

Abstract
CCAAT enhancer binding protein alpha (C/EBPα) is the founding member of a family of basic region/leucine zipper (bzip) transcription factors and is a master regulator of granulopoiesis. It is expressed at high levels throughout myeloid differentiation and binds to the promoters of multiple myeloid- specific genes at different stages of myeloid maturation. Profound hematopoietic abnormalities occur in mice nullizygous for C/EBPα̤ including a selective early block in the differentiation of granulocytes. Mutations in C/EBPα are present in a subset of patients with AML presenting with a normal karyotype. These mutations can result in the expression of a 30kD dominant negative C/EBPα isoform, which contributes to loss of C/EBPα function. The molecular basis for this observation remains unknown. In addition to phoshorylation, C/EBPα is modified, post-translationally by a small ubiquitin-related modifier (SUMO) at a lysine residue (K159), which lies within the growth inhibitory region of the C/EBPα protein. Sumoylation at K159 in the C/EBPα protein prevents association of the SWI/SNF chromatin remodeling complex with C/EBPα, thereby hampering transactivation. In this review, the functional implications of post-translational modification, particularly sumoylation, of C/EBPα in normal granulopoiesis and leukemia are considered.
Keywords: transcription factor, myeloid leukemia, post-translational modification
Introduction
CCAAT enhancer binding protein (C/EBP) family of transcription factors
CCAAT enhancer binding proteins represent a family of basic region-leucine zipper (bZIP) class of transcription factors that recognize the consensus DNA-binding sequence 5′TKNNGYAAK3′ (Y=C or T, K=T or G) within the regulatory regions of target genes. C/EBP family proteins bind DNA either as homo- or hetero- dimers. This family of transcription factors, which plays a crucial role in hematopoiesis, includes C/EBPα,β,γ,δ,ε and CHOP-GADD 153, each of which contain highly homologous C-terminal dimerization (leucine-zipper) domains and DNA-binding (basic-region) motifs, but differ in their N-terminal transactivation domains, with the exception of CHOP-GADD 153, which lacks this domain altogether [1,2]. CHOP-GADD 153 can dimerize with and inhibit transactivation by C/EBPα,β and ε, and is found at a breakpoint in the gene associated with liposarcomas [3]. With the exception of C/EBPε, which is expressed at high levels mainly in the late stages of granulopoiesis and in T-lymphocytes, the other C/EBP members are expressed in a wide variety of cells including liver, adipose tissue, lung, intestine, adrenal gland, blood mononuclear cells and placenta [2]. The C/EBP family members exert pleiotropic effects in the tissues in which they are expressed. This may be due to their tissue and stage-specific expression, their ability to dimerize with members of their own family and of the Fos/Jun and ATF/CREB families of transcription factors, and their ability to interact with other transcription factors such as NF-κB and Sp-1 [4,5]. The C/EBP factors regulate the differentiation of a variety of cellular systems.
CCAAT enhancer binding protein alpha (C/EBPα)
C/EBPα is the founding member of a family of basic region/leucine zipper (bzip) transcription factors and is a master regulator of granulopoiesis.[6–8]‥ It is expressed at high levels throughout myeloid differentiation and binds to the promoters of multiple myeloid- specific genes at different stages of myeloid maturation [6,7][8]. C/EBPα also plays a role in adipocyte differentiation:;inhibition of C/EBPα blocks adipocyte differentiation and overexpression of this C/EBP family member induces adipocyte differentiation[9]. Regulation of constitutive hepatic genes as well as acute phase response genes in the liver involves several C/EBP family members, in particular C/EBPα [10].
Profound hematopoietic abnormalities occur in mice nullizygous for C/EBPα[11]. C/EBPα−/− mice die perinatally due to defects in gluconeogenesis resulting in hypoglycemia [12]; they also have a selective early block in the differentiation of granulocytes. Since the expression of C/EBPα is reduced in many myeloid leukemias, this transcription factor has been hypothesized to serve, in some settings, as a tumor suppressor.
