

Abstract
Cultured vascular endothelial cell (EC) exposure to steady laminar shear stress results in peroxynitrite (ONOO−) formation intramitochondrially and inactivation of the electron transport chain. We examined whether the “hyperoxic state” of 21% O2, compared with more physiological O2 tensions (Po2), increases the shear-induced nitric oxide (NO) synthesis and mitochondrial superoxide (O2·−) generation leading to ONOO− formation and suppression of respiration. Electron paramagnetic resonance oximetry was used to measure O2 consumption rates of bovine aortic ECs sheared (10 dyn/cm2, 30 min) at 5%, 10%, or 21% O2 or left static at 5% or 21% O2. Respiration was inhibited to a greater extent when ECs were sheared at 21% O2 than at lower Po2 or left static at different Po2. Flow in the presence of an endothelial NO synthase (eNOS) inhibitor or a ONOO− scavenger abolished the inhibitory effect. EC transfection with an adenovirus that expresses manganese superoxide dismutase in mitochondria, and not a control virus, blocked the inhibitory effect. Intracellular and mitochondrial O2·− production was higher in ECs sheared at 21% than at 5% O2, as determined by dihydroethidium and MitoSOX red fluorescence, respectively, and the latter was, at least in part, NO-dependent. Accumulation of NO metabolites in media of ECs sheared at 21% O2 was modestly increased compared with ECs sheared at lower Po2, suggesting that eNOS activity may be higher at 21% O2. Hence, the hyperoxia of in vitro EC flow studies, via increased NO and mitochondrial O2·− production, leads to enhanced ONOO− formation intramitochondrially and suppression of respiration.
Keywords: shear stress, endothelium, mitochondria, reactive oxygen species
mitochondria are the subcellular organelles for the production of cellular energy, but they also activate intracellular signaling pathways that modulate cell proliferation or promote cell cycle arrest and apoptosis by oxidative or nitrosative reactions. These reactions are regulated by the matrix concentration of nitric oxide (NO), which diffuses to the mitochondria from the cytosolic NO synthase (NOS) isoforms and is thought to be produced also locally by a mitochondrial NOS variant (11). In rat heart, skeletal muscle and liver mitochondria (15, 57, 58), isolated rat hearts (59), and whole animals (68), NO at physiological concentrations binds reversibly and in competition with oxygen (O2) to the reduced binuclear center CuB/a3 of cytochrome-c oxidase (complex IV) of the electron transport chain (ETC) and inhibits mitochondrial O2 uptake. At slightly higher concentrations, NO oxidizes ubiquinol of ubiquinol-cytochrome c reductase (complex III) to increase unstable ubisemiquinone, which, by univalent electron transfer to O2, produces the reactive O2 species (ROS) superoxide (O2·−) (57, 58). O2·− generated from that location is released both into the matrix and intermembrane space (51), where it is dismutated to hydrogen peroxide (H2O2) by manganese superoxide dismutase (MnSOD) and copper zinc SOD (CuZnSOD), respectively (29). Hence, mitochondrial NO metabolism involves regulation of O2 consumption, H2O2 production, and the effects of the freely diffusible (to the cytosol) H2O2 on cell signaling and gene expression (11).
NO and O2·− combine at a diffusion-limited reaction rate to form peroxynitrite (ONOO−) (40), which is able to nitrate and/or oxidize amino acid side chains of mitochondrial proteins and result in irreversible inhibition of NADH-ubiquinone reductase (complex I), succinate-ubiquinone reductase (complex II), complex IV, aconitase, and ATPase (12, 13, 18, 65). In agreement with its effects on ETC components, ONOO− potentiated the inhibition of respiration in isolated cardiac muscle compared with the inhibition elicited by NO alone and made it irreversible with time (79). Under stress conditions, such as those at reperfusion (RP) following ischemia (I), where NO production is upregulated in endothelial cells (ECs) and cardiomyocytes, either because of increases in Ca2+-dependent NOS activity via changes in shear stress and intracellular Ca2+ concentration or because of expression of inducible NOS isoforms, ONOO− formation predominates over O2·− dismutation (24, 76). ONOO−-mediated inactivation of complex I increases O2·− production from that location, and O2·− generated by complex I is released exclusively into the matrix (16, 51, 63–65). Animal I/RP models showed that, upon RP, the ETC complex activities are reduced, complex I is the main source of mitochondrial ROS (2, 31, 54), and NO derived from endothelial NOS (eNOS) is the one responsible for ONOO− formation, inhibition of complex I activity, and suppression of tissue O2 consumption (84).
We recently showed that cultured human umbilical vein EC (HUVEC) exposure to steady laminar shear stress results in inactivation of the ETC complexes I, II/III, and IV, which is, at least in part, due to ONOO− formation in the mitochondria (30). Specifically, when ECs were preincubated and sheared in the presence of either the eNOS inhibitor NG-nitro-l-arginine methyl ester (l-NAME) or the ONOO− scavenger uric acid (UA), the flow effect on complexes I and II/III was attenuated and the effect on complex IV was abolished. Inhibition of complex IV is expected in NO-producing cells (17). However, shear-induced inhibition affected all complexes (including complex I), was accompanied by nitrotyrosine staining in the mitochondria, and was prevented by UA, indicating that endogenous eNOS-derived NO diffuses to the mitochondria where it reacts with O2·− generated by the ETC resulting in mitochondrial ONOO− formation, and the latter is responsible for the inhibition of ETC at multiple sites (30).
All flow studies with cultured ECs, as well as the reoxygenation (RO) phase of hypoxia (H)/RO studies, including work from our group (30, 37, 48, 52, 80, 81), have been carried out at the atmospheric O2 concentration (21% O2), which gives rise to a partial pressure of O2 (Po2) of ∼160 mmHg, above the levels in arterial blood (75–100 mmHg; 10–13% O2) and well above the levels in venous blood (35 mmHg; 5% O2) or the levels to which ECs are exposed in tissue capillaries (mean capillary blood Po2 in the heart is ∼20 mmHg; 2.6% O2) (72). Since it is known that 1–4% of O2 reacting with the ETC is incompletely reduced to O2·−, and the O2·− formation rate by the ETC complexes III and I increases linearly with O2 concentration (42, 74), we hypothesized that, under flow, the relative “hyperoxic state” of 21% O2 raises the O2 concentration in EC mitochondria and enhances the formation of O2·−, which, via the reaction with shear-induced NO, increases the intramitochondrial ONOO− levels and potentiates the inhibition of respiration. Although it is known that 1) either hyperoxia (21–100% O2) or H (1–3% O2)/RO (21% O2) increases the mitochondrial O2·− generation and inhibits respiration (9, 21, 33, 67, 71) and 2) shear at 21% O2 increases the EC ONOO− formation intramitochondrially (30), the respiratory function of sheared ECs at 21% O2 and at lower, but physiological, O2 levels has not been examined.
In the present study, cultured bovine aortic ECs (BAECs) were exposed to steady laminar flow (shear stress of 10 dyn/cm2) for different periods of time at O2 levels of either 21% (atmospheric level), 10% (close to the 12.5% O2 that accurately simulates the arterial O2 level for people in cities with elevation close to sea level), or 5% (venous O2 level). Corresponding static controls were maintained at the same O2 levels. Electron paramagnetic resonance (EPR) oximetry, a technique capable of measuring Po2 changes vs. time from a relatively small number of cells (34, 53), was used to measure EC O2 consumption rates. Since ONOO− is formed in mitochondria of ECs sheared at 21% O2 (30) and ONOO− is known to greatly suppress respiration (79), we hypothesized that the O2 consumption rates would be lower for sheared vs. static ECs and would be modulated by the Po2 during flow. Since the matrix O2·− concentration is controlled by MnSOD, the effect of MnSOD overexpression on EC respiration was also examined. Determination of intracellular and mitochondrial O2·− production, and the dependence of the latter on NO, by ECs static or sheared at different Po2 were based on the oxidation of the fluorophore dihydroethidium (DHE) and its mitochondria-targeted derivative MitoSOX red, respectively. A possible mechanism that would explain why shear-induced inhibition of respiration may be less at O2 levels <21% is that eNOS activity may be lower under shear at <21% O2, leading to reduced NO production and mitochondrial ONOO− generation. Hypoxia is known to attenuate NO production by ECs from different vascular beds (44, 70, 78), whereas hyperoxia was shown to increase eNOS expression and activity in bovine pulmonary artery ECs (44). To test whether low, but physiological, Po2 affects eNOS activity during shear, NO production and eNOS phosphorylation at a key regulatory site were examined in ECs sheared at different O2 levels.
EXPERIMENTAL PROCEDURES
EC culture.
Primary BAECs were purchased from Cambrex (East Rutherford, NJ) and cultured in Dulbecco's modified Eagle's medium (DMEM) with l-glutamine and NaHCO3 supplemented with 10% fetal bovine serum (FBS) and antibiotics. ECs (passages 3–6) were seeded onto glass slides (75 × 38 mm; Fisher Scientific, Pittsburgh, PA) that were sterilized, air-dried, and coated with a 0.5% gelatin subbing solution that contained 0.05% potassium chromium sulfate (Sigma, St. Louis, MO). EC monolayers were used within 24 h upon confluence.
EC exposure to shear stress.
