

Abstract
Differences between the crystal structures of inhibitor-bound and uninihibited forms of phosphofructokinase (PFK) from B. stearothermophilus have led to a structural model for allosteric inhibition by phosphenolpyruvate (PEP) wherein a dimer-dimer interface within the tetrameric enzyme undergoes a quaternary shift. We have developed a labeling and hybridization technique to generate a tetramer with subunits containing two different extrinsic fluorophores simultaneously in known subunit orientations. This construct has been utilized in the examination of the effects of allosteric ligand and substrate binding on the subunit affinities of tetrameric PFK using several biophysical and spectroscopic techniques including 2-photon, dual-channel Fluorescence Correlation Spectroscopy (FCS). We demonstrate that PEP-binding at the allosteric site is sufficient to reduce the affinity of the active site interface from beyond the limits of experimental detection to nanomolar affinity, while conversely strengthening the interface at which it is bound. The reduced interface affinity is specific to inhibitor-binding, as binding the activator ADP at the same allosteric site causes no reduction in subunit affinity. With inhibitor bound, the weakened subunit affinity has allowed the kinetics of dimer association to be elucidated.
Phosphofructokinase catalyzes the MgATP-dependent phosphorylation of fructose 6-phosphate (F6P), producing fructose 1,6-bisphosphate and MgADP. This reaction represents the first committed step of glycolysis and hence is judiciously regulated (1). Most prokaryotic PFK isoforms are allosterically activated by ADP and are inhibited by phosphoenolpyruvate (PEP). The K-type allosteric response of the enzyme is characterized by the reduction of F6P binding affinity with PEP bound in the allosteric site, and increased F6P affinity when ADP is bound in the allosteric site (2-9).
Phosphofructokinase from the moderate thermophile Bacillus stearothermophilus (BsPFK) is a homotetramer consisting of 4 identical subunits, the orientation of which provides 4 identical active sites and 4 allosteric sites along respective dimer-dimer interfaces. Each binding site is composed of residues from both subunits at the interface, such that each subunit contributes complementary halves of 2 active sites and complementary halves of 2 allosteric sites, and the complete tetramer is required for all 8 sites (10-13).
The chemical denaturant KSCN has been utilized to examine the subunit associations of E. coli PFK (EcPFK) and based on these denaturation data, the active site interface is hypothesized to have lower affinity than the allosteric site interface (14-16). The sensitivity of enzyme activity to chemical perturbation is higher than that of the intrinsic tryptophan fluorescence of the enzyme. The differential sensitivities to denaturant are attributed to disruption of the active interface, leading to loss of activity and disruption of the allosteric interface, leading to solvent-relaxation of tryptophan fluorescence, respectively (15). In the presence of ligands, the active site interface is stabilized to KSCN dissociation by F6P, and is weakly stabilized by bound allosteric inhibitor, PEP.
The crystal structures of BsPFK have been solved in both an uninhibited form in the absence of regulatory ligands (12), and in an inhibited form with phosphoglycolate, a PEP analog, bound in the allosteric site (13). The most striking structural difference between the two structures is a 7° rotation of the subunits relative to one another at the active site interface. In this examination we attribute a significant loss of subunit affinity to this inhibitor-bound conformation.
Fluctuation Correlation Spectroscopy (FCS) is a technique that interrogates fluctuations in fluorescence intensity to determine diffusion and other temporally-dependent characteristics (17-20). Autocorrelation analysis of fluorescence intensity fluctuations arising from changes in number density of fluorescent particles diffusing through a small, precisely-defined laser focus yield the apparent diffusion coefficient and particle number, from which association data can be derived (17-23). In complementary fashion, the deconvolution of the photon count probability density yields two parameters; the average particle number, N, and the molecular brightness, ε (24-26).
Placing multiple extrinsic fluorophores in known locations within the PFK tetramer has afforded us experimental control over both the identification and quantification of the subunit affinities of the specific dimer interfaces within the PFK tetramer. Two fluorophores, AlexaFluor488 and Texas Red, are used to label the tetramer such that two subunits associated at the allosteric site interface are modified with identical fluorophores and two subunits associated at the active site interface are modified by distinct fluorophores. The broad two-photon cross sections of these two fluorophores permit their simultaneous excitation using a single Titanium: Sapphire pulsed laser source (23, 27). Dissociation at the active site interface is anticipated to separate a dimer of AlexaFluor488-modified subunits from a dimer of Texas Red-modified subunits resulting in loss of cross-correlation, but constant apparent particle number and molecular brightness. Conversely, dissociation at the allosteric site interface separates dimers that consist of an AlexaFluor488-labeled subunit and a Texas Red-labeled subunit. In this case, dissociation causes loss of cross-correlation and molecular brightness but an increase in apparent particle number.
We examine the impact of ligand and substrate-binding on the thermodynamics and kinetics of the subunit interactions of BsPFK, and demonstrate that the inhibited conformation of the enzyme has significantly reduced affinity along the active-site interface.
Materials and Methods
Materials
Spectral grade EPPS was purchased from Research Organics. MgCl2, NH4Cl and EDTA were spectral or reagent grade and were purchased from Sigma. AlexaFluor 488 C2-maleimide, Texas Red C2-maleimide and Rhodamine-6G were purchased from Molecular Probes (Eugene, Oregon). Absolute, punctilious grade ethanol was purchased from AAPER and stored in glass bottles. P-tertphenyl was purchased from Kodak Chemicals, Rochester, NY.
Enzyme purification and labeling
Enzyme purification was accomplished as described previously with several modifications. The K90,91E mutant BsPFK has been generated by site-directed mutagenesis and its purification described (5, 8). In this BsPFK variant, surface lysine residues K90 and K91, which are located well removed from any binding site or subunit interface, have been converted to aspartate, conferring an altered electrostatic surface to the enzyme. These modifications do not alter the catalytic or regulatory properties of the enzyme (5). Following Mimetic blue dye (Amicon) affinity chromatography and overnight dialysis, peak fractions were applied to a 10 mL Pharmacia MonoQ anion exchange column and a linear gradient of NaCl from 0 − 1 M was used in elution. Activity was assayed as described (5). Peak fractions were extensively dialyzed into buffer F (50 mM EPPS, 10 mM MgCl2, 10 mM NH4Cl, 0.2 mM EDTA). Purity was confirmed by SDS-PAGE and enzyme was quantified as described (28). PFK samples were thiol-labeled with AlexaFluor488 C2-maleimide or Texas Red C2-maleimide as described (29). Labeling reactions contained 20−50 μM PFK, 1 mM F6P, 10 % glycerol, and approximately 10-fold excess label in buffer F or DMSO. 2 mL-labeling reactions were incubated for 2 hours at room temperature with slow stirring. Labeled protein was isolated from free fluorophore by repeated size exclusion chromatography using a 20 mL G-50 Sephadex column. Elimination of free fluorophore was considered complete when additional size exclusion chromatography produced no change in fluorescence anisotropy. Greater than 90 % of the subunits were labeled with a stoichiometry of 1:1 (fluorophore:subunit), and the extrinsic fluorophore reduced the specific activity of the enzyme less than 30 %.