The C/EBPα gene
The C/EBPα gene is intronless and generates two isoforms as a result the differential utilization of alternate translational start codons. The resultant p42kD (full length) and p30kD (truncated) C/EBPα proteins differ from each other at the N-terminus, which is abbreviated in the p30kD protein (see Figure 1). Three activation domains referred to as TE-I, TE-II and TE-III have been described in the full length p42 protein (see Figure 1). The p30kD C/EBPα isoform lacks the TE-I and TEII domains, which are known to bind to the basal transcriptional machinery (e.g., TATA box binding protein, TBP), thus making p30 a less efficient transactivator as compared to its full length counterpart. P30 is a dominant negative form of C/EBPα. This isoform heterodimerizes with p42 C/EBPα, thereby inhibiting the ability of the latter to transactivate target genes. Conditional ablation of C/EBPα in the marrow of mice results in a block at the transition from the common myeloid progenitors (CMPs) to granulocyte-monocyte progenitors (GMPs), resulting in the accumulation of immature myeloid blast cells in the marrow, similar to that observed in acute myeloid leukemia (AML). Additionally, mice in which the dominant-negative p30 kDa isoform is expressed but the p42 kDa isoform is disrupted, develop an AML-like disease (C. Nerlov, unpublished data). However, neither conventional nor conditional C/EBPα-knockout mice develop AML. The functional role of p30kD C/EBPα in myeloid cells is, therefore, more complex than previously thought, as the p30kD isoform rescues the p42kD C/EBPα null phenotype from early mortality, suggesting that an exclusive dominant negative role ascribed to this isoform may be an insufficient explanation[6].
Figure 1. A map of the structural features of the human C/EBPα protein.
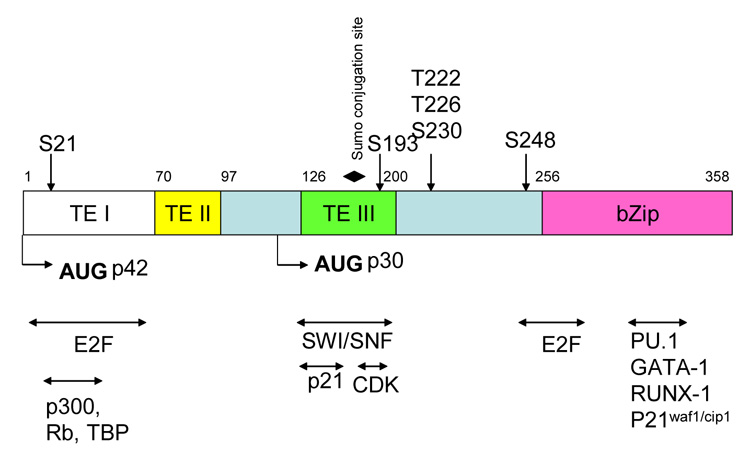
The transactivation domains TE-I, TE-II and TE-III are shown. Translation start sites for the two C/EBPα isoforms p42 and p30 are indicated by arrows. Phosphorylation sites (indicated by vertical arrows) and the single sumoylation site (indicated by a solid diamond) are shown. Protein-protein interacting partners of C/EBPα are indicated by horizontal arrows. Bzip marks the basic leucine zipper motifs.
Growth inhibition associated with C/EBPα
In addition to its ability to transactivate myeloid-specific genes, C/EBPα also arrests mitotic growth. This growth arrest is thought occur by three main mechanisms. First, C/EBPα can induce the expression of p21waf1/cip1, a CDK (cyclin-dependent kinase) inhibitor, and disrupt E2F transcriptional complexes during the G1 phase of the cell cycle[13–16]. Second, C/EBPα can mediate growth control through interaction with the cyclin-dependent kinases CDK2 and CDK4[17]. Third, C/EBPα can interact with the SWI/SNF chromatin-remodeling complex[8]. Thus C/EBPα acts to facilitate exit from the cell cycle as well as to upregulate gene expression specific to the terminally differentiated state. This transcription factor is thereby positioned to play a vital role in orchestrating normal differentiation.
C/EBPα and leukemogenesis
Several groups have reported mutations in the C/EBPα gene in a subset of patients with AML presenting with normal karyotypes [6]. These mutations can be broadly classified into two main categories. The first includes in-frame mutations clustered in the C-terminus of the C/EBPα protein. This domain harbors the highly conserved basic-region leucine-zipper responsible for DNA binding, as well as the dimerization domain. Often additional mutations in the second C/ebpα allele accompany these mutations resulting in functional loss of C/EBPα in the cells of these patients. The second category of mutations involves the N-terminus of C/EBPα. These frame-shift mutations are responsible for the premature termination of the full length p42 C/EBPα isoform while keeping the truncated p30C/EBPα protein intact. The remaining p42C/EBPα is rendered inactive by the dominant -negative activity of the p30 isoform[6]. Additionally, knock-in mice with a targeted mutation in the C/EBPα basic region that disrupts the C/EBPα–E2F association also develop myeloid leukemia.