Three glass slides with EC monolayers were assembled side-by-side into a parallel-plate flow chamber, and the chamber was connected at both ends to a reservoir forming a flow loop (26). ECs were exposed to a constant gravity-driven laminar shear stress of 10 dyn/cm2 (low arterial range). Flow rate through the chamber was monitored by an ultrasonic flow sensor (Transonic Systems, Ithaca, NY). The Po2 in the system was controlled by injecting a gas mixture of air, N2, and CO2 to effect the desired final medium O2 concentrations of either 5%, 10%, or 21%, while maintaining 5% CO2. The balance of the gases was controlled by a manual gas proportioner (Aalborg, Orangeburg, NY), so that the O2 concentration, which was measured immediately downstream of the flow chamber, remained at target. Recirculating culture medium was constantly exposed to a counter-current flow of the sterile-filtered gas mixture that was warmed and humidified by bubbling through water; this permitted the use of protein-rich medium (with 10% FBS) without foaming. Medium O2 concentration and temperature were monitored real-time by inline optical O2 and temperature sensors (World Precision Instruments, Sarasota, FL). The temperature of the entire system was kept at 37°C. Corresponding static incubations of EC monolayers were placed in preequilibrated media in humidified incubators (Heraeus Instruments, Tewksbury, MA) preset for specified desired O2 concentrations of either 5% or 21% O2 for the same time periods as the sheared monolayers. Some ECs were preincubated with either 100 μM of l-NAME for 4 h or 50 μM of UA for 30 min (both from Sigma) and either subjected to shear or left static in medium containing the same concentration of the respective drug. Cell viability was determined by trypan blue exclusion at the end of treatment, and it was ≥90%. The chosen concentration of the eNOS inhibitor is known to inhibit NO production by sheared BAECs (77). The UA concentration was chosen on the basis of the scavenging of ONOO− formed by either chemically stimulated BAECs (85) or sheared HUVECs (30).
EC infection with adenoviral vectors.
The replication-deficient adenovirus Ad.MnSOD, which contains cDNA coding for human MnSOD including that for the mitochondrial leader sequence cloned into pAd.cytomegalovirus-link, expresses MnSOD in mitochondria and was purchased from the Vector Core Facility of the University of Iowa (86). HUVEC infection with Ad.MnSOD is known to produce an increase in expression of the 22-kDa protein and in total SOD activity, and colocalization experiments using confocal microscopy have shown that MnSOD protein is localized in the mitochondria (23). The infectious units were at 1 × 1010 plaque-forming units (pfu)/ml and were determined by assessing pfu on 293 cells. ECs were infected for 48 h before shear exposure (∼80% confluency) at a multiplicity of infection (MOI) of 100 in culture medium. MnSOD expression was tested in transfected ECs by assaying protein content using Western blot analysis. Simultaneously with treatment groups, control ECs were treated with Ad.empty, which contains no foreign cDNA (also purchased from the Vector Core Facility of the University of Iowa).
Measurement of O2 consumption.
At the end of shear exposure, ECs were harvested and centrifuged, and the pellet was resuspended at a density in the order of 107 cells/ml in modified Krebs-Henseleit buffer (in mM: 117.3 NaCl, 4.7 KCl, 1.3 MgSO4, 1.2 CaCl2, 1.2 KH2PO4, and 25 NaHCO3, pH 7.4) supplemented with 20 mM glucose, in agreement with earlier respirometry studies (17). EC respiration was measured immediately using EPR oximetry with lithium phthalocyanine (LiPc) as the probe in a Bruker ER-300 X-band EPR spectrometer fitted with a TM110 microwave cavity, as described previously (34, 36, 53). This technique is based on the principle of EPR line broadening by O2 and is capable of determining O2 concentration with a resolution of submicromolar concentrations in small volumes. The EPR line width vs. O2 calibration curve was constructed using known ratios of premixed O2 and N2 gases (36). LiPc crystals were added to EC suspensions, incubated for 10 min at 37°C, and sampled in a 50-μl glass capillary sealed at both ends. Po2 (mmHg) was computed from the EPR line width using the calibration curve. Plots of Po2 vs. time were obtained, and the slope of the linear section represented the O2 consumption rate (V̇o2, mmHg/min). Po2 and V̇o2 data were used to fit the equation (27):
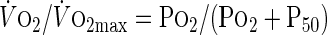 |
(1) |
where V̇o2 is the respiration rate at a given Po2, V̇o2 max is the maximum respiration rate, and P50 is the Po2 at which the respiration rate is 50% of maximum. The parameters V̇o2 max and P50 were obtained using nonlinear least-squares fitting of the data. Knowing the solubility of O2 in H2O (1.22 nmol/mmHg at 37°C) and the cell density, V̇o2 max was expressed in nmol O2·min−1·10−6 cells (53).
Fluorescence detection of intracellular and mitochondrial O2·−.
DHE and MitoSOX red (514 nm excitation/585 nm emission) were used to detect intracellular and mitochondrial O2·− production, respectively, and 4′,6-diamidino-2-phenylindole (DAPI; 359 nm excitation/461 nm emission) was used to label cell nuclei (Molecular Probes, Eugene, OR). Although DHE is widely used for the detection of intracellular O2·−, it is known that the hydroethidine moiety of DHE (and its mitochondria-targeted derivative, MitoSOX) reacts with O2·− to specifically produce hydroxyethidium (OH-Etd+) and can react with different oxidants (such as H2O2 in the presence of heme proteins or peroxidases) to produce ethidium (Etd+). Both products form a complex with DNA, which greatly enhances their fluorescence when excited at 514 nm (83). While investigating the MitoSOX oxidation by O2·−, it was shown that OH-Etd+ presents an excitation maximum at 396 nm that is not present in the excitation spectrum of Etd+ (66). Hence, in the present study, MitoSOX was excited by either the 514 or the 405 nm laser line of confocal microscopes to more accurately assess O2·− production in mitochondria of sheared ECs, as was recently described for hyperglycemic ECs (61). Specifically, at the end of shear, EC monolayers were incubated with either DHE or MitoSOX red (10 μM) and DAPI (1 μM) for 10 min in the dark at 37°C, washed with PBS, and mounted with Fluoromount-G (Southern Biotech, Birmingham, AL), and images (20× for DHE, 62× for MitoSOX red and DAPI) were obtained by confocal microscopy (Zeiss LSM 510, Peabody, MA, for excitation at 514 or 359 nm; Olympus Fluoview 1000, Center Valley, PA, for excitation at 405 nm) and overlaid using LSM software. Digital images from three fields of view were collected per experiment and corrected for autofluorescence, and the background fluorescence was excluded from calculations by thresholding. The mean fluorescence intensity per image was calculated and averaged over the three images, using MetaMorph software (Universal Imaging, Downingtown, PA). Some EC monolayers were preincubated with either l-NAME (100 μM for 4 h), the ETC complex III inhibitor antimycin A (10 μM for 30 min), or both and were either left static or sheared in the presence of the respective drug. Image acquisition conditions were kept constant for comparison between treatments.
Measurement of NO production.
Medium samples were drawn at different time points during flow exposure (with replacement of same volume of fresh medium to maintain circulating medium volume), and the accumulation of NO metabolites [nitrite (NO2−) and nitrate (NO3−); (NOx)] was measured using a chemiluminescence analyzer (Sievers 270B; Sievers Instruments, Boulder, CO), as described previously (30). For each experiment, a standard curve was constructed using different concentrations of NaNO3 for calculation of NOx content per sample. The background signal in perfusion medium was subtracted from each measured value, and values were mathematically corrected for the dilution effect of medium replacement.
Western blot analysis.
ECs were scraped into ice-cold lysis buffer (0.06 M Tris·HCl, 2% SDS, and 5% glycerol, pH 6.8). Cell extracts were homogenized by sonication, boiled for 5 min, and centrifuged at 10,000 rpm for 10 min at 4°C to remove the insoluble material. Protein concentration in cell lysates was determined using the bicinchoninic acid assay (Pierce, Rockford, IL). Supernatant containing 30 μg of protein was mixed with 2× sample buffer and applied in duplicate on 4–20% Tris·HCl gels (Bio-Rad Laboratories, Hercules, CA) for protein separation by sodium dodecyl sulfate-polyacrylamide gel electrophoresis at 100-V constant voltage. Proteins were electrophoretically transferred at 100-V constant voltage at room temperature for 1 h onto nitrocellulose membranes (Bio-Rad). After incubation in blocking solution (10 mM Tris·HCl, 0.2% nonfat milk, 100 mM NaCl, and 0.1% Tween 20, pH 7.5), duplicate membranes were hybridized with primary antibodies against either eNOS and phospho-eNOS (Ser1179), respectively, or MnSOD and β-actin, respectively. The anti-eNOS antibody was from Transduction Laboratories (Lexington, KY), anti-p-eNOS (Ser1179; based on bovine eNOS sequence and equivalent to human Ser1177) from Upstate Biotechnology (Lake Placid, NY), anti-MnSOD from Abcam (Cambridge, MA), and anti-β-actin from Sigma. Membrane-bound primary antibodies were detected using secondary antibodies conjugated with alkaline phosphatase. Immunoblots were developed on films using the enhanced chemiluminescence method (Bio-Rad).
Statistical analysis.
