

Abstract
int-6 is one of the frequent integration sites for mouse mammary tumor viruses. Although its product is the e-subunit of translation initiation factor eIF3, other evidence indicates that it interacts with proteasomes or other proteins to regulate protein stability. Here we report that the fission yeast int6+ is required for overcoming stress imposed by histidine starvation, using the drug 3-aminotriazole (3AT). Microarray and complementary Northern studies using wild-type, int6Δ or gcn2Δ mutants indicate that 3AT-treated wild-type yeast induces core environmental stress response (CESR) genes in addition to typical general amino acid control (GAAC) genes whose transcription depends on the eIF2 kinase, Gcn2. In agreement with this, Sty1 MAPK and its target transcription factor Atf1, which signal the CESR, are required for overcoming 3AT-induced starvation. We find that Int6 is required for maintaining the basal level of Atf1 and for rapid transcriptional activation of the CESR on 3AT-insult. Pulse labeling experiments indicate that int6Δ significantly slows down de novo protein synthesis. Moreover, Atf1 protein half-life was reduced in int6Δ cells. These effects would account for the compromised Atf1 activity on 3AT-induced stress. Thus, the robust protein synthesis promoted by intact eIF3 appears to be a part of the requisites for sound Sty1 MAPK-dependent signaling governed by the activity of the Atf1 transcription factor.
Mouse mammary tumor virus integrates into the mouse genome, altering
proteins or their expression and thereby eliciting mammary tumors. The sites
of mouse mammary tumor virus integration were identified to delineate the
molecular basis of the mammary tumorigenesis (see Refs.
1 and
2 for reviews). These so-called
int sites include members of the Wnt (Wnt-1/int-1,
Wnt-3, and Wnt-10b) and Fgf families (Fgf-3/int-2,
Fgf-4/hst, and Fgf-8/AIGF), Notch-4/int-3 and
eIF3e/int-6. Thus, all of the Int proteins except Int-6 are a well
characterized growth factor or transmembrane receptor. Int-6 is an abundant
protein and found to be identical to the e (p48)-subunit of eukaryotic
translation initiation factor-3
(eIF3),3 a
multisubunit protein (of 11–13 subunits in mammals and plants) required
for initiating protein synthesis
(3). The eIF3 directly binds to
the 40 S ribosomal subunit and mediates
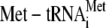 and mRNA
binding to the ribosome (4).
Int-6/eIF3e was recently determined to be a part of the functional core of
mammalian eIF3, comprising only 6 subunits
(5). Curiously, eIF3 isolated
from the budding yeast Saccharomyces cerevisiae does not contain the
homologue of Int-6/eIF3e (6,
7). Yet, eIF3 isolated from the
fission yeast Schizosaccharomyces pombe includes its homologue Int6
(also known as Yin6) (8,
9). In addition to its role in
translation, human Int-6 appears to regulate protein stability by directly
binding to HIF2α (10)
and MCM7 (11). In fission
yeast, Int6 interacts with the 26 S proteasome to promote ubiquitin-dependent
degradation of cyclin/Cdc13 and securin/Cut2
(12). However, the mechanism
of gene regulation change and ultimately, cancer formation, by int-6
gene disruption is not yet understood.
and mRNA
binding to the ribosome (4).
Int-6/eIF3e was recently determined to be a part of the functional core of
mammalian eIF3, comprising only 6 subunits
(5). Curiously, eIF3 isolated
from the budding yeast Saccharomyces cerevisiae does not contain the
homologue of Int-6/eIF3e (6,
7). Yet, eIF3 isolated from the
fission yeast Schizosaccharomyces pombe includes its homologue Int6
(also known as Yin6) (8,
9). In addition to its role in
translation, human Int-6 appears to regulate protein stability by directly
binding to HIF2α (10)
and MCM7 (11). In fission
yeast, Int6 interacts with the 26 S proteasome to promote ubiquitin-dependent
degradation of cyclin/Cdc13 and securin/Cut2
(12). However, the mechanism
of gene regulation change and ultimately, cancer formation, by int-6
gene disruption is not yet understood.
To investigate the biological function of Int6 in gene regulation, we have utilized cDNA microarray analysis and biochemical and genetic tools available in fission yeast S. pombe. In this report, we focus on stress response induced by amino acid starvation. In mammalian cells and the budding yeast S. cerevisiae subjected to amino acid depletion, phosphorylation of the α subunit of eIF2 at Ser-51 by an eIF2 kinase (eIF2K) termed GCN2 alters this translation factor, resulting in a block in global translation (for review, see Ref. 13). Coincident with reduced protein synthesis, eIF2 phosphorylation promotes preferential translation of key transcription factors by a mechanism involving alternative ribosomal re-initiation at short upstream open reading frame elements located in the 5′-leader of mRNAs. Accordingly, GCN2 activation induces translation of distinct basic leucine zipper (bZIP)-type transcription factors, mammalian ATF4, and S. cerevisiae Gcn4p, respectively, for directing stress responsive genes involved in amino acid metabolism and nutrient salvaging (the general amino acid control or GAAC response). In mammals, four eIF2Ks, including GCN2, HRI, PKR, and PERK/PEK, function to regulate translation in response to different stress arrangements (14). S. pombe encodes only GCN2 and HRI orthologues, Gcn2 and Hri1/Hri2, that are activated by amino acid starvation and oxidative stress, respectively (15, 16).
In the present study, we show that histidine starvation caused by the drug 3-aminotriazole (3AT) induces Sty1 mitogen-activated protein kinase (MAPK)-dependent signaling pathway in S. pombe, in addition to the GAAC response as typically induced in S. cerevisiae. Moreover, we show that Int6 stimulates the Sty1-dependent response by enhancing Atf1 expression in response to histidine starvation. The Sty1 MAPK (also known as Spc1 and Phh1) transduces the stress-activated signaling pathway in response to a number of conditions including oxidative stress, osmotic stress, and DNA damaging conditions (17, 18). The mechanism of Sty1 activation is best studied in the H2O2-induced oxidative stress response, which involves two different pathways. In the first pathway, three related membrane-spanning histidine kinases, Mak1, Mak2, and Mak3, are believed to directly sense the stress condition, transducing the signal to the Wis4 and Win1 MAPK kinase kinases (MAPKKK), the Wis1 MAPK kinase (MAPKK), and then to Sty1 MAPK (19). In the second pathway, the oxidized cysteine of Sty1 makes a direct disulfide linkage with a cysteine residue of Tpx1, the sole 2-Cys peroxiredoxin; this interaction is additionally required for full Sty1 activation (20). The active Sty1 then moves to the nucleus, and phosphorylates Atf1 therein (17), resulting in increased Atf1 stability and transcriptional activity (21).
Activated Sty1 is also recruited to the chromosomes, in a manner dependent on Atf1 but independent of its phosphorylation status (22). Together, Sty1-mediated signaling induces transcription of Atf1-dependent genes, termed the core environmental stress response (CESR) (18). Yet, in the presence of H2O2, the peroxidase activity of Tpx1 catalyzes a disulfide linkage formation within a second bZIP transcription factor Pap1, activating transcription of an overlapping but distinct set of genes compared with the CESR (23). It is proposed that a high dose of H2O2 activates Sty1 and hence Atf1, whereas a low dose of H2O2 activates Pap1, but not Sty1 or Atf1 (24). In this way, fission yeast can express similar but distinct transcriptional profiles appropriate for the level of H2O2 exposure.
By studying the genome-wide effect of Int6 on the 3AT-induced starvation response, we not only link the Sty1 MAPK pathway to the metabolic stress that is well characterized in S. cerevisiae, but also suggest that the robust translation initiation promoted by intact eIF3 is one of the requisites for sound MAPK-depending signaling governed by the activity of the Atf1 transcription factor. Thus, the present study provides an important insight into the mechanism of gene regulation changes caused by the compromised activity of Int6/eIF3e-directed translation.
MATERIALS AND METHODS
Yeast S. pombe Strains—S. pombe strains used in this study are listed in Table 1. Strains WY764, KAY252, KAY406, KAY309, KAY311, KAY296, KAY329, KAY415, KAY609, and KAY665 were all derived from SP223 (h- ade6-M216 leu1–32 ura4-D18). All the strains used in this study to test the ability to induce the amino acid starvation response are Leu+ (see below).
TABLE 1.