Non-denaturing PAGE
Non-denaturing PAGE was performed in 9 % polyacrylamide gels in Tris:glycine, pH 8.3. Samples were run at 120 V for 2 hours on ice and were visualized using UV excitation or were Coomassie-stained as described (5). Kinetic time-points were obtained by incubation of the labeled protein with the specified ligand for the indicated time prior to quenching of dimer-exchange by addition of 1 mM F6P and gel-loading buffer. Densitometry was performed on the UV-exposed gels using an Alphaimager 950 (Alpha Innotech Corporation, San Leandro CA) and NIH Image V1.62f software, (NIH, Maryland).
Label hybrid formation
Multi-labeled enzyme was created by allowing dimer exchange between Texas Red labeled wild type BsPFK and AlexaFluor488 labeled BsPFK-K90,91E. Wild type-Texas Red and K90,91E-AlexaFluor488 were incubated for 30 minutes in 0.4 M KSCN and 1 mM PEP and dialyzed into buffer A (25 mM EPPS, 5 mM MgCl2, 5 mM NH4Cl, 0.2 mM EDTA) overnight. Creation of the mixed tetramer was confirmed by the presence of three bands on a non-denaturing PAGE gel corresponding to each parent enzyme and a single hybrid tetramer. Complete randomization of the tetramer is characterized by 5 distinct electrophoretic bands, which was not observed under dimer exchange conditions (5, 8). The solution of mixed tetramers was applied to a Pharmacia MonoQ column and a linear salt gradient from 15 mM to 50 mM NaCl in 200 mL total volume was used to elute each species. Peak fractions were identified by UV-VIS spectroscopy, assayed for activity and concentrated using Amicon centrifuge concentrators (Millipore, Billerica MA). In later experiments, dimer exchange was induced by incubation with 4 mM PEP for 4 hours at room temperature.
Steady-state fluorescence
Steady-state fluorescence spectra, intensity and anisotropy were performed in L-format using either an SLM-8000 with an ISS Phoenix upgrade or an ISS Koala spectrofluorometer. Excitation wavelengths were 495 nm for AlexaFluor488 or 592 nm for Texas Red-labeled enzyme. Intensity was determined by integration of the blank-subtracted emission spectra. Spectra were taken with 1 sec integration time over 1 nm wavelength intervals. Experiments using the ISS Koala used excitation slit widths of 0.5 mm and emission slit widths of 1 mm, corresponding to bandwidths of 4 and 8 nm respectively. The SLM modifications included a xenon lamp and power supply upgrade to ISS components, ISS X-1 photon counting detection and ISS Vinci acquisition and analysis software. Excitation slit widths of 2 − 4 nm as appropriate and emission slit widths of 8 nm were used. Anisotropy was determined using an appropriate cut-on filter: a Schott OG-530 for experiments using AlexaFluor488 labeled enzyme, or a Corning 5−71 colored glass filter for experiments using Texas Red labeled enzyme. Intensity was attenuated using partially-silvered neutral density filters in the excitation path in all anisotropy titrations. The anisotropy of unlabeled protein was determined using an SLM 8000 modified as described above with excitation wavelength of 300 nm, and emission was measured through a Schott 345 nm long-pass filter. In each case, data were collected until the absolute error in anisotropy was ≤ 0.002.
Frequency-domain fluorescence lifetime measurements
Frequency domain lifetime measurements were performed using an ISS Koala spectrofluorometer in L-format. A frequency-doubled, mode-locked Spectra Physics Tsunami Titanium-Sapphire (Ti: Sapph) pulsed-laser with <100 fs pulse-length and 80 MHz pulse frequency was used as the excitation source. The Ti: Sapph was optically pumped using a 10 W Spectra Physics Millenia-X 532 nm Nb:YVO4 CW laser. The pulse frequency was reduced to 4 MHz using a Spectra Physics Model 3980 pulse selector. A Spectra Physics flexible harmonic generator was used to generate the second harmonic for visible fluorophore excitation and the third harmonic for intrinsic tryptophan excitation. Parasitic fundamental wavelength excitation was eliminated using a Corion FR-400-S bandpass filter. A Spectra Physics model 3930 “lok-to-clock” electronics module was used to provide phased-lock loop stabilization to the Ti: Sapph emission. The “lok-to-clock” module was coupled to a Marconi Instruments model 2022D frequency synthesizer and an ISS linear broadband RF amplifier allowing synchronization of the Hamamatzu R928 PMT detectors with the laser emission. The cuvette temperature was maintained at 25°C using a circulating water bath. Data were collected at frequencies from 4 MHz to 320 MHz, and were fit using Globals software (University of Illinois, Champaign, IL). At each frequency, data were collected until the standard error was less than 0.1 in phase and 0.002 in modulation. The reference standard for each measurement was Rhodamine-6G in absolute ethanol. Data collected on the extrinsically labeled proteins were fit to a 1 gaussian distribution and 1-discrete lifetime model, with the discrete species representing a “zero-lifetime” scatter component. Intrinsic tryptophan fluorescence data were fit to a 2-discrete lifetime model, as described previously (28) using p-tertphenyl (Eastman Kodak, Rochester NY) as a reference standard.
Fluorescence correlation spectroscopy
Correlation spectroscopy was performed using a first-generation ISS ALBA FCS instrument, including a Nikon Eclipse TE300 inverted microscope with a 60 X, 0.7 NA Nikon air-gap objective lens. A mode-locked Spectra Physics Tsunami Ti:Sapph pulsed-laser with <100 fs pulse-length at 800 nm was used as the two-photon excitation source. The Ti: Sapph was optically pumped using a 10 W Spectra Physics Millenia-X 532 nm Nb:YVO4 CW laser operating at 7.6 W, resulting in average output power of 1.1 W. The excitation beam was reduced to 10 % intensity using a 90/10 broadband dielectric beam splitter (Newport) and was collimated using a Newport HB-4XAR.16 beam expander. The excitation power was approximately 20 mW at the sample. The excitation and emission paths were separated using a Chroma Technologies E700SP-2P dichroic mirror. In multi-channel experiments, emission wavelengths were separated using a Chroma Technologies Q570LP dichroic onto parallel APD detectors and filtered using Chroma Technologies 545/30 or 610/75 bandpass filters in each channel respectively. 400 μl samples were examined in Labtech 8-well borosilicate culture cuvettes. Data were collected at 50 kHz for 30−60 seconds and each acquisition was repeated 5 times. Data were analyzed using ISS-ALBA correlation software and were plotted using Kaleidagraph (Synergy Software, Reading, PA).