The V̇o2 max and P50 values are expressed as means ± SE of n ≥ 3 independent experiments. Significant differences between treatments (static vs. shear at 21% O2, 30 min; 5% O2, 30 min; 21% O2, 16 h) were determined using the SAS 9.1 software (Cary, NC) to perform one-way analysis of variance (ANOVA) followed by Bonferroni's test for pairwise comparisons. V̇o2 max values of ECs that were either kept static (21% O2) or sheared (21% O2, 30 min) with or without l-NAME or UA were analyzed similarly. Means ± SE of normalized (to corresponding controls) fluorescence from n ≥ 3 independent experiments were estimated for ECs labeled with either DHE (excitation at 514 nm) or MitoSOX (excitation at 514 and 405 nm) and either left static (21% O2) or sheared (5% or 21% O2, 30 min). Means ± SE of normalized (to corresponding controls) fluorescence from n ≥ 3 independent experiments were estimated for ECs labeled with MitoSOX (excitation at 514 nm) and either left static (21% O2) or sheared (21% O2, 30 min) with or without l-NAME, antimycin A, or both. Significant differences of normalized fluorescence data between treatments were determined using one-way ANOVA followed by Bonferroni's test for pairwise comparisons. In the case of NOx production measurements vs. time from either static or sheared ECs at different O2 levels, NOx production data from each treatment and at each time point were expressed as means ± SE of n independent experiments. Data were analyzed using a mixed-effect model with treatment (5 levels: static, 5% O2; static 21% O2; shear, 5% O2; shear, 10% O2; shear, 21% O2), time (4 levels: 5, 15, 30 and 60 min), and their interaction as fixed effects and trial and replicate (nested within trial) as random effects. Data from time zero were not used in the analysis, since NOx production was uniformly zero at that time point. Comparisons were made between treatment conditions at each time point using a Bonferroni correction for multiple comparisons. P values <0.05 were considered significant.
RESULTS
Shear stress inhibits EC respiration, and inhibition depends on Po2.
Since NO is known to reduce the V̇o2, either via competitive binding to complex IV or via formation of ONOO− that irreversibly inhibits the ETC at multiple sites (15, 57, 58, 79), and EC exposure to steady laminar flow increases the NO production resulting in ONOO− formation in the mitochondria (30), we tested whether respiration is decreased in sheared ECs. BAECs were sheared (10 dyn/cm2) for 30 min at either 5%, 10%, or 21% O2, and corresponding controls were left static at either 5% or 21% O2 for the same period. The Po2 measurements vs. time (Fig. 1A) were performed on EC suspensions (2.4 × 107 cells/ml) at the end of treatment by EPR oximetry and are representative ones of three experiments with similar results. The slope of the linear portion of the Po2 vs. time data represents the V̇o2. V̇o2 was reduced in sheared ECs compared with static controls, and the inhibition of respiration was greater at the highest Po2 tested (21% O2) (Fig. 1A). No difference in respiration was observed between ECs that were left static at different O2 concentrations or between ECs that were sheared at 5% or 10% O2 (Fig. 1A). The inhibition observed after 30 min shear at 21% O2 was still present when ECs were sheared for prolonged time (16 h) (Fig. 1B). The Po2 measurements vs. time (Fig. 1B) were performed on EC suspensions (1.3 × 107 cells/ml) at the end of treatment and are representative of three experiments with similar results. The V̇o2 of ECs treated identically was significantly higher in 2.4 × 107 vs. 1.3 × 107 cells/ml, in agreement with a previous report (35) that V̇o2 is proportional to the cell density.
Fig. 1.

Effects of Po2 during shear exposure (A) and duration of shear exposure at a specific Po2 (B) on endothelial cell (EC) respiration. A: bovine aortic ECs (BAECs) were exposed to a wall shear stress of 10 dyn/cm2 for 30 min at either 5%, 10%, or 21% O2, and corresponding controls were left static at either 5% or 21% O2 for the same period. Po2 measurements vs. time were performed on EC suspensions (2.4 × 107 cells/ml) at the end of treatment by electron paramagnetic resonance (EPR) oximetry and are representative of 3 experiments with similar results. B: BAECs were sheared (10 dyn/cm2) for either 30 min or 16 h at 21% O2, and corresponding controls were left static at 21% O2 for the same periods. Po2 measurements vs. time were performed on EC suspensions (1.3 × 107 cells/ml) at the end of treatment as before and are representative of 3 experiments with similar results.
Analysis of O2 consumption kinetics: V̇o2 max of sheared ECs is lower at the highest Po2 tested.
Since there was no difference in respiratory function between ECs sheared at 5% vs. 10% O2, all subsequent studies focused on ECs sheared at either 5% or 21% O2. The Po2 measurements vs. time (Fig. 2A) were performed on EC suspensions (0.8 × 107 cells/ml) at the end of treatment by EPR oximetry and are representative of six experiments with similar results. Data in the low Po2 range were analyzed using Eq. 1, and V̇o2 vs. Po2 lines were plotted (Fig. 2B). It is important to notice that, even under the lowest Po2 tested (5% O2; 35 mmHg), static and sheared ECs respire at their maximum rates (V̇o2 = V̇o2 max at either 5% or 21% O2). For this characteristic run, V̇o2 max of static ECs was the same at 5% and 21% O2 (3.5 and 3.7 mmHg/min, respectively), whereas, for sheared ECs, V̇o2 max at 5% O2 was 2.7 mmHg/min and at 21% O2 was 1.4 mmHg/min. P50 of static ECs was the same at 5% and 21% O2 (4.0 and 4.2 mmHg), remained the same when ECs were sheared at 5% O2 (3.9 mmHg), but declined when ECs were sheared at 21% O2 (1.6 mmHg). To quantify the effect of flow at different O2 levels on the respiratory parameters, V̇o2 max (in nmol O2·min−1·10−6 cells) and P50 (in mmHg) values from n = 3–6 independent experiments were expressed as means ± SE of 6 different treatments (static vs. shear at either 21% or 5% O2 for 30 min; static vs. shear at 21% O2 for 16 h) (Table 1). V̇o2 max of sheared ECs at 21% O2 for either 30 min or 16 h was significantly different from V̇o2 max of corresponding static controls and of sheared ECs at 5% O2 for 30 min. P50 of sheared ECs at 21% O2 for either 30 min or 16 h was significantly different from P50 of corresponding static controls (Table 1).
Fig. 2.

Analysis of O2 consumption kinetics. A: BAECs were sheared (10 dyn/cm2) for 30 min at either 5% or 21% O2, and corresponding controls were left static at either 5% or 21% O2 for the same period. Po2 measurements vs. time were performed on EC suspensions (0.8 × 107 cells/ml) as before and are representative of 6 experiments with similar results. B: data in A were analyzed using Eq. 1, and O2 consumption rate (V̇o2) vs. Po2 lines were plotted.
Table 1.
V̇o2max and P50 values for static versus sheared bovine aortic endothelial cells
| Treatment | Po2, %O2 | Time | V̇o2max, nmol O2·min−1·10−6 cells | P50, mmHg |
|---|---|---|---|---|
| Static | 21 | 30 min | 9.8±0.8 | 4.9±0.8 |
| Shear | 21 | 30 min | 4.1±0.7*† | 2.6±0.4* |
| Static | 5 | 30 min | 9.3±1.4 | 4.6±0.6 |
| Shear | 5 | 30 min | 8.1±0.6 | 4.3±0.7 |
| Static | 21 | 16 h | 9.6±0.4 | 4.7±0.7 |
| Shear | 21 | 16 h | 4.6±0.7*† | 2.5±0.4* |
Values are means ± SE; n = 3–6 experiments. Plots of Po2 versus time were obtained from electron paramagnetic resonance (EPR) oximetry measurements on endothelial cell (EC) suspensions. Before EPR oximetry, ECs were either static or sheared at 10 dyn/cm2 for either 30 min or 16 h. Maximum respiration rate (V̇o2max) and Po2 at which V̇o2 is 50% of maximum (P50) values were determined, as described. Using the solubility of O2 in H2O at 37°C and the known cell number of each experiment, V̇o2max was expressed in nmol O2·min−1·10−6 cells.
P < 0.05 versus corresponding static control.
P < 0.05 versus shear at 5% O2 for 30 min.
NO and mitochondrial ONOO− are responsible for the inhibitory effect of flow on EC respiration.
To determine the role of shear-induced NO and the resultant ONOO− formation on the observed reduction of V̇o2, ECs were preincubated and sheared at 21% O2 for 30 min in the presence of either 100 μM of the eNOS inhibitor l-NAME or 50 μM of the ONOO− scavenger UA. The Po2 measurements vs. time (Fig. 3A) were performed on EC suspensions (2.4 × 107 cells/ml) at the end of treatment and are representative of three experiments with similar results. Either l-NAME or UA abolished the inhibitory effect of shear at 21% O2 on EC respiration: V̇o2 max (means ± SE of n = 3 independent experiments) of sheared ECs (4.3 ± 0.3 nmol O2·min−1·10−6 cells) was significantly different from V̇o2 max of corresponding static controls (9.5 ± 0.6 nmol O2·min−1·10−6 cells) and of sheared ECs in the presence of either l-NAME (9.3 ± 0.7 nmol O2·min−1·10−6 cells) or UA (9.1 ± 0.2 nmol O2·min−1·10−6 cells). There was no difference in respiration between static ECs that were preincubated and left static at 21% O2 for 30 min with or without l-NAME or UA (only data from static ECs without any drug are shown; Fig. 3A). The NO fraction that forms ONOO− in the mitochondria is determined by the local concentration of O2·−, which is controlled by MnSOD (47). To test whether the inhibitory mechanism is mitochondrial ONOO−-dependent, ECs were transfected with either Ad.MnSOD that expresses MnSOD in the mitochondria or Ad.empty, either exposed to shear at 21% O2 for 30 min or left static under the same conditions, and EC respiration was measured as before. Western blot analysis verified an increase in MnSOD protein levels in static Ad.MnSOD-transfected ECs when compared to static ECs either nontransfected or transfected with the control virus (no changes were observed in β-actin expression, and it served as protein loading control; inset of Fig. 3B). The Po2 measurements vs. time (Fig. 3B) were performed on EC suspensions (2.4 × 107 cells/ml) at the end of treatment and are representative of two experiments with identical results. Transfection with Ad.MnSOD, and not with the control virus, almost completely protected the sheared ECs from inhibition of respiration, whereas it had no effect on the respiration of static ECs (Fig. 3B). There was no difference between static nontransfected and static transfected with the control virus ECs (data not shown).