Strains used in this study
| Strains | Genotype (reference if constructed previously) |
|---|---|
| WY764 | h– ade6-M216 leu1–32::leu1+ ura4-D18::ura4+ (15) |
| KAY252 | h– ade6-M216 leu1–32::leu1+ ura4-D18::ura4+ int6Δ::kanMX |
| KAY406 | h– ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 gcn2Δ::ura4+ |
| KAY309 | h– ade6-M216 leu1–32::leu1+ ura4-D18 hri1Δ::ura4+ |
| KAY311 | h– ade6-M216 leu1–32 ura4-D18::ura4+ hri2Δ::leu1+ |
| KAY296 | h– ade6-M216 leu1–32 ura4-D18 hri1::ura4+ hri2Δ::leu1+ gcn2Δ::ura4+ |
| KAY329 | h– ade6-M216 leu1–32 ura4-D18 hri1Δ::ura4+ hri2Δ::leu1+ gcn2Δ::ura4+ int6Δ::kanMX |
| KAY415 | h– ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 gcn2Δ::ura4+ int6Δ::kanMX |
| KAY609 | h– ade6-M216 leu1–32 ura4-D18 hri1Δ::ura4+ hri2Δ::leu1+ int6Δ::kanMX |
| KAY665 | h– ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 pap1Δ::ura4+ |
| K340 | h– ade1–40 (31) |
| TY34 | h– ade1–40 gcn5Δ::kanMX (31) |
| KAY456 | h90 ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18::ura4+ |
| KAY508 | h90 ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18::ura4+ int6::kanMX |
| KAY464 | h90 ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 atf1::ura4+ |
| KAY484 | h90 ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 pcr1::ura4+ |
| KAY584 | h90 ade6-M216 or 210 leu1–32::leu1+ 〈pJK148〉 ura4-D18 atf1::ura4+ pcr1::ura4+ |
| KAY569 | h90 ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 atf1::ura4+ int6Δ::kanMX |
| KAY606 | h90 ade6-M216 leu1–32::leu1+ 〈pJK148〉 ura4-D18 pcr1::ura4+ int6Δ::kanMX |
| KAY672 | h? ade6-M216 or 210 leu1–32::leu1+ 〈pJK148〉 ura4-D18 atf1::ura4+ pap1::ura4+ |
| KAY641 | h– leu1–32::leu1+ 〈pJK148〉 ura4-D18::ura4+ |
| KAY647 | h– leu1–32::leu1+ 〈pJK148〉 ura4-D18::ura4+ int6Δ::kanMX |
| KAY640 | h– leu1–32::leu1+ 〈pJK148〉 ura4-D18 sty1Δ::ura4+ |
| KAY645 | h– leu1–32::leu1+ 〈pJK148〉 ura4-D18 sty1Δ::ura4+ int6Δ::kanMX |
| KAY655 | h– sty1-His6HA::ura4+ |
| KAY658 | h– sty1-His6HA::ura4+ int6Δ::kanMX |
To construct KAY252, the leu1–32 locus of SP223 was converted to leu1+ by transformation with pJK148 (25) that had been linearized by NdeI digestion. The ura4-D18 locus of the resulting strain was altered to ura4+ by transformation with DNA amplified by PCR using oligos 5′-GTGTGTACTTTGAAAGTCTAGCTTTACAGC-3′ and 5′-GACAAAACAGCTTGTATAGATTTTAATATG-3′ and total DNA obtained from a ura4+ strain as template. Finally, int6+ was deleted by transformation with the int6Δ::kanMX module generated by PCR as described previously (8).
To generate KAY406, the leu1–32 locus of SP223 was converted to leu1+ with pJK148 as described above, and then the gcn2+ locus was deleted by transformation with gcn2Δ::ura4+, PCR-amplified using oligos 5′-CAAAGAGTAAAATTAGAAAATAAAC-3′ and 5′-CATTTGCCTTTACCCGCTGATAATG-3′, and total DNA from WY766 (15) as template. KAY309, KAY311, and KAY296 were created by crossing WY764 and its isogenic derivative WY766 (15). The resulting diploid strains were allowed to sporulate to produce haploid progenies, which were then used to screen for strains with the desired genotypes. hri1 and hri2 deletions were confirmed by PCR as described (15). KAY329, KAY415, and KAY609 were constructed by transformation of KAY296, KAY406, and WY459 (15), respectively, with the int6::kanMX module, as described (8).
To construct KAY665, SP223 was transformed with the pap1::ura4+ disruption construct that was amplified by PCR using oligos 5′-CATTGCTTTGTGAATTACATT-3′ and 5′-CGATGAGGGGTTGTCTTTGT-3′ and DNA of TP108-03 (h- leu1 ura4 pap1Δ::ura4+, a gift of C. Shimoda) as template, and then the leu1–32 locus was converted into leu1+ using pJK148, as described above.
KAY456, KAY508, KAY464, KAY484, KAY584, KAY569, and KAY606 were all derived from isogenic strains JY878 (h90 ade6-M216 leu1–32 ura4-D18), JX303 (h90 ade6-M216 leu1–32 ura4-D18 atf1Δ::ura4+), and JX25 (h90 ade6-M216 leu1–32 ura4-D18 pcr1Δ::ura4+), hence isogenic to each other. The three parental strains are kind gifts from M. Yamamoto. KAY456 was constructed by converting the leu1–32 and ura4-D18 loci of JY878 into leu1+ and ura4+, respectively, as described above. KAY508 was prepared by introducing int6Δ::kanMX into KAY456, as described above. Strains KAY464 and KAY484 were constructed by converting the leu1–32 locus of JX303 and JX25, respectively, into leu1+, as described above. KAY569 was constructed by transforming KAY464 with the int6Δ::kanMX module DNA. To create KAY584, we first constructed KAY485 (h90 ade6-M210 leu1–32::leu1+ 〈pJK148〉 ura4-D18 pcr1Δ::ura4+) by converting the leu1–32 locus of JX26 (h90 ade6-M210 leu1–32 ura4-D18 pcr1Δ::ura4+; isogenic to JX25 and a gift of M. Yamamoto) into leu1+ using pJK148, as described above. Then a diploid strain from crossing KAY485 and KAY464 was allowed to sporulate, producing haploid progenies. A clone, later named KAY584, had the desired genotype with pcr1Δ and atf1Δ markers, which were confirmed by PCR.
To create KAY606, initial attempts to introduce int6Δ::kanMX into KAY484 by transformation were unsuccessful. Thus a diploid strain from the cross of KAY508 and KAY485 was allowed to sporulate and a haploid strain with the desired genotypes was selected and named KAY606. KAY672 is a haploid progeny from a cross between KAY465 (h90 ade6-M210 leu1–32 ura4-D18 atf1Δ::ura4+) and KAY665.
KAY641, KAY647, KAY640, and KAY645 were all derived from isogenic strains PR109 (h- leu1–32 ura4-D18) and MR1366 (h- leu1–32 ura4-D18 spc1::ura4+) (26), kindly provided by P. Russell. KAY641 was prepared by converting the leu1–32 and ura4-D18 loci of PR109, as described above. Then KAY647 was created by introducing int6Δ::kanMX as described above. KAY640 was prepared by converting MR1366 to leu+ as described above. Then KAY645 was made by introducing int6Δ::kanMX into KAY640, as described above. KAY655 was a haploid progeny from a cross between a sty1-His6HA strain and a leu1+ wild-type strain. The int6+ gene of KAY655 was deleted as mentioned above to generate KAY658.
Yeast Media and Growth Conditions—Cells were grown at 30 °C in YES media containing 225 mg/liter of adenine, leucine, histidine, lysine, and uracil or EMM media containing the appropriate auxotrophic requirements. For amino acid starvation, EMM media were supplemented with 100 mg/liters of para-amino benzoic acid and inositol, 225 mg/liter of all 20 amino acids lacking histidine and leucine, adenine, and uracil, and 1125 mg/liter of leucine (EMM-C), with or without 10 mm 3AT.
Microarray Analysis—Yeast strains were cultured in liquid EMM-C at 30 °C at an early exponential growth, and treated with 30 mm 3AT. To avoid perturbation from stress response induced by higher cell density,4 we maintained the culture density at A600 ≤ 0.5 by diluting the culture when harvesting cells at 1-h post 3AT treatment if necessary. 50 ml of cells were harvested before or 1 and 3 h after the 3AT treatment and then total RNA was isolated using a hot-phenol protocol (27). We used DNA microarrays carrying all known and predicted S. pombe genes. 20 μg of total RNA was labeled by either Cy3-dCTP (reference RNA: wild-type time 0 RNA) or Cy5-dCTP (sample RNAs). Hybridization and initial data analysis were performed as previously described (27). The normalized data were analyzed with GeneSpring software (Agilent). Data will be submitted to ArrayExpress. All processed data are available online.
Northern Blotting—30 μg of total RNAs, which were extracted as described above, were separated by MOPS-formaldehyde gel electrophoresis, transferred to Hybond-XL membrane (Amersham Biosciences) by conventional capillary blotting, and fixed on the membrane with UV. Probes were prepared by PCR from genomic DNA using the gene-specific primers, listed in Table 2, with radiolabeled nucleotide. The RNAs transferred to the membrane were hybridized with the probe at 65 °C for 20 h. The membrane was then washed, exposed to imaging plate for an appropriate time, and analyzed by Cyclone™ Storage Phosphor System (Packard).
TABLE 2.
List of oligodeoxynucleotides used to prepare radiolabeled probes for Northern blotting in this study The following primers were designed by the Bähler group in their microarray studies (27).
| Name | Nucleotide sequences (5′ to 3′) | Probe | |
|---|---|---|---|
| 1 | 53-F9_atf1 mts1 sss1 gad7_F | ACCCCTACTGGAGCTGGATT | atf1 |
| 2 | 53-F9_atf1 mts1 sss1 gad7_R | TACCTGTAACAGCTTGGGGG | atf1 |
| 3 | 27-H10_pcr1 mts2_F | CGCTTCTAAATTTCGCCAGA | pcr1 |
| 4 | 27-H10_pcr1 mts2_R | GGTGTTGATTTGGAGGGAGA | pcr1 |
| 5 | 33-G9_gpd1_F | GGTGTAGTTGGCTCCGGTAA | gpd1 |
| 6 | 33-G9_gpd1_R | TCTCACAGAATTGCTCACGG | gpd1 |
| 7 | 12-F1_cta1_F | TTCGTGATCCCGCTAAATTC | cta1 |
| 8 | 12-F1_cta1_R | AAGGTAAAGCGTCCAACCCT | cta1 |
Immunoblotting—Yeast strains were cultured, treated with 100 μg/ml cycloheximide, if necessary, and collected as described in the microarray section except that 5 ml of culture was collected at each time point. 30–50 μl of wet cells were collected, washed, and resuspended in 50 μl (one or more wet cell volume) of modified buffer K containing 20 mm Tris-HCl, pH 7.5, 100 mm KCl, 5 mm MgCl2, 0.1% Triton X-100, 0.05% SDS, 7 mm β-mercaptoethanol, 5 mm sodium fluoride, 0.2 mm sodium vanadate, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml each of aprotinin, leupeptin, and pepstatin, and Complete™ protease inhibitors (Roche). Resuspended cells were lysed by vortex with 0.5-mm glass beads in 4 cycles of 30-s spin and 30-s cooling. The supernatant was collected by centrifugation at 14,000 × g at 4 °C and employed as WCE. The protein concentration of the WCE was determined using the Protein assay kit I (Bio-Rad). WCEs were resolved by 8 or 15% SDS-PAGE, and transferred to Hybond ECL nitrocellulose membrane (Amersham Biosciences) by electroblotting. The immobilized samples were incubated in phosphate-buffered saline/Tween containing 5% nonfat dry milk, followed by immunoblotting using the ECL chemiluminescent system (Amersham Biosciences).