Results
Formation of mixed tetramers
To address the issue of symmetry inherent to homotetrameric PFK, we have introduced multiple fluorophores in known positions within the tetramer. In this manner, the subunit interfaces are uniquely identified by their orientation with respect to the various fluorophores and a specific interface undergoing dissociation can be identified and its affinity quantified.
Incubation of BsPFK-Texas Red with K90,91E-AlexaFluor488 in 0.4 M KSCN and 1 mM PEP induces dimer exchange between these two enzymes, and following dialysis, produces a mixed population of tetramers (Figure 1). The additional surface charges on K90,91E subunits impart higher electrophoretic mobility, with the increased mobility corresponding to the relative number of K90,91E subunits in each tetramer. Nondenaturing PAGE confirms the formation of mixed tetramers (Figure 2). The heterogeneous mixture following dialysis is shown in lane 8 of the UV-visualized gel. Three bands are observed: tetramers containing four K90,91E-AlexaFluor488 subunits appear green, tetramers containing four wild type-Texas Red subunits appear red and those tetramers consisting of two of each type of subunit appear orange and as expected migrate at an intermediate rate. Following dialysis, the mixed solution of PFK tetramers was resolved by anion exchange chromatography. A linear salt gradient is used to elute each species, and increasing stoichiometry of K90,91E subunits yields a higher elution volume as is evident from the native PAGE analysis (Figure 2). Correspondingly, low elution volume fractions contain wild type-Texas Red tetramers, (lane 2), intermediate elution volume fractions contain the mixed tetramer and appear orange (lanes 3−4) and higher elution volume fractions contain the K90,91E – AlexaFluor488 tetramers and appear green (lanes 5−6). Chromatography fractions containing the mixed tetramer yield a single band of intermediate electrophoretic mobility in native PAGE analysis, confirming that these fractions represent a mixed tetramer containing Texas Red and AlexaFluor488-labeled subunits, rather than a heterogeneous mixture of tetramers (Figure 2, lanes 2−6). UV-VIS spectroscopy confirms that these fractions contain absorbance corresponding to both Texas Red and Alexa488, whereas those fractions with either red or green emission show absorbance corresponding to those respective fluorophores alone. Multi-channel Fluorescence Correlation Spectroscopy was used to further confirm the homogeneity of the mixed tetramers (Figure 3). Cross-correlation analysis can be used to determine the concentration of the double-labeled species, specifically the mixed tetramer in this case, and its diffusion coefficient. The wild type-Texas Red tetramer shows minimal cross-correlation amplitude relative to background (Figure 3A) whereas the mixed tetramer has high relative cross-correlation as expected (Figure 3B).
Figure 1.
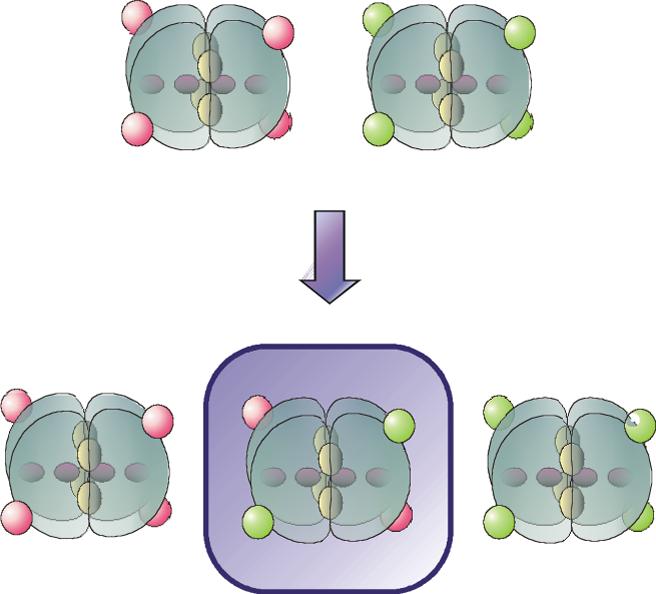
Labeling and hybridization strategy. Blocks: PFK subunits, red ovals: Texas Red, green ovals: AlexaFluor488, “interior” red ovals: active sites, “interior” yellow ovals: allosteric sites. Incubation of wild type-Texas Red and K90,91E-AlexaFluor488 with 4mM PEP and KSCN, followed by dialysis to remove the KSCN, yields a mixed population of tetramers, which are then separated by anion exchange chromatography.
Figure 2.

Formation of mixed tetramers. A 9−12% gradient, non-denaturing polyacrylamide gel of the mixed tetramer is used to visualize the fluorescent-labeled proteins under UV excitation. Lanes 2−6 result from anion exchange chromatography as described in the text. Lane 1: wild type BsPFK-Texas Red, lane 2: fraction 5, lane 3: fraction 19, lane 4: fraction 25, lane 5: fraction 33, lane 6: fraction 42, lane 7: K90,91EAlexaFluor488, lane 8: dimer exchange mixture following dialysis.
Figure 3.

Correlation curves for mixed tetramers. Panel A: autocorrelation and cross correlation curves for BsPFK-Texas Red. Panel B: autocorrelation and cross correlation curves for the mixed tetramer. Panel C: PEP-induced loss of cross correlation in the mixed tetramer. 15 nM mixed-tetramer is incubated with 4 mM PEP, resulting in significant reduction of cross correlation amplitude. Circles: red channel autocorrelation, squares: green channel autocorrelation, diamonds: cross correlation.
Subunit dissociation by Fluorescence Correlation Spectroscopy
FCS was used to examine subunit dissociation in the mixed tetramer. Within the accessible range of concentrations with our experimental arrangement, no dissociation at either interface is observed. Both the particle number and the G(0) values for autocorrelation and cross-correlation scale linearly with concentration between ∼500 pM and 100 nM, suggesting that the two interfaces have subnanomolar affinities under these conditions (data not shown).
