

Key Points
Interferons (IFNs) are key components of the innate immune response and the first line of defence against virus infection.
Among the hundreds of IFN-induced genes, only a few have been ascribed direct antiviral activity in vivo: ISG15 (IFN-stimulated protein of 15 kDa), the Mx (myxovirus resistance) proteins, 2′,5′-oligoadenylate synthetase (OAS)-regulated ribonuclease L (RNaseL) and protein kinase R (PKR).
These proteins separately block viral transcription, degrade viral RNA, inhibit translation or modify the proteasome to control all steps of viral replication.
ISG15 is part of a ubiquitin-like pathway that modulates the function of numerous protein targets.
The Mx proteins seem to survey exocytic events and mediate vesicle trafficking to trap viral components.
The OAS-regulated RNaseL pathway degrades single-stranded RNA in virus-infected cells.
PKR inhibits translation and participates in signal transduction.
Additional functions of each of these proteins are still being uncovered, suggesting they have broader roles in the host immune response.
Type I interferons (IFNs) provide the first line of defence against viral infection. As discussed in this Review, the IFN-induced antiviral effector proteins, such as ISG15, Mx proteins, ribonuclease L and protein kinase R, are important components of this response.
Abstract
Since the discovery of interferons (IFNs), considerable progress has been made in describing the nature of the cytokines themselves, the signalling components that direct the cell response and their antiviral activities. Gene targeting studies have distinguished four main effector pathways of the IFN-mediated antiviral response: the Mx GTPase pathway, the 2′,5′-oligoadenylate-synthetase-directed ribonuclease L pathway, the protein kinase R pathway and the ISG15 ubiquitin-like pathway. As discussed in this Review, these effector pathways individually block viral transcription, degrade viral RNA, inhibit translation and modify protein function to control all steps of viral replication. Ongoing research continues to expose additional activities for these effector proteins and has revealed unanticipated functions of the antiviral response.
Main
Interferon (IFN) was discovered more than 50 years ago as an agent that inhibited the replication of influenza virus1. The IFN family of cytokines is now recognized as a key component of the innate immune response and the first line of defence against viral infection. Accordingly, IFNs are currently used therapeutically, with the most noteworthy example being the treatment of hepatitis C virus (HCV) infection, and they are also used against various other disorders, including numerous malignancies and multiple sclerosis (reviewed in Ref. 2).
Three classes of IFN have been identified, designated types I to III, and are classified according to the receptor complex they signal through (Fig. 1). The type II class of IFN comprises the single IFNγ gene product that binds the IFNγ receptor (IFNGR) complex, and mediates broad immune responses to pathogens other than viruses. The more recently described type III IFNs include three IFNλ gene products that signal through receptors containing IFNLR1 (IFNλ receptor 1; also known as IL-28Ra) and IL-10R2 (also known as IL-10Rβ). So far, little is known about the type III IFNs, although they are known to regulate the antiviral response and have been proposed to be the ancestral type I IFNs3. Type I IFNs, which in humans comprise 13 IFNα subtypes, IFNβ, IFNκ, IFNɛ, IFNο, IFNτ and IFNδ, engage the ubiquitously expressed IFNAR (IFNα receptor) complex that is composed of IFNAR1 and IFNAR2. The function of type I IFNs is well characterized and they are known to be essential for mounting a robust host response against viral infection. Accordingly, IFNAR-deficient mice have increased susceptibility to numerous viruses but maintain resistance to other microbial pathogens, such as Listeria monocytogenes4,5. Similarly, humans with genetic defects in components of the IFNR signalling pathway (STAT1 (signal transducer and activator of transcription 1), TYK2 (tyrosine kinase 2) or UNC93B) die of viral disease, with the defect in IFNAR (rather than IFNGR) signalling having the more significant role6,7,8,9.
Figure 1. Interferon receptor signalling.
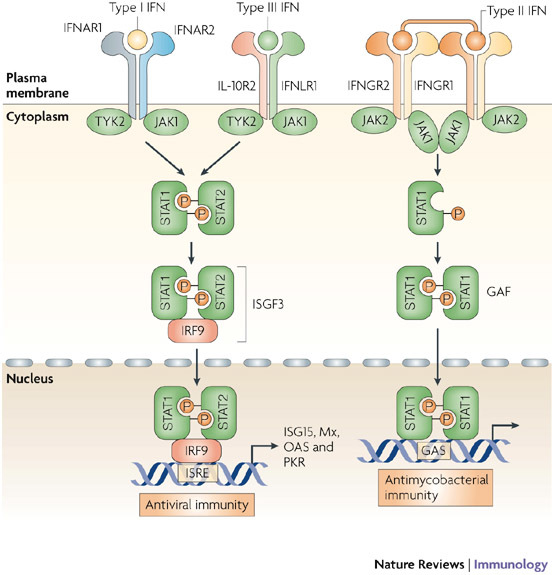
The action of the interferons (IFNs) is mediated through three receptor complexes: a heterodimer of IFNα receptor 1 (IFNAR1) and IFNAR2 binds type I IFNs; the interleukin-10 receptor 2 (IL-10R2) associates with IFNLR1 (IFNλ receptor 1) to bind the three IFNλ subtypes; and a tetramer consisting of two IFNGR2 (IFNγ receptor 2) chains and two IFNGR1 chains binds dimers of the type II IFNγ. Following binding by type I IFNs, signal transduction is initiated by pre-associated tyrosine kinases (JAK1 and TYK2 (tyrosine kinase 2)), which phosphorylate IFNAR1 and leads to the recruitment and phosphorylation of the signal transducers and activators of transcription (STATs). STAT heterodimers associate with IFN-regulatory factor 9 (IRF9) to form IFN-stimulated gene factor 3 (ISGF3), or STAT homodimers to form the IFNγ activation factor (GAF). These complexes translocate to the nucleus to induce IFN-stimulated genes from IFN-stimulated response elements (ISREs) or GAS promoter elements, for type I and type III, or type II IFN responses, respectively. Divergence from this simplified signalling pathway can occur, for example, type I IFNs are reported to elicit STAT homodimers, and more complicated interplay, with activation of other STAT proteins, occurs than is shown here. ISG15, IFN-stimulated protein of 15 kDa; Mx, myxovirus resistance; OAS, 2′,5′-oligoadenylate synthetase; PKR, protein kinase R.
The binding of type I IFNs to the IFNAR initiates a signalling cascade, which leads to the induction of more than 300 IFN-stimulated genes (ISGs)10. However, relatively few of these ISGs have been directly implicated in instigating the antiviral state. Instead, many of the gene products encode pattern-recognition receptors (PRRs) that detect viral molecules and modulate signalling pathways, or transcription factors that form an amplification loop resulting in increased IFN production and protection from virus spread to limit disease. Nevertheless, some ISGs encode proteins with potential for direct antiviral activity, including proteins that catalyse cytoskeletal remodelling, that induce apoptosis, that regulate post-transcriptional events (splicing, mRNA editing, RNA degradation and the multiple steps of protein translation) and proteins that are involved in subsequent post-translational modification. Indeed, several such proteins, ISG15 (IFN-stimulated protein of 15 kDa), the GTPase Mx1 (myxovirus resistance 1), ribonuclease L (RNaseL) and protein kinase R (PKR; also known as EIF2αK2), have been shown to function as antiviral effectors in studies of knockout mice. Mice with mutations or deficiencies in key steps of the pathways that are triggered by these proteins have increased susceptibility to viral infection.
In this Review, we summarize our current understanding of the role of ISG15, Mx1, RNaseL and PKR in the antiviral host immune response. However, although these four proteins are known to be important in the antiviral response, they do not represent the complete repertoire of antiviral effectors. Additional ISGs that probably also have important roles in antiviral activities include: the deaminases ADAR1 (adenosine deaminase, RNA-specific 1) and APOBEC (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide) proteins; the exonuclease ISG20; members of the tripartite-motif-containing (TRIM) proteins such as TRIM19 (also known as PML); the putative S-adenosyl-L-methionine viperin; and the highly IFN-induced translation regulators IFIT1 (IFN-induced protein with tetratricopeptide repeats 1) and IFIT2. All of these ISGs have been reported to function as antiviral proteins in vitro but further investigation using the appropriate gene knockout mouse models is needed for a better understanding of their relative importance in the antiviral response. In addition, responses elicited by IFN-induced microRNAs are just emerging as regulators of viral infection11.