Pulse Labeling Experiments—Yeast were grown to A600 ∼ 1 in 50 ml of EMM + adenine with a constant aeration at 30 °C and then supplemented with 30 mm 3AT. At appropriate times, 5 ml of the culture was transferred to a 50-ml conical tube, to which 300 μCi of [35S]methionine (PerkinElmer Life Sciences, 1175 Ci/mmol) was added. After 20 min, 1 mm non-radiolabeled methionine was added to the culture, and cells were harvested immediately for WCE preparation as described under “Immunoblotting.” WCE samples containing 10 μg of total protein were resolved by SDS-PAGE, followed by Coomassie staining. The dried gel was analyzed with the STORM PhosphorImager.
RESULTS
Gcn2 and Int6 Are Required for Histidine Starvation Response Induced by 3AT—Previous studies by others implicated eIF3i/Sum1 and eIF3e/Int6 in stress-activated signaling by pathways not yet identified (28, 29). In addition, translational control induced by amino acid starvation has been well characterized using yeast S. cerevisiae as a model system. Thus, we studied the effect of int6+ deletion (int6Δ) on the amino acid starvation response in fission yeast. A specific measure of the activity of S. cerevisiae Gcn2p eIF2 kinase and its Gcn2-dependent GAAC response is resistance to growth on 3AT. 3AT is a competitive inhibitor of histidine biosynthesis, and loss of GCN2 prevents induced expression of amino acid biosynthetic genes required for growth in the presence of this drug. To test if Int6 is required for the histidine starvation response, we examined 3AT sensitivity of the int6Δ mutant in leu+ backgrounds. The plate assay showed that the int6 deletion confers sensitivity of S. pombe to 3AT (Fig. 1A, middle panel, row 2), a phenotype analogous to the Gcn- (general control non-derepressible) phenotype in S. cerevisiae. We also confirmed that the deletion of gcn2 (Fig. 1A, middle panel, row 3), but not of the other S. pombe eIF2K genes hri1 or hri2 (rows 4 and 5), conferred 3AT sensitivity (15). Combining gcn2Δ with hri1Δ hri2Δ did not enhance 3AT sensitivity compared with gcn2Δ alone (rows 3 and 6), indicating that among the three eIF2Ks Gcn2 is solely responsible for the starvation response. In S. cerevisiae, the histone acetyltransferase Gcn5 promotes different gene transcription programs, including the GAAC (30). Deletion of its S. pombe orthologue gcn5 (31) resulted in 3AT sensitivity (Fig. 1A, middle panel, rows 10 and 11). These results implicate Int6, Gcn2, and Gcn5 in the histidine starvation response. 3AT sensitivity displayed by the mutants was relaxed by addition of histidine in the medium (Fig. 1A, right panel), confirming that 3AT imposes histidine starvation to mutant yeasts.
FIGURE 1.

Int6, Gcn2, and Gcn5 are positive regulators of 3AT-induced starvation response in fission yeast. A, S. pombe strains WY764 (row 1), KAY252 (row 2), KAY406 (row 3), KAY309 (row 4), KAY311 (row 5), KAY296 (row 6), KAY329 (row 7), KAY415 (row 8), KAY609 (row 9), K340 (row 10), TY34 (row 11) containing the indicated mutations were grown overnight in rich medium (YES) and diluted to A600 = 0.15. 5 μl of each sample and its 10-fold serial dilutions were spotted onto EMM-C agar medium with supplements indicated to the top, and incubated for 4 days (left and right panels) or 7 days (middle panel) at 30 °C. The supplements were none (left panel), 10 mm 3AT (middle panel), and 10 mm 3AT and 10 mm histidine (right panel). B, eIF2 is phosphorylated in response to 3AT-induced stress. WY764 (WT), KAY252 (int6Δ), KAY406 (gcn2Δ), and KAY296 (gcn2Δ hri1Δ hri2Δ) cells were grown in EMM-C to an early exponential phase and then 30 mm 3AT was added. Cells were harvested for WCE preparation followed by immunoblotting with anti-phospho-eIF2α (top panel) or total eIF2α (bottom panel) antibodies, as described (16). The graph to the right shows relative levels of phospho-eIF2, normalized for total eIF2 levels in wild-type (black symbols) or gcn2Δ (gray symbols) cells. Bars indicate S.D. (n = 2). C, histidine reverses 3AT-induced eIF2 phosphorylation. Experiments in panel B were repeated with wild-type yeast (WY764) grown in the presence (lanes 5–7, gray symbols in the graph) or absence (lanes 2–4, black symbols in the graph) of histidine (1 mm) added to the medium together with 30 mm 3AT. The graph to the right shows relative levels of phospho-eIF2, normalized for total eIF2 levels with S.D. indicated by bars (n = 2).
Genome-wide Expression Profiles of Fission Yeast in Response to 3AT-induced Histidine Starvation—Next, we wished to address the relationship between Int6/eIF3e and the components of the Gcn2 eIF2K pathway. As shown in Fig. 1A, combining int6Δ with gcn2Δ or hri1Δ hri2Δ marginally increased 3AT sensitivity of the parental int6Δ strain (middle panel, rows 2–3 and 7–9), suggesting that Int6 and eIF2Ks are involved in parallel pathways. However, the phenotypic difference is minor and its interpretation awaited further examination such as gene transcription profiling.
Accordingly, we performed transcriptional profiling using cDNA microarray to compare mRNA levels among wild-type, int6Δ, gcn2Δ, and triple eIF2K knock-out strains. These strains were cultured to an early log phase and then 30 mm 3AT was added to elicit histidine starvation. To determine the time of 3AT treatment for microarray experiments, we first examined the timing of eIF2 phosphorylation in response to 3AT-induced stress. As shown in Fig. 1B, eIF2 was phosphorylated in wild-type cells within 1 h after the addition of 30 mm 3AT, and a high level of phosphorylated eIF2 was sustained for an additional period of 2 h (lanes 1–5). This phosphorylation was almost eliminated by gcn2Δ (lanes 11–15), although a modest amount of eIF2 phosphorylation was detected in the Gcn2-deficient cells at 1 h, which was dependent on Hri1 or Hri2 (compare lanes 13 and 18). Likewise, the addition of 1 mm histidine to the 3AT medium eliminated eIF2 phosphorylation for the initial period of 3 h (Fig. 1C, compare lanes 2–3 versus 5–6). These results together suggest that histidine starvation resulted in Gcn2 activation. Following 5 h of 3AT treatment, we did detect eIF2 phosphorylation even with the addition of histidine (Fig. 1C, lane 7). This suggests that, in S. pombe, longer periods of exposure to 3AT can trigger stress arrangements that induce eIF2 phosphorylation, extending beyond histidine starvation. Importantly, the finding of similar (or slightly stronger) eIF2α phosphorylation observed in int6Δ cells during 3AT-induced stress (Fig. 1B, lanes 6–10) excludes the model that Int6 activates Gcn2, and rather supports the model that Gcn2 and Int6 regulate parallel pathways.
Based on these results, we harvested cultures after 1 and 3 h of 3AT treatment to monitor their immediate and longer term effects on genome-wide transcription, respectively, as described under “Materials and Methods.” Fig. 2A indicates the expression profiles of genes induced (red) or repressed (green) by ≥2-fold after both 1 and 3 h of the 3AT treatment in wild-type cells. These genes are clustered using a hierarchical algorithm, using the datasets obtained with int6Δ, gcn2Δ, and the triple eIF2K deletion mutants (see below).
FIGURE 2.

Microarray studies. A, strains used in Fig. 1B were cultured in EMM-C and treated with 30 mm 3AT for the indicated times, followed by RNA extraction and cDNA microarray analysis (27). Shown is the clustering of genes whose transcripts were changed ≥2-fold by 3AT-induced starvation in wild-type cells. Red/green scale bar at the top indicates the fold changes in transcription. Numbers beside the clustering pattern indicate the groups of coregulated genes. B, model for 3AT-induced histidine starvation response pathways in fission yeast. 3AT-induced histidine starvation creates at least two signals that activate the indicated signal transduction pathways. In the first pathway, histidine starvation activates Sty1 MAPK via unidentified signaling molecule(s) (denoted by “?”) by an unknown mechanism (see “Discussion”). Active Sty1 inhibits proteolysis of Atf1 (stopped bars) and also activates transcriptional function of Atf1 (a vertical arrow) (21). Atf1 and Pcr1 together activate CESR (cluster 2 in panel A). Atf1-dependent transcription of atf1+ and pcr1+ (Fig. 5, A and B) may help to enhance CESR transcription (curved arrows). Here we suggest that Int6/eIF3e promotes Atf1 protein translation (and stability) (an arrow). In the second pathway, histidine starvation activates Gcn2 and downstream GAAC response (cluster 3 in panel A) by the mechanism characterized for S. cerevisiae GAAC (13). “?” under Gcn2 indicates the unidentified transcription factor, which would be analogous to Gcn4p in S. cerevisiae.
