eight genes were smaller than the genome-wide mean genomic length, five of which were in cases associated with an abnormal phenotype. Details of the individuals in whom both breakpoints could be fully categorised are given in Table 5.
Table 4.
Characteristics of the Genes Disrupted by Breakpoints
| With Phenotype | Controls | Genome-wide | |
|---|---|---|---|
| Number Genes Disrupted | 25 | 20 | |
| Mean Number Exons | 13 | 12.75 | 8.8 |
| Mean Transcript Size | 2.992 | 3.119 | 1.34 |
| Mean Genomic Length (kb) | 179.9 | 278.93 | 27 |
| Mean Exon Size (kb) | 0.23 | 0.245 | 0.145 |
| Mean Intron Size (kb) | 14.74 | 23.47 | 3.36 |
Table 5.
Individuals in whom both Breakpoints are Fully Characterized
| Case (%) n = 32 | Control (%) n = 16 | |
|---|---|---|
| Both breakpoints disrupt gene(s) | 4 (12.5%) | 2 (12.5%) |
| One breakpoint disrupts gene(s) | 14 (43.7%) | 10 (62.5%) |
| Neither BP disrupts a gene | 14 (43.7%) | 4 (25%) |
Among the cases with a phenotype, 3/25 (12%) of the disrupted genes have previously be shown to have pathogenic mutations associated with specific Mendelian disease: In ROOJA with t(5;7)(q21;q33), the 5q breakpoint interrupts RASA1, mutations of which cause Capillary Malformation-Arteriovenous Malformation (OMIM 608354). ROOJA is a complex case with severe neurocognitive impairment and short stature. On reexamination she was found to have several small capillary malformations compatible with this diagnosis. F03-0432 was identified antenatally with multiple renal cysts, somatic overgrowth, and a de novo t(X;8)(q26.1;p23.1). In this case the X breakpoint disrupts GPC3, the causative gene for the X-linked recessive disorder Simpson-Golabi-Behmel syndrome (OMIM 312870). The morphological abnormalities in this fetus were compatible with this diagnosis, although this diagnosis was not made until the mapping results were available. NCL-B04-2316 was a male infant with severe cardiac malformations and t(3;6)(q23;p21.1), the 6p breakpoint interrupted RUNX2, which when heterozygously mutated causes cleidocranial dysplasia (OMIM 119600). Radiological review of this case showed previously unsuspected absence of the clavicles. In controls, 1/20 of the disrupted genes (5%) has associated Mendelian diseases; in BL03-2725, the 14q21 breakpoint interrupts GALC, mutations in which cause the recessive neurometabolic disorder Krabbe disease (OMIM 245200).
The only biological-process-category GO terms associated with more than one gene were those for “transcription/regulation of transcription” and “signal transduction/intracellular signaling.” We note that 4/25 (0.16, 95% CI 0.06–0.35) genes disrupted in cases with a phenotype were associated with GO terms for transcription and regulation of transcription (SATB2, RUNX2, RXRA, and CUTL1), while 0/20 (0.0, 95% CI 0.0-0.16) genes disrupted in controls had these GO terms.
Cytogenetic Features of Breakpoints
Chromosomal bands were assigned to each breakpoint with the cytogenetic band track in Ensembl. Figure 1 shows the distribution of breakpoints across all of the chromosomes. No breakpoints are found on 10p, 16p, 17p, 19p, 19q, or 21. No bias toward telomeric bands was identified. Figure 2 shows the distribution of breakpoints in the various chromosome bands: G-positive/R-negative (G+ve) bands (G1 darkest, G4 palest), R-positive/G-negative (R+ve) bands, and centromeric bands.21 R+ve bands make up ∼45% of genome.22,23 We found that 35/86 (0.41, 95% CI 0.31–0.51) of breakpoints from cases with a phenotype localize to R+ve bands, compared with 13/60 (0.22 95% CI 0.13–0.34) in controls (p < 0.05). We also found that 17/24 (0.71 95% CI 0.51–0.85) of the genes disrupted by breakpoints in the cases with phenotype mapped to R+ve bands, compared to 5/20 (0.25 95% CI 0.11–0.47) in controls (p < 0.01). Classifying the disrupted genes as larger or smaller than the genome-wide mean of 27 Kb, we found that 12/16 (0.75, 95% CI 0.51–0.90) of the large genes in cases with a phenotype mapped to R+ve bands, while only 3/14 (0.21, 95% CI 0.08–0.48) did so in controls (p < 0.01).