ISG15, the Mx proteins, the 2′,5′-oligoadenylate synthetase (OAS)-directed RNaseL pathway and PKR have varying responsiveness to type I IFNs: ISG15 is one of the most highly induced ISGs and, when coupled to protein substrates, it modulates numerous cellular activities; Mx proteins are also highly induced by type I IFNs and then assemble into oligomers that are constitutively active; OAS proteins are expressed at low levels in unstimulated cells, and are considerably induced by type I IFNs produced in response to viral RNA; and PKR is constitutively expressed as an inactive kinase that is activated by viral RNA, then further upregulated by type I IFNs. This variable responsiveness to IFN also underlies the function of each protein as solely an IFN effector, or as PRRs to enhance the IFN response.
ISG15
One of the most prominent ISGs to be induced during viral infection and the ensuing type I IFN response is the ∼15 kDa protein ISG15. Although the ISG15 gene was cloned over 20 years ago12, an antiviral function of the encoded protein has only recently been established and considerable work is still required to detail all of its actions and to resolve contradictory findings.
ISG15 was identified soon after the landmark discovery of ubiquitin, and was immediately recognized as a ubiquitin homologue13 (Fig. 2). Protein ubiquitylation regulates many aspects of the innate immune response, including intracellular signal transduction (for example, the activation of nuclear factor-κB (NF-κB)), and functions of the adaptive immune system, such as initiating tolerance (reviewed in Ref.14). Given the importance of ubiquitylation in the immune response, it is perhaps not surprising that there is an IFN-regulated ubiquitin-like protein response. This response, as mediated by ISGs, is referred to as ISGylation.
Figure 2. Domain structure of antiviral proteins.
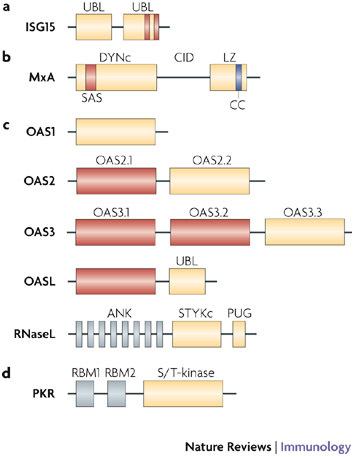
a | Interferon-stimulated protein of 15 kDa (ISG15) contains two ubiquitin-like (UBL) domains. The C-terminal UBL encodes conserved hydrophobic patches, charged pockets (indicated by vertical bars) and diglycine residues similar to ubiquitin and other ubiquitin-like proteins. Accordingly, the C-terminal UBL is sufficient for activation and transfer of ISG15 to E1 and E2 enzymes. However, the N-terminal UBL seems to be necessary for E3-mediated conjugation to protein substrates. b | The distinguishing features of the dynamin protein family of Mx GTPases are the large GTP-binding domain (DYNc) that contains three consensus GTP-binding elements (not indicated) and a self-assembly sequence (SAS), the central interactive domain (CID), and the C-terminal leucine zipper (LZ). The LZ region contains a coiled-coil (CC) domain at its C terminus that forms bundles of α-helices involved in protein–protein interactions. c | The 2′,5′-oligoadenylate synthetase 1 (OAS1), OAS2 and OAS3 proteins contain 1, 2 or 3 copies respectively, of the OAS domains. Duplicated OAS domains in OAS2 and OAS3, and the single domain in OASL (OAS-like) are catalytically inactive (shown in red). The C-termini of all the OAS proteins are variable, with OASL also encoding a unique UBL domain. The OAS domain produces 2′,5′-oligoadenylates that activate ribonuclease L (RNaseL). RNaseL has eight N-terminal ankyrin repeats (ANK), which mediate protein–protein interactions, and two kinase-like domains, a Ser/Thr/Tyr kinase (STYKc) and a PUG domain that are found in some kinases and several other nuclear proteins. However, neither domain has functional kinase activity. d |Protein kinase R (PKR) contains two repeated RNA-binding motifs (RBMs), which together constitute the N-terminal RNA-binding domain. On binding of viral RNA, steric inhibition of enzyme activity is relieved, leading to the activation of the C-terminal serine/threonine protein kinase domain (S/T-kinase).
ISG15 is expressed as a 165 amino-acid precursor that is subsequently processed to expose the C-terminal sequence LRLRGG. The equivalent diglycine residues within this motif in ubiquitin are adenylated and conjugated by a thiolester bond to cysteine residues of three enzymes, a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase enzyme (E3), before being transferred to lysine residues on protein substrates. As the ubiquitin E1 enzyme (UBE1) is unable to form a thiolester bond with ISG15, ISGylation was initially thought to require a parallel and distinct pathway15. However, having identified the enzymes that catalyse ISGylation, it is now becoming clear that there is direct interplay between ubiquitylation and ISGylation. The enzyme UBE1L (E1-like ubiquitin-activating enzyme) was shown to be the specific ISG15-activating enzyme (Ref. 16). Challenging this specificity, two E2 ubiquitin-conjugating enzymes, UBCH6 (also known as UBE2E1) and UBCH8 (also known as UBE2L6) were also shown to serve as ISG15 carriers17,18. Downregulation of UBCH8 expression by RNA interference indicated that UBCH8 functions as the main E2 ISG15-conjugating enzyme in HeLa cells19. Finally, two E3 ubiquitin ligases, HERC5 (homologous to the E6-associated protein C terminus (HECT) domain and RCC1-like domain containing protein 5) and TRIM25, have also been shown to conjugate ISG15 to protein substrates, through their respective HECT or RING (really interesting new gene) domains20,21. Appropriately, all enzymes identified in the ISGylation pathway are coordinately induced by type I IFNs (Fig. 3). As with ubiquitylation, ISGylation is reversible and several enzymes that catalyse the hydrolysis of ISG15 (termed deISGylation) have been identified, including ubiquitin-specific protease 18 (USP18, also known as UBP43), USP2, USP5, USP13 and USP14 (Refs 22,23).
Figure 3. Mechanism of action of ISG15.
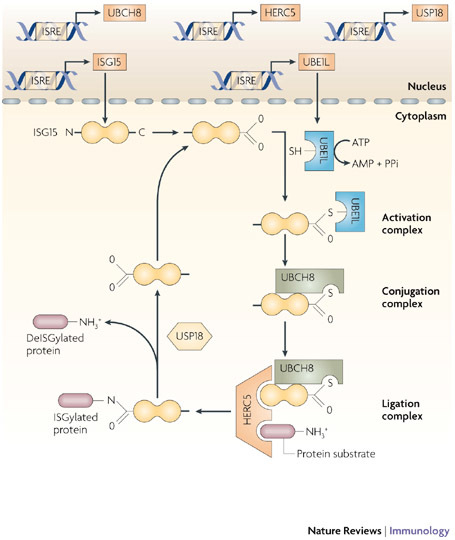
The expression of interferon (IFN)-stimulated protein of 15 kDa (ISG15), the E1-activating enzyme UBE1L (E1-like ubiquitin-activating enzyme) and multiple E2-conjugating enzymes (shown here as an example is UBCH8) and E3-ligase enzymes (such as HERC5 (homologous to the E6-associated protein C terminus domain and RCC1-like domain containing protein 5)) is coordinately induced by type I IFNs through IFN-stimulated response elements (ISREs) in their respective gene promoter regions. E1, E2 and E3 proteins sequentially catalyse the conjugation of ISG15 to numerous protein substrates to modulate pleiotropic cellular responses to inhibit virus production. This process (known as ISGylation) is reversibly regulated by proteases (such as ubiquitin-specific protease 18 (USP18)), which are also induced by IFNs.