Table 3 summarizes the statistics of these experiments, by presenting the number of genes whose expression responded to 3AT at different levels. Within 1 and 3 h of 3AT-induced histidine starvation, 7.2 and 10.5%, respectively, of all measured genes displayed ≥2-fold induction in wild-type cells (Table 3, third and fourth columns). Gene repression was also elicited upon histidine depletion, with 2.4 and 8.1% of all the genes showing ≥2-fold reduction following 1 or 3 h of 3-AT addition, respectively.
TABLE 3.
Summary of expression profiles after 1 and 3 hours of 3AT-induced histidine starvation
|
Type of expression
|
-Fold change
|
No. of responding genes
|
|||||||
|---|---|---|---|---|---|---|---|---|---|
|
WT
|
int6–
|
gcn2–
|
hri1– hri2–
gcn2–
|
||||||
| 1 h | 3 h | 1 h | 3 h | 1 h | 3 h | 1 h | 3 h | ||
| Induced | ≥10 | 24 | 52 | 15 | 97 | 23 | 76 | 22 | 75 |
| ≥4 | 106 | 131 | 125 | 381 | 98 | 185 | 80 | 179 | |
| ≥2 | 356 | 502 | 521 | 1056 | 406 | 593 | 377 | 472 | |
| ≥2 (% of total) | 7.2 | 10.5 | 10.4 | 21.0 | 8.0 | 11.7 | 7.5 | 9.4 | |
| <2 | 2098 | 1915 | 2011 | 1409 | 2279 | 1907 | 1976 | 1797 | |
| Repressed | ≥10 | 0 | 1 | 3 | 7 | 0 | 0 | 1 | 4 |
| ≥4 | 9 | 31 | 11 | 94 | 6 | 24 | 12 | 31 | |
| ≥2 | 121 | 385 | 244 | 872 | 345 | 333 | 169 | 221 | |
| ≥2 (% of total) | 2.4 | 8.1 | 4.9 | 17.3 | 6.8 | 6.6 | 3.3 | 4.4 | |
| <2 | 2395 | 1974 | 2236 | 1695 | 2049 | 2239 | 2535 | 2557 | |
| Total open reading frames considered | 4970 | 4776 | 5012 | 5032 | 5079 | 5072 | 5057 | 5047 | |
To compare this 3AT-induced response with that reported in S. cerevisiae (32), we looked for genes that were induced ≥2-fold after 1 and 3 h of 3AT treatment in S. pombe and showed clear homology to S. cerevisiae genes. Of 133 such S. pombe genes, 55 were also induced ≥2-fold in S. cerevisiae subjected to histidine starvation. Likewise, 18 of 62 S. pombe genes with S. cerevisiae orthologues were ≥2-fold down-regulated in both yeasts. Thus, similar sets of genes are transcribed in response to histidine starvation in fission and budding yeasts.
Genes activated ≥2-fold in both yeasts subjected to histidine limitation include 9 amino acid biosynthesis genes including C56E4.03, his4, leu3, and lys4 (aro8, his7, leu4, and lys21, respectively, in S. cerevisiae), whereas those repressed ≥2-fold include rpl14 (large ribosomal protein), pac1 (rRNA and small nuclear RNA processing), and mrpl44 (mitochondrial ribosomal protein) (rpl14, YMR239c, mrpl44, respectively, in S. cerevisiae). These results support the idea that stress in general induces amino acid synthetic enzymes and represses ribosome synthesis in fission yeast (18).
That a relatively small number of genes were similarly regulated in both yeasts may be due in part to the relatively strict threshold of ≥2-fold changes at both 1 and 3 h. As shown in Fig. 3, 3AT induces the expression of additional amino acid biosynthetic genes in S. pombe, covering ∼40% of the genes under this category (see below). Likewise, most other ribosomal proteins are repressed in response to 3AT-induced starvation albeit to a smaller degree (data not shown). A second reason for the difference between the two yeasts is that fission yeast induces a novel set of genes termed CESR during histidine starvation, in addition to a typical GAAC response dependent on Gcn2 eIF2K, as described below (also see Fig. 2B).
FIGURE 3.

The induction of amino acid synthesis genes is largely dependent on Gcn2. Relative mRNA levels from amino acid biosynthetic genes in wild-type and gcn2 mutant were taken from microarray data in this study. Levels are shown by logarithmic scale with the time course of 0 (white bar), 1 (gray bar), and 3 h (black bar) after 3AT treatment. The amino acid biosynthetic genes in S. cerevisiae were defined in (32). Amino acid enzymes were identified by homology using YOGY (48) and listed in the figure as follows: row 1, his1; 2, his5; 3, his2; 4, his7; 5, his3; 6, his6; 7, his4; 8, gln1; 9, PB1E7.07; 10, C132.04c; 11, C662.12c; 12, gst1; 13, gst2; 14, C17H9.13c; 15, pro1; 16, PYUG7.05; 17, C428.05c; 18, C725.14; 19, arg3; 20, argx; 21, arg7; 22, arg11; 23, C1271.14; 24, arg1; 25, arg5; 26, arg4; 27, map1; 28, lys3; 29, lys1; 30, lys2; 31, lys7; 32, C3B8.03; 33, C31G5.04; 34, lys4; 35, aro1; 36, C1223.14; 37, P8A3.07c; 38, C24H6.10c; 39, C16E8.04c; 40, C569.07; 41, C1773.13; 42, C56E4.03; 43, trp3; 44, trp1; 45, trp4; 46, trp2; 47, C30D10.16; 48, C1494.04c; 49, C364.97; 50, C3H7.07c; 51, gly1; 52, cit1; 53, C24C9.06c; 54, C1683.11c; 55, gcv1; 56, C13G6.06c; 57, P19A11.01; 58, dld1; 59, shm2; 60, C24C9.12c; 61, C725.01; 62, C10F6.13c; 63, C119.10; 64, C1827.06c; 65, C19F5.04; 66, C776.03; 67, C4C3.03; 68, thrc; 69, C3H7.02; 70, C869.05c; 71, C1739.06c; 72, C106.17c; 73, met6; 74, sua1; 75, met26; 76, C584.01c; 77, met5; 78, met14; 79, met16; 80, C428.11; 81, tol1; 82, sam1; 83, C1677.03c; 84, ilv1; 85, ilv3; 86, ilv5; 87, C14C8.04; 88, leu2; 89, leu1; 90, leu3; 91, eca39; 92, C582.08. In column a, the genes whose orthologs in S. cerevisiae are defined as Gcn4 targets are shown in gray. - indicates the gene for which insufficient data exist to assess Gcn4 dependence (32). Columns b and c are based on the present studies using S. pombe.In column b, gray indicates the genes whose expression in wild-type S. pombe cells is induced after 3AT treatment (genes whose expression in S. pombe wild type is induced more than 1.5-fold at 1 and/or 3 h after 3AT treatment). In column c, gray indicates the genes whose induction is dependent on Gcn2 (genes that are induced in wild type and whose average mRNA level at 1 and 3 h is higher in wild type than in gcn2Δ>1.4-fold).
Transcriptional Profiling of int6Δ and gcn2Δ Mutants Identifies Three Gene Clusters That Are Differentially Regulated—The transcription profiles obtained with gcn2Δ and hri1Δ hri2Δ gcn2Δ triple mutants were similar (Fig. 2A, columns 7–12), confirming that Gcn2 is the primary eIF2K required for the histidine starvation response. The hierarchical clustering analysis shown in Fig. 2A indicated that the genes regulated by histidine starvation can be divided into three clusters (see gene lists in supplemental Table S1).
Cluster 1 includes 91 genes that are repressed in both wild-type and mutant int6Δ, gcn2Δ, and hri1Δ hri2Δ gcn2Δ strains, although to a lesser degree in the three mutant strains. This cluster overlaps significantly with those genes whose expression is consistently reduced in response to diverse environmental stresses, including cadmium, heat shock, DNA damaging conditions, oxidative treatment, and osmotic stresses (repressed CESR) (18). These genes include rpl14, pac1, and mrpl44 as mentioned above.
Cluster 2 represents 169 genes whose expression was induced in wild-type cells and often delayed among the three mutants, but most notably in int6Δ (also see supplemental Fig. S1 for the profiles of 8 well characterized genes). This group overlaps significantly with the induced CESR in response to the five stress conditions mentioned above (18). Because the induced CESR depends on Sty1 and Atf1, this result raises the possibility that Int6 modulates expression of these genes as part of the Sty1 MAPK pathway. This point is the major focus of this report and will be substantiated in the experiments presented below.