Fig. 3.

Effects of an endothelial nitric oxide (NO) synthase (eNOS) inhibitor, a ONOO− scavenger, and manganese superoxide dismutase (MnSOD) overexpression on the inhibitory effect of shear stress at 21% O2 on EC respiration. A: BAECs were preincubated with either 100 μM of NG-nitro-l-arginine methyl ester (l-NAME) for 4 h or 50 μM of uric acid (UA) for 30 min and then sheared at 21% O2 for 30 min or left static in the incubator with or without the same concentration of the respective drug. Po2 measurements vs. time were performed on EC suspensions (2.4 × 107 cells/ml) as before and are representative of 3 experiments with similar results. Since there was no difference in respiration between static ECs with or without l-NAME or UA, only data from static ECs without any drug are shown. B: BAECs were transfected with either Ad.MnSOD (multiplicity of infection of 100) or a control virus (Ad.empty) before either shear exposure at 21% O2 for 30 min or static incubation under the same conditions. Po2 measurements vs. time were performed on EC suspensions (2.4 × 107 cells/ml) as before and are representative of 2 experiments with identical results. Western blot analysis verified an increase in MnSOD protein levels in static Ad.MnSOD-transfected ECs when compared with static ECs either nontransfected or transfected with the control virus (inset).
Mitochondrial O2·− production by sheared ECs increases with Po2 and depends on NO.
Shear exposure (at either 5% or 21% O2 for 30 min) caused an increase in fluorescence of either DHE (excitation at 514 nm) or MitoSOX red (excitation at 514 and 405 nm), as monitored by confocal microscopy, and the increase was higher when ECs were sheared at 21% than at 5% O2 (Fig. 4A). No noticeable increase in fluorescence occurred in static controls maintained at either Po2 (only static control at 21% O2 is shown; Fig. 4A). The MitoSOX fluorescence signals of sheared ECs were significantly different from those of their corresponding static controls at either excitation, and signals at 405 nm excitation were only slightly lower than signals at 514 nm excitation, suggesting that MitoSOX oxidation in ECs sheared at either 5% or 21% O2 is predominantly O2·−-specific (Fig. 4B). The fluorescence signals of ECs labeled with either DHE or MitoSOX (excitation at 514 and 405 nm) and sheared at 5% O2 were significantly different from the respective signals of ECs sheared at 21% O2 (Fig. 4B). Since there was no appreciable difference in MitoSOX fluorescence between the two excitations, the rest of the fluorescence experiments were performed at an excitation of 514 nm. MitoSOX red is known to not get oxidized by NO or ONOO− (66). l-NAME (100 μM) did not affect the MitoSOX fluorescence signal in static ECs, probably because of the fact that the relatively low amount of NO produced is not sufficient to significantly inhibit complex IV and enhance O2·− generation by the mitochondrial ETC (Fig. 5, A and B). Shear (21% O2 for 30 min) as before, increased the MitoSOX fluorescence due to increased NO generation and intramitochondrial ONOO− formation, which are expected to increase the mitochondrial O2·− generation at the level of ETC complexes III and I. Preincubation and shear at 21% O2 in the presence of l-NAME (100 μM) significantly decreased the MitoSOX fluorescence signal compared with sheared ECs, suggesting that NO and/or its product, ONOO−, is a major contributor to the mitochondrial generation of O2·− (Fig. 5, A and B). To test whether MitoSOX fluorescence of sheared ECs in the presence of l-NAME is restored when a respiratory inhibitor is supplied, antimycin A (10 μM) was used. Antimycin A, by binding to cytochrome b, increases the concentration of ubisemiquinone, resulting in increased O2·− generation from complex III (73). As expected, antimycin A significantly increased the fluorescence of static ECs (Fig. 5, A and B). However, it significantly blocked the shear-induced increase in fluorescence, possibly because shear exposure inhibits complex III activity by ∼70% (30) and decreased complex III activity reduces the enzyme-mediated O2·− production (14). Last, when either static or sheared ECs were treated with both l-NAME and antimycin A, the signal was significantly increased compared with either static or sheared ECs treated with just l-NAME, respectively, suggesting that MitoSOX fluorescence of ECs in the absence of NO is sensitive to O2·− generation by the ETC complex III (Fig. 5, A and B).
Fig. 4.

Intracellular and mitochondrial O2·− production by ECs sheared at different Po2. A: BAECs were sheared at either 5% or 21% O2 for 30 min, and corresponding controls were left static at each Po2 for the same period. At the end of shear or static exposure, ECs were incubated with either dihydroethidium (DHE) or MitoSOX red (10 μM) for 10 min, and images (DHE, 20×, 514 nm/585 nm; MitoSOX, 62×, either 514 or 405 nm/585 nm) were obtained by confocal microscopy. Since no difference in DHE and MitoSOX fluorescence was detected between static controls maintained at either 5% or 21% O2, only static controls at 21% O2 are shown. B: digital images from 3 fields of view were collected per experiment, and the average fluorescence intensity was calculated. Normalized (to corresponding static controls) fluorescence data from n ≥ 3 independent experiments for ECs labeled with either DHE (514 nm) or MitoSOX (514 and 405 nm) and either left static (21% O2) or sheared (5% or 21% O2) are shown as means ± SE. *P < 0.05 vs. static control. †P < 0.05 vs. shear (21% O2).
Fig. 5.

Regulation of mitochondrial O2·− production by shear-induced NO. A: BAECs were sheared at 21% O2 for 30 min, and corresponding controls were left static under the same conditions. BAECs were preincubated with either l-NAME (100 μM for 4 h), antimycin A (10 μM for 30 min), or both and either left static or sheared in the presence of the respective drug. At the end of shear or static exposure, ECs were incubated with MitoSOX red (10 μM) and DAPI (1 μM) for 10 min, and images (MitoSOX, 62×, 514 nm/585 nm; DAPI, 62×, 359 nm/461 nm) were obtained by confocal microscopy and merged. B: digital images from 3 fields of view were collected per experiment, and the average fluorescence intensity was calculated. Normalized (to corresponding static controls) fluorescence data from n ≥ 3 independent experiments for ECs labeled with MitoSOX (514 nm) and either left static (21% O2) or sheared (21% O2) with or without l-NAME, antimycin A, or both are shown as means ± SE. *P < 0.05 vs. static control. †P < 0.05 vs. shear (21% O2).
NOx production by sheared ECs increases with Po2.
Since less NO production by ECs sheared at either 5% or 10% O2, compared with ECs sheared at 21% O2, would lead to less mitochondrial ONOO− formation and partially restore respiration, NOx production in culture media of ECs exposed to static or shear conditions at different Po2 was measured vs. time using a chemiluminescence analyzer. Data at each time point and treatment are means ± SE of five independent experiments (Fig. 6A). Shear stress at 21% O2 elicited a transient burst in NOx production within the first 15 min from the onset of flow followed by a lower sustained release, in agreement with earlier studies (41, 77). When ECs were sheared at either 5% or 10% O2, the initial burst occurred, but it was smaller in magnitude, so during the rest of the time (15–60 min), the NOx production by sheared ECs at 21% O2 was significantly higher than the production by ECs either sheared at lower Po2 or left static at 5% or 21% O2. At 5 min from the flow onset, the NOx production by sheared ECs at 21% O2 was significantly higher than that by ECs in all other conditions except from shear at 10% O2 (Fig. 6A). Since Ser1179 (based on bovine eNOS sequence and equivalent to human Ser1177) phosphorylation is considered the most important of the regulatory eNOS phosphorylation sites and diverse stimuli, including shear stress, cause early phosphorylation of this site (7, 50), we examined whether shear at different Po2 alters eNOS activity via Ser1179 phosphorylation. ECs were either sheared at 5%, 10%, or 21% O2 or left static at 5% or 21% O2 for 30 min, and cell lysates were analyzed by Western blot with antibodies specific for phospho-eNOS (Ser1179) or total eNOS (protein loading control). Shear greatly increased Ser1179 phosphorylation at each Po2 tested compared with corresponding static controls, but there was no difference in the ratio of phospho-eNOS (Ser1179) to total eNOS among ECs sheared at either 5%, 10%, or 21% O2 (Fig. 6B).
Fig. 6.