At least 158 putative ISG15 target proteins have been identified so far24,25,26. Many of these substrates have important roles in the type I IFN response, including the signalling components JAK1 (Janus kinase 1) and STAT1, the PRRs, such as RIG-I (retinoic-acid-inducible gene I), and the antiviral effector proteins MxA, PKR and RNaseL24. Unlike ubiquitylation, ISGylation does not promote degradation of the target protein (as occurs following K48-linked ubiquitin), but instead parallels the activating effects of ubiquitylation (mediated by K63-linked ubiquitin). Accordingly, ISG15 has been reported to prevent virus-mediated degradation of IFN-regulatory factor 3 (IRF3), thereby increasing the induction of IFNβ expression27. ISGylation has also been shown to modulate the function of enzymes. An example of this is the increased affinity of ISGylated eukaryotic translation initiation factor 4E family member 2 (EIF4E2; also known as 4EHP) for the 5′ cap structure of RNA28. Conversely, conjugation of ISG15 to protein phosphatase 1B (PPM1B) suppressed the activity of this enzyme, thereby enhancing NF-κB signalling29.
In addition to its intracellular role, ISG15 is secreted in large amounts and has been shown to act as a cytokine to modulate immune responses30. The mechanism by which extracellular ISG15 functions is unresolved. Ubiquitin is also secreted from cells and has immunomodulatory effects that are not understood31, although it might be involved in extracellular ubiquitylation, as suggested by analysis of surface proteins on spermatozoa during post-testicular maturation32. It is therefore possible that secreted ISG15 might function in extracellular ISGylation, a possibility that could be tested by studying the effect of treating cells from Ube1l−/− mice with ISG15.
Consistent with its designation as an antiviral protein, mice deficient in ISG15 have increased susceptibility to infection with several viruses, including the influenza A and B viruses, Sindbis virus, herpes simplex virus 1 (HSV-1) and murine γ-herpesvirus33,34. In addition, infection of Ifnar1−/− mice with a recombinant chimeric Sindbis virus expressing ISG15 protected against the Sindbis-virus-induced lethality that occurs following infection with wild-type virus33. Compellingly, this protective effect of ISG15 expression required the conserved LRLRGG sequence at the C terminus of ISG15 (Ref. 35). In contrast to these reports, however, a similar chimeric ISG15-expressing Sindbis virus did not rescue Ifnar1−/− mice from Sindbis-virus-induced lethality, although it did provide modest protection in vitro36. Ablation of the deISGylation enzyme USP18 in mice increased their resistance to viral infection, notably to vesicular stomatitis virus (VSV) infection. However, the expected reciprocal increased sensitivity to VSV has not been observed in either Isg15−/− or Ube1l−/− mice37. Isg15−/− mouse embryonic fibroblasts were, however, more susceptible to VSV infection, although this sensitivity was lost after type I IFN treatment, suggesting that circulating IFNs in vivo may obscure some viral resistance mechanisms27.
Other in vitro experiments support a role for ISG15 in mediating resistance to Ebola virus, through ISGylation of the E3 ubiquitin ligase NEDD4 (neural precursor cell expressed, developmentally downregulated 4), thereby blocking the activity of NEDD4 (Ref. 38). Similarly, ubiquitylation of the HIV-1 proteins Gag and Tsg101 is inhibited by ISG15 (Ref. 39). Ubiquitylation of NEDD4 and Gag or Tsg101 mediate the release of virions from cells. Further corroboration of an antiviral role for ISG15 comes from the identification of viral proteins that have evolved to target different steps of ISGylation. Nonstructural protein 1 (NS1) of influenza B virus binds ISG15 at the N-terminus and blocks ISGylation16,40. Finally, several viral proteases from severe acute respiratory syndrome (SARS)-associated coronavirus, Crimean-Congo haemorrhagic fever virus, equine arteritis virus, porcine respiratory and reproductive syndrome virus, and Sindbis virus have been found to mediate deISGylation41,42,43.
Therefore, an important role for ISGylation in the IFN-mediated antiviral response, through the modification of components of the host immune response or viruses, is emerging.
Mx GTPases
In addition to components of the ISGylation pathway, type I IFNs induce the expression of several guanine-hydrolysing proteins. This class of protein is involved in scission to mediate vesicle budding, organogenesis and cytokinesis. There is evidence that four families within this protein class are involved in host resistance to pathogens, and these include the p47 guanylate-binding proteins (GBPs), the p65 GBPs, the very large inducible GTPases and the Mx proteins44. However, of these four protein families, only the Mx proteins have a well-characterized antiviral role and show a strict dependence on type I and III IFNs for their expression45.
The Mx family GTPases, which comprise MxA and MxB in humans and Mx1 and Mx2 in mice, were first identified as antiviral proteins by the observation that the sensitivity of many inbred mouse strains to orthomyxomavirus was solely due to mutations within the Mx locus on chromosome 16 (Refs 46,47,48). This sensitivity could be rescued by restoration of Mx1 expression49. Strikingly, constitutive expression of the human equivalent of mouse Mx1, MxA, in IFNAR-deficient mice confers full resistance to otherwise fatal infection with Thogoto virus, LaCrosse virus or Semliki Forest virus49.
The two human Mx proteins are encoded on chromosome 21, in a region syngeneic to the Mx region on mouse chromosome 16 (Refs 50,51). The human proteins and mouse Mx2 are cytoplasmic, whereas mouse Mx1 localizes to the nucleus. This differential distribution in mice is thought to allow each protein to target viruses that replicate in either cell compartment52. However, of the Mx family of proteins, only human MxA has been shown to have antiviral activity, and this is targeted against both nuclear and cytoplasmic viruses.
Viruses that are susceptible to the activities of Mx proteins include orthomyxomaviruses, paramyxomaviruses, rhabdoviruses, togaviruses and bunyaviruses. Similarly, human MxA has been shown to inhibit all infectious genera of the Bunyaviridae family (orthobunyavirus, hantavirus, phlebovirus and dugbe virus)53. Members of other virus families, such as the clinically significant coxsackie virus (from the Picornaviridae family) and hepatitis B virus (HBV; from the Hepadnaviridae family) are also susceptible to human MxA antiviral activity54,55. In addition, genetic studies of human populations have shown that a polymorphism in the MXA gene correlates with increased susceptibility to HCV56, HBV57 and measles virus, with the latter associated with higher rates of subacute sclerosing panencephalitis58. Appropriately, Mx proteins are expressed by various cell types in peripheral tissues, for example, by hepatocytes, endothelial cells and immune cells, including peripheral blood mononuclear cells, plasmacytoid dendritic cells and myeloid cells59.
The Mx proteins have a large (relative to many other GTPases) N-terminal GTPase domain, a central interacting domain (CID) and a C-terminal leucine zipper (LZ) domain (Fig. 2). Both the CID and the LZ domain are required to recognize target viral structures. The main viral target seems to be viral nucleocapsid-like structures60. By virtue of their location near the smooth endoplasmic reticulum, Mx proteins can survey exocytic events and mediate vesicle trafficking to trap essential viral components, and in so doing, they prevent viral replication at early time points61 (Fig. 4). Both MxA and Mx1 associate with subunits of the influenza virus polymerase (PB2 and nucleocapsid protein) to block viral gene transcription62. This is a potent antiviral measure, which effectively prevents the generation of viral mutants that escape Mx-mediated antiviral mechanisms. As a result, few viral countermeasures against Mx proteins have been identified.
Figure 4. Mechanism of action of MxA.
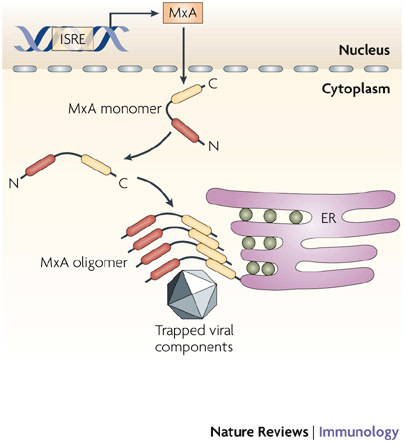
Following stimulation with type I interferons (IFNs), MXA (myxovirus-resistance A) gene expression is induced through an IFN-stimulated response element (ISRE) in the gene promoter. The MxA protein accumulates in the cytoplasm on intracellular membranes (such as the endoplasmic reticulum, ER) as oligomers formed by association between the leucine zipper (LZ) domain and central interactive domain of the protein. Following viral infection, MxA monomers are released and bind viral nucleocapsids or other viral components, to trap and then degrade them.