No mutations in the C/EBPα locus have been reported in patients with FAB-M2 leukemia associated with either the t(8;21) (AML1-ETO) or inv(16) (CBFb-MYH11) translocation, despite the fact that leukemic cells derived from these patients express decreased levels of C/EBPα mRNA. Leukemias involving the internal tandem duplication (ITD) in FLT-3 (Fms-like tyrosine kinase 3) that constitutively activates FLT-3 kinase activity have been reported to block C/EBPα function via phosphorylation at Serine 21 (S21) in the C/EBPα protein. Hence, the increased FLT-3 activity appears to both increase cellular proliferation and increase cell viability and also prevent C/EBPα from inducing granulocytic differentiation [18]. Decreased expression of C/EBPα has also been described in AML with the t(8;21) translocation resulting in the AML1-ETO fusion protein. AML1-ETO is thought to have a negative effect on the positive autoregulation of C/EBPα. Other subtypes of AML including the t(3;21), resulting in AML-1-MDS-1-Evi-1 fusion protein and the inv(16) mutation are also associated with reduced expression of C/EBPα [8]. Downregulation of C/EBPα expression is a vital correlate in the development of some myeloid leukemias. Based on evidence from mouse models and from patients with AML, complete loss of C/EBPα is not necessary for AML development. However, mutations resulting in the loss of one or more vital function of C/EBPα, such as cell cycle exit or growth arrest, may contribute to the development of AML.
A study of six patients with B-cell precursor acute lymphoblastic leukemia (BCP-ALL), harboring the t(14;19)(q32;q13) translocation, demonstrated over expression of C/EBPα at the mRNA and protein levels suggesting that C/EBPα overexpression may also be leukemogenic. Here activation of C/EBPα is thought to result from the juxtaposition of the immunoglobin gene enhancer upstream of the C/EBPα gene [19]. Together these data suggest that increasing or decreasing the level of C/EBPα expression can contribute to leukemogenesis.
Post-translational modifications of C/EBPα
Phosphorylation and sumoylation appear to be important post-translational modifications affecting C/EBPα activity.
Phosphorylation
C/EBPα is phosphorylated at several sites, indicating a significant role for posttranslational modifications in mediating C/EBPα activity (Figure 1, Table 1). At S21, phosphorylation is mediated by extracellular signal receptor kinase (Erk1/2).[20]. In leukemias harboring the FLT-3-ITD mutation, constitutive FLT-3 kinase activity results in sustained phosphorylation at S21 in the C/EBPα protein. Phosphorylation at S21 in turn has been shown to interfere with the ability of C/EBPα to induce granulocytic differentiation, an observation that may explain the differentiation block associated with leukemia [18].
Table 1.
Effects of Phosphorylation on the function of C/EBPα.
| Pathway |
Phosphorylated residue |
Biological effect |
Refs |
|---|---|---|---|
| Flt-3 (activated) via ERK1/2 kinase | S21 | Blocks C/EBPα-induced granulocytic differentiation | [18][20] |
| Glycogen synthase kinase (GSK3) | T222, T226 (rodent residue numbers) S230 | Effects preadipocye differentiation | [24] |
| RAS signaling | S248 | Increases transactivation of the G-CSF receptor thereby upregulating granulocytic differentiation | [21] |
| ? | S193 | Phosphorylation at S193 promotes binding of CDK2 and Brm1 to C/EBPα, thereby inhibiting proliferation in hepatocytes | [22] |
| PI3K/Akt activation increases PP2A | Dephosphorylation at S193 | Accererates proliferation of hepatocytes via Rb:E2F interactions | [23] |
Phosphorylation of C/EBPα at S248 is thought to be mediated by activated Ras in a manner stimulating in vitro granulopoeisis by enhancing the ability of C/EBPα to upregulate the expression of the granulocyte-colony stimulating factor receptor(G-CSFR)[21] via the C/EBP binding site in its promoter.