NOx production and eNOS phosphorylation (Ser1179) by ECs sheared at different Po2. A: BAECs were sheared for 30 min at either 5%, 10%, or 21% O2, and corresponding controls were left static at either 5% or 21% O2 for the same period. Medium samples were drawn at different time points after either the onset of flow or static incubation, and NOx accumulation was measured using a chemiluminescence analyzer. Data are means ± SE of 5 independent experiments. *P < 0.05 vs. all other conditions (shear, 5% O2; shear, 10% O2; static, 5% O2; static, 21% O2) at the same time point. †P < 0.05 vs. all other conditions except shear, 10% O2 at the same time point. B: BAECs were treated as in A, and lysates were analyzed by Western blot for phospho (p)-eNOS (Ser1179) and total eNOS.
DISCUSSION
The present study provides the first evidence that shear-induced NO, via formation of ONOO− in the mitochondria, reduces the rate of O2 consumption by cultured vascular ECs exposed to steady laminar flow (Figs. 1–3 and Table 1). The degree of inhibition of EC respiration depends on the Po2 during shear exposure; the effect is greater when flow is carried out at the atmospheric O2 concentration (Figs. 1A and 2 and Table 1). Exposure of cultured ECs to 21% O2 is mildly hyperoxic compared with the in vivo normoxia, and, hence, in the present study, BAECs were sheared at an arterial level of shear stress under either 5%, 10%, or 21% O2. Our choice of 10% O2 is within the normal O2 level for the arterial wall (accurate for arterial blood Po2 of people who live at high altitude), whereas 5% O2 is mildly hypoxic for arterial ECs (72). Greater inhibition of respiration for ECs sheared under the relative hyperoxic 21% O2 is consistent with our hypothesis that higher Po2 may result in increased shear-induced NO production and O2·− generation by the ETC leading to higher ONOO− formation in the mitochondria and enhanced inhibition of respiration. Even if shear-induced NO production remained the same at 21% vs. 5% O2 (according to Fig. 6A, it is increased at 21% O2), O2·− formation by the ETC is known to increase linearly with Po2 (42, 74) and, since O2·− does not diffuse across cell membranes, the fraction of shear-induced NO that forms ONOO− intramitochondrially would be expected to be higher at 21% vs. 5% O2.
ONOO− is known to suppress respiration to a greater degree than the suppression due to NO alone (79). In agreement with this, either the eNOS inhibitor l-NAME or the ONOO− scavenger UA, at concentrations known to, at least in part, prevent the shear (21% O2)-induced inhibition of complex I activity (30) [complex I is inactivated by ONOO− (12)], blocked the shear (21% O2)-induced inhibition of respiration (Fig. 3A). In an earlier study, shear stress (21% O2) was shown to increase nitrotyrosine staining, a “footprint” of ONOO− generation, in HUVEC mitochondria, and the signal was inhibited by preincubation and shear exposure in the presence of UA (30). According to a report (69), UA does not scavenge ONOO− itself, as is mostly stated in the literature, but the nitrogen dioxide radical (NO2·), a product of the reaction between ONOO− and CO2. The finding that EC transfection with Ad.MnSOD, which expresses MnSOD in the mitochondria, before flow treatment blocked the shear-induced inhibition of respiration (Fig. 3B) provides evidence that the enhanced suppression of respiration for ECs sheared at 21% O2 is due to mitochondrial ONOO− formation. O2·− is known to react with NO at a faster rate [k1 = 1.9 × 1010 M−1 s−1 (38)] than with MnSOD [k2 = 2.3 × 109 M−1 s−1 (39)]. Although there is a 10-fold difference in the rate constants and NO levels may be in the low μM range inside the mitochondria, it is the local concentration of MnSOD that determines whether NO will outcompete MnSOD and react with O2·− to generate ONOO− or MnSOD will outcompete NO and prevent ONOO− formation. MnSOD concentration in the mitochondria is ∼10 μM (61), and EC infection with Ad.MnSOD (MOI of 100) was found to cause a fivefold increase in MnSOD levels/activity (23), thus making possible the prevention of shear-induced ONOO− formation in the mitochondria of ECs infected with Ad.MnSOD.
MitoSOX fluorescence (excitation at 405 nm) showed that the mitochondrial O2·− levels are increased in parallel with the Po2 during shear exposure, and a comparison between the two excitation wavelengths showed that, even when the excitation is at 514 nm, most of the fluorescence is due to O2·−-specific oxidation products (Fig. 4). MitoSOX red fluorescence (excitation at 514 nm) suggested that the shear-induced NO is, at least in part, responsible for the generation of O2·− by the ETC (Fig. 5). To accurately demonstrate mitochondrial O2·− generation, the oxidation product OH-Etd+ in mitochondrial cell fractions should be detected by HPLC, as described for whole EC lysates (83). However, MitoSOX fluorescence (excitation at 514 nm) from l-NAME-treated sheared ECs responded to mitochondrial O2·− generation induced by antimycin A (Fig. 5). In general, our findings using fluorescence microscopy agree with the literature that mitochondrial utilization of excess NO involves ubiquinol oxidation that increases the O2·− production rate, and formation of ONOO− that, via inhibition of the ETC at multiple sites, amplifies the O2·− generation (11, 57, 58).
One novelty of the present study is the technique used to measure O2 consumption rates by the relatively low number of ECs exposed to flow in a perfusion chamber: EPR oximetry with LiPc as the probe, a technique capable of yielding high-resolution Po2 vs. time data, similar to the data normally obtained with respirometry (27), in small volumes of cell suspensions containing as few as 104 cells for certain cell types (36). EPR oximetry has been used to measure the V̇o2 of static ECs from mouse aorta (∼4 nmol O2·min−1·10−6 cells) (53), but it has never been used to measure the V̇o2 of sheared ECs from any species or vascular bed (note that V̇o2 = V̇o2 max at ≥ 3% O2; Fig. 2B). Using EPR oximetry, we found that static BAECs have a V̇o2 of ∼10 nmol O2·min−1·10−6 cells (sheared at 21% O2 BAECs have ∼50% of that; Table 1), which is higher but in the same order of V̇o2 values measured for static HUVECs or porcine aortic ECs (2–3 nmol O2·min−1·10−6 cells) using standard respirometry with a Clark-type O2 electrode (10, 17). The estimated P50 for static BAECs (∼5 mmHg) is in the same range of P50 values reported for HUVECs (60), and sheared at 21% O2 BAECs have ∼50% of that (Table 1). P50 is analogous to the apparent Michaelis-Menten constant Km in purified enzymatic reactions. However, since the O2-dependence of respiration is sensitive to each component of the ETC and not only to complex IV, the mitochondrial P50 (reciprocal value of O2 affinity) is potentially a function of each element in the ETC, in contrast to the terminological implications of a Km constant. The lower P50 value in the case of sheared at 21% O2 ECs shows that there is a shift in O2 binding (toward higher affinity) in the mitochondria of sheared ECs, possibly because of oxidative/nitrosative modifications of ETC proteins. In general, P50 increases with the rate of oxidative phosphorylation, which is mediated by the ETC (27). This agrees with our findings that P50 values were decreased when ECs were sheared at 21% compared with static conditions, since shear-induced ONOO− formation in the mitochondria leads to inhibition of the ETC (30).
In agreement with a study by Moncada's group (17), who showed that the V̇o2 of cultured ECs is dependent on Po2 only when the cells are activated to synthesize NO, no difference in V̇o2 was found between static BAECs at either 5% or 21% O2 (Figs. 1A and 2 and Table 1). In their study, EC treatment with the neuropeptide bradykinin, which activates eNOS, induced production of NO that interacted at the level of the ETC complex IV and inhibited respiration; inhibition was greater at lower Po2 that resembled the physiological range in tissues (17). In the present study, endogenous NO production due to shear exposure inhibited respiration, but the effect was more pronounced at 21% O2 compared with that at lower Po2 levels, because of ONOO− formation in the mitochondria of ECs sheared at 21% O2. Although both studies examined the effect of endogenous NO on EC respiration at various Po2 levels, the difference in their findings is probably due to the difference in the EC NO response elicited by diverse stimuli, a mechanical (shear stress) and a chemical one (bradykinin): EC exposure to steady laminar flow increases NO production in a biphasic manner with an initial rapid production that is Ca2+/calmodulin-dependent followed (after ∼15 min; Fig. 6A) by a less rapid, sustained production that is independent of further changes in Ca2+/calmodulin (41). In contrast, bradykinin-mediated NO production is comparable to the initial shear stress-mediated, Ca2+/calmodulin-dependent NO production without the sustained component (41). Sustained shear-induced NO production involves phosphorylation of eNOS at several sites and its interaction with proteins, such as caveolin and heat shock protein 90 (HSP90) (6, 8), and may be the missing link for the accumulation of high NO levels in mitochondria and the resultant ONOO− formation. The same extent of inhibition of respiration was present even after prolonged (16 h) shear exposure (Fig. 1B), which agrees with the decreased activities of ETC complexes measured in an earlier study (30).