Figure 1.
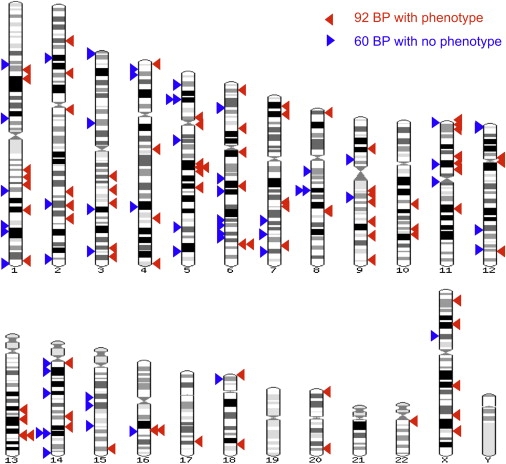
Breakpoint Distribution
The genome-wide distribution of the 152 breakpoints mapped in this study. The breakpoints in cases are shown as red arrowheads on the right-hand side of each ideogram, and the breakpoints in controls are shown as blue arrowheads on the left-hand side. This figure was made with the Karyoview facility on the Ensembl Genome Browser.
Figure 2.
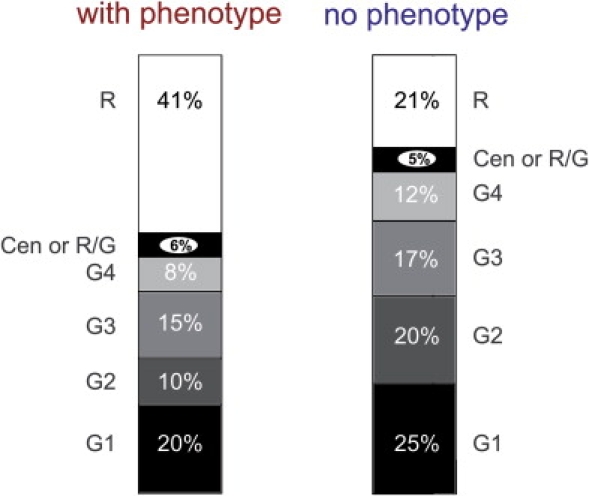
Band-Type Distribution
Graphical representations of the cytogenetic band distribution of the molecularly characterized breakpoints. The left-hand bar diagram shows the distribution in the Case group, and the right-hand diagram shows the Control-group distribution. R indicates R+ve bands (taken to be synonymous with G-light bands here). Cen or R/G indicates bands assigned as centromeric or existing at the junction between an R+ve band and a G+ve band. G1–G4 indicate G-dark bands, with G1 the darkest and G4 the lightest.
Genomic Context of Breakpoint Regions
The level of cross-species conservation in the ∼200Kb window around the breakpoints was significantly higher in cases than in controls (mean conservation score ± standard error of the mean [SE] 0.191 ± 0.003 cf 0.180 ± 0.004). No significant differences between cases and controls were identified in GC content (Mean % GC content ± SE cases 39.99 ± 0.51 compared with controls 40.14 ± 0.65) or in the number of genes (200 kb window mean numbers of genes in cases and in controls were 1.45 and 1.80, respectively).
No differences between cases or controls were found in the number of segmental duplications around the breakpoints (data not shown). On metaphase FISH analysis, most breakpoint BAC clones showed little or no significant crosshybridization, suggesting that they contain only a small amount of sequence duplicated elsewhere in the genome. Several breakpoints mapped to sites of large-scale segmental duplications that can mediate deletion and duplication rearrangements.24 In the case BL02-3104, the 22q breakpoint region could not be mapped to less than 500 kb given the LCR-B low copy repeat. Three breakpoint-spanning BACs contain clusters of olfactory receptor genes, two in cases with no phenotype (B97-0349 3p14; EDINN 14q11.2) and one in a case with phenotype (CV1222 1q32). None of the segmental duplications have paralogous sequences at the breakpoint in the other chromosome involved in the translocation or inversion.