Most viral escape mechanisms that have been described target type I IFN signalling: for example, highly virulent strains of influenza virus increase their replicative fitness to effectively 'out run' the IFN response63. More directly, the HBV precore or core protein has been reported to interact with the MXA promoter to prevent MXA gene expression64. Also, West Nile virus (WNV) produces what seem to be decoy cytoplasmic membrane structures that 'hide' crucial viral replication components from Mx proteins65.
In contrast to ISG15 and Mx, the OAS and RNaseL pathway and PKR are expressed ubiquitously at low levels but their level of expression can be increased by exposure to type I IFNs. Constitutive levels of OAS and PKR mean that these proteins function not only as effector proteins but also as double-stranded RNA (dsRNA)-specific PRRs to trigger the antiviral response.
The OAS and RNaseL pathway
Initially identified as IFN-induced proteins that generate low-molecular-weight inhibitors of cell-free protein synthesis, the OAS proteins are distinguished by their capacity to synthesize 2′,5′-linked phosphodiester bonds to polymerize ATP into oligomers of adenosine66,67. These unique 2′,5′-oligomers specifically activate the latent form of RNaseL, which can then mediate RNA degradation68. In this way, OAS in combination with RNaseL constitutes an antiviral RNA decay pathway. As the OAS proteins are constitutively expressed at low levels, they can act as PRRs for the detection of viral dsRNA in the cytoplasm65. RNA degraded by RNaseL can activate the other cytoplasmic PRRs, such as RIG-I and MDA5 (melanoma differentiation-associated gene 5), resulting in the induction of type I IFN gene expression. This accounts for the observation that RNaseL-deficient cells show markedly decreased IFNβ production owing to reduced signalling through these PRRs69.
The four OAS genes identified in humans, termed OAS1, OAS2, OAS3 and OASL (OAS-like), have been mapped to chromosome 12 (chromosome 5 in mice) (reviewed in Ref. 70). OAS1 has two spliced forms in humans (eight in mice) that produce two proteins of 40 and 46 kDa that differ at their C termini by 18 and 54 amino acids, respectively. OAS2 produces four alternatively spliced transcripts that encode two proteins of 69 and 71 kDa. OAS3 encodes a single transcript that produces a 100 kDa protein. These proteins have considerable homology to each other, with OAS1, OAS2 and OAS3 encoding one, two and three OAS domains, respectively (Fig. 2). The most distinctive of the OAS proteins is OASL. Two OASL transcripts are expressed, which produce two proteins of 30 and 59 kDa. The higher molecular weight OASL protein contains a putative nuclear-localization signal (RKVKEKIRRTR) at its C terminus that probably accounts for its unique distribution in the cell compared with the other OAS isoforms. The OASL protein also has an OAS domain, but mutations at key residues disable its catalytic function. Interestingly, one of the two mouse homologues of OASL retains its 2′,5′-polymerase activity. In addition to the OAS domain, OASL has a unique 160 amino-acid C terminus that encodes a ubiquitin-like domain that is homologous to ISG15. Accordingly, OASL becomes conjugated through ISGylation to cellular proteins following the treatment of cells with type I IFNs71.
There seems to be differential expression and induction of each form of the human OAS proteins72. Also, each of the three functional OAS proteins has unique biological functions. A tripeptide motif (CFK) within the OAS domains of OAS1 and OAS2 mediates oligomerization, so the catalytically active form of these enzymes is a tetramer and dimer for OAS1 and OAS2, respectively73. This tripeptide motif is not conserved in the OAS domains of OAS3 and OASL and therefore these proteins function as monomers. The polymerization of OAS monomers influences their processivity; OAS3 synthesizes dimeric molecules of 2′,5′-linked oligomers, whereas OAS1 and OAS2 can synthesize trimeric and tetrameric oligomers74,75. The dimeric 2′,5′-linked oligomers are not efficient activators of RNaseL76 and, consequently, are thought to regulate alternative processes, with one report suggesting a role in gene expression by regulating DNA topoisomerase I77.
The 2′,5′-dependent RNaseL is expressed as an 80 kDa protein with two kinase-like domains (PUG and STYKc) and eight ankyrin repeats (Fig. 2) (reviewed in Ref. 78). The enzyme is constitutively expressed as an inactive monomer and is activated through binding of 2′,5′-linked oligomers (generated by OAS proteins) to the ankyrin repeats, which subsequently leads to homodimerization79. The active dimeric enzyme then degrades single-stranded RNA (ssRNA)80,81 (Fig. 5). Owing to their perceived common effect on RNaseL, the antiviral function of the OAS proteins has been investigated using RNaseL-deficient mice82. These mice show increased susceptibility to RNA viruses from the Picornaviridae, Reoviridae, Togaviridae, Paramyxoviridae, Orthomyxoviridae, Flaviviridae and Retroviridae families78. An antiviral role for RNaseL against DNA viruses is less directly established, although as these viruses produce dsRNA replicative intermediates they can induce the production of 2′,5′-linked oligomers. However, activation of RNaseL following binding of 2′,5′-linked oligomers is less commonly observed in response to DNA viruses, presumably because of virally encoded inhibitory factors, such as the E3L protein from vaccinia virus83. The direct importance of OAS proteins in the antiviral response in humans is highlighted by genetic studies showing that polymorphisms within a splice-acceptor site of the OAS1 gene (producing two isoforms of the enzyme with different activities) significantly correlate with the antiviral response to the yellow fever vaccine in immunization clinical trials84.
Figure 5. The OAS1–RNaseL antiviral pathway.
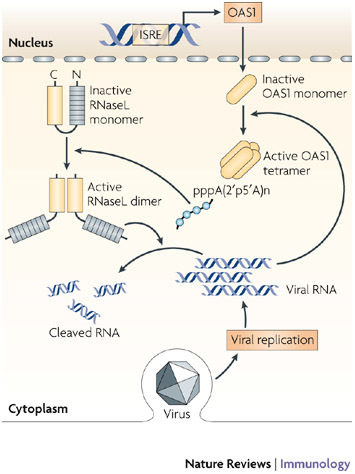
2′,5′-oligoadenylate synthetase 1 (OAS1) is expressed at low constitutive levels and is upregulated by type I interferons (IFNs). OAS1 protein accumulates in the cell cytoplasm as an inactive monomer. Following activation by viral double-stranded RNA (dsRNA), the enzyme oligomerizes to form a tetramer that synthesizes 2′,5′-oligoadenylates that, in turn, activate the constitutively expressed inactive ribonuclease L (RNaseL). The binding of 2′,5′-oligoadenylates to RNaseL triggers the dimerization of enzyme monomers, through their kinase-like domains, and this then enables RNAseL to cleave cellular (and viral) RNAs. ISRE, IFN-stimulated response element.
It has recently become apparent that the OAS proteins have additional antiviral functions that are independent of RNaseL activity. The precise mechanisms of RNaseL independence remain to be elucidated. Nevertheless, single nucleotide polymorphisms (SNPs) at a splice enhancer site in OASL have been correlated with susceptibility to infection with WNV85. Intriguingly, the capacity to accept GTP implies a potential role for OAS in RNA splicing, whereby the enzyme generates a 2′,5′-phosphodiester bond between the guanine at the 5′ end of an intron, and the adenine of a 3′ splice signal in the splicing intermediate structure86.