Since the rate of formation of ONOO− is first order in the concentrations of both NO and O2·−, our findings of ONOO− formation intramitochondrially (Fig. 3B and Ref. 30) suggest that, at the relative hyperoxic 21% O2, either the NO concentration increases (primarily due to eNOS activation and possibly due to changes in mitochondrial NOS activity) or the mitochondrial O2·− concentration increases, or both (the latter seems to be the case, on the basis of Figs. 4, 5, and 6A) (62). There is evidence in the literature that hyperoxia increases both the NO synthesis and the ROS generation from the ETC: Short-term BAEC incubation at Po2 levels in the physiological range led to a decrease in the rate of thapsigargin-induced NOx production, compared with that at atmospheric Po2, suggesting that eNOS activity declines at lower Po2 (78). At prolonged times (24 h), changes in O2 concentrations from 95% to 3% caused a progressive decrease in eNOS mRNA and protein levels, which correlated with changes in enzyme activity, in cultured BAECs, and bovine pulmonary artery ECs (44). Sheep pulmonary microvascular EC exposure to hyperoxia (100% O2, 30 min) increased the cellular O2·− production, as detected by EPR spectroscopy, and blocking the ETC with rotenone decreased the O2·− signal (67). Detection of ROS in capillary ECs by in situ imaging of 2′,7′-dichlorofluorescein fluorescence in perfused rat lungs showed that ROS formation increases with Po2 (21–100% O2, 90 min) and originates from mitochondria (9).
Our NOx measurements in media of BAECs sheared at different Po2 levels (Fig. 6A) suggest that shear at 21% O2 may increase eNOS activity to a greater extent than shear at lower Po2 levels, leading to enhanced ONOO− formation and suppression of respiration. Since NO3− is the degradation product of ONOO− and its nitrating intermediates (43) and ONOO− formation is enhanced under shear at 21% O2 (Fig. 3 and Ref. 30), ONOO− may contribute to the elevated NOx production measurement. Last, the degree of eNOS phosphorylation at Ser1179 (equivalent to human Ser1177), a critical requirement for eNOS activation (50), was found to be the same among ECs sheared at either 5%, 10%, or 21% O2 (Fig. 6B). Increased eNOS activity under shear at 21% O2 compared with shear at lower Po2 levels may be due to differences in posttranslational protein modifications [nitrosylation, acylation, or phosphorylation of other sites, such as phosphorylation at Ser635 (5, 8)] and/or alterations in interactions between eNOS and its regulatory proteins calmodulin, caveolin, HSP90, and platelet EC adhesion molecule-1 (3, 22, 25, 50).
In eNOS-transfected EC lines, the dynamic regulation of respiration by endogenous NO was shown to provide protection against H2O2-mediated injury and death (55), probably by maintaining mitochondrial membrane potential and preventing apoptosis (4). Although the functional significance of mitochondria-derived ROS in ECs has received little attention, it is well documented that EC mitochondrial O2·− production from ETC complexes I and III is responsible for the shear-induced dilation in human coronary arterioles (46, 82). It is also known that, during EC exposure to cyclic strain, mitochondrial ROS mediate the activation of nuclear factor-κB and upregulation of vascular cell adhesion molecule-1 (1). During EC exposure to flow, ONOO− was found to mediate the activation of c-Jun NH2-terminal kinase (JNK), a mitogen-activated protein kinase that determines cell survival in response to environmental stress (28). Formation of ONOO− in the mitochondria of sheared ECs (Fig. 4 and Ref. 30) suggests that the intramitochondrial ONOO− is probably responsible for activation of JNK, which is a signaling molecule in the pathway leading to growth arrest by laminar shear stress (45).
Our finding that flow at hyperoxic Po2 causes nitrosative stress to the EC mitochondria and inhibition of respiration agrees with studies in reperfused isolated heart models, where lowering the Po2 during early RP had a beneficial effect on mitochondrial bioenergetic parameters such as O2 consumption, ETC complex activities, mitochondrial ROS production and lipid peroxidation, and improved cardiac performance (49, 56). I/RP-induced EC injury is believed to be self-inflicted, resulting from a burst of endogenous ONOO− upon the onset of RP, and, due to decreased bioavailable NO, it predisposes ECs to increased neutrophil adhesion (24, 76). In agreement with that, bolus injections of ONOO− into isolated hearts were shown to acutely inhibit EC-dependent coronary vasodilation (75). Furthermore, in a rat model of splanchnic artery occlusion/RP, use of the ONOO− decomposition catalyst 5,10,15,20-tetrakis(2,4,6-trimethyl-3,3-disulfonatophenyl)porphyrinato iron reduced P-selectin staining localized mainly in vascular ECs and improved bowel survival, suggesting that its beneficial effect may be due to inhibition of neutrophil-mediated damage in the later stages of RP (19). Although minimization of intracellular ONOO− was shown to limit the postischemic EC inflammatory response, the contribution of mitochondrial ONOO− in the EC dysfunction can only be derived from in vitro studies with H/RO-treated cells. These studies showed that the mitochondria are a source of EC ROS production following H or anoxia (1–2 h)/RO (1 h), the ROS release site is the ETC complex III, and the neutrophil-EC adhesion at prolonged RO (10 h) is mediated by the postanoxic EC mitochondrial ROS production (32, 33, 71). RO following H (4 h) induced loss of mitochondrial membrane potential and cytochrome c release, suggesting the mitochondria as the initiation site of EC apoptosis (20). However, H/RO studies do not include the flow component that is essential in the RP phase of I/RP, and, as a result, do not provide information on the shear-induced NO generation upon RP and the resultant mitochondrial ROS/RNS production.
In summary, our data collectively demonstrate that endogenous NO, via formation of ONOO− in the mitochondria, significantly reduces the rate of O2 consumption by cultured vascular ECs exposed to steady laminar flow at the atmospheric Po2. When flow experiments are performed at lower, more physiological Po2 levels, the endogenous NO production is modestly reduced and the mitochondrial O2·− generation (in part, due to the decline in NO) is decreased, leading to less ONOO− formation intramitochondrially and, hence, insignificant suppression of respiration. The present study points to the importance of conducting in vitro flow studies at physiological O2 concentrations, since the dynamic regulation of respiration by endogenous NO, the NO synthesis, and the generation of mitochondrial ROS are all, at least in part, regulated by Po2.
GRANTS
This work was supported by National Institutes of Health Grants HL-91417 (to B. R. Alevriadou), EB-04658 and HL-78796 (to G. Ilangovan), and HL-38324, HL-63744, and HL-65608 (to J. L. Zweier), and by the Samuel J. Roessler Fellowship (to C. I. Jones III).
Acknowledgments
The authors thank Dr. Yeong-Renn Chen, Davis Heart and Lung Research Institute (DHLRI), Ohio State University (OSU), for critical review of the manuscript; Dr. Dmitry Terentyev, DHLRI, OSU, for expert technical assistance with confocal microscopy; and Dr. Christopher H. Holloman, Statistical Consulting Service, OSU, for expert statistical analysis.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- 1.Ali MH, Pearlstein DP, Mathieu CE, Schumacker PT. Mitochondrial requirement for endothelial responses to cyclic strain: implications for mechanotransduction. Am J Physiol Lung Cell Mol Physiol 287: L486–L496, 2004. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosio G, Zweier JL, Duilio C, Kuppusamy P, Santoro G, Elia PP, Tritto I, Cirillo P, Condorelli M, Chiariello M, Flaherty JT. Evidence that mitochondrial respiration is a source of potentially toxic oxygen free radicals in intact rabbit hearts subjected to ischemia and reflow. J Biol Chem 268: 18532–18541, 1993. [PubMed] [Google Scholar]
- 3.Bagi Z, Frangos JA, Yeh JC, White CR, Kaley G, Koller A. PECAM-1 mediates NO-dependent dilation of arterioles to high temporal gradients of shear stress. Arterioscler Thromb Vasc Biol 25: 1590–1595, 2005. [DOI] [PubMed] [Google Scholar]
- 4.Beltran B, Mathur A, Duchen MR, Erusalimsky JD, Moncada S. The effect of nitric oxide on cell respiration: a key to understanding its role in cell survival or death. Proc Natl Acad Sci USA 97: 14602–14607, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boo YC, Hwang J, Sykes M, Michell BJ, Kemp BE, Lum H, Jo H. Shear stress stimulates phosphorylation of eNOS at Ser(635) by a protein kinase A-dependent mechanism. Am J Physiol Heart Circ Physiol 283: H1819–H1828, 2002. [DOI] [PubMed] [Google Scholar]