The phosphorylation status of C/EBPα at S193, which is located within the growth inhibitory region or synergy control region (Figure 1), plays a role in liver cell regeneration. Phosphorylation at S193 blocks proliferation of hepatocytes as a result of C/EBPα binding to cyclin-dependent kinase-2 (cdk-2) and to the ATPase subunit of the SWI/SNF chromatin remodeling complex [22]. Dephosphorylation of S193 on the other hand, promotes proliferation by preferentially binding the retinoblastoma protein (Rb) in the repressive Rb-E2F complex, thereby freeing E2F, a transcription factor essential for cell cycle progression, to activate the expression of genes required for G1/S progression. In hepatic tumors, activation of the PI3K/Akt signaling pathway leads to the accumulation of the protein phosphatase PP2A, which in turn dephosphorylates C/EBPα at S193 thereby promoting proliferation [23] [22]. A role for S193 in modulating granulopoiesis has not been reported in the literature thus far.
Finally, phosphorylation at T222, T226 and S230 (numbering as per rodent sequence), by GSK3 kinase (glycogen synthase kinase 3) influences preadipocyte differentiation[24]. Studies addressing the in vivo function of these phosphorylation sites are now beginning to emerge.
Sumoylation
SUMO (small ubiquitin –like modifier) reversibly and post-translationally modifies target proteins thereby altering protein function and fate. The list of sumoylated proteins is increasing and includes several that play pivotal roles in processes as diverse as transcriptional regulation, nuclear targeting, sub-nuclear targeting, chromatin structure and genome stability.
The pathway leading to SUMO conjugation involves an evolutionarily conserved three step process similar to ubiquitin conjugation (Figure 2). The SUMO peptide is first activated by the formation of a thioester bond between its C-terminal glycine and the catalytic cysteine of the E1 activating enzyme heterodimer (Aos/Uba2) in a process requiring ATP hydrolysis. Next, activated SUMO is transferred to the catalytic cysteine of the E2 conjugating enzyme Ubc9. The final step involves the transfer of the SUMO moiety from E2 to the specific substrate in the presence of an E3 ligases. Thus, sumoylation results in the formation of an isopeptide bond between the C-terminal carboxy group of SUMO and the ε-amino group of a lysine residue in the target protein which generally harbors the consensus sequence ϕKXE, where ϕ is a hydrophobic amino acid, K is the sumoylated lysine, X is any amino acid and E is glutamic acid. Three families of E3 ligases have been described thus far. These include the PIAS (protein inhibitor of activated STAT) family of proteins; RanBP2, which is localized in the nuclear pore complex; and Pc2, which is a component of the polycomb protein complex [25] and Mms1. In addition, SUMO peptidases,or members of the Sentrin specific proteases (SENP) family, complete the cycle by deconjugating SUMO from target proteins (see Figure 2).
Figure 2. The Sumoylation pathway.
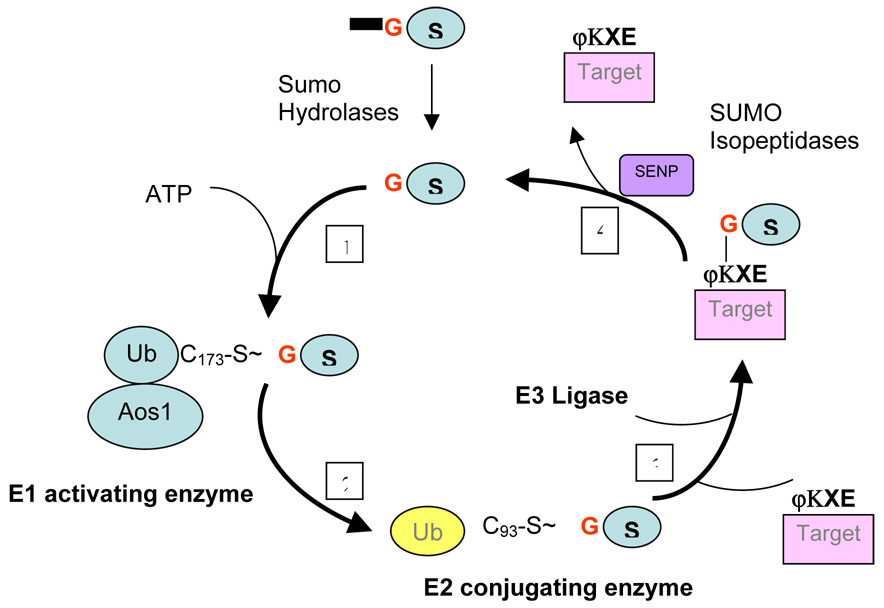
A general schema of the multi-step process that leads to sumoylation of a target protein. 1. The SUMO peptide is activated by the formation of a thioester bond between its C-terminal glycine and the catalytic cysteine of the E1 activating enzyme heterodimer (Aos/Uba2) in a process requiring ATP hydrolysis. 2. activated SUMO is transferred to the catalytic cysteine of the E2 conjugating enzyme Ubc9. 3. The SUMO moiety is next transferred from E2 to the specific substrate often harboring a lysine residue within the consensus sequence ΨKXE, where Ψ is a large hydrophobic residue and K the modified lysine, in the presence of an E3 ligases. 4. Sumo-specific peptidases (SENPs) complete the cycle by removing the Sumo moiety from target substrates.