Influence of ligands on subunit dissociation and enzyme stability
The effects of ligand-binding on the subunit affinities of BsPFK were examined using non-denaturing PAGE analysis. If dynamic dissociation/re-association occurs between wild type-Texas Red and K90,91E-Alexa488 subunits, the mixed tetramer will be observed, providing a rigorous assay for exchange. 1 μM of each of the two parent proteins was therefore incubated with various substrates and ligands for varying lengths of time. Native PAGE was then used to analyze samples from each time point. In the absence of ligands or in the presence of saturating F6P, no mixed tetramer is observed (Figure 4A). In the presence of saturating PEP concentrations, a orange band is observed at intermediate electrophoretic mobility and the intensity of this band increases with time (Figure 4B), indicating subunit exchange. Interestingly, both allosteric effectors bind the same allosteric sites on the tetramer, yet binding of the activator, ADP, does not stimulate exchange (Figure 4C).
Figure 4.
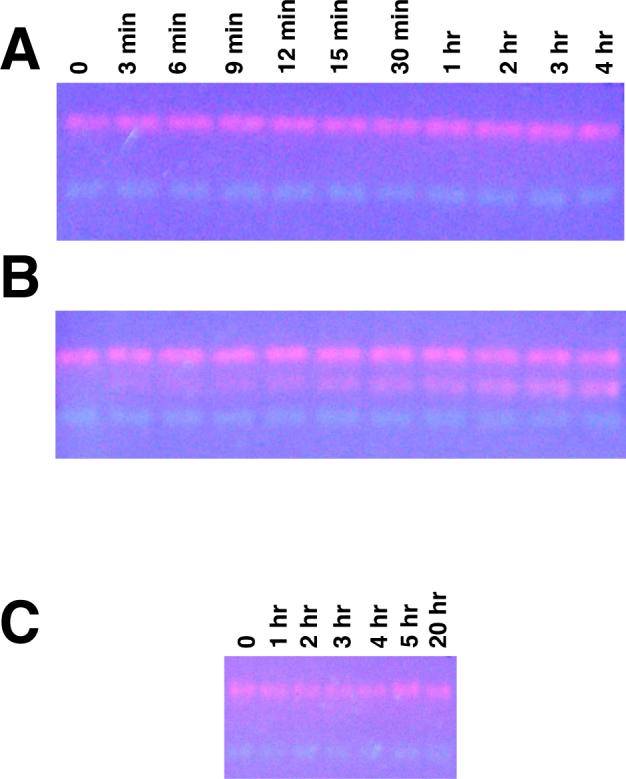
Ligand-induced subunit exchange of 1μM BsPFK. Panel A: incubation time points from 0 − 4 hrs, no ligands. Panel B: incubation time points, 4 mM PEP. Panel C: incubation time points, 2 mM ADP.
While re-hybridization was observed using the native PAGE analysis, an independently stable dimer could not be detected with this technique. Native PAGE with saturating PEP in both the gel and the running buffer resulted in band smearing, indicating that dissociation had occurred within the gel matrix over the course of electrophoresis (data not shown). Under these conditions, however, a stable population of dimer was not observed even after pre-incubation with PEP, suggesting that during the observed subunit exchange, the lifetime of the dimer is short relative to the experimental electrophoresis time.
To further confirm these ligand-binding effects, urea denaturation was utilized to determine the relative stability of the enzyme in various ligand-bound states (Figure 5). In the presence of saturating PEP, approximately 0.8 M urea is sufficient to reduce enzyme activity by 50%. In contrast, approximately 8M urea is required to achieve 50% of the urea-induced change in intrinsic tryptophan fluorescence. In the absence of ligands, however, 4M urea is required to produce a 50% change in both activity and intrinsic fluorescence intensity.
Figure 5.
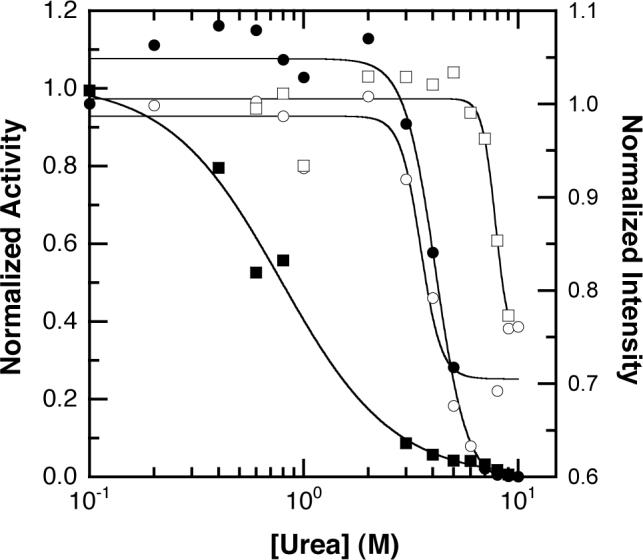
Ligand-binding effects on the structural stability of PFK. A titration of urea with 330 nM PFK is shown. Circles: no ligands, squares: 4 mM PEP. Open symbols: intrinsic tryptophan fluorescence intensity, solid symbols: enzyme activity.
Kinetics of dimer exchange
To quantify the rates of dimer exchange, densitometry was performed on the mixed tetramer band on the native PAGE gels shown in Figure 4. The concentration of BsPFK loaded onto each lane was 1 μM. In the absence of ligands, no mixed tetramer is observed; hence the intensity of the band is negligible and constant with time (Figure 6). In the presence of PEP, this band appears within minutes and saturates in approximately four hours. The band intensity vs. time data are fit best by a double exponential, with apparent rate constants for exchange equal to kapp1 = 1.7 ± 0.3×10−4s−1 and kapp2 =1.0 ± 0.7×10−2 s−1.
Figure 6.
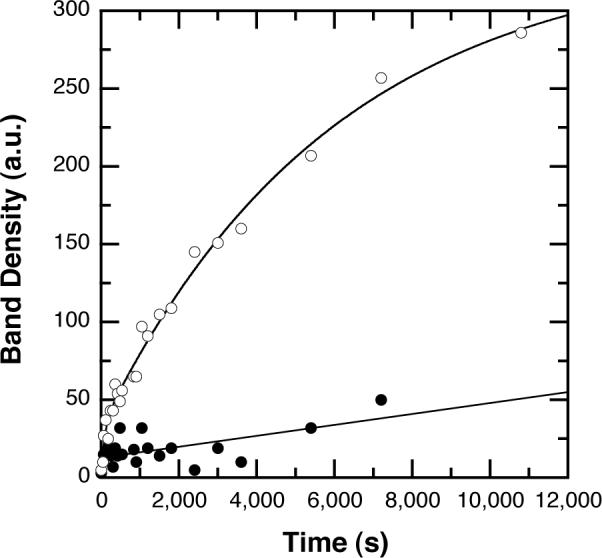
Quantitative native-PAGE kinetic analysis of subunit exchange. Intensity of the fluorescence of the orange band corresponding to the mixed tetramer obtained from densitometry of the UV-exposed gel is plotted as a function of time. Open circles: 4 mM PEP, closed circles: no ligands. Data from the PEP-treated sample are fit to a double-exponential model.