Cluster 3 contains 68 genes that are induced in both wild-type and int6Δ S. pombe cells, but not in gcn2Δ and eIF2K triple knock-out mutants. Expression of these genes is clearly dependent on Gcn2 in a fashion analogous to the Gcn2-dependent GAAC response described in budding yeast (32) (Fig. 3). Earlier it was shown that Gcn2 facilitates phosphorylation of eIF2 during 3AT exposure (Fig. 1B). Thus, 3AT stress appears to trigger Gcn2- and Sty1-dependent pathways, corresponding to genes comprising clusters 3 and 2, respectively, as described schematically in Fig. 2B.
Gcn2-dependent Response Genes (Cluster 3) Include Amino Acid Synthetic Genes—The cluster 3 genes, whose 3AT-induced transcription depends on Gcn2, include each of the 9 amino acid enzymes mentioned above. Interestingly, they also include 8 transcripts encoded by the Tf2 retrotransposons (see “Discussion”). The genes of this group overlap significantly with the group of genes whose expression was increased in a minimal medium (EMM) compared with a rich medium (YE),4 and the amino acid metabolism module defined as genes coregulated in both S. cerevisiae and S. pombe (33). These findings confirm and expand the reported function of fission yeast Gcn2 in amino acid response (15). In addition, the cluster 3 genes overlap with genes induced during oxidative stress that are independent of the CESR (34). These non-Sty1-dependent genes included 6 of the 9 amino acid biosynthetic enzymes and 7 of the 8 Tf2 transcripts. These data provide experimental evidence that eIF2Ks activate a common set of genes between histidine starvation and oxidative stress responses.
Because amino acid synthetic genes are characteristic of the Gcn2-dependent cluster 3 genes as observed with the GAAC response in S. cerevisiae (32) and a similar eIF2K-dependent response in mammals (35), we examined the expression profiles of S. pombe genes in this category. Fig. 3 lists all the known or predicted S. pombe genes involved in amino acid biosynthesis, with their 3AT-induced expression levels. In S. cerevisiae, 76% of these genes are induced upon 3AT treatment in a Gcn4p-dependent manner (Fig. 3, column a) (32). In S. pombe, ∼40% of them are induced ≥1.5-fold during 3AT treatment (Fig. 3, column b) and the majority (32 of 36) are dependent on Gcn2 (Fig. 3, column c). Thus, amino acid synthesis genes are the common targets of the Gcn2-dependent pathway in S. cerevisiae, S. pombe, as well as in mammals.
However, we noted that genes involved in methionine synthesis were repressed, rather than induced as in S. cerevisiae, in response to 3AT treatment in S. pombe (Fig. 3, rows 69–82). The bZIP factor Zip1 promotes cadmium-induced expression of methionine synthesis genes via a mechanism involving inhibition of ubiquitin-dependent proteolysis of Zip1 (36). By contrast in S. cerevisiae, the homologous bZIP factor Met4p activates methionine biosynthetic genes in response to histidine starvation by a mechanism dependent on Gcn4p (32) (Fig. 3, column a). Thus, our results indicate that methionine biosynthetic genes under Zip1 control are not induced in S. pombe during histidine starvation, in contrast to those under Met4p control in S. cerevisiae.
Fission Yeast Activates Sty1-dependent Signaling Pathway during 3AT-induced Histidine Starvation—The specific induction of the Sty1 MAPK-dependent CESR during 3AT-induced starvation is a novel aspect of the fission yeast response, lacking in the budding yeast response (Fig. 2B). Thus, we examined whether the Sty1-dependent pathway is indeed active in the 3AT-induced response. First, we tested 3AT sensitivity of the yeast mutant deleted for Sty1 and its transcription factor target Atf1. Atf1 forms a heterodimer with another bZIP factor Pcr1 to activate the CESR transcription (37, 38). Thus, we also tested a pcr1Δ mutant. As shown in Fig. 4A, sty1Δ, atf1Δ, and pcr1Δ each conferred sensitivity to 3AT (rows 3, 7, and 8) in a manner reversible by histidine added to the medium (supplemental Fig. S2). These results genetically implicate the Sty1-signaling components in 3AT-induced histidine starvation response. The atf1 pcr1 double mutant showed similar growth defects as the single atf1Δ and pcr1Δ mutants in 3AT medium (Fig. 4A, rows 7–9), consistent with the idea that an Atf1/Pcr1 heterodimer has the strongest potential to activate 3AT-induced CESR. Interestingly, yeast cells deleted for both sty1+ and int6+ grew less rapidly in the 3AT medium than yeast deleted for either sty1+ or int6+ (Fig. 4A, rows 2–4). In agreement with this, the int6Δ atf1Δ and int6Δ pcr1Δ double mutants grew more slowly than the cells deleted for only int6+ in 3AT medium (Fig. 4A, rows 6–11). These observations together with the microarray analysis suggest that Int6 is required for full induction of the CESR by the Sty1/Atf1/Pcr1 pathway.
FIGURE 4.

Sty1 is activated during the 3AT-induced starvation response. A, haploid S. pombe strains KAY641 (row 1), KAY647 (row 2), KAY640 (row 3), KAY645 (row 4), KAY456 (row 5), KAY508 (row 6), KAY464 (row 7), KAY484 (row 8), KAY584 (row 9), KAY569 (row 10), and KAY606 (row 11) carrying the indicated mutations were spotted onto EMM-C medium with (+) or without (-) 10 mm 3AT as described in legend to Fig. 1A. B, KAY655 (sty1-His6HA int6+) and KAY658 (sty1–6hisHA int6Δ) were treated with 30 mm 3AT or 1 mm H2O2 for the indicated times and used to prepare WCE. Western blots of the WCE were probed using anti-phospho-p38 antibodies. The blot was then stripped and probed with anti-HA antibodies to detect total levels of Sty1 protein. The graphs at the bottom indicate the time course of Sty1 activation in wild-type (int6+) and int6Δ cells. The relative reactivities against anti-phospho p38 antibodies compared with the value at time 0 were measured by NIH Image and plotted against time. Bars indicate standard deviations (n = 6 for int6+; n = 3 for int6Δ).
We next tested whether Sty1 is activated during the 3AT-induced starvation. Activation of Sty1 can be monitored by the status of the threonine and tyrosine residues that are phosphorylated by the MAPKK Wis1. These phosphorylation sites are well conserved between yeasts and the mammalian homologue p38 (39). As shown in Fig. 4B, Sty1 activation, as measured by reactivity to anti-phopho-p38 antibody, peaked at 15 min after 3AT treatment (top gels, lanes 1–5) without altering the cellular abundance of Sty1 as examined with antibodies specific to the HA epitope tag introduced at the C terminus of Sty1 (bottom gels). As expected, we confirmed that H2O2-induced oxidative stress strongly activated Sty1 (Fig. 4B, lane 6). We also noted that Sty1 was rapidly activated in both 3AT- and H2O2-treated int6Δ cells (Fig. 4B, lanes 7, 8, and 12). However, Sty1 was activated for a longer duration in int6Δ cells (lanes 7–11) (see “Discussion”).
We next examined if Sty1 activation is accompanied by changes in levels of Atf1-dependent transcription. Northern blotting experiments in Fig. 5A, panels under WT, confirmed that the transcript levels for the known downstream targets under the CESR, pcr1+, gpd1+, cta1+ (ctt1+) and atf1+ itself peaked at 0.5 to 1 h of 3AT treatment (see quantitation in Fig. 5B, panels 1–4, black symbols). Of the CESR genes tested, gpd1+ encodes a glycerol synthesis enzyme induced by osmotic stress, and cta1+ encodes a catalase. In sty1Δ cells, 3AT addition did not induce the transcripts of these downstream target genes (Fig. 5, A, panel under sty1Δ, and B, panels 1–4, gray symbols). These results suggest that sty1+ is required for 3AT-induced, Atf1-dependent transcription.
FIGURE 5.

3AT-induced histidine starvation induces transcription of genes under the CESR in Sty1 MAPK- and Int6/eIF3e-dependent manners. A, isogenic pairs of wild-type (WT) and its sty1Δ or int6Δ derivatives, KAY641 (WT, lanes 1–5) and KAY640 (sty1Δ, lanes 6–10) and WY764 (WT, lanes 11–14) and KAY252 (int6Δ, lanes 15–18) were grown in EMM-C and collected at the indicated time after addition of 30 mm 3AT for total RNA preparation, as described exactly for microarray studies. Then equal amounts of total RNA samples were analyzed by Northern blotting using radiolabeled probes specific for the genes indicated to the left. B, the intensity of the bands of the gels shown in panel A is compared with the value for wild type at time 0 and plotted against the time after 3AT treatment. Black lines and symbols, wild-type. Gray lines and symbols, sty1Δ or int6Δ. Bars indicate standard deviations (n = 3 for panels 5–6; n = 2 for all others). C, the wild-type and sty1Δ or int6Δ isogenic strains used in panel A were grown in 20 ml of EMM-C medium and collected for WCE preparation. 5, 10, and 20 μg of total protein amounts of WCE were separated by SDS-PAGE and subjected to immunoblotting with affinity-purified anti-Atf1 (21) and α-tubulin (Sigma) antibodies. WCE from strain KAY584 (atf1Δ, Table 1) was used as a negative control. D, graphs in panel 1 show the levels of Atf1 normalized for that of tubulin in sty1Δ or int6Δ cells, compared with those in wild-type cells. Bars indicate standard deviations (n = 3 for sty1Δ, n = 5 for int6Δ). Graphs in panel 2 show the atf1 mRNA levels in sty1Δ or int6Δ cells, taken from the values in panel 1 or 5, respectively, of B, before 3AT treatment (time 0).