Four SUMO family members, SUMO1.2,3 and 4 have been described, all of which are approximately 100 amino acids in length. SUMO 2 and 3 are 95% identical, whereas SUMO 1 is approximately 45% identical to the other two family members. SUMO 2 and 3 probably share similar functions. In addition, seven mammalian SENP family members have been recognized thus far [25].
RanGAP1 was the first protein found to be modified by sumoylation. While unmodified RanGAP1 is found to be dispersed in the cytoplasm, sumoylation of this protein targets it to the nuclear pore complex[26]. Additionally, sumoylation targets promyelocytic leukemia (PML) and the homeodomain-interacting protein kinase 2 (HIPK2) to specific domains within the nucleus[27]. By knocking out the essential E2 conjugating Ubc9 enzyme, the sumoylation pathway iwas shown to be essential for nuclear integrity and normal segregation of chromosomes[28].
Sumoylation and Transcription
In general, sumoylation of transcription factors leads to inhibition of transcriptional activity. However, there are exceptions to this rule. For example, sumoylation of T-cell factor 4 (Tcf-4) and Ikaros results in enhanced transcriptional activation[29,30]. A number of mechanisms have been proposed to explain the impact of sumoylation on target proteins. SUMO likely competes with other post-translational modifications such as ubiquination and acetylation, which compete for specific lysine residues within target proteins. This competition may lead to an alternate fate for the target protein as is the case with the inhibitor of NFkB and murine double minute 2. Both these proteins are targeted for degradation by ubiquination. However, competition for the target lysine residue by SUMO stabilizes these factors. Sumoylation of transcription factors is also thought to confer new protein-protein interaction properties, which in turn can significantly alter transcription activity. Histone deacetylases (HDACs) are generally correlated with repression of transcription and can be recruited to sumoylated transcription factor complexes. For example, SUMO modification of the transcription factor Elk-1 has been correlated with increased association with HDAC2[31]. Additionally, sumoylation of the co-activator p300 is associated with increased levels of HDAC6[32]. HDACs are thus thought to contribute a broad, but context dependent role in SUMO-associated repression. Proteins which themselves do not undergo SUMO modification but are capable of interacting with sumoylated substrates do so via a SUMO-interacting motif (SIM). Deletion of the SIM in the SUMO E3 ligase PIASxa disrupts the non-covalent binding of this protein to SUMO[33]. Thus, proteins with SIMs may contribute to the repressive effects of SUMO-modification via non-covalent interactions, which are most likely context dependent.
Sumoylation of C/EBPα
C/EBPα is modified by SUMO-1 at lysine residue 159, which lies within a conserved negative subdomain within in the TE-III domain (see Figure 1) referred to as the synergy control motif [34]. Synergy control motifs have been described in the negative regulatory domains of several transcription factors, and have been found to be sites of sumoylation [35]. Sumoylation of C/EBPα dramatically decreases its ability to transactivate the liver specific albumin gene in the presence of BRG1, the core subunit of the ATPase dependent SWI/SNF chromatin remodeling complex. In addition, sumoylated C/EBPα fails to induce proliferation arrest because its interaction with BRG1 is inhibited. SUMO modification alters C/EBPα transcriptional activity by affecting its repertoire of protein-protein interactive partners[36].