PKR
Similar to OAS, PKR was initially identified as a regulator of the antiviral response through studies of protein synthesis in cell-free lysates from IFN- and dsRNA-treated cells66,87. PKR belongs to a small family of protein kinases that respond to environmental stresses to regulate protein synthesis (the other members are EIF2αΚ1 (also known as HRI), EIF2αK3 (also known as PERK) and EIF2αK4 (also known as GCN2)). Members of this kinase family phosphorylate EIF2α at serine residue 51, resulting in sequestration of the limiting guanine-nucleotide exchange factor EIF2β (Ref. 88). This prevents recycling of GDP, halting translation, and therefore allows the cell to reconfigure gene expression. Much of the antiviral and antiproliferative activities of PKR can be attributed to its phosphorylation of EIF2α. Moreover, structural determination of the complex of EIF2α and PKR argues against the existence of alternative substrates89. However, there is extensive biological and biochemical evidence for alternative PKR targets, although the consequences of PKR-mediated phosphoregulation of other proteins have not been well characterized. As well as directly regulating proteins by phosphorylation, PKR induces cellular responses by modulating cell-signalling pathways (discussed below).
PKR is constitutively expressed in all tissues at a basal level and is upregulated by type I and type III IFNs90. Under normal circumstances, PKR is maintained as an inactive monomer, through steric hindrance of the kinase domain by its N terminus91,92 (Fig. 2). This repression is released by activating ligands, including viral RNAs, polyanionic molecules such as heparin93 or ceramide94, and protein activators95, which elicit a conformational change that allows binding of ATP to the C-terminal kinase domain. The kinase domain consists of two lobes that separately regulate the interaction between protein monomers and the substrate. The active PKR enzyme consists of a homodimer orientated in a parallel, back-to-back arrangement, with the active sites of the enzyme facing outwards89. Dimerization induces, and requires, autophosphorylation at several key residues96,97,98. Activation of PKR following dimerization then leads to the phosphorylation of EIF2α to halt translation (Fig. 6).
Figure 6. Mechanism of action of PKR.
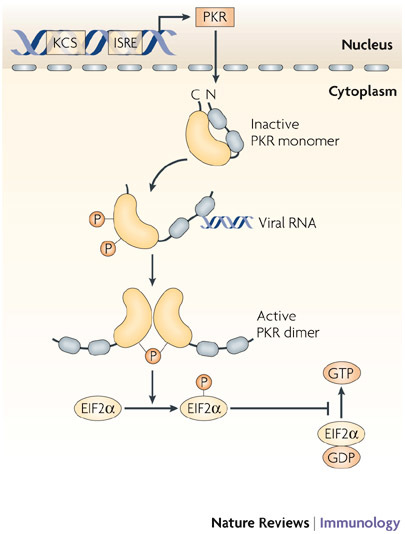
Protein kinase R (PKR) is constitutively expressed, and is also induced by type I interferons (IFNs) under the control of a kinase conserved sequence (KCS) and IFN-stimulated response element (ISRE) in the promoter of PKR. The kinase accumulates in the nucleus and cytoplasm as an inactive monomer, which is activated directly by viral RNAs, and by several other ligands, such as ceramide or the protein activator PACT (protein activator of the IFN-inducible protein kinase). Following activation, PKR monomers are phosphorylated and dimerize to form the active enzyme. Activated PKR regulates several cell signalling pathways through mechanisms that have not been fully explained, but a crucial function of PKR in viral defence is the inhibition of translation by phosphorylation of eukaryotic translation initiation factor 2α (EIF2α).
Direct activation of PKR has been shown with various RNAs by virtue of the two RNA-binding motifs (RBMs) in the N-terminal of PKR. All RBMs that have been tested bind dsRNA independently of sequence, but recognize a specific higher ordered structure. Accordingly, PKR, similar to the OAS–RNaseL pathway, functions as a PRR. Although RBMs have been shown to bind to just 16 base pairs of RNA, longer RNA moieties are required to engage both of the RBMs in PKR and to activate the kinase function99. Consequently, dsRNA that is longer than 30 base pairs activates PKR most effectively. Also, ssRNAs of 47 bases that have limited ternary structure activate the kinase if they contain 5′-triphosphates100. As cellular RNA transcripts predominantly have 5′-monophosphates, this enables PKR to specifically target viral RNAs.
Other pathogen-associated molecules, such as lipopolysaccharide (LPS), which is a ligand of Toll-like receptor 4 (TLR4), can also activate PKR101, but this is probably indirect activation via another protein. Indeed, three key protein interactions have been identified. PKR interacts with the tumour-necrosis factor (TNF)-receptor-associated factor (TRAF) family of adaptor molecules that are integral to TLR signalling pathways102. The protein activator of PKR PACT (protein activator of the IFN-inducible protein kinase; also known as PRKRA) responds to stress-inducing molecules, such as hydrogen peroxide, ceramide and cytokines (including IFNγ, IL-3 and TNF)95. However, the consequences of activation of PKR by PACT in an antiviral context awaits the characterization of PACT-deficient mice. Finally, PKR can be activated by cleavage of its inhibitory N terminus by caspases (caspase-3, caspase-7 and caspase-8), which generate a constitutively active, truncated, kinase domain103,104.
The role of PKR in antiviral responses has been investigated in mutant and transgenic mouse models. Mice expressing PKR with deletion mutations that target both functional domains of the enzyme105,106, transgenic mice expressing a trans-dominant negative mutant of human PKR that is defective in kinase activity (Lys296Arg)107, and transgenic mice overexpressing wild-type human PKR, have all been generated108. These transgenic mice have impaired antiviral responses and show increased susceptibility to otherwise innocuous infections with viruses, such as VSV109,110, influenza virus and bunyawera virus111. Experiments in PKR-deficient mouse embryonic fibroblasts show that PKR is involved in protection against infection with several RNA viruses, including HCV112, hepatitis D virus113, WNV114, HIV-1 (Ref. 115), Sindbis virus116, encephalomyocarditis virus117, and foot-and-mouth disease virus118, as well as some DNA viruses such as HSV-1 (Ref. 119). As with MxA and OAS1, genetic analysis of human populations show that polymorphisms in the PKR gene correlate with the outcome of HCV infection120.
Additional immune functions
Studies of the ISG15, OAS–RNaseL and PKR pathways suggest additional functions for each of these proteins. The main activating enzyme in ISGylation, UBE1L, was shown to be deleted in almost all small-cell lung cancers, and so the gene product was speculated to be a tumour suppressor121,122. Given the specificity of UBE1L in ISGylation, this presents an intriguing possible mechanism by which type I IFNs could regulate proliferation.
RNaseL-deficient mice have an enlarged thymus and spleen due to suppressed apoptosis82. The significance of the ability of RNaseL to induce apoptosis has been illustrated by genetic analysis that identified a polymorphism that generates an amino-acid substitution at position 462 (Arg462Gly) that was associated with reduced enzyme activity and increased incidence of prostate cancer123. Intriguingly, this SNP was then shown to be associated with a putative oncolytic xenotropic murine leukaemia-related virus124. Other nonviral functions of RNaseL are implied in experiments that show delayed skin-graft rejection in RNaseL-deficient mice125. A polymorphism identified in OAS1 has also been associated with type I diabetes, which is consistent with a viral aetiology for this disease126,127.
Given that PKR modulates several signalling pathways, a broader function than that determined solely by regulation of translation would be expected. As mentioned above, PKR is required for TLR4-mediated apoptosis in macrophages. Although PKR-mediated apoptosis is in part attributable to inhibition of translation through EIF2α phosphorylation, alternative signalling through IRF3 is also important101. PKR also promotes degradation of the inhibitor IκB, thereby activating the potent transcription factor NF-κB128. Regulation of NF-κB by PKR accounts for the diminished expression of nitric-oxide synthase 2 and IFNβ by PKR-deficient cells129. In addition, PKR was shown to be required for LPS-induced STAT-mediated inflammatory signalling130. There is also a defect in IFN-induced phosphorylation of serine 727 in STAT1 (Ref. 131), which was shown to be necessary for the basal expression of caspase-3. Accordingly, PKR-deficient fibroblasts are variably resistant to apoptosis that is induced by different stimuli, including dsRNA, LPS and TNF132. Conversely, overexpression of PKR in NIH3T3 fibroblasts sensitizes them to apoptosis103. PKR may also influence the adaptive immune response by negatively regulating CD8+ T-cell function, as PKR-deficient mice have increased contact hypersensitivity responses and stimulus-dependent T-cell proliferation133. PKR has also been implicated in IgE class switching in B cells. This points to a mechanism by which viral infection might induce IgE-mediated disorders, such as allergy and asthma134,135. Surprisingly, these signalling events are not wholly mediated by direct phosphorylation by PKR, suggesting that it might act as a scaffold to bridge signalling pathways from alternative PRRs136. The mechanisms underlying these links to adaptive immunity are intriguing but remain to be explained.