Copy-number variants were found at 4/89 breakpoints (4.6%) in cases with a phenotype (CV345 6p23; BL02-3104 22q11; BL03-1791 7q32.2; B00213 7p21.1) and at 5/60 (8.3%) breakpoints in the control group (B97-0349 11q12.2; EDINN 14q11.2; BL03-1979 6q23.2; B01-2804 4p16.1; BL03-2435 6q21). It was observed that 16/28 (57%) synteny breaks occur between chicken and human, and 7/28 (25%) occur between mouse and human. No significant differences were seen between cases and controls.
Multivariate Analysis and Positive Predictive Values
Binary logistic-regression analysis was carried out in an attempt to identify those variables of most importance for distinguishing the breakpoints in the normal and abnormal groups of patients. In the first set of calculations, the variables considered were: whether an interrupted gene had a GO ontology suggesting that it encoded a DNA-binding protein or transcriptional regulator (Gene Class), whether the mapped breakpoint was in a G+ve or R+ve band (G/R), whether the breakpoint disrupted a gene (GeneBroken), whether there was a deletion or duplication at the breakpoint (Del/Dup), and the level of conservation in a 200Kb window around the breakpoint (cons200).
If all of these variables were included in the regression equation, only the coefficient of G/R was statistically significant (p < 0.01). With Forward Stepwise Conditional Method, only the G/R term was included in the equation. The analysis was repeated, including the variables calculated for a 200Kb window around the breakpoint: percent GC content (GC200), number of CpG islands (CpG200), number of genes (Genes200), and number of exons (Exons200). The results were essentially unchanged, with only the coefficient of G/R statistically significant.
There were 27 normal and 40 abnormal individuals for whom both breakpoints were classified as being in a G+ve or R+ve band. Among the control group, 15 individuals had two G+ve breakpoints, 11 individuals had one G+ve and one R+ve breakpoint, and one individual had two R+ve breakpoints. Among the cases, the numbers were 13 (G+ve/G+ve),18 (G+ve/R+ve), and 9 (R+ve/R+ve). The positive predictive value of abnormality for an individual who has one breakpoint in an R+ve band was 27/39 (0.69, 95% CL 0.54–0.81), and for individual who has two breakpoints in an R+ve band the value was 9/10 (0.90, 95% CL 0.60–0.98).
Discussion
This study was motivated by a specific and common clinical problem in prenatal genetic counseling: a fetus identified with a de novo apparently balanced chromosomal rearrangement (DN-ABCR). We asked the question, “Can we identify breakpoint characteristics associated with phenotypic abnormality in DN-ABCR cases that might be of predictive use in prenatal cases?” We chose to FISH map breakpoints of a large series of DN-ABCR cases for which adequate clinical-assessment and follow-up data were available. This enables comparison of individuals with and without phenotypic abnormality, somewhat analogous to a case-control study, and has resulted in the largest series of molecularly characterized DN-ABCR breakpoints reported to date. From the reported molecular pathology in ABCR cases associated with a Mendelian disease, we hypothesized that abnormal outcome would be associated with the following breakpoint characteristics: the presence of aneuploidy in cis or trans, direct interruption of a transcription unit, disruption of cis-regulation of developmentally critical genes, or, more speculatively, alteration in chromatic state.
As predicted, the presence of a deletion or duplication in cis was statistically more common in the case group. In two cases, the identification of a deletion, either at the breakpoint or in cis with the breakpoint, confirmed suspected clinical diagnoses of SOX2 anophthalmia syndrome25 and Mowat-Wilson syndrome,26 respectively. In the other four cases, the finding of aneuploid segments provided a plausible explanation for the clinical phenotype, although unambiguous assignment of pathogenesis was not possible given the complex and/or unique nature of the mutations. In all aneuploidy, useful prediction of clinical consequences is possible only if consistent clinical features are documented in individuals with overlapping aneuploid regions. Such clinical interpretation will be considerably aided by the continuing growth of publicly accessible repositories of phenotypes associated with molecularly-characterized structural chromosome anomalies, such as DECIPHER.