Both C/EBPα p42 and p30 transcativate the myeloid-specific lactoferrin gene promoter harboring a C/EBP site, by approximately 2.5–3 fold above the activity of LF89 (a reporter gene plasmid harboring a C/EBP-responsive motif in the lactoferrin promoter) alone. Cotransfection of p42 with an expression plasmid for Sumo-1 however, significantly reduces LF89 driven luciferase activity compared to p42 alone. In comparison, Sumo-1 has a much less profound effect on the reduction of p30 driven LF89 luciferase activity (manuscript in preparation). Overexpression of the p30 isoform of C/EBPα in an erythroleukemia cell line results in upregulation of Ubc9, which is essential for sumoylation (Figure 2)(37). Upregulation of Ubc9 sumoylates the p42 isoform, thereby rendering it inactive with respect to transactivation of a C/EBP responsive reporter plasmid in transactivation assays. Sumoylation of p42 C/EBPα in AML may be the result of increased expression of the p30 isoform(37). It is unclear if upregulation of Ubc9 by the p30 isoform of C/EBPα acts exclusively through sumoylation of C/EBPα p42, or whether other sumoylation targets also come into play. In this context, sumoylation of other C/EBP family members also occurs and the functional impact on these transcription factors is documented. Also, the issue of sumoylation of p30 itself was not addresssed. Our laboratory has demonstrated that C/EBPαp30 can be sumoylated. We have shown that a) mutations of the sumoylated lysine residue in both p42 and p30 (lysine to alanine) renders both C/EBPα isoforms resistant to Sumo-1 mediated repression of LF89 and b) that sumoylated levels of C/EBPα decrease upon neutrophil maturation and that expression of the lactoferrin gene in the developing neutrophil correlates with loss of binding of sumoylated C/EBPα p42 to the LF promoter (manuscript in preparation).
Since the ratio of p42:p30 is altered in acute myeloid leukemias (AML) we hypothesize that the differential effect of Sumo-1 on p42 versus p30 may contribute to blocking preferentially the activity of p42, which in turn may contribute to the leukemic phenotype. In this context we have shown that sumoylated p42 remains persistently bound to the LF promoter in the acute promyelocytic leukemia cell line NB4 following ATRA-induction (manuscript in preparation). This observation is correlated with lack of lactoferrin expression in these cells. We also show that regardless of its sumoylation status, the p30 C/EBPα isoform binds poorly to the LF promoter in a C/EBP binding site pulldown assay, compared to its full length counterpart. This suggests that sumoylation of the two C/EBPα isoforms has functionally disparate effects, possibly due to differential protein-protein binding properties of each sumoylated isoform.
Thus, upregulation of p30 in leukemic cells may result in increased levels of Ubc9, which in turn may aid in sumoylating both C/EBPα p42 and p30, the functional implications of which add an additional layer of complexity to the altered function of C/EBPα in AML.
Acknowledgements
The author would like to thank Drs Miyake and Johnson for valuable reagents and Dr Nancy Berliner and members of her laboratory for continued support and helpful discussions. This study was funded in part by NIH PO1HL63357.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lekstrom-Himes J, Xanthopoulos KG. Biological role of CCAAT/enhancer-binding protein family of transcription factors. J.Biol.Chem. 1998;273:28545. doi: 10.1074/jbc.273.44.28545. [DOI] [PubMed] [Google Scholar]
- 2.Ramji D, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ron D, Habener JF. CHOP, a novel develpomentally regulated nuclear protein that dimerizes with the transcription factors C/EBP and LAP and functions as a dominant negative regulator of gene transcription. Genes Dev. 1992;6:439. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- 4.Tenen DG, Hromas R, Licht JD, Zhang D-E. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489. [PubMed] [Google Scholar]