Based on the kinetic data obtained from native gel electrophoresis, dissociation was further examined in solution. If addition of PEP to the mixed tetramer induces dissociation of the active site interface, loss of cross-correlation is expected. Alternatively, if the disruption occurs at the allosteric site, cross-correlation is retained on dissociation. Following the addition of PEP, the cross-correlation amplitude of the mixed tetramer decreases relative to its initial amplitude (Figure 3C), confirming the veracity of the electrophoretic mobility data and firmly establishing the active site interface as the site of PEP-induced dissociation. This phenomenon is observed in the absence of chemical chaotrope, confirming that PEP-binding is sufficient to induce the dissociation.
Within a mixed tetramer, two Texas Red-labeled and two AlexaFluor488-labeled subunits are in close proximity, with cysteine residues separated by 30 Å to 70 Å, hence Förster Resonance Energy Transfer (FRET) is predicted (30, 31). The fluorescence emission of the hybrid tetramer from Texas Red is higher and the AlexaFluor488 emission is lower than that of an equivalent concentration of an equimolar mixture of donor or acceptor-labeled PFK (Figure 7A) or free dye. The dynamic fluorescence properties of the donor in the mixed tetramer are perturbed as well. The frequency domain data obtained for the donor in the mixed tetramer were fit best by a continuous gaussian distribution of lifetimes with a center of 4.1 ns and a width of 1.2 ns (χ2 = 1.23) while K90,91E-AlexaFluor488 is described by a distribution centered at 4.30 ns and a width of 0.8 ns (χ2 = 1.86). Based on these observations, we conclude that inter-subunit FRET is occurring and can be exploited to confirm the kinetics of PEP-induced subunit exchange. Addition of PEP to a solution of the mixed tetramer results in an increase in donor emission and a decrease in acceptor emission with time (Figure 7B). The mixed tetramer is expected to segregate 1:2:1, resulting in a ∼50 % loss of FRET as outlined in scheme 1.
Figure 7.

Förster Resonance Energy Transfer in the mixed tetramer. Panel A: FRET within the mixed tetramer. Spectra represent the K90,91E-AlexaFluor488 (green), wild type-Texas Red (red), K90,91E-AlexaFluor488 + wild type-Texas Red (black), and the mixed tetramer (orange). Spectra are normalized to concentration. Panel B: the kinetics of dimer exchange following addition of PEP to equimolar concentrations of K90,91EAlexaFluor488 + wild type-Texas Red. The ratio of donor to acceptor emission (λ = 520 nm / 605 nm) is plotted vs. time, and the solid lines represent single exponential fits. The curves correspond to total protein concentrations of 100 nM, 200 nM, 400 nM and 800 nM from top to bottom. Inset: Replot of the dependence of the apparent rate constants on BsPFK concentration, emphasizing low concentration values.
scheme 1.
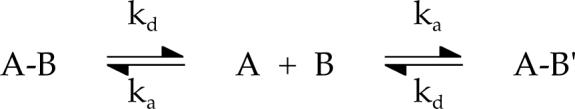
In this scheme, A-B and A-B' represent the mixed and single-colored tetramer, respectively. The loss of FRET can be fit to a single exponential decay. This was repeated at multiple enzyme concentrations and the association and dissociation rate constants were derived from a linear fit to the rate-concentration replot (Figure 7B inset), using the simplifying assumption that labeling is quantitative. The resulting rate constants are equal to ka = 4.7 ± 0.6×10−7 nM−1s−1 and kd = 5.3 ± 0.9×10−5 s−1 which predict a Kd of 110 ± 20 nM (PFK concentration expressed as subunit concentration). It is noteworthy that the apparent association rate constant is lower than typically observed for bimolecular protein-protein associations. The low rate constant may reflect a relatively rapid conformational adjustment of the PEP-bound dissociated form that interferes with the ability of the subunits to re-associate.
Subunit dissociation using fluorescence anisotropy
Fluorescence anisotropy has been used to examine the subunit affinities in a direct titration of the Texas Red-modified wild type tetramer (Figure 8). At high protein concentrations, the anisotropy of all the ligand-bound states of PFK-Texas Red is approximately 0.14. Incubation of BsPFK-Texas Red with an excess of unlabeled PFK under conditions of dimer exchange increases the observed anisotropy from 0.14 to 0.21, and exchange to the level of monomers further increases the anisotropy to 0.23. To induce dimer exchange, KSCN and PEP were added as described. Monomer exchange was induced by addition of KSCN to a final concentration of 2 M. In both cases, exchange was followed by extensive dialysis prior to fluorescence measurements. The fluorescence lifetime of wild type-Texas Red is described by a gaussian distribution with a center of 3.9 ns and width of 2.0 ns (χ2 = 11.6), low relative to the 4.21 ± 0.01 ns lifetime of the dye (32). These data suggest that the low anisotropy values are the result of homotransfer (homo-FRET) between fluorophores on adjacent subunits. No change in anisotropy is observed at any concentration of PFK examined in the absence of ligands or in the presence of F6P. In the presence of saturating PEP, an increase in anisotropy is observed, yielding an apparent affinity of 110 ± 20 nM. This value closely matches the apparent Kd calculated from kinetic exchange rates. The increase in anisotropy is attributed to dissociation at the active site interface with concomittant loss of homotransfer. A similar increase in anisotropy is observed for PFK in the presence of saturating PEP and F6P, with an apparent Kd of 0.81 ± 0.12 nM. F6P-binding to the free enzyme thus stabilizes the active site interface to PEP-induced dissociation by 130-fold, or approximately 2.9 kcal/mol.
Figure 8.