As shown in Fig. 5, C, lanes 1–9, and D, columns 1 and 2, deletion of sty1+ strongly diminished the steady-state abundance of the Atf1 protein during growth in the control EMM-C medium, when measured by immunoblotting with affinity purified anti-Atf1 antibodies (21). Under our gel electrophoretic conditions, we detected two prominent protein bands of 75 and 78 kDa, which migrated slower than the predicted size of Atf1 (59,711 Da), but both bands were absent in the atf1Δ strains (lanes 7–9), indicating that they represent authentic Atf1. The size of the lower band (75 kDa) matches that of the band reported previously as the unphosphorylated Atf1 (21). The 78-kDa species appeared to comigrate with the phospho-Atf1 band induced by H2O2 treatment (21).5 Currently, we do not understand the difference between these two Atf1 proteins. However, we propose that both the bands are unphosphorylated Atf1, because we observed them in sty1Δ cells (lanes 4–6). The diminished Atf1 abundance in sty1Δ cells is in agreement with the finding that loss of sty1+ decreased the atf1+ transcript level (17), and our observation that the basal transcript levels of the four CESR genes were lower in sty1Δ cells than in wild-type cells (Fig. 5, A and B, see the following values of transcript levels at time 0, compared with wild-type; 53 ± 0.3% for atf1, 42 ± 1.6% for pcr1, 42 ± 4.7% for gpd1, 27 ± 15% for cta1).
Given that 3AT activates the Sty1/Atf1 pathway, we wished to address whether amino acid depletion also stimulates the H2O2-senser transcription factor Pap1. Pap1 and Atf1 activate similar (although distinct) sets of genes under oxidative stress conditions, and cta1+ for instance, is activated by both. Thus, we focused on two well characterized Pap1-dependent genes, trr1+ (23) and tpx1+ (24), encoding a thioredoxin reductase and the 2-Cys peroxiredoxin, respectively, which are specifically regulated by Pap1, but not by Atf1 or Sty1, at very low levels of H2O2. As shown in Fig. 6A, our microarray data indicated that expression of these genes was not at all increased during the 3AT-induced stress in wild-type cells (interestingly, tpx1+ is induced specifically in gcn2Δ cells, see “Discussion”). In addition, we found that pap1+ is dispensable for growth in the presence of 3AT (Fig. 6B, row 3). Furthermore, pap1Δ did not enhance 3AT sensitivity when combined with the atf1Δ mutant, excluding a redundant role played by Pap1 (Fig. 6B, rows 2–4). Thus, Pap1 is not activated under the 3AT-induced stress conditions, nor is it required for overcoming this nutritional stress.
FIGURE 6.

Pap1 is not activated during 3AT-induced stress response or required for it. A, relative transcript levels for tpx1+ and trr1+ were taken from microarray data in Fig. 1C and plotted against time in hours after 3AT treatment. Filled diamonds, wild-type. Open squares, int6Δ cells. Filled triangles, gcn2Δ cells. B, cultures of KAY456 (wild-type, WT, row 1), KAY464 (row 2), KAY665 (row 3), and KAY672 (row 4) were spotted onto EMM-C medium with (+) or without (-) 10 mm 3AT, as described in the legend to Fig. 1A.
CESR Gene Transcription Depends on Int6/eIF3e—In int6Δ cells, Sty1 was activated by 3AT within the first 15 min as in wild-type (Fig. 4B, lanes 7–11), suggesting that Int6 controls events downstream of Sty1, rather than directly activating Sty1. Our microarray data supports the model that Int6 modulates the Sty1 stress response. Transcription of 52% of cluster 2 genes was significantly reduced or delayed in the int6Δ mutant during 3AT-induced starvation as compared with wild-type cells. Of the cluster 2 genes that require Int6, 62% are known targets of the Sty1/Atf1 pathway (18). These results support the idea that Int6 is an important element of the CESR expression.
To further test this idea, we performed Northern blotting. We again confirmed that the mRNA levels expressed from the known Sty1/Atf1-target genes were increased shortly (within 1 h) after 3AT treatment (Fig. 5, A and B, panels 5–8, black symbols, in the graph). Strikingly, the increase in the abundance of these messages was significantly delayed and the levels of induced transcripts were reduced in int6Δ cells (Fig. 5, A and B, panels 5–8, and gray symbols in the graph), confirming the microarray data. Thus, CESR gene transcription depends on Int6.
Fig. 5, C, lanes 10–18, and D, columns 3–4, indicate that deletion of int6+ lowers the steady-state Atf1 abundance before 3AT treatment, as observed with the sty1Δ mutant. Because the Atf1 mRNA level was unaltered in non-stressed int6Δ cells compared with that in wild-type (Fig. 5D, columns 7 and 8), the observed reduction in Atf1 protein abundance is due to changes at the translational and/or post-translational levels (see below). The 5-fold decrease in Atf1 abundance (Fig. 5D, columns 3 and 4) explains why the initial rate of Atf1-dependent transcription appeared to be diminished after 3AT treatment, as observed in Figs. 2A and 5, A and B, without altering the initial Sty1 activation (Fig. 4B). However, the fact that int6Δ diminished, but did not completely eliminate, CESR transcription contrasts with a strong Gcn- (3AT sensitive) phenotype observed with the mutant (Fig. 1A). These results suggest that Int6 is responsible for additional mechanism(s) beyond transcriptional regulation to assist cells to overcome growth inhibition caused by 3AT, as we address next.
Robust General Protein Synthesis and Atf1 Protein Stability Mediated by Int6/eIF3e Underlie the Rapid Atf1-dependent Response—Given the important role of Int6 in eIF3 and translation, we hypothesized that loss of Int6 reduces general protein synthesis, or Atf1-specific synthesis. Either would contribute to the reduced steady-state abundance of Atf1 (Fig. 5, C and D), which then would compromise the initial Atf1-dependent response upon 3AT-induced stress. In support of the idea that int6Δ reduces the general translation initiation rate, we previously reported that this mutation significantly decreased the polysome abundance when the cells were grown in a similar EMM medium (8). To directly measure the effect of int6Δ on the de novo protein synthesis rate, we performed in vivo pulse-labeling experiments with [35S]methionine. To maximize [35S]methionine incorporation, we used the minimal EMM + adenine medium, rather than EMM-C (supplemented with complete amino acids) that was used in the experiments described earlier (Figs. 1, 2, 3, 4, 5). However, as shown in Fig. 7A, we confirmed that int6Δ conferred sensitivity to 3AT in this medium, although its effect on yeast growth was more severe than in EMM-C, as reported previously (8). In addition, we observed that Atf1 abundance was strongly diminished by int6Δ in this medium as well (Fig. 7B).
FIGURE 7.

Int6/eIF3e promotes de novo protein synthesis. A, cultures of haploid S. pombe strains WY764 (WT, row 1) and KAY252 (int6Δ, row 2) were spotted onto EMM + adenine medium with (+) or without (-) 4 mm 3AT, as described in the legend to Fig. 1A, and incubated at 30 °C for 6 and 5 days, respectively. B, Atf1 abundance was monitored using strains in A, with affinity-purified anti-Atf1 antibodies (21), exactly as described in the legend to Fig. 5, A and B, but using EMM + adenine medium. The graph indicates relative abundances for Atf1 after correction by tubulin abundance with S.D. in lines (n = 3). C, WY764 (WT, lanes 1–4) and KAY252 (int6Δ, lanes 5–8) were cultured in the presence of 3AT for up to 3 h, as indicated. Prior to harvesting, cultures were pulse-labeled with [35S]methionine, and the total proteins in the harvested cells were analyzed by SDS-PAGE. Radiolabeled proteins were visualized by phosphorimaging (bottom panel). To ensure normalization of total proteins analyzed in the SDS-PAGE, the top panel shows Coomassie staining of this gel. Lane M, size standards. D, quantitation of radiolabeled proteins in response to 3AT treatment from panel C is presented with the value in lane 1 (no 3AT treatment) indicated as 100%. Averages from three independent experiments are shown with S.D. in lines. Column numbers corresponds to lane numbers in C.
As shown in Fig. 7C, int6Δ significantly reduced 35S radiolabel incorporation into total proteins (bottom panel, compare lanes 1 and 5), and the quantification of the total radioactivity from the gel indicated that the de novo protein synthesis rate under nonstarved conditions was reduced to 37% (±15) (n = 3) in int6Δ cells compared with wild-type (Fig. 7D). The observed reduction in general protein synthesis before 3AT treatment accounts at least partially for the low abundance of Atf1 in non-starved int6Δ cells (Figs. 4C and 7B), leading to diminished Atf1-dependent transcription during the initial phase of 3AT insult (Figs. 2A, 5A, and 5B). We also found that 3AT treatment strongly reduced the de novo protein synthesis rate both in wild-type and int6Δ cells, with the levels of translation in the int6Δ strain being undetectable after 1 h of 3AT treatment (Fig. 7D). These latter results confirm that 3AT-induced histidine starvation leads to significantly lowered general translation.