- 5.Tenen D. Abnormalities of C/EBP alpha transcription factor: a major target in acute myeloid leukemia. Leukemia. 2001;15:688. doi: 10.1038/sj.leu.2402088. [DOI] [PubMed] [Google Scholar]
- 6.Muller B, Pabst T. C/EBPα and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol. 2006;13:7. doi: 10.1097/01.moh.0000190110.08156.96. [DOI] [PubMed] [Google Scholar]
- 7.Schuster M, Porse B. C/EBPa: a tumour suppressor in multiple tissues? Biochim. Biophys. Acta. 2006;1766:88. doi: 10.1016/j.bbcan.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Fuchs O. Growth-inhibiting activity of transcription factor C/EBPa, its role in hematopoiesis and its tumour suppressor or oncogenic properties in leukaemias. Folia Biologica (Praha) 2007;53:97. [PubMed] [Google Scholar]
- 9.Darlington GJ, Rossi SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. J. Biol. Chem. 1998;273:30057. doi: 10.1074/jbc.273.46.30057. [DOI] [PubMed] [Google Scholar]
- 10.Diehl AM. Roles of CCAAT/enhancer-binding proteins in regulation of liver regenerative growth. J. Biol. Chem. 1998;273:30843. doi: 10.1074/jbc.273.47.30843. [DOI] [PubMed] [Google Scholar]
- 11.Zhang P, Iwama A, Datta MW, Darlington GJ, Link DC, Tenen DG. Granulocyte development in C/EBP-alpha−/−mice: The role of expression of the IL-6 and G-CSF receptors; 39th Annual Meeting of the American Society of Hematology; San Diego, California, USA. 1997. Dec, [Google Scholar]
- 12.Wang N-D, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD, Taylor LR, Wilson DR, Darlington GJ. Impaired energy homeostasis in C/EBP-alpha knockout mice. Science. 1995;269:1108. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- 13.Porse B, Pedersen T, Xu X, Lindberg B, Wewer U, Friis-Hansen L, Nerlov C. E2F repression by C/EBPα is required for adipogenesis and granulopoiesis. Cell. 2001;107:247. doi: 10.1016/s0092-8674(01)00516-5. [DOI] [PubMed] [Google Scholar]
- 14.Timchenko N, Harris T, Wilde M, et al. CAAT/enhancer binding protein alpha regulates p21 protein and hepatocyte proliferation in newborn mice. Mol Cell Biol. 1997;17:7353. doi: 10.1128/mcb.17.12.7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Timchenko N, Wilde M, Darlington G. C/EBPalpha regulates formation of S-phase-specific E2F-p107 complexes in livers of newborn mice. Mol Cell Biol. 1999;19:2936. doi: 10.1128/mcb.19.4.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ. CCAAT/enhancer binding protein alpha (C/EBPa) inhibits cell proliferation through the p21(WAF-1/CIP-1/SDI-1) protein. Genes Dev. 1996;10:804. doi: 10.1101/gad.10.7.804. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, Iakova P, Wilde M. C/EBP[alpha] arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell. 2001;8:817–828. doi: 10.1016/s1097-2765(01)00366-5. eaCEaacptdioCaCMC. [DOI] [PubMed] [Google Scholar]
- 18.Radomska H, Basseres D, Zheng R, Zhang P, Dayaram T, Yamamoto Y, Sternberg D, Lokker N, Giese N, Bohlander S, Schnittger S, Delmotte M-H, Davis R, Small D, Hiddemann W, Gilliland D, Tenen D. Block of C/EBPa function by phosphorylation in acute myeloid leukemia with FLT-3 activating mutations. J.Expt. Med. 2006;203 doi: 10.1084/jem.20052242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapiro E, Russell L, Radford-Weiss L, Bastard C, Lessard M, Struski S, Cave H, Fert-Ferrer S, Barin C, Maarek O, Della-Valle V, Strefford J, Berger R, Harrison C, Bernard O, Ngugen-Khac F. Overexpression of C/EBPA resulting from the translocation t(14;19)(q32;q31) of human precursor B-cell acute lymphoblastic leukemia. Blood. 2006;108:3560. doi: 10.1182/blood-2006-03-010835. [DOI] [PubMed] [Google Scholar]
- 20.Ross S, Radomska H, Wu B, Zhang P, Winnay, Bajnok N, Wright W, Schaufele F, DG T, MacDougald O. Phosphorylation of C/EBPalpha inhibits granulopoiesis. Mol Cell Biol. 2004;24:675. doi: 10.1128/MCB.24.2.675-686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Behre G, Singh S, Liu H, Bortolin L, Christopeit M, Radomska H, Rangatia J, Hiddemann W, Friedman A, Tenen D. Ras signaling enhances the activity of C/EBP alpha to induce granulocytic differentiation by phosphorylation of serine 248. J. Biol. Chem. 2002;277:26293. doi: 10.1074/jbc.M202301200. [DOI] [PubMed] [Google Scholar]