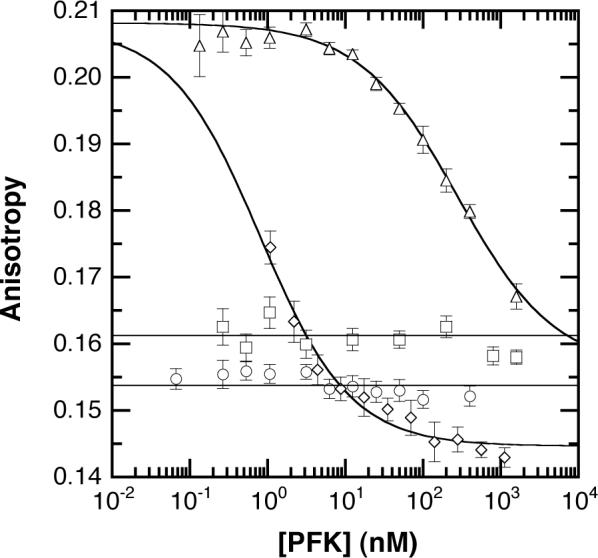
Anisotropy vs. protein concentration. Anisotropy of wild type labeled with Texas Red is plotted vs. enzyme concentration for the various ligand-bound states of BsPFK. Circles: free enzyme, squares: PFK + 1 mM F6P, triangles: PFK + 4 mM PEP, diamonds: PFK + 4 mM F6P + 20 mM PEP.
Discussion
Homotetrameric PFK is symmetrically arranged as a dimer of dimers. The interfacial nature of the ligand and substrate-binding sites permits the careful examination of the effects of ligand-binding on the relative subunit affinities. The incorporation of multiple fluorophores in known positions within the tetramer provides a different fluorescence response for each of the theoretically possible dimer dissociation events. This asymmetry of fluorophores affords, therefore, the possibility of quantitative assessment of the different dimer associations within the tetramer and the effect of ligand-binding and allosteric interactions thereon.
The introduction of the multiple fluorophores is facilitated via the use of the K90,91E variant, in which two positively charged surface residues have been changed to negatively charged residues via site-directed mutagenesis. These modifications impart a charge difference of −4 with respect to the wild type enzyme, which conveys additional electrophoretic mobility and unique ion exchange chromatographic characteristics. The two enzyme species have been differentially labeled, wild type with Texas Red, and K90,91E with AlexaFluor488 such that the catalytic and regulatory properties of the labeled enzymes are minimally affected. The two labeled enzymes were then hybridized utilizing a procedure that results in a dimer containing two Texas Red-labeled, wild type subunits separated by the active site interface from a dimer containing two K90,91E-AlexaFluor488 subunits. These hybrids contain two wild type and two K90,91E subunits and hence have electrophoretic and chromatographic mobility intermediate between tetramers containing either 4 wild type or 4 mutant subunits. Following hybridization, a mixture of the 3 tetrameric species is present, and native PAGE analysis of this mixture reveals 3 bands – a red (wild type), green (K90,91E) and orange (mixed tetramer). Anion exchange chromatography was used to separate the population of mixed tetramer, and native PAGE was used to confirm its purity. The presence of a single orange band on the PAGE gel indicates that the orange fractions contain a single, mixed-label species rather than a mixed population of Texas Red and AlexaFluor488-labeled enzyme. The 3 bands on the gel indicate that randomization has occurred between dimers, but no farther. Exchange at the monomer level is characterized by the presence of 5 bands on this gel (5).
The identities of the labeled species are further confirmed by UV-VIS spectroscopy. The absorbance spectrum of each fraction is consistent with the categorization of each species. For the intermediate fractions consisting of the mixed tetramer, the ratio of absorbances of Texas Red and AlexaFluor488 confirm a stoichiometry of 1:1, as expected. FCS was then utilized to demonstrate the linkage between the fluorophores – a wild type-Texas Red tetramer has minimal cross-correlation, whereas the mixed tetramer shows a high degree of cross-correlation. This cross-correlation can be used to monitor the dissociation of the tetramer.
The development of the hybridization technique also provides a unique tool in the analysis of subunit exchange phenomena of PFK. The occurrence of the orange mixed tetramer band on the native PAGE gel is characteristic for dynamic association/dissociation in a mixed solution of labeled wild type and K90,91E, hence this technique was exploited to examine the effects of ligation state on the stability of the enzyme. The two parent enzymes were incubated in the presence of the various allosteric effectors and substrates for various times up to several hours, then were subjected to native PAGE analysis. In the absence of ligand, an intermediate mobility orange band is not observed at any length of time. In the presence of saturating PEP, rehybridization occurs, as is evident from the time-dependent development of a orange band of intermediate electrophoretic mobility. The presence of the mixed tetramer is observed after a few minutes and its concentration saturates in ∼4 hrs. These data are evidence that PEP-binding is sufficient to induce dimer exchange and suggest that the allosteric inhibitor weakens the active site interface, permitting this exchange. While it is conceivable that dissociation occurs at the allosteric site interface, it is unlikely given that this disruption of the allosteric site interface would abrogate PEP-binding. ADP, an allosteric activator that associates with the same enzyme regulatory ligand-binding site, causes no such effect. Attempts to isolate an independently stable dimer in the native gel using PEP in both gel and buffer were unsuccessful, resulting in a broad distribution of fluorescence throughout the gel lane, suggesting that dissociation of the tetramer had occurred during electrophoresis. PEP-binding is thus insufficient to stabilize the dimer independently of the tetramer, suggesting that the dimer is a short-lived species under these conditions. Interestingly, PEP has been shown to modestly stabilize E. coli PFK to KSCN-induced dissociation (15).
To further examine the PEP-induced destabilization of the BsPFK tetramer, the enzyme's sensitivity to urea denaturation under various ligation conditions was established. In the absence of PEP, 4 M urea is sufficient to decrease both intrinsic tryptophan fluorescence and catalytic activity by 50%. These data suggest that in the absence of ligands, the transition from tetramer to monomer occurs in an apparently concerted dissociation since the changes in activity and intrinsic fluorescence each result from the dissociation of a different interface (15). We attribute this result to comparable relative affinities between the allosteric site interface of the dimer and active site interface of the tetramer. In the presence of saturating PEP, the activity was reduced, as expected in the presence of inhibitor, and was 50% disrupted by approximately 0.8 M urea. On the other hand, 50% of the perturbation in the intrinsic tryptophan fluorescence required 8 M urea. These data suggest that PEP is strongly weakening the active site interface while increasing the stability of the allosteric site interface.
The time-dependent emergence of the mixed tetramer from the mixture of the two parent enzymes in the presence of PEP provides a structure for the kinetic analysis of dimer exchange. Densitometry was performed on the fluorescence arising from the orange, mixed tetramer band on the gels. In the absence of PEP, no band is observed. In the presence of PEP, the emergence of the band is evident as an increase in fluorescence intensity. This intensity can be fit to a double exponential, suggesting the possibility of a slow conformational adjustment following association.