To examine posttranslational regulation by Int6, we also addressed the difference in the half-life of Atf1 in wild-type and int6Δ cells grown in the EMM-C medium. We found that the treatment of wild-type cells with cycloheximide, which inhibits translation, did not reduce Atf1 abundance to half following 3 h of cycloheximide treatment (Fig. 8A). By contrast, in int6Δ cells, Atf1 abundance was reduced to half within 3 h of cycloheximide treatment (Fig. 8, A and B). This and other experiments indicated that the Atf1 half-life in int6Δ cells was 2.6 ± 1.3 h (n = 5) in the EMM-C medium. As a control, we also measured tubulin levels, and found that this protein was stable in both wild-type and int6Δ cells within the time frame examined (Fig. 8). Thus, in addition to synthesis of the Atf1 protein, Int6 appears to contribute to maintaining the Atf1 protein stability. We do note that the reduced Atf1 half-life in int6Δ cells is still much longer than that typically reported for ubiquitin-regulated proteins, such as the bZIP transcription factor Zip1, which has a half-life of <30 min (36). This would suggest that Int6 has a primary role in Atf1 synthesis, with a secondary role in Atf1 stabilization.
FIGURE 8.

Atf1 protein half-life is reduced in int6Δ cells. A, immunoblot analyses of proteins from cycloheximide-treated cells. 30-ml cultures of WY764 (WT; lanes 1–4) and KAY252 (int6Δ, lanes 5–8) were grown in the EMM-C medium and supplemented with 100 mg/ml cycloheximide (CHX) at A600 = 0.7–0.8. 5 ml of the culture was harvested for WCE preparation at the indicated time, as described under “Materials and Methods.” 10 μg of WCE sample was analyzed for immunoblotting with antibodies indicated to the left, as in Fig. 5C. WCE from strain KAY584 (atf1Δ, Table 1) was used as a negative control (lane 9). B, measurement of Atf1 protein half-life in int6Δ cells. Densities of Atf1 (panel 1) and tubulin (panel 2) protein bands, which were found in the immunoblot shown in A, were measured by NIH Image software and compared with the value at time 0 in wild-type and plotted against time after cycloheximide treatment at a semilogarithmic scale. Thick lines indicate exponential trendlines for each dataset (wild-type, black symbols; int6Δ, gray symbols). Equation y = A e-Bx for Atf1 in int6Δ cells is indicated in panel 1. Atf1 protein half-life (t½) was calculated from ln2/B. Graph was drawn with the averages from two independent but equivalent exposures of the same immunoblot membrane, with bars representing S.D.
DISCUSSION
Int6/eIF3e Facilitates MAPK/Atf1 Signaling—In this study, we showed that two major protein kinases, Sty1 MAPK and Gcn2 eIF2K (Figs. 1, B and C, and 4B), and their downstream target transcription (Figs. 2 and 5, A and B) are activated rapidly during 3AT-induced histidine starvation, and that Int6 plays an important role in modulating the Sty1-depndent response during this nutritional insult (Fig. 5). Our pulse-labeling experiment (Fig. 7), together with polysome profiling reported previously (8), indicated that int6Δ compromises general translation in the minimal medium. We also found that int6Δ decreased Atf1 protein half-life (Fig. 8). We propose that these effects led to a lower abundance of Atf1 before 3AT-induced stress (Figs. 5, C and D, and 7B), which significantly diminishes Atf1-directed transcription in the initial phase of 3AT-induced response (Figs. 2A and 5, A and B).
Our results showing ∼60% reduction in general translation by int6Δ in the minimal medium (Fig. 7, C and D) indicate that Int6/eIF3e is a general translation factor. This idea agrees with recent findings that its mammalian homologue, Int-6/eIF3e, is one of the six subunits constituting the functional core of eIF3 (5) and interacts with eIF4G to mediate mRNA binding to the 40 S subunit (40).
Activation of the Sty1 MAPK Pathway during 3AT-induced Histidine Starvation—We also found that 3AT-induced starvation activates the Sty1 MAPK (Fig. 4). Although 3AT is known to inhibit many enzymes including catalase (reviewed in Ref. 41) whose inhibition may generate H2O2, histidine reversed the 3AT sensitivity caused by all the tested mutants deleted for Int6/eIF3e and the Sty1 signaling components (Fig. 1A and supplemental S2). Thus, the 3AT-induced response is primarily caused by histidine starvation. At present, however, it is unclear how this stimulus activates Sty1. Because Pap1-dependent transcription was not induced (Fig. 6), Sty1 activation may be independent of H2O2 as an intracellular signaling molecule (42), or if H2O2 is involved, its transient production level may be kept high enough to prevent Pap1 activation (24). The H2O2-independent mechanism could involve one or two of the membrane-spanning kinases Mak1/2/3 (19), which might be activated by changes in plasma membrane activities, such as inhibition of amino acid uptake, potentially caused by immediate inhibition of protein synthesis on 3AT treatment (Fig. 7, C and D). The H2O2-dependent mechanism would be consistent with recent studies highlighting the positive role of H2O2 in cell signaling (reviewed in Ref. 43). A subtle perturbation in the cellular metabolism by 3AT-induced starvation can potentially produce H2O2 in excess, for instance, by perturbation of mitochondrial respiration (the major source of reactive oxygen species) (43), by oxidative decomposition of 3AT or histidine synthesis intermediates, or by negative regulation of antioxidents, such as Tpx1 (42). Thus, it would be important to determine which signaling components known to activate Sty1 are required for overcoming the 3AT-induced stress.
As discussed above, the effect of int6Δ on the Sty1-dependent response includes diminished Atf1 activity at the initial phase of 3AT-induced response. We also observed that Sty1 is activated for a longer duration on 3AT insult in int6Δ cells (Fig. 4B). The mechanism of this phenomenon is unclear, but we suggest, among other possibilities, that this is due to delay in transcription of Sty1 regulators, such as pyp2+ encoding the Sty1 phosphatase (44), during the initial phase of 3AT-induced response. Indeed, our microarray data indicates that int6Δ delays pyp2+ transcription as a cluster 2 gene (supplemental Table S1 and Fig. S1). In any event, we predict that the prolonged Sty1 activation enhances the later phase of 3AT-induced response in int6Δ cells. In agreement with this prediction, we observed that expression of some cluster 2 genes (CESR) is higher in int6Δ cells than in wild-type after 3 h of 3AT treatment, although at 1 h of 3AT treatment, their expression is lower in int6Δ (Fig. 2A).6 This explains how initial perturbation of transcriptional activities affects the later phase of activities governed by the same transcription factor.
The S. pombe Sty1/Atf1 pathway is highly homologous to a mammalian stress-induced pathway dependent on p38 MAPK and activating transcription factor 2 (ATF2) (17). Forced expression of ATF2 is suggested to facilitate formation of malignant tumors (45, 46). It is possible that the p38/ATF2 signaling pathway is altered in mammalian cells deficient in int-6 in a manner similar to the Sty1/Atf1 pathway in fission yeast int6Δ cells.
Role of Gcn2 eIF2K in the 3AT-induced Stress Response of Fission Yeast—Here we reported that the Gcn2 eIF2K derepresses transcription of Tf2 retrotransposons on 3AT insult, besides confirming that it promotes transcription of a set of genes akin to the GAAC response described in S. cerevisiae (32) (Fig. 2A and Table S1). It was recently reported that a very similar set of genes, amino acid biosynthetic genes and Tf2 retrotransposons, is up-regulated in the strain deleted for hst4+ encoding a histone deacetylase. In fission yeast, different chromatin modifiers appear to silence transcription of defined heterochromatin areas, and Hst4 mediates the silencing of retrotransposons (47). Together, these results raise an interesting possibility that Gcn2 regulates retrotransposon silencing, as well as amino acid biosynthetic gene transcription, by regulating Hst4 or its cofactor.
Chen et al. (34) recently showed that a group of genes coregulated between S. cerevisiae and S. pombe termed the amino acid metabolism module (33) overlap with Pap1-dependent genes induced by oxidative stress, leading to the suggestion that Pap1 plays a similar role to S. cerevisiae Gcn4p (34). This study does not support this idea, as the 3AT-induced, Gcn2-dependent genes (cluster 3) did not overlap with the Pap1-dependent genes, nor was Pap1 required for overcoming the 3AT-induced stress (Fig. 6). If this is true, the amino acid metabolism module may include a broader set of genes, one dependent on Pap1 and the other, Gcn2.
It was interesting to find that the transcription of tpx1+, encoding the 2-Cys peroxiredoxin, is rapidly induced in gcn2Δ cells (Fig. 6). Our microarray data presented here and complementary studies indicated that tpx1+ is among ∼200 genes that are induced in gcn2Δ cells during 3AT insult, but not in wild-type cells, and moreover, that the initial rate of CESR transcription is in general greater in gcn2Δ cells.7 The group of genes specifically induced in gcn2Δ cells included many other antioxidants and heat shock proteins, but did not overlap with genes whose expression depends on Pap1 that also activates tpx1+ transcription on oxidative stress (24). Thus, Gcn2 appears to repress this new set of genes as well as the CESR during 3AT-induced response. This suggests that there is coordination between the two major stress response pathways, and that cells compensate for the loss of the eIF2K stress response by further inducing the MAPK pathway.
Supplementary Material
Acknowledgments
We are indebted to Gavin Burns for help with microarray processing and Paul Russell, Takashi Toda, Kunihoro Ohta, and Chikashi Shimoda for timely gifts of materials. We also thank Beth Montelone and Chris Thorpe for critical reading of the manuscript, and Y. “Taro” Watanabe and members of Masayuki Yamamoto lab for technical advice. Special thanks are also due to T. Toda for comments on ubiquitin-regulated proteolysis and Daniel Lackner, Kouji Hirota, and K. Ohta for sharing results prior to publication.