- 22.Wang G, Timchenko T. Dephosphorylated C/EBPalpha accelerates cell proliferation through sequestering retinoblastoma protein. Mol Cell Biol. 2005;25:1325. doi: 10.1128/MCB.25.4.1325-1338.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang G, Iakova P, Wilde M, Awad S, Timchenko T. Liver tumors escape negative control of proliferation via PI3K/Akt-mediated block of C/EBP alpha growth inhibitory activity. Genes Dev. 2004;18:912. doi: 10.1101/gad.1183304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu HK, Perrier S, Lipina C, Finlay D, McLauchlan H, Hastie CJ, Hundal HS, C S. Functional characterisation of the regulation of CAAT enhancer binding protein alpha by GSK-3 phosphorylation of Threonines 222/226. BMC Mol Biol. 2006;6:7. doi: 10.1186/1471-2199-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukhopadhyay D, Dasso M. Modification in reverse:the SUMO proteases. Trends Biochem Sci. 2007;32:286. doi: 10.1016/j.tibs.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Saitoh H, Pu R, Cavenagh M, Dasso M. RanBP2 associates with Ubc9p and a modified form of RanGAP1. Proc. Natl.Acad.Sci USA. 1997;94:3736. doi: 10.1073/pnas.94.8.3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim Y, Choi C, Kim Y. Covalent modification of the homeodomain-interacting protein kinase 2 (HIPK2) by the ubiquitin-like protein SUMO-1. Proc. Natl.Acad.Sci USA. 1999;96:12350. doi: 10.1073/pnas.96.22.12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nacerddine K, Lehembre F, Bhaumik M, Artus j, Cohen-Tannoudji M, Babinet C, Pandolfi P, Dejean A. The SUMO pathway is essential for nuclear integrity and chromosome segregation in mice. Dev Cell. 2005;9:769. doi: 10.1016/j.devcel.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto H, Ihara M, Matsuura Y, Kikuchi A. Sumoylation is involved in B-catenin-dependent activation of Tcf-4. EMBO J. 2003;22:2047. doi: 10.1093/emboj/cdg204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez-del Arco P, Koipally J, Georgopoulos K. Ikaros SUMOylation: switching out of repression. Mol Cell Biol. 2005;25:2688. doi: 10.1128/MCB.25.7.2688-2697.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang S, Sharrocks A. SUMO promotes HDAC-mediated transcriptional repression. Mol Cell. 2004;13:611. doi: 10.1016/s1097-2765(04)00060-7. [DOI] [PubMed] [Google Scholar]
- 32.Gridwood D, Bumpass D, Vaughan O, Thain A, Anderson L, Snowden A, Garcia-Wilson E, Perkins N, Hay R. p300 transcriptional repression is mediated by SUMO modification. Mol Cell. 2003;11:1043. doi: 10.1016/s1097-2765(03)00141-2. [DOI] [PubMed] [Google Scholar]
- 33.Song J, Durrin L, Wilkinson T, Krontiris T, Chen Y. Identification of a SUMO-binding motif that recognizes SUMO-modfied proteins. Proc. Natl.Acad.Sci USA. 2004;101:14373. doi: 10.1073/pnas.0403498101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian L, Benson M, Iniguez-Lluhi J. A synegy control motif within the attenuator domain of CCAAT/enhancer binding protein alpha inhibits transcriptional synergy through its PIASy-enhanced modification by SUMO-1 or Sumo-3. J. Biol. Chem. 2003;278:9134. doi: 10.1074/jbc.M210440200. [DOI] [PubMed] [Google Scholar]
- 35.Kim J, Cantwell C, Johnson P, Pfarr C, Williams S. Transcriptional activity of CCAAT/enhancer-binding proteins is controlled by a conserved inhibitory domain that is a target for sumoylation. J. Biol. Chem. 2002;277:38037–38044. doi: 10.1074/jbc.M207235200. JBC. [DOI] [PubMed] [Google Scholar]
- 36.Sato Y, Miyake K, Kaneoka H, Iijima S. Sumoylation of CCAAT/enhancer binding protein a and its functional roles in hepatocyte differentiation. J. Biol. Chem. 2006;281:21629. doi: 10.1074/jbc.M600852200. [DOI] [PubMed] [Google Scholar]
- 37.Geletu M, Balkhi M, Peer Zada A, Christopeit M, Pulikkan J, Trivedi A, Tenen D, Behre G. Target proteins of C/EBPalphap30 in AML: C/EBPalphap30 enhances sumoylation of C/EBPalphap42 via up-regulation of Ubc9. Blood. 2007;110:3301. doi: 10.1182/blood-2007-01-071035. [DOI] [PubMed] [Google Scholar]