Under conditions of dynamic association, the mixed tetramer would be expected to re-hybridize to form the two original single-labeled tetramers over time. Specifically, the mixed tetramer is expected to form Texas Red labeled wild type, mixed tetramer and K90,91E-AlexaFluor488 in a ratio of 1:2:1. Consequently, if the dissociation occurs at the active interface separating the two different fluorophores, cross-correlation will decrease. Alternatively, if the dissociation occurs at the allosteric site interface separating a K90,91E – wild type dimer from a similar K90,91E – wild type dimer, cross-correlation will persist during dissociation. Following the relative cross-correlation as a function of time after addition of PEP yields a time-dependent loss of cross-correlation, confirming that the PEP-induced re-hybridization is due to dissociation at the active site interface. Furthermore, this solution measurement is a true equilibrium measurement, eliminating the product separation that occurs during an electrophoretic separation.
Within the mixed tetramer, fluorescently labeled subunits are in close proximity, leading to inter-subunit energy transfer between fluorophores. This FRET has been exploited to obtain kinetic data. Starting with the mixed tetramer, addition of PEP induces re-hybridization and at equilibrium the population of mixed tetramer is expected to represent approximately 50% of the total enzyme. Monitoring the ratio of donor to acceptor emission after PEP addition yields a time-dependent decay in the FRET signal, as the mixed tetramer re-hybridizes to a mixed population of tetramers. When the decay is fit to a single-exponential, its concentration dependence yields an apparent dissociation constant of 110 nM.
The steady-state anisotropy of the Texas Red-labeled enzyme has been used to determine the subunit affinities of the various ligand-bound states of the enzyme. The anisotropy of all these enzyme forms is low at high concentration, and increases following hybridization to an excess of unlabeled enzyme. These data suggest that the Texas Red is undergoing homotransfer between subunits. In the absence of ligands or in the presence of F6P, no change in anisotropy is observed at any protein concentration examined. Conversely, PEP-bound PFK undergoes dissociation with Kd = 110 nM and F6P/PEP-bound PFK undergoes dissociation with Kd = 0.81 nM. The enzyme consequently undergoes a ∼130-fold or ∼3 kcal/mol stabilization to PEP-induced dissociation by the simultaneous binding of F6P. It is relevant that the BsPFK with the inhibitor phosphoglycolate bound exhibits an altered structure of the allosteric site interface as determined by X-ray crystallography (13). To the extent that these altered structural features are also present when PEP is bound, they are likely to be involved in the weakened dimer-dimer affinity. Unfortunately no data are available that might suggest how both F6P and PEP together influence the structure of the enzyme.
Protein-ligand interactions that mediate or regulate protein-protein associations are ubiquitous in nature. We have demonstrated conclusively that the binding of the allosteric inhibitor PEP causes a significant reduction of protein-protein affinity at a site distant from the binding site in BsPFK, across the active-site dimer-dimer interface. Ligand-mediated loss of protein affinity of this type is common and is observed in such generic biochemical phenomena as transcription factor association with nucleic acids. Ligand-binding has been shown to cause eukaryotic PFK aggregation to high order oligomers (33) and inhibitor-induced dissociation has been demonstrated for both Thermus thermophilus PFK (34) and for site mutants of B. stearothermophilus PFK (35). These data represent the first direct correlation between ligand-binding and protein association for wild type B. stearothermophilus PFK. Since F6P promotes the stability of the tetramer and PEP weakens both the active site dimer-dimer interface and the binding of F6P to the active site, thermodynamic linkage requires that all these equilibria be involved in establishing the overall magnitude of the apparent influence of PEP on F6P binding. However, the precise molecular role that the subunit interactions play in the coupling between PEP and F6P remains to be elucidated.
Acknowledgements
The authors wish to acknowledge the assistance and expertise of Dr. Mauricio Lasagna in the collection and analysis of frequency-domain fluorescence lifetime data.
Abbreviations
- EPPS
N-[2-hydroxyethyl]piperazine-N′-[3-propanesulfonic acid]
- Tris
Tris(hydroxymethyl)aminomethane
- MgCl2
magnesium chloride
- NH4Cl
ammonium chloride
- EDTA
ethylenediaminetetraacetic acid
- BsPFK
Bacillus stearothermophilus phosphofructokinase
- F6P
fructose-6-phosphate
- PEP
phospho(enol)pyruvate
- ADP
adenosine-5′-diphosphate
- FCS
fluorescence correlation spectroscopy
- PCH
photon counting histogram
- 2PE
two photon excitation
- Ti: Sapph
Titanium: Sapphire
- Nb:YVO4
Neobium: yttrium vanadate laser
- PAGE
polyacrylamide gel electrophoresis
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
Footnotes
This work was supported by National Institutes of Health grant GM33216 and Robert A. Welch Foundation grant A1543.