This work was supported, in whole or in part, by National Institutes of Health Grants GM67481 (to K. A.) and GM49164 (to R. W.). This work was also supported by NCRR K-INBRE Pilot Grant P20 RR016475, an Innovative Award from the Kansas State University Terry Johnson Cancer Center (to K. A.) and Cancer Research UK Grant C9546/A6517 (to J. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S2 and Table S1.
Footnotes
The abbreviations used are: eIF, eukaryotic initiation factor; 3AT, 3-aminotriazole; MAPK, mitogen-activated protein kinase; CESR, core environmental stress response; MOPS, 4-morpholinepropanesulfonic acid; WCE, whole cell extract; EMM, Earle's minimal medium; HA, hemagglutinin.
S. Watt and J. Bahler, unpublished data.
N. Nemoto and K. Asano, unpublished observations.
T. Udagawa, K. Hitota, N. Nemoto, J. Bahler, K. Ohta, and K. Asano, manuscript in preparation.
T. Udagawa and K. Asano, unpublished observations.
References
- 1.Li, Y., Hively, W. P., and Varmus, H. E. (2000) Oncogene 19 1002-1009 [DOI] [PubMed] [Google Scholar]
- 2.Callahan, R., and Smith, G. H. (2000) Oncogene 19 992-1001 [DOI] [PubMed] [Google Scholar]
- 3.Asano, K., Merrick, W. C., and Hershey, J. W. B. (1997) J. Biol. Chem. 272 23477-23480 [DOI] [PubMed] [Google Scholar]
- 4.Hinnebusch, A. (2006) Trends Biochem. Sci. 31 553-562 [DOI] [PubMed] [Google Scholar]
- 5.Masutani, M., Sonenberg, N., Yokoyama, S., and Imataka, H. (2007) EMBO J. 26 3373-3383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Asano, K., Phan, L., Anderson, J., and Hinnebusch, A. G. (1998) J. Biol. Chem. 273 18573-18585 [DOI] [PubMed] [Google Scholar]
- 7.Phan, L., Zhang, X., Asano, K., Anderson, J., Vornlocher, H. P., Greenberg, J. R., Qin, J., and Hinnebusch, A. G. (1998) Mol. Cell. Biol. 18 4935-4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akiyoshi, Y., Clayton, J., Phan, L., Yamamoto, M., Hinnebusch, A. G., Watanabe, Y., and Asano, K. (2001) J. Biol. Chem. 276 10056-10062 [DOI] [PubMed] [Google Scholar]
- 9.Zhou, C., Arslan, F., Wee, S., Krishnan, S., Ivanov, A. R., Oliva, A., Leatherwood, J., and Wolf, D. A. (2005) BMC Biol. 3 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, L., Uchida, K., Endler, A., and Shibasaki, F. (2007) J. Biol. Chem. 282 12707-12716 [DOI] [PubMed] [Google Scholar]
- 11.Buchsbaum, S., Morris, C., Bochard, V., and Jalinot, P. (2007) Oncogene 26 5132-5144 [DOI] [PubMed] [Google Scholar]
- 12.Yen, H.-C., Gordon, C., and Chang, E. C. (2003) Cell 112 207-217 [DOI] [PubMed] [Google Scholar]
- 13.Hinnebusch, A. G. (2005) Annu. Rev. Microbiol. 59 407-450 [DOI] [PubMed] [Google Scholar]
- 14.Dever, T. E. (2002) Cell 108 545-556 [DOI] [PubMed] [Google Scholar]
- 15.Zhan, K., Narasimhan, J., and Wek, R. C. (2004) Genetics 168 1867-1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhan, K., Vattem, K. M., Bauer, B. N., Dever, T. E., Chen, J.-J., and Wek, R. C. (2002) Mol. Cell. Biol. 22 7134-7146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilkinson, M. G., and Millar, J. B. A. (1998) Genes Dev. 12 1391-1397 [DOI] [PubMed] [Google Scholar]
- 18.Chen, D., Toone, W. M., Mata, J., Lyne, R., Burns, G., Kivinen, K., Brazma, A., Jones, N., and Bähler, J. (2003) Mol. Biol. Cell 14 214-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikner, A., and Shiozaki, K. (2005) Mutat. Res. 569 13-27 [DOI] [PubMed] [Google Scholar]
- 20.Veal, E. A., Findlay, V. J., Day, A. M., Bozonet, S. M., Evans, J. M., Quinn, J., and Morgan, M. A. (2004) Mol. Cell 15 129-139 [DOI] [PubMed] [Google Scholar]
- 21.Lawrence, C. L., Maekawa, H., Worthington, J. L., Reiter, W., Wilkinson, C. R. M., and Jones, N. (2007) J. Biol. Chem. 282 5160-5170 [DOI] [PubMed] [Google Scholar]
- 22.Reiter, W., Watt, S., Dawson, K., Lawrence, C. L., Bähler, J., Jones, N., and Wilkinson, C. R. M. (2007) J. Biol. Chem. 283 9945-9956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bozonet, S., Findlay, V. J., Day, A. M., Cameron, J., Veal, E. A., and Morgan, B. A. (2005) J. Biol. Chem. 280 23319-23327 [DOI] [PubMed] [Google Scholar]
- 24.Quinn, J., Findlay, V. J., Dawson, K., Millar, J. B. A., Jones, N., Morgan, B. A., and Toone, W. M. (2002) Mol. Biol. Cell 13 805-816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keeney, J. B., and Boeke, J. D. (1994) Genetics 136 849-856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiozaki, K., and Russell, P. (1995) Nature 378 739-743 [DOI] [PubMed] [Google Scholar]
- 27.Lyne, R., Burns, G., Mata, J., Penkett, C. J., Rustici, G., Chen, D., Langford, C., Vetrie, D., and Bähler, J. (2003) BMC Genomics 4 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Humphrey, T., and Enoch, T. (1998) Genetics 148 1731-1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crane, R., Craig, R., Murray, R., Dunand-Sauthier, I., Humphrey, T., and Norbury, C. (2000) Mol. Biol. Cell 11 3993-4003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holstege, F. C. P., Jennings, E. G., Wyrick, J. J., Lee, T. I., Hengartner, C. J., Green, M. R., Goulb, T. R., Lander, E. S., and Young, R. A. (1998) Cell 95 717-728 [DOI] [PubMed] [Google Scholar]
- 31.Yamada, T., Mizuno, K.-I., Hirota, K., Kon, N., Wahls, W. P., Hertsuiker, E., Murofushi, H., Shibata, T., and Ohta, K. (2004) EMBO J. 23 1792-1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Natarajan, K., Meyer, M. R., Jackson, B. M., Slade, D., Roberts, C., Hinnebusch, A. G., and Marton, M. J. (2001) Mol. Cell. Biol. 21 4347-4368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanay, A., Regev, A., and Shamir, R. (2005) Proc. Acad. Natl. Sci. U. S. A. 102 7203-7208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen, D., Wilkinson, C. R. M., Watt, S., Penkett, C. J., Toone, W. M., Jones, N., and Bähler, J. (2008) Mol. Biol. Cell 19 308-317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding, H. P., Zhang, Y., Zeng, H., Novoa, I., Lu, P. D., Calfon, M., Sadri, N., Yun, C., Popko, B., Paules, R., Stojdl, D. F., Bell, J. C., Hettmann, T., Leiden, J. M., and Ron, D. (2003) Mol. Cell 11 619-633 [DOI] [PubMed] [Google Scholar]
- 36.Harrison, C., Katayama, S., Dhut, S., Chen, D., Jones, N., Bähler, J., and Toda, T. (2005) EMBO J. 24 599-610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanoh, J., Watanabe, Y., Ohsugi, M., Iino, Y., and Yamamoto, M. (1996) Genes Cells 1 391-408 [DOI] [PubMed] [Google Scholar]
- 38.Watanabe, Y., and Yamamoto, M. (1996) Mol. Cell. Biol. 16 704-711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith, D. A., Toone, W. M., Chen, D., Bähler, J., Jones, N., Morgan, B. A., and Quinn, J. (2002) J. Biol. Chem. 277 33411-33421 [DOI] [PubMed] [Google Scholar]
- 40.LeFebvre, A. K., Korneeva, N. L., Trutschl, M., Cvek, U., Duzan, R. D., Bradley, C. A., Hershey, J. W. B., and Rhoads, R. E. (2006) J. Biol. Chem. 281 22917-22932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ueda, M., Kinoshita, H., Yoshida, T., Kamasawa, N., Osumi, M., and Tanaka, A. (2003) FEMS Microbiol. Lett. 219 93-98 [DOI] [PubMed] [Google Scholar]
- 42.Wood, Z. A., Poole, L. B., and Karplus, P. A. (2003) Science 300 650-653 [DOI] [PubMed] [Google Scholar]
- 43.Veal, E. A., Day, A. M., and Morgan, B. A. (2007) Mol. Cell 26 1-14 [DOI] [PubMed] [Google Scholar]
- 44.Degols, G., Shiozaki, K., and Russell, P. (1996) Mol. Cell. Biol. 16 2870-2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huguier, S., Baguet, J., Perez, S., van Dam, H., and Castellazzi, M. (1998) Mol. Cell. Biol. 18 7020-7029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bhoulik, A., Jones, N., and Ronai, Z. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4222-4227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Durand-Dubief, M., Sinha, I., Fagerstrom-Billai, F., Bonilla, C., Wright, A., Grunstein, M., and Ekwell, K. (2007) EMBO J. 26 2477-2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Penkett, C. J., Morris, J. A., Wood, V., and Bähler, J. (2006) Nucleic Acids Res. 34 W330-W334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.