- 6.Boo YC, Jo H. Flow-dependent regulation of endothelial nitric oxide synthase: role of protein kinases. Am J Physiol Cell Physiol 285: C499–C508, 2003. [DOI] [PubMed] [Google Scholar]
- 7.Boo YC, Sorescu G, Boyd N, Shiojima I, Walsh K, Du J, Jo H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at Ser1179 by Akt-independent mechanisms: role of protein kinase A. J Biol Chem 277: 3388–3396, 2002. [DOI] [PubMed] [Google Scholar]
- 8.Boo YC, Sorescu GP, Bauer PM, Fulton D, Kemp BE, Harrison DG, Sessa WC, Jo H. Endothelial NO synthase phosphorylated at SER635 produces NO without requiring intracellular calcium increase. Free Radic Biol Med 35: 729–741, 2003. [DOI] [PubMed] [Google Scholar]
- 9.Brueckl C, Kaestle S, Kerem A, Habazettl H, Krombach F, Kuppe H, Kuebler WM. Hyperoxia-induced reactive oxygen species formation in pulmonary capillary endothelial cells in situ. Am J Respir Cell Mol Biol 34: 453–463, 2006. [DOI] [PubMed] [Google Scholar]
- 10.Carlisle R, Rhoads CA, Aw TY, Harrison L. Endothelial cells maintain a reduced redox environment even as mitochondrial function declines. Am J Physiol Cell Physiol 283: C1675–C1686, 2002. [DOI] [PubMed] [Google Scholar]
- 11.Carreras MC, Poderoso JJ. Mitochondrial nitric oxide in the signaling of cell integrated responses. Am J Physiol Cell Physiol 292: C1569–C1580, 2007. [DOI] [PubMed] [Google Scholar]
- 12.Cassina A, Radi R. Differential inhibitory action of nitric oxide and peroxynitrite on mitochondrial electron transport. Arch Biochem Biophys 328: 309–316, 1996. [DOI] [PubMed] [Google Scholar]
- 13.Castro L, Rodriguez M, Radi R. Aconitase is readily inactivated by peroxynitrite, but not by its precursor, nitric oxide. J Biol Chem 269: 29409–29415, 1994. [PubMed] [Google Scholar]
- 14.Chen YR, Chen CL, Yeh A, Liu X, Zweier JL. Direct and indirect roles of cytochrome b in the mediation of superoxide generation and NO catabolism by mitochondrial succinate-cytochrome c reductase. J Biol Chem 281: 13159–13168, 2006. [DOI] [PubMed] [Google Scholar]
- 15.Cleeter MW, Cooper JM, Darley-Usmar VM, Moncada S, Schapira AH. Reversible inhibition of cytochrome c oxidase, the terminal enzyme of the mitochondrial respiratory chain, by nitric oxide. Implications for neurodegenerative diseases. FEBS Lett 345: 50–54, 1994. [DOI] [PubMed] [Google Scholar]
- 16.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA 95: 7631–7636, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Clementi E, Brown GC, Foxwell N, Moncada S. On the mechanism by which vascular endothelial cells regulate their oxygen consumption. Proc Natl Acad Sci USA 96: 1559–1562, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cooper CE, Davies NA, Psychoulis M, Canevari L, Bates TE, Dobbie MS, Casley CS, Sharpe MA. Nitric oxide and peroxynitrite cause irreversible increases in the Km for oxygen of mitochondrial cytochrome oxidase: in vitro and in vivo studies. Biochim Biophys Acta 1607: 27–34, 2003. [DOI] [PubMed] [Google Scholar]
- 19.Cuzzocrea S, Misko TP, Costantino G, Mazzon E, Micali A, Caputi AP, Macarthur H, Salvemini D. Beneficial effects of peroxynitrite decomposition catalyst in a rat model of splanchnic artery occlusion and reperfusion. FASEB J 14: 1061–1072, 2000. [DOI] [PubMed] [Google Scholar]
- 20.Dhar-Mascareno M, Carcamo JM, Golde DW. Hypoxia-reoxygenation-induced mitochondrial damage and apoptosis in human endothelial cells are inhibited by vitamin C. Free Radic Biol Med 38: 1311–1322, 2005. [DOI] [PubMed] [Google Scholar]
- 21.Du G, Mouithys-Mickalad A, Sluse FE. Generation of superoxide anion by mitochondria and impairment of their functions during anoxia and reoxygenation in vitro. Free Radic Biol Med 25: 1066–1074, 1998. [DOI] [PubMed] [Google Scholar]
- 22.Dudzinski DM, Michel T. Life history of eNOS: partners and pathways. Cardiovasc Res 75: 247–260, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fennell JP, Brosnan MJ, Frater AJ, Hamilton CA, Alexander MY, Nicklin SA, Heistad DD, Baker AH, Dominiczak AF. Adenovirus-mediated overexpression of extracellular superoxide dismutase improves endothelial dysfunction in a rat model of hypertension. Gene Ther 9: 110–117, 2002. [DOI] [PubMed] [Google Scholar]
- 24.Ferdinandy P, Schulz R. Nitric oxide, superoxide, and peroxynitrite in myocardial ischaemia-reperfusion injury and preconditioning. Br J Pharmacol 138: 532–543, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci 118: 4103–4111, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production in cultured human endothelial cells. Science 227: 1477–1479, 1985. [DOI] [PubMed] [Google Scholar]
- 27.Gnaiger E Bioenergetics at low oxygen: dependence of respiration and phosphorylation on oxygen and adenosine diphosphate supply. Respir Physiol 128: 277–297, 2001. [DOI] [PubMed] [Google Scholar]
- 28.Go YM, Patel RP, Maland MC, Park H, Beckman JS, Darley-Usmar VM, Jo H. Evidence for peroxynitrite as a signaling molecule in flow-dependent activation of c-Jun NH2-terminal kinase. Am J Physiol Heart Circ Physiol 277: H1647–H1653, 1999. [DOI] [PubMed] [Google Scholar]
- 29.Gutteridge JM, Halliwell B. Free radicals and antioxidants in the year 2000. A historical look to the future. Ann NY Acad Sci 899: 136–147, 2000. [DOI] [PubMed] [Google Scholar]
- 30.Han Z, Chen YR, Jones CI 3rd, Meenakshisundaram G, Zweier JL, Alevriadou BR. Shear-induced reactive nitrogen species inhibit mitochondrial respiratory complex activities in cultured vascular endothelial cells. Am J Physiol Cell Physiol 292: C1103–C1112, 2007. [DOI] [PubMed] [Google Scholar]
- 31.Hardy DL, Clark JB, Darley-Usmar VM, Smith DR. Reoxygenation of the hypoxic myocardium causes a mitochondrial complex I defect. Biochem Soc Trans 18: 549, 1990. [DOI] [PubMed] [Google Scholar]
- 32.Ichikawa H, Flores S, Kvietys PR, Wolf RE, Yoshikawa T, Granger DN, Aw TY. Molecular mechanisms of anoxia/reoxygenation-induced neutrophil adherence to cultured endothelial cells. Circ Res 81: 922–931, 1997. [DOI] [PubMed] [Google Scholar]
- 33.Ichikawa H, Kokura S, Aw TY. Role of endothelial mitochondria in oxidant production and modulation of neutrophil adherence. J Vasc Res 41: 432–444, 2004. [DOI] [PubMed] [Google Scholar]
- 34.Ilangovan G, Manivannan A, Li H, Yanagi H, Zweier JL, Kuppusamy P. A naphthalocyanine-based EPR probe for localized measurements of tissue oxygenation. Free Radic Biol Med 32: 139–147, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Ilangovan G, Osinbowale S, Bratasz A, Bonar M, Cardounel AJ, Zweier JL, Kuppusamy P. Heat shock regulates the respiration of cardiac H9c2 cells through upregulation of nitric oxide synthase. Am J Physiol Cell Physiol 287: C1472–C1481, 2004. [DOI] [PubMed] [Google Scholar]
- 36.Ilangovan G, Zweier JL, Kuppusamy P. Microximetry: simultaneous determination of oxygen consumption and free radical production using electron paramagnetic resonance spectroscopy. Methods Enzymol 381: 747–762, 2004. [DOI] [PubMed] [Google Scholar]
- 37.Jones CI 3rd Zhu H, Martin SF, Han Z, Li Y, Alevriadou BR. Regulation of antioxidants and phase 2 enzymes by shear-induced reactive oxygen species in endothelial cells. Ann Biomed Eng 35: 683–693, 2007. [DOI] [PubMed] [Google Scholar]
- 38.Kissner R, Nauser T, Bugnon P, Lye PG, Koppenol WH. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem Res Toxicol 10: 1285–1292, 1997. [DOI] [PubMed] [Google Scholar]
- 39.Klug D, Rabani J, Fridovich I. A direct demonstration of the catalytic action of superoxide dismutase through the use of pulse radiolysis. J Biol Chem 247: 4839–4842, 1972. [PubMed] [Google Scholar]
- 40.Koppenol WH, Moreno JJ, Pryor WA, Ischiropoulos H, Beckman JS. Peroxynitrite: a cloaked oxidant from superoxide and nitric oxide. Chem Res Toxicol 5: 834–842, 1992. [DOI] [PubMed] [Google Scholar]
- 41.Kuchan MJ, Frangos JA. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am J Physiol Cell Physiol 266: C628–C636, 1994. [DOI] [PubMed] [Google Scholar]
- 42.Lenaz G Role of mitochondria in oxidative stress and ageing. Biochim Biophys Acta 1366: 53–67, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Lewis RS, Deen WM. Kinetics of the reaction of nitric oxide with oxygen in aqueous solutions. Chem Res Toxicol 7: 567–574, 1994. [DOI] [PubMed] [Google Scholar]
- 44.Liao JK, Zulueta JJ, Yu FS, Peng HB, Cote CG, Hassoun PM. Regulation of bovine endothelial constitutive nitric oxide synthase by oxygen. J Clin Invest 96: 2661–2666, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin K, Hsu PP, Chen BP, Yuan S, Usami S, Shyy JY, Li YS, Chien S. Molecular mechanism of endothelial growth arrest by laminar shear stress. Proc Natl Acad Sci USA 97: 9385–9389, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res 93: 573–580, 2003. [DOI] [PubMed] [Google Scholar]