Concluding remarks and future directions
The analyses of mice with targeted deletions in the genes encoding ISG15, Mx1, PKR and RNaseL have helped to elucidate specific roles for the gene products in the antiviral response. Further details of the mechanisms of action of each of these effectors are still to be elucidated. Considerable work is still required to decipher the processes of ISGylation, to characterize protein substrates and to detail the consequence for the antiviral response. Also, the precise function of the Mx proteins remains uncertain. The contribution of alternative PKR substrates (besides EIF2α) to the immune response is poorly explored, and questions remain about specific roles for PKR in regulating inflammatory responses, in particular whether this requires kinase function or its putative role as an adaptor protein. The precise roles of the different OAS proteins, especially relating to RNaseL-independent antiviral effects, are ill-defined. These alternative functions will be better addressed in vivo by the analysis of mice with more subtle targeted mutations, and in vitro by detailed biochemical analyses (for example, by identification of specific phosphorylation sites on substrates for PKR) and by more detailed structural investigation of the mechanism of activation of each protein.
Strategies used by viruses to escape these antiviral pathways are also informative and a large number of viral countermeasures to block ISG15, PKR, RNaseL and Mx have been reported (reviewed in Refs 78,137,138). It should be noted, however, that many of the mechanisms attributed to virus-evolved avoidance of antiviral effectors and enhanced virulence have not been rigorously proven. An exception to this is the mechanism used by the HSV-1 protein ICP34.5 for targeting PKR. In this case, a virus that has been attenuated by the deletion of ICP34.5 only replicates as efficiently as wild-type virus when it infects PKR-deficient mice. Restoration of virulence was shown to depend on ICP34.5 by using both host and viral mutants139. Although this approach is not trivial to replicate, it remains the most valid method for defining effective viral avoidance of type I IFN activity.
Another important approach currently underway is to catalogue SNPs in each of these genetic loci in human populations and other animal species. Correlation of genetic polymorphisms in putative antiviral genes with susceptibility to viruses, as well as with incidence of other immune responses, is emerging as a powerful means of confirming gene function and measuring their contribution to disease.
Finally, the most interesting developments in this field are likely to come from further insights into the broader role of each of these antiviral proteins, particularly in mediating adaptive immunity. Although the mechanisms underlying these links to adaptive immunity are yet to be explained, progress in this area has great potential to manipulate immunity and therefore to enhance resistance to pathogens and disease or, alternatively, to diminish deleterious autoimmune responses. This will probably also require parallel advancement in our understanding of the specificities of type I and type III IFN signalling.
Acknowledgements
The work in our laboratory is supported by grants from the US National Institutes of Health (P01 CA062220 and R01 AI034039) and the Australian National Health and Medical Research Council (436814).
Glossary
- Pattern-recognition receptors
(PRRs). Host receptors that can sense pathogen-associated molecular patterns and initiate signalling cascades that lead to an innate immune response. These can be membrane bound (such as Toll-like receptors) or soluble cytoplasmic receptors (such as RIG-I, MDA5 and NLRs).
- GTPase
A signalling protein that binds guanine nucleotides. A GTPase is active in the GTP-bound state and is inactivated when it hydrolyses GTP into GDP.
- RNaseL
(Ribonuclease L). A constitutively expressed, latent, nonspecific endoribonuclease that is activated by 2′,5′-oligoadenylate. Type I interferons induce RNaseL expression as part of the host antiviral response.
- APOBEC
(Apolipoprotein B mRNA-editing enzyme, catalytic polypeptide). A cytidine deaminase enzyme family that includes: APOBEC1, an RNA editor involved in lipid metabolism; APOBEC3G and APOBEC3F, two DNA editors with antiretroviral activity; and activation-induced cytidine deaminase (AID), a DNA editor mediating immunoglobulin gene diversification.
- Ubiquitin
A small protein that is attached to other proteins by ubiquitin ligases. Depending on the mode of attachment, ubiquitin can either activate signalling function or target a protein for destruction by the proteasome.
- Ubiquitylation
The attachment of the small protein ubiquitin to lysine residues that are present in other proteins. Protein ubiquitylation occurs in three enzymatic steps requiring a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3), which catalyses the ligation of an isopeptide bond between the C-terminal domain of ubiquitin and an amino group belonging to a lysine residue of the target protein.
- RNA interference
The use of double-stranded RNAs with sequences that match a given gene, to 'knockdown' the expression of that gene by directing RNA-degrading enzymes to destroy the encoded mRNA transcript.
- Plasmacytoid dendritic cells
(pDCs). A unique family of dendritic cells with a morphology that resembles that of a plasmablast. These cells are also known as interferon (IFN)-producing cells because they are the main source of type I IFNs during viral infections.
- TNF-receptor-associated factor family
(TRAF family). A family of conserved scaffold proteins that link receptors of the tumour-necrosis factor (TNF) and Toll/interleukin-1 receptor family to various protein kinases (such as IRAK1). These proteins encode RING domains that, by association with E3-ubiquitin ligases, catalyse polyubiquitylation of proteins (such as IKKs to activate the transcription factor NF-κB) to transduce signals in the cell.
Biographies
Anthony J. Sadler is a research fellow at the Centre for Cancer Research at the Monash Institute of Medical Research, Monash University, Melbourne, Australia. Prior to this, he worked on microbial pathogens and the invertebrate immune response, receiving his Ph.D. from the Department of Microbiology at the University of Otago, Dunedin, New Zealand, followed by postdoctoral studies at the Department of Cancer Biology, Lerner Research Institute, Cleveland Clinic Foundation, USA. His scientific interests are currently the vertebrate immune response to pathogens, particularly viruses. His focus is on host receptors for RNA, including Toll-like receptors, protein kinase R and the cytoplasmic helicases, and the signalling pathways that are triggered by these receptors.
Bryan R. G. Williams is Director of the Monash Institute of Medical Research and is a professor in the Faculty of Medicine, Nursing and Health Sciences at Monash University. He received his Ph.D. from the Department of Microbiology at the University of Otago. Following postdoctoral training at the National Institute for Medical Research, Mill Hill, London, he held faculty positions at the University of Toronto and the Hospital for Sick Children, Toronto, Canada, and was Chairman of the Department of Cancer Biology, Lerner Research Institute, USA. His scientific achievements are in the fields of interferons, genetics and molecular biology of tumour suppression. His research focuses on understanding the mechanisms of signal transduction by interferons and cytokines. He is internationally recognized for his studies of protein–RNA interactions in the innate immune system.