Previous studies using array-based comparative genomic hybridization (aCGH) in ABCR cases27,28 have reported frequent abnormal copy-number variants on chromosomes not involved in the breakpoints. We found no examples of this in the 12 cases studied with aCGH. This discrepancy could be explained by the different exclusion criteria; most significantly, we excluded all complex rearrangements and cases with uncertainty about whether the karyotype was balanced. However, it is clear that a genome-wide technique to assess copy-number variation may be indicated in ABCR cases with an abnormal phenotype. Whether this investigation will provide useful predictive information in the prenatal context must be the subject of future studies.
The most surprising result from the study was the lack of difference in the proportions of breakpoints that resulted in disruption of a gene between cases and controls. In three cases, the pathogenic nature of one breakpoint was clear because mapping demonstrated that a known autosomal-dominant or X-linked recessive disease gene was interrupted: Capillary Malformation-Arteriovenous Malformation syndrome (RASA1; OMIM 608354) and Cleidocranial Dysostosis (RUNX2; OMIM 119600) were identified as part of more complex malformation syndromes, and Simpson Golabi Behmel (GPC3; OMIM 312870) syndrome was identified in a prenatally ascertained case. Only one known disease gene was interrupted in the control group, and this gene causes an autosomal-recessive disorder, Krabbe disease (OMIM 245200). The carrier frequency of this rare condition is low; thus, it is not surprising that this individual was unaffected. The gene-ontology classifications of the disrupted genes were not clearly different between the groups but did suggest that signal transduction and DNA binding might be overrepresented among cases. As expected, the physical size of the interrupted genes in both cases and controls was several times larger than the genome average, presumably due to the larger “target size” for mutation.
The only significant “predictor” of phenotypic abnormality in the group as a whole was whether one or both breakpoints mapped to R+ve bands (taken in this study to be synonymous with G-negative bands).29 R+ve bands account for almost half of the genome, and they are considered to be GC-rich, be early replicating, be methylcytosine-rich, and to contain most housekeeping and tissue-specific genes.22 Nagakome and Chiyo30 noted that the breakpoints in structural chromosome rearrangements were mostly in R-bands. This observation was supported by subsequent reports.31 Savage, however, suggested that the “Nakagome phenomenon” could be an artifact caused by the natural tendency of the human brain to interpret patterns with dark bands and thus overassign breakpoints to light bands.32 Our study was FISH based and used only molecular assignment of R-band status; it was thus immune to this artifact.
The predictive effect in ABCR breakpoints cannot be accounted for by any of the reported features of R+ve bands. In particular, the GC content, gene density, and CpG-island density are very similar between cases and controls in a 200Kb window around the breakpoints. The proportion of breakpoints that interrupt genes is similar in both groups, suggesting no clear mechanism for this strong predictive effect. It is possible that one of the major drivers for the abnormal phenotypic outcome is disruption of cis-regulation of developmentally critical genes. This is difficult to prove, because the mechanism of cis-regulatory effects and the size of the domains of action are not yet clear. In this regard, it is interesting that the mean conservation score in the 200 Kb window around the breakpoints was significantly higher in cases compared to controls.
There are several limitations to this study. The most obvious is that it is impossible to be certain that the DN-ABCRs in our “control” group are truly benign, given that individuals in this class may develop future disease. We have tried to minimize this with careful clinical assessment. However, age-specific defects might be impossible to identify depending on the age of the subject; e.g., infertility in a child. Most of the cases were ascertained for two UK clinical-genetics departments and were not chosen for any specific phenotypic characteristic. However, in the course of the study several DN-ABCR cases associated with eye malformation or orofacial clefts were sent to us because of our long-standing research interest in these birth defects. This could introduce a bias in the case group. We have sought to address this by including only the first cases that involved a specific disease gene (e.g., SOX2) in the analysis. We have also reanalyzed the data, removing these additional cases—this made no significant difference to the results (data not shown but available on request).