- 47.Macmillan-Crow LA, Cruthirds DL. Invited review: manganese superoxide dismutase in disease. Free Radic Res 34: 325–336, 2001. [DOI] [PubMed] [Google Scholar]
- 48.Martin SF, Chatterjee S, Parinandi N, Alevriadou BR. Rac1 inhibition protects against hypoxia/reoxygenation-induced lipid peroxidation in human vascular endothelial cells. Vascul Pharmacol 43: 148–156, 2005. [DOI] [PubMed] [Google Scholar]
- 49.Massoudy P, Mempel T, Raschke P, Becker BF. Reduction of oxygen delivery during post-ischemic reperfusion protects the isolated guinea pig heart. Basic Res Cardiol 94: 231–237, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Mount PF, Kemp BE, Power DA. Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J Mol Cell Cardiol 42: 271–279, 2007. [DOI] [PubMed] [Google Scholar]
- 51.Muller FL, Liu Y, Van Remmen H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: 49064–49073, 2004. [DOI] [PubMed] [Google Scholar]
- 52.Ng CK, Deshpande SS, Irani K, Alevriadou BR. Adhesion of flowing monocytes to hypoxia-reoxygenation-exposed endothelial cells: role of Rac1, ROS, and VCAM-1. Am J Physiol Cell Physiol 283: C93–C102, 2002. [DOI] [PubMed] [Google Scholar]
- 53.Pandian RP, Parinandi NL, Ilangovan G, Zweier JL, Kuppusamy P. Novel particulate spin probe for targeted determination of oxygen in cells and tissues. Free Radic Biol Med 35: 1138–1148, 2003. [DOI] [PubMed] [Google Scholar]
- 54.Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94: 53–59, 2004. [DOI] [PubMed] [Google Scholar]
- 55.Paxinou E, Weisse M, Chen Q, Souza JM, Hertkorn C, Selak M, Daikhin E, Yudkoff M, Sowa G, Sessa WC, Ischiropoulos H. Dynamic regulation of metabolism and respiration by endogenously produced nitric oxide protects against oxidative stress. Proc Natl Acad Sci USA 98: 11575–11580, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Petrosillo G, Di Venosa N, Ruggiero FM, Pistolese M, D'Agostino D, Tiravanti E, Fiore T, Paradies G. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen-free radicals and cardiolipin. Biochim Biophys Acta 1710: 78–86, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Poderoso JJ, Carreras MC, Lisdero C, Riobo N, Schopfer F, Boveris A. Nitric oxide inhibits electron transfer and increases superoxide radical production in rat heart mitochondria and submitochondrial particles. Arch Biochem Biophys 328: 85–92, 1996. [DOI] [PubMed] [Google Scholar]
- 58.Poderoso JJ, Lisdero C, Schopfer F, Riobo N, Carreras MC, Cadenas E, Boveris A. The regulation of mitochondrial oxygen uptake by redox reactions involving nitric oxide and ubiquinol. J Biol Chem 274: 37709–37716, 1999. [DOI] [PubMed] [Google Scholar]
- 59.Poderoso JJ, Peralta JG, Lisdero CL, Carreras MC, Radisic M, Schopfer F, Cadenas E, Boveris A. Nitric oxide regulates oxygen uptake and hydrogen peroxide release by the isolated beating rat heart. Am J Physiol Cell Physiol 274: C112–C119, 1998. [DOI] [PubMed] [Google Scholar]
- 60.Presley T, Kuppusamy P, Zweier JL, Ilangovan G. Electron paramagnetic resonance oximetry as a quantitative method to measure cellular respiration: a consideration of oxygen diffusion interference. Biophys J 91: 4623–4631, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quijano C, Castro L, Peluffo G, Valez V, Radi R. Enhanced mitochondrial superoxide in hyperglycemic endothelial cells: direct measurements and formation of hydrogen peroxide and peroxynitrite. Am J Physiol Heart Circ Physiol 293: H3404–H3414, 2007. [DOI] [PubMed] [Google Scholar]
- 62.Quijano C, Romero N, Radi R. Tyrosine nitration by superoxide and nitric oxide fluxes in biological systems: modeling the impact of superoxide dismutase and nitric oxide diffusion. Free Radic Biol Med 39: 728–741, 2005. [DOI] [PubMed] [Google Scholar]
- 63.Radi R, Cassina A, Hodara R, Quijano C, Castro L. Peroxynitrite reactions and formation in mitochondria. Free Radic Biol Med 33: 1451–1464, 2002. [DOI] [PubMed] [Google Scholar]
- 64.Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch Biochem Biophys 308: 89–95, 1994. [DOI] [PubMed] [Google Scholar]
- 65.Riobo NA, Clementi E, Melani M, Boveris A, Cadenas E, Moncada S, Poderoso JJ. Nitric oxide inhibits mitochondrial NADH:ubiquinone reductase activity through peroxynitrite formation. Biochem J 359: 139–145, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA 103: 15038–15043, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sanders SP, Zweier JL, Kuppusamy P, Harrison SJ, Bassett DJ, Gabrielson EW, Sylvester JT. Hyperoxic sheep pulmonary microvascular endothelial cells generate free radicals via mitochondrial electron transport. J Clin Invest 91: 46–52, 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen W, Xu X, Ochoa M, Zhao G, Wolin MS, Hintze TH. Role of nitric oxide in the regulation of oxygen consumption in conscious dogs. Circ Res 75: 1086–1095, 1994. [DOI] [PubMed] [Google Scholar]
- 69.Squadrito GL, Cueto R, Splenser AE, Valavanidis A, Zhang H, Uppu RM, Pryor WA. Reaction of uric acid with peroxynitrite and implications for the mechanism of neuroprotection by uric acid. Arch Biochem Biophys 376: 333–337, 2000. [DOI] [PubMed] [Google Scholar]
- 70.Su Y, Block ER. Role of calpain in hypoxic inhibition of nitric oxide synthase activity in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol 278: L1204–L1212, 2000. [DOI] [PubMed] [Google Scholar]
- 71.Therade-Matharan S, Laemmel E, Duranteau J, Vicaut E. Reoxygenation after hypoxia and glucose depletion causes reactive oxygen species production by mitochondria in HUVEC. Am J Physiol Regul Integr Comp Physiol 287: R1037–R1043, 2004. [DOI] [PubMed] [Google Scholar]
- 72.Tsai AG, Johnson PC, Intaglietta M. Oxygen gradients in the microcirculation. Physiol Rev 83: 933–963, 2003. [DOI] [PubMed] [Google Scholar]
- 73.Turrens JF Mitochondrial formation of reactive oxygen species. J Physiol 552: 335–344, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Turrens JF, Freeman BA, Levitt JG, Crapo JD. The effect of hyperoxia on superoxide production by lung submitochondrial particles. Arch Biochem Biophys 217: 401–410, 1982. [DOI] [PubMed] [Google Scholar]
- 75.Villa LM, Salas E, Darley-Usmar VM, Radomski MW, Moncada S. Peroxynitrite induces both vasodilatation and impaired vascular relaxation in the isolated perfused rat heart. Proc Natl Acad Sci USA 91: 12383–12387, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang P, Zweier JL. Measurement of nitric oxide and peroxynitrite generation in the postischemic heart. J Biol Chem 271: 29223–29230, 1996. [DOI] [PubMed] [Google Scholar]
- 77.Wang W, Diamond SL. Does elevated nitric oxide production enhance the release of prostacyclin from shear stressed aortic endothelial cells? Biochem Biophys Res Commun 233: 748–751, 1997. [DOI] [PubMed] [Google Scholar]
- 78.Whorton AR, Simonds DB, Piantadosi CA. Regulation of nitric oxide synthesis by oxygen in vascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 272: L1161–L1166, 1997. [DOI] [PubMed] [Google Scholar]
- 79.Xie YW, Wolin MS. Role of nitric oxide and its interaction with superoxide in the suppression of cardiac muscle mitochondrial respiration. Involvement in response to hypoxia/reoxygenation. Circulation 94: 2580–2586, 1996. [DOI] [PubMed] [Google Scholar]
- 80.Yeh LH, Kinsey AM, Chatterjee S, Alevriadou BR. Lactosylceramide mediates shear-induced endothelial superoxide production and intercellular adhesion molecule-1 expression. J Vasc Res 38: 551–559, 2001. [DOI] [PubMed] [Google Scholar]
- 81.Yeh LH, Park YJ, Hansalia RJ, Ahmed IS, Deshpande SS, Goldschmidt-Clermont PJ, Irani K, Alevriadou BR. Shear-induced tyrosine phosphorylation in endothelial cells requires Rac1-dependent production of ROS. Am J Physiol Cell Physiol 276: C838–C847, 1999. [DOI] [PubMed] [Google Scholar]
- 82.Zhang DX, Gutterman DD. Mitochondrial reactive oxygen species-mediated signaling in endothelial cells. Am J Physiol Heart Circ Physiol 292: H2023–H2031, 2007. [DOI] [PubMed] [Google Scholar]
- 83.Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA 102: 5727–5732, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao X, He G, Chen YR, Pandian RP, Kuppusamy P, Zweier JL. Endothelium-derived nitric oxide regulates postischemic myocardial oxygenation and oxygen consumption by modulation of mitochondrial electron transport. Circulation 111: 2966–2972, 2005. [DOI] [PubMed] [Google Scholar]
- 85.Zmijewski JW, Moellering DR, Le Goffe C, Landar A, Ramachandran A, Darley-Usmar VM. Oxidized LDL induces mitochondrially associated reactive oxygen/nitrogen species formation in endothelial cells. Am J Physiol Heart Circ Physiol 289: H852–H861, 2005. [DOI] [PubMed] [Google Scholar]
- 86.Zwacka RM, Zhou W, Zhang Y, Darby CJ, Dudus L, Halldorson J, Oberley L, Engelhardt JF. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat Med 4: 698–704, 1998. [DOI] [PubMed] [Google Scholar]