Related links
FURTHER INFORMATION
References
- 1.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc. R. Soc. Lond. [Biol.] 1957;147:258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 2.Borden EC, et al. Interferons at age 50: past, current and future impact on biomedicine. Nature Rev. Drug Discov. 2007;6:975–990. doi: 10.1038/nrd2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levraud JP, et al. Identification of the zebrafish IFN receptor: implications for the origin of the vertebrate IFN system. J. Immunol. 2007;178:4385–4394. doi: 10.4049/jimmunol.178.7.4385. [DOI] [PubMed] [Google Scholar]
- 4.O'Connell RM, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J. Exp. Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 6.Casrouge A, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 7.Dupuis S, et al. Impaired response to interferon-α/β and lethal viral disease in human STAT1 deficiency. Nature Genet. 2003;33:388–391. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 8.Jouanguy E, et al. Human primary immunodeficiencies of type I interferons. Biochimie. 2007;89:878–883. doi: 10.1016/j.biochi.2007.04.016. [DOI] [PubMed] [Google Scholar]
- 9.Minegishi Y, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–755. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 10.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc. Natl Acad. Sci. USA. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pedersen IM, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–922. doi: 10.1038/nature06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blomstrom DC, Fahey D, Kutny R, Korant BD, Knight E., Jr. Molecular characterization of the interferon-induced 15-kDa protein. Molecular cloning and nucleotide and amino acid sequence. J. Biol. Chem. 1986;261:8811–8816. [PubMed] [Google Scholar]
- 13.Loeb KR, Haas AL. The interferon-inducible 15-kDa ubiquitin homolog conjugates to intracellular proteins. J. Biol. Chem. 1992;267:7806–7813. [PubMed] [Google Scholar]
- 14.Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nature Rev. Immunol. 2005;5:941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narasimhan J, Potter JL, Haas AL. Conjugation of the 15-kDa interferon-induced ubiquitin homolog is distinct from that of ubiquitin. J. Biol. Chem. 1996;271:324–330. doi: 10.1074/jbc.271.1.324. [DOI] [PubMed] [Google Scholar]
- 16.Yuan W, Krug RM. Influenza B virus NS1 protein inhibits conjugation of the interferon (IFN)-induced ubiquitin-like ISG15 protein. EMBO J. 2001;20:362–371. doi: 10.1093/emboj/20.3.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takeuchi T, Iwahara S, Saeki Y, Sasajima H, Yokosawa H. Link between the ubiquitin conjugation system and the ISG15 conjugation system: ISG15 conjugation to the UbcH6 ubiquitin E2 enzyme. J. Biochem. (Tokyo) 2005;138:711–719. doi: 10.1093/jb/mvi172. [DOI] [PubMed] [Google Scholar]
- 18.Zhao C, et al. The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN-αβ-induced ubiquitin-like protein. Proc. Natl Acad. Sci. USA. 2004;101:7578–7582. doi: 10.1073/pnas.0402528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim KI, Giannakopoulos NV, Virgin HW, Zhang DE. Interferon-inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol. Cell Biol. 2004;24:9592–9600. doi: 10.1128/MCB.24.21.9592-9600.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wong JJ, Pung YF, Sze NS, Chin KC. HERC5 is an IFN-induced HECT-type E3 protein ligase that mediates type I IFN-induced ISGylation of protein targets. Proc. Natl Acad. Sci. USA. 2006;103:10735–10740. doi: 10.1073/pnas.0600397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou W, Zhang DE. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006;281:3989–3994. doi: 10.1074/jbc.M510787200. [DOI] [PubMed] [Google Scholar]
- 22.Catic A, et al. Screen for ISG15-crossreactive deubiquitinases. PLoS ONE. 2007;2:e679. doi: 10.1371/journal.pone.0000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ, Zhang DE. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 24.Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc. Natl Acad. Sci. USA. 2005;102:10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giannakopoulos NV, et al. Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem. Biophys. Res. Commun. 2005;336:496–506. doi: 10.1016/j.bbrc.2005.08.132. [DOI] [PubMed] [Google Scholar]
- 26.Takeuchi T, Yokosawa H. Detection and analysis of protein ISGylation. Methods Mol. Biol. 2008;446:139–149. doi: 10.1007/978-1-60327-084-7_10. [DOI] [PubMed] [Google Scholar]
- 27.Lu G, et al. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell. Mol. Biol. 2006;52:29–41. [PubMed] [Google Scholar]
- 28.Okumura F, Zou W, Zhang DE. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev. 2007;21:255–260. doi: 10.1101/gad.1521607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takeuchi T, Kobayashi T, Tamura S, Yokosawa H. Negative regulation of protein phosphatase 2Cβ by ISG15 conjugation. FEBS Lett. 2006;580:4521–4526. doi: 10.1016/j.febslet.2006.07.032. [DOI] [PubMed] [Google Scholar]
- 30.D'Cunha J, et al. In vitro and in vivo secretion of human ISG15, an IFN-induced immunomodulatory cytokine. J. Immunol. 1996;157:4100–4108. [PubMed] [Google Scholar]
- 31.Majetschak M, et al. Extracellular ubiquitin inhibits the TNF-α response to endotoxin in peripheral blood mononuclear cells and regulates endotoxin hyporesponsiveness in critical illness. Blood. 2003;101:1882–1890. doi: 10.1182/blood-2002-03-0918. [DOI] [PubMed] [Google Scholar]
- 32.Sakai N, Sawada H, Yokosawa H. Extracellular ubiquitin system implicated in fertilization of the ascidian, Halocynthia roretzi: isolation and characterization. Dev. Biol. 2003;264:299–307. doi: 10.1016/j.ydbio.2003.07.016. [DOI] [PubMed] [Google Scholar]
- 33.Lenschow DJ, et al. Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J. Virol. 2005;79:13974–13983. doi: 10.1128/JVI.79.22.13974-13983.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osiak A, Utermohlen O, Niendorf S, Horak I, Knobeloch KP. ISG15, an interferon-stimulated ubiquitin-like protein, is not essential for STAT1 signaling and responses against vesicular stomatitis and lymphocytic choriomeningitis virus. Mol. Cell Biol. 2005;25:6338–6345. doi: 10.1128/MCB.25.15.6338-6345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenschow DJ, et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl Acad. Sci. USA. 2007;104:1371–1376. doi: 10.1073/pnas.0607038104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Y, Burke CW, Ryman KD, Klimstra WB. Identification and characterization of interferon-induced proteins that inhibit alphavirus replication. J. Virol. 2007;81:11246–11255. doi: 10.1128/JVI.01282-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim KI, et al. Ube1L and protein ISGylation are not essential for α/β interferon signaling. Mol. Cell Biol. 2006;26:472–479. doi: 10.1128/MCB.26.2.472-479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malakhova OA, Zhang DE. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J. Biol. Chem. 2008;283:8783–8787. doi: 10.1074/jbc.C800030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okumura A, Lu G, Pitha-Rowe I, Pitha PM. Innate antiviral response targets HIV-1 release by the induction of ubiquitin-like protein ISG15. Proc. Natl Acad. Sci. USA. 2006;103:1440–1445. doi: 10.1073/pnas.0510518103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chang YG, et al. Different roles for two ubiquitin-like domains of ISG15 in protein modification. J. Biol. Chem. 2008;283:13370–13377. doi: 10.1074/jbc.M800162200. [DOI] [PubMed] [Google Scholar]
- 41.Arguello MD, Hiscott J. Ub surprised: viral ovarian tumor domain proteases remove ubiquitin and ISG15 conjugates. Cell Host Microbe. 2007;2:367–369. doi: 10.1016/j.chom.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 42.Frias-Staheli N, et al. Ovarian tumor domain-containing viral proteases evade ubiquitin- and ISG15-dependent innate immune responses. Cell Host Microbe. 2007;2:404–416. doi: 10.1016/j.chom.2007.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lindner HA, et al. Selectivity in ISG15 and ubiquitin recognition by the SARS coronavirus papain-like protease. Arch. Biochem. Biophys. 2007;466:8–14. doi: 10.1016/j.abb.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacMicking JD. IFN-inducible GTPases and immunity to intracellular pathogens. Trends Immunol. 2004;25:601–609. doi: 10.1016/j.it.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 45.Haller O, Arnheiter H, Lindenmann J, Gresser I. Host gene influences sensitivity to interferon action selectively for influenza virus. Nature. 1980;283:660–662. doi: 10.1038/283660a0. [DOI] [PubMed] [Google Scholar]