An important secondary aim of the study was to determine if FISH mapping of individual ABCR cases was practical as an antenatal clinical test. Generally, identifying a breakpoint BAC clone required two rounds of BAC FISH analysis, each with ten probes per breakpoint. The first round was for identification of 1Mb flanking clones. The second round was for identification of a breakpoint clone from a contig of clones that spanned the flanking clones. A third round of FISH analysis with PCR probes was performed if the genetic pathology was not clear from the BAC FISH. The requirement for a third round of FISH was a hindrance to the timely completion of the analysis. Another source of delay resulted from ordering of the BAC clones with which to fill in the contig between the 1Mb clones, but this could be solved by having all BAC tiling-path clones available locally. In many cases it would be possible to complete the analyses within two weeks. This makes “real-time” clinical analysis a realistic option with chorionic villus sampling, but it would be restricted to laboratories with specialized molecular cytogenetic capabilities. It thus seems unlikely that such clinical analysis will become routine in the short term because of both the practical difficulties in the high-throughput FISH analysis and the uncertainty regarding the genetic counseling of cases without Mendelian disease-gene involvement or significant aneuploidy.
In conclusion, the molecular characteristics of the breakpoints do have apparent predictive value. However, there is significant heterogeneity in the pathogenic mechanism. Indeed, the genetic mechanism underlying with strongest predictor of abnormal phenotypic outcome, localization to R+ve bands, remains obscure.
Supplemental Data
Two additional tables are available with this paper online at http://www.ajhg.org/.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
DECIPHER database, decipher.sanger.ac.uk
Ensembl genome browser, www.ensembl.org
Online Mendelian Inheritance in Man, http://www.ncbi.nlm.nih.gov/Omim/
Primer3 program, www-genome.wi.mit.edu/cgi-bin/primer/primer3_www.cgi
UCSC genome resources, genome.ucsc.edu
Accession Numbers
Breakpoint-mapping data included in this study have been entered into the DECIPHER database with the following accession numbers, each referring to an individual with an ABCR: EDB00001779, EDB00001778, EDB00001777, EDB00001776, EDB00001775, EDB0000174, EDB00001773, EDB00001765, EDB00001764, EDB00001763, EDB00001759, EDB00001757, EDB00001752, EDB00001739, EDB00001737, EDB00001713, EDB00001712, EDB00001711, EDB00001710, EDB00001709, EDB00001708, EDB00001386, EDB00001385, EDB00001384, EDB00001383, EDB00001382, EDB00001381, EDB00001380, EDB00001368, EDB00001367, EDB00001364, EDB00001363, EDB00001362, EDB00001361, EDB00001360.
Acknowledgments
First and foremost, we thank the children and adults who provided information and samples for this study. We thank the Wellcome Trust Sanger Institute for the kind gift of almost all of the BAC and PAC clones used in this study. We thank Frances Flinter and R. Curtis Rogers for referring cases included in this study. Funding for this study was provided by PPP Health Care Foundation and the UK Medical Research Council (MRC).