References
- 1.Bloxham DP, Lardy HA. Phosphofructokinase. In: Boyer P, editor. The Enzymes. 3rd ed. Academic Press; New York: 1973. pp. 239–278. [Google Scholar]
- 2.Blangy D, Buc H, Monod J. Kinetics of the allosteric interactions of phosphofructokinase from Escherichia coli. J. Mol. Biol. 1968;31:13–35. doi: 10.1016/0022-2836(68)90051-x. [DOI] [PubMed] [Google Scholar]
- 3.Changeux JP, Edelstein SE. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 4.Reinhart GD. Quantitative analysis and interpretation of allosteric behavior. Methods Enzymol. 2004;380:187–203. doi: 10.1016/S0076-6879(04)80009-0. [DOI] [PubMed] [Google Scholar]
- 5.Kimmel JL, Reinhart GD. Isolation of an individual allosteric interaction in tetrameric phosphofructokinase from Bacillus stearothermophilus. Biochemistry. 2001;40:11623–11629. doi: 10.1021/bi010844a. [DOI] [PubMed] [Google Scholar]
- 6.Fenton AW, Paricharttanakul NM, Reinhart GD. Identification of substrate contact residues important for the allosteric regulation of phosphofructokinase from Escherichia coli. Biochemistry. 2003;42:6453–6459. doi: 10.1021/bi034273t. [DOI] [PubMed] [Google Scholar]
- 7.Kimmel JL, Reinhart GD. Reevaluation of the accepted allosteric mechanism of phosphofructokinase from Bacillus stearothermophilus. Proc. Natl. Acad. Sci. USA. 2000;97:3844–3849. doi: 10.1073/pnas.050588097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ortigosa AD, Kimmel JL, Reinhart GD. Disentangling the web of allosteric communication in a homotetramer: heterotropic inhibition of phosphofructokinase from Bacillus stearothermophilus. Biochemistry. 2004;43:577–586. doi: 10.1021/bi035077p. [DOI] [PubMed] [Google Scholar]
- 9.Reinhart GD, Hartleip SB, Symcox MM. Role of coupling entropy in establishing the nature and magnitude of allosteric response. Proc. Natl. Acad. Sci. USA. 1989;86:4032–4036. doi: 10.1073/pnas.86.11.4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rypniewski WR, Evans PR. Crystal structure of unliganded phosphofructokinase from Escherichia coli. J. Mol. Biol. 1989;207:805–521. doi: 10.1016/0022-2836(89)90246-5. [DOI] [PubMed] [Google Scholar]
- 11.Shirikihara Y, Evans PR. Crystal structure of the complex of phosphofructokinase from Escherichia coli with its reaction products. J. Mol. Biol. 1988;204:973–994. doi: 10.1016/0022-2836(88)90056-3. [DOI] [PubMed] [Google Scholar]
- 12.Hudson PJ, Hengartner H, Harris JI. Protein: Structure, Function and Industrial Applications. Oxford University Press; New York: 1979. The primary structure of phosphofructokinase from Bacillus stearothermophilus; pp. 341–347. [Google Scholar]
- 13.Evans PR, Hudson PJ. Structure and control of phosphofructokinase from Bacillus stearothermophilus. Nature. 1979;279:500–504. doi: 10.1038/279500a0. [DOI] [PubMed] [Google Scholar]
- 14.LeBras G, Auzat I, Garel JR. Tetramer-dimer equilibrium of phosphofructokinase and formation of hybrid tetramers. Biochemistry. 1995;34:13203–13210. doi: 10.1021/bi00040a036. [DOI] [PubMed] [Google Scholar]
- 15.Deville-Bonne D, LeBras G, Teschner W, Garel JR. Ordered disruption of subunit interfaces during the stepwise reversible dissociation of Escherichia coli phosphofructokinase with KSCN. Biochemistry. 1989;28:1917–1922. doi: 10.1021/bi00430a073. [DOI] [PubMed] [Google Scholar]
- 16.Johnson JL, Lasagna MD, Reinhart GD. Influence of a sulfhydryl cross-link across the allosteric-site interface of E. coli phosphofructokinase. Prot. Sci. 2001;10:2186–2194. doi: 10.1110/ps.02401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Elson E, Magde D. Fluorescence correlation spectroscopy. I. Conceptual basis and theory. Biopolymers. 1974;13:1–27. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 18.Magde D, Elson EL, Webb WW. Fluorescence correlation spectroscopy. II. An experimental realization. Biopolymers. 1974;13:29–60. doi: 10.1002/bip.1974.360130103. [DOI] [PubMed] [Google Scholar]
- 19.Thompson NL. Fluorescence correlation spectroscopy. In: Lakowicz JR, editor. Topics in Fluorescence Spectroscopy, Volume 1: Techniques. Plenum Press; New York: 1991. pp. 337–378. [Google Scholar]
- 20.Chen Y, Muller JD, Berland KM, Gratton E. Fluorescence Fluctuation Spectroscopy. Methods. 1999;19:234–252. doi: 10.1006/meth.1999.0854. [DOI] [PubMed] [Google Scholar]
- 21.Schwille P, Meyer-Almes FJ, Rigler R. Dual color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 1997;72:1878–1886. doi: 10.1016/S0006-3495(97)78833-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kettling U, Koltermann A, Schwille P, Eigen M. Real-time enzyme kinetics monitored by dual-color fluorescence cross-correlation spectroscopy. Proc. Natl. Acad. Sci. USA. 1998;95:1416–1420. doi: 10.1073/pnas.95.4.1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinze KG, Koltermann A, Schwille P. Simultaneous two-photon excitation of distinct labels for dual-color fluorescence crosscorrelation analysis. Proc. Nat. Acad. Sci. USA. 2000;97:10377–10382. doi: 10.1073/pnas.180317197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Müller JD, So PTC, Gratton E. The photon counting histogram in fluorescence fluctuation spectroscopy. Biophys. J. 1999;77:553–567. doi: 10.1016/S0006-3495(99)76912-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang B, Perroud TD, Zare RN. Photon Counting Histogram: One Photon Excitation. ChemPhysChem. 2004;5:1523–1531. doi: 10.1002/cphc.200400176. [DOI] [PubMed] [Google Scholar]
- 26.Müller JD, Chen Y, Gratton E. Resolving heterogeneity on the single molecule level with the photon counting histogram. Biophys. J. 2000;78:474–486. doi: 10.1016/S0006-3495(00)76610-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Callis PR. Two-photon induced fluorescence. Ann. Rev. Phys. Chem. 1997;48:271–297. doi: 10.1146/annurev.physchem.48.1.271. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JL, Reinhart GD. Failure of a two-state model to describe the influence of phospho(enol)pyruvate on phosphofructokinase from Escherichia coli. Biochemistry. 1997;36:12814–12822. doi: 10.1021/bi970942p. [DOI] [PubMed] [Google Scholar]
- 29.Haugland RP. 9th Edition Molecular Probes, Inc.; Eugene OR: 2002. Handbook of Fluorescent Probes and Research Products. [Google Scholar]
- 30.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd Edition Plenum Press; New York: 1999. [Google Scholar]
- 31.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Methods Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 32.Brismar H, Trepte O, Ulhafke B. Spectra and fluorescence lifetimes of lissamine rhodamine, tetramethylrhodamine isothiocyanate, texas red and cyanine 3.18 fluorophores: influences of some environmental factors recorded with a scanning confocal microscopy. J. Hist. Cytochem. 1995;43:699–707. doi: 10.1177/43.7.7608524. [DOI] [PubMed] [Google Scholar]
- 33.Reinhart GD, Lardy HA. Rat liver phosphofructokinase: use of fluorescence polarization to study aggregation at low protein concentrations. Biochemistry. 1980;19:1484–1490. doi: 10.1021/bi00548a035. [DOI] [PubMed] [Google Scholar]
- 34.Xu J, Oshima T, Yoshida M. Tetramer-dimer conversion of phosphofructokinase from Thermus thermophilus induced by its allosteric effectors. J. Mol. Biol. 1990;215:597–606. doi: 10.1016/S0022-2836(05)80171-8. [DOI] [PubMed] [Google Scholar]
- 35.Riley-Lovingshimer MR, Ronning DR, Sacchettini JC, Reinhart GD. Reversible ligand-induced dissociation of a tryptophan-shift mutant of phosphofructokinase from Bacillus stearothermophilus. Biochemistry. 2002;41:12967–12974. doi: 10.1021/bi0263412. [DOI] [PubMed] [Google Scholar]