References
- 1.Jacobs P.A., Browne C., Gregson N., Joyce C., White H. Estimates of the frequency of chromosome abnormalities detectable in unselected newborns using moderate levels of banding. J. Med. Genet. 1992;29:103–108. doi: 10.1136/jmg.29.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ray P.N., Belfall B., Duff C., Logan C., Kean V., Thompson M.W., Sylvester J.E., Gorski J.L., Schmickel R.D., Worton R.G. Cloning of the breakpoint of an X;21 translocation associated with Duchenne muscular dystrophy. Nature. 1985;318:672–675. doi: 10.1038/318672a0. [DOI] [PubMed] [Google Scholar]
- 3.Mercer J.F., Livingston J., Hall B., Paynter J.A., Begy C., Chandrasekharappa S., Lockhart P., Grimes A., Bhave M., Siemieniak D. Isolation of a partial candidate gene for Menkes disease by positional cloning. Nat. Genet. 1993;3:20–25. doi: 10.1038/ng0193-20. [DOI] [PubMed] [Google Scholar]
- 4.Nishimura D.Y., Swiderski R.E., Alward W.L., Searby C.C., Patil S.R., Bennet S.R., Kanis A.B., Gastier J.M., Stone E.M., Sheffield V.C. The forkhead transcription factor gene FKHL7 is responsible for glaucoma phenotypes which map to 6p25. Nat. Genet. 1998;19:140–147. doi: 10.1038/493. [DOI] [PubMed] [Google Scholar]
- 5.Kurotaki N., Imaizumi K., Harada N., Masuno M., Kondoh T., Nagai T., Ohashi H., Naritomi K., Tsukahara M., Makita Y. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat. Genet. 2002;30:365–366. doi: 10.1038/ng863. [DOI] [PubMed] [Google Scholar]
- 6.Breg W.R., Miller D.A., Allderdice P.W., Miller O.J. Identification of translocation chromosomes by quinacrine fluorescence. Am. J. Dis. Child. 1972;123:561–564. doi: 10.1001/archpedi.1972.02110120085007. [DOI] [PubMed] [Google Scholar]
- 7.Fryns J.P., Kleczkowska A., Kubien E., Van Den Berghe H. On the excess of mental retardation and/or congenital malformations in apparently balanced reciprocal translocations. A critical review of the Leuven data 1966–1991. Genet. Couns. 1991;2:185–194. [PubMed] [Google Scholar]
- 8.Funderburk S.J., Spence M.A., Sparkes R.S. Mental retardation associated with “balanced” chromosome rearrangements. Am. J. Hum. Genet. 1977;29:136–141. [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs P.A. Correlation between euploid structural chromosome rearrangements and mental subnormality in humans. Nature. 1974;249:164–165. doi: 10.1038/249164a0. [DOI] [PubMed] [Google Scholar]
- 10.Jacobs P.A., Matsuura J.S., Mayer M., Newlands I.M. A cytogenetic survey of an institution for the mentally retarded: I. Chromosome abnormalities. Clin. Genet. 1978;13:37–60. doi: 10.1111/j.1399-0004.1978.tb04127.x. [DOI] [PubMed] [Google Scholar]
- 11.Nielsen J., Krag-Olsen B. Follow-up of 32 children with autosomal translocations found among 11,148 consecutively newborn children from 1969 to 1974. Clin. Genet. 1981;20:48–54. doi: 10.1111/j.1399-0004.1981.tb01806.x. [DOI] [PubMed] [Google Scholar]
- 12.Speed R.M., Johnston A.W., Evans H.J. Chromosome survey of total population of mentally subnormal in North-East of Scotland. J. Med. Genet. 1976;13:295–306. doi: 10.1136/jmg.13.4.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tharapel A.T., Summitt R.L., Wilroy R.S.J., Martens P. Apparently balanced de novo translocations in patients with abnormal phenotypes: report of 6 cases. Clin. Genet. 1977;11:255–269. doi: 10.1111/j.1399-0004.1977.tb01310.x. [DOI] [PubMed] [Google Scholar]
- 14.MacGregor D.J., Imrie S., Tolmie J.L. Outcome of de novo balanced translocations ascertained prenatally. J. Med. Genet. 1989;26:590–591. doi: 10.1136/jmg.26.9.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warburton D. De novo balanced chromosome rearrangements and extra marker chromosomes identified at prenatal diagnosis: clinical significance and distribution of breakpoints. Am. J. Hum. Genet. 1991;49:995–1013. [PMC free article] [PubMed] [Google Scholar]
- 16.Fantes J., Ragge N.K., Lynch S.A., McGill N.I., Collin J.R., Howard-Peebles P.N., Hayward C., Vivian A.J., Williamson K., van Heyningen V. Mutations in SOX2 cause anophthalmia. Nat. Genet. 2003;33:461–463. doi: 10.1038/ng1120. [DOI] [PubMed] [Google Scholar]
- 17.Fiegler H., Carr P., Douglas E.J., Burford D.C., Hunt S., Scott C.E., Smith J., Vetrie D., Gorman P., Tomlinson I.P., Carter N.P. DNA microarrays for comparative genomic hybridization based on DOP-PCR amplification of BAC and PAC clones. Genes Chromosomes Cancer. 2003;36:361–374. doi: 10.1002/gcc.10155. [DOI] [PubMed] [Google Scholar]
- 18.RG N . DG A. Proportions and their differences. In: Altman D.G., Machin D., Bryant T.N.M.J.G., editors. Statistics with Confidence. BMJ Books; London: 2000. pp. 45–56. [Google Scholar]
- 19.Boland E., Clayton-Smith J., Woo V.G., McKee S., Manson F.D., Medne L., Zackai E., Swanson E.A., Fitzpatrick D., Millen K.J. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am. J. Hum. Genet. 2007;81:292–303. doi: 10.1086/519999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogic S., Mackworth A.K., Ouellette F.B. Evaluation of gene-finding programs on mammalian sequences. Genome Res. 2001;11:817–832. doi: 10.1101/gr.147901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Francke U. High-resolution ideograms of trypsin-Giemsa banded human chromosomes. Cytogenet. Cell Genet. 1981;31:24–32. doi: 10.1159/000131622. [DOI] [PubMed] [Google Scholar]
- 22.Craig J.M., Bickmore W.A. Chromosome bands–flavours to savour. Bioessays. 1993;15:349–354. doi: 10.1002/bies.950150510. [DOI] [PubMed] [Google Scholar]
- 23.Furey T.S., Haussler D. Integration of the cytogenetic map with the draft human genome sequence. Hum. Mol. Genet. 2003;12:1037–1044. doi: 10.1093/hmg/ddg113. [DOI] [PubMed] [Google Scholar]
- 24.Lupski J.R., Stankiewicz P. Genomic disorders: molecular mechanisms for rearrangements and conveyed phenotypes. PLoS Genet. 2005;1:e49. doi: 10.1371/journal.pgen.0010049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragge N.K., Lorenz B., Schneider A., Bushby K., de Sanctis L., de Sanctis U., Salt A., Collin J.R., Vivian A.J., Free S.L. SOX2 anophthalmia syndrome. Am. J. Med. Genet. A. 2005;135:1–7. doi: 10.1002/ajmg.a.30642. [DOI] [PubMed] [Google Scholar]
- 26.Mowat D.R., Croaker G.D., Cass D.T., Kerr B.A., Chaitow J., Ades L.C., Chia N.L., Wilson M.J. Hirschsprung disease, microcephaly, mental retardation, and characteristic facial features: delineation of a new syndrome and identification of a locus at chromosome 2q22-q23. J. Med. Genet. 1998;35:617–623. doi: 10.1136/jmg.35.8.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Astbury C., Christ L.A., Aughton D.J., Cassidy S.B., Kumar A., Eichler E.E., Schwartz S. Detection of deletions in de novo “balanced” chromosome rearrangements: further evidence for their role in phenotypic abnormalities. Genet. Med. 2004;6:81–89. doi: 10.1097/01.gim.0000117850.04443.c9. [DOI] [PubMed] [Google Scholar]
- 28.Gribble S.M., Prigmore E., Burford D.C., Porter K.M., Ng B.L., Douglas E.J., Fiegler H., Carr P., Kalaitzopoulos D., Clegg S. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J. Med. Genet. 2005;42:8–16. doi: 10.1136/jmg.2004.024141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dutrillaux B., Lejeune J. C R Acad Sci Hebd Seances Acad Sci D. 1971;272:2638–2640. [PubMed] [Google Scholar]
- 30.Nakagome Y., Chiyo H. Nonrandom distribution of exchange points in patients with structural rearrangements. Am. J. Hum. Genet. 1976;28:31–41. [PMC free article] [PubMed] [Google Scholar]
- 31.Nakagome Y., Matsubara T., Fujita H. Distribution of break points in human structural rearrangements. Am. J. Hum. Genet. 1983;35:288–300. [PMC free article] [PubMed] [Google Scholar]
- 32.Savage J.R. Assignment of aberration breakpoints in banded chromosomes. Nature. 1977;270:513–514. doi: 10.1038/270513a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.