- 46.Haller O, Arnheiter H, Gresser I, Lindenmann J. Genetically determined, interferon-dependent resistance to influenza virus in mice. J. Exp. Med. 1979;149:601–612. doi: 10.1084/jem.149.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lindenmann J. Resistance of mice to mouse adapted myxoviruses. Virology. 1962;16:203–204. doi: 10.1016/0042-6822(62)90297-0. [DOI] [PubMed] [Google Scholar]
- 48.Lindenmann J. Inheritance of resistance to influenza virus in mice. Proc. Soc. Exp. Biol. Med. 1964;116:506–509. doi: 10.3181/00379727-116-29292. [DOI] [PubMed] [Google Scholar]
- 49.Arnheiter H, Skuntz S, Noteborn M, Chang S, Meier E. Transgenic mice with intracellular immunity to influenza virus. Cell. 1990;62:51–61. doi: 10.1016/0092-8674(90)90239-B. [DOI] [PubMed] [Google Scholar]
- 50.Aebi M, et al. cDNA structures and regulation of two interferon-induced human Mx proteins. Mol. Cell Biol. 1989;9:5062–5072. doi: 10.1128/MCB.9.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Horisberger MA, et al. cDNA cloning and assignment to chromosome 21 of IFI-78K gene, the human equivalent of murine Mx gene. Somat. Cell. Mol. Genet. 1988;14:123–131. doi: 10.1007/BF01534397. [DOI] [PubMed] [Google Scholar]
- 52.Haller O, Frese M, Rost D, Nuttall PA, Kochs G. Tick-borne thogoto virus infection in mice is inhibited by the orthomyxovirus resistance gene product Mx1. J. Virol. 1995;69:2596–2601. doi: 10.1128/jvi.69.4.2596-2601.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Andersson I, et al. Human MxA protein inhibits the replication of Crimean-Congo hemorrhagic fever virus. J. Virol. 2004;78:4323–4329. doi: 10.1128/JVI.78.8.4323-4329.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chieux V, et al. Inhibition of coxsackievirus B4 replication in stably transfected cells expressing human MxA protein. Virology. 2001;283:84–92. doi: 10.1006/viro.2001.0877. [DOI] [PubMed] [Google Scholar]
- 55.Gordien E, et al. Inhibition of hepatitis B virus replication by the interferon-inducible MxA protein. J. Virol. 2001;75:2684–2691. doi: 10.1128/JVI.75.6.2684-2691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hijikata M, Ohta Y, Mishiro S. Identification of a single nucleotide polymorphism in the MxA gene promoter (G/T at nt -88) correlated with the response of hepatitis C patients to interferon. Intervirology. 2000;43:124–127. doi: 10.1159/000025035. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki F, et al. Single nucleotide polymorphism of the MxA gene promoter influences the response to interferon monotherapy in patients with hepatitis C viral infection. J. Viral Hepat. 2004;11:271–276. doi: 10.1111/j.1365-2893.2004.00509.x. [DOI] [PubMed] [Google Scholar]
- 58.Torisu H, et al. Functional MxA promoter polymorphism associated with subacute sclerosing panencephalitis. Neurology. 2004;62:457–460. doi: 10.1212/01.WNL.0000106940.95749.8E. [DOI] [PubMed] [Google Scholar]
- 59.Fernandez M, et al. In vivo and in vitro induction of MxA protein in peripheral blood mononuclear cells from patients chronically infected with hepatitis C virus. J. Infect. Dis. 1999;180:262–267. doi: 10.1086/314859. [DOI] [PubMed] [Google Scholar]
- 60.Kochs G, Haller O. Interferon-induced human MxA GTPase blocks nuclear import of Thogoto virus nucleocapsids. Proc. Natl Acad. Sci. USA. 1999;96:2082–2086. doi: 10.1073/pnas.96.5.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Accola MA, Huang B, Al Masri A, McNiven MA. The antiviral dynamin family member, MxA, tubulates lipids and localizes to the smooth endoplasmic reticulum. J. Biol. Chem. 2002;277:21829–21835. doi: 10.1074/jbc.M201641200. [DOI] [PubMed] [Google Scholar]
- 62.Turan K, et al. Nuclear MxA proteins form a complex with influenza virus NP and inhibit the transcription of the engineered influenza virus genome. Nucleic Acids Res. 2004;32:643–652. doi: 10.1093/nar/gkh192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimm D, et al. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc. Natl Acad. Sci. USA. 2007;104:6806–6811. doi: 10.1073/pnas.0701849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fernandez M, Quiroga JA, Carreno V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J. Gen. Virol. 2003;84:2073–2082. doi: 10.1099/vir.0.18966-0. [DOI] [PubMed] [Google Scholar]
- 65.Hoenen A, Liu W, Kochs G, Khromykh AA, Mackenzie JM. West Nile virus-induced cytoplasmic membrane structures provide partial protection against the interferon-induced antiviral MxA protein. J. Gen. Virol. 2007;88:3013–3017. doi: 10.1099/vir.0.83125-0. [DOI] [PubMed] [Google Scholar]
- 66.Kerr IM, Brown RE, Hovanessian AG. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature. 1977;268:540–542. doi: 10.1038/268540a0. [DOI] [PubMed] [Google Scholar]
- 67.Rebouillat D, Hovanessian AG. The human 2′,5′-oligoadenylate synthetase family: interferon-induced proteins with unique enzymatic properties. J. Interferon Cytokine Res. 1999;19:295–308. doi: 10.1089/107999099313992. [DOI] [PubMed] [Google Scholar]
- 68.Clemens MJ, Williams BRG. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell. 1978;13:565–572. doi: 10.1016/0092-8674(78)90329-X. [DOI] [PubMed] [Google Scholar]
- 69.Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448:816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hovanessian AG, Justesen J. The human 2′-5′oligoadenylate synthetase family: unique interferon-inducible enzymes catalyzing 2′-5′ instead of 3′-5′ phosphodiester bond formation. Biochimie. 2007;89:779–788. doi: 10.1016/j.biochi.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 71.Andersen JB, Strandbygard DJ, Hartmann R, Justesen J. Interaction between the 2′-5′ oligoadenylate synthetase-like protein p59 OASL and the transcriptional repressor methyl CpG-binding protein 1. Eur. J. Biochem. 2004;271:628–636. doi: 10.1046/j.1432-1033.2003.03966.x. [DOI] [PubMed] [Google Scholar]
- 72.Marie I, Svab J, Robert N, Galabru J, Hovanessian AG. Differential expression and distinct structure of 69- and 100-kDa forms of 2–5A synthetase in human cells treated with interferon. J. Biol. Chem. 1990;265:18601–18607. [PubMed] [Google Scholar]
- 73.Ghosh A, Sarkar SN, Guo W, Bandyopadhyay S, Sen GC. Enzymatic activity of 2′-5′-oligoadenylate synthetase is impaired by specific mutations that affect oligomerization of the protein. J. Biol. Chem. 1997;272:33220–33226. doi: 10.1074/jbc.272.52.33220. [DOI] [PubMed] [Google Scholar]
- 74.Hartmann R, Justesen J, Sarkar SN, Sen GC, Yee VC. Crystal structure of the 2′-specific and double-stranded RNA-activated interferon-induced antiviral protein 2′-5′-oligoadenylate synthetase. Mol. Cell. 2003;12:1173–1185. doi: 10.1016/S1097-2765(03)00433-7. [DOI] [PubMed] [Google Scholar]
- 75.Sarkar SN, Pal S, Sen GC. Crisscross enzymatic reaction between the two molecules in the active dimeric P69 form of the 2′-5′ oligodenylate synthetase. J. Biol. Chem. 2002;277:44760–44764. doi: 10.1074/jbc.M207126200. [DOI] [PubMed] [Google Scholar]
- 76.Dong B, et al. Intrinsic molecular activities of the interferon-induced 2-5A-dependent RNase. J. Biol. Chem. 1994;269:14153–14158. [PubMed] [Google Scholar]
- 77.Castora FJ, Erickson CE, Kovacs T, Lesiak K, Torrence PF. 2′,5′-oligoadenylates inhibit relaxation of supercoiled DNA by calf thymus DNA topoisomerase I. J. Interferon Res. 1991;11:143–149. doi: 10.1089/jir.1991.11.143. [DOI] [PubMed] [Google Scholar]
- 78.Silverman RH. Viral encounters with 2′,5′-oligoadenylate synthetase and RNase L during the interferon antiviral response. J. Virol. 2007;81:12720–12729. doi: 10.1128/JVI.01471-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nakanishi M, Goto Y, Kitade Y. 2–5A induces a conformational change in the ankyrin-repeat domain of RNase L. Proteins. 2005;60:131–138. doi: 10.1002/prot.20474. [DOI] [PubMed] [Google Scholar]
- 80.Floyd-Smith G, Slattery E, Lengyel P. Interferon action: RNA cleavage pattern of a (2′-5′)oligoadenylate-dependent endonuclease. Science. 1981;212:1030–1032. doi: 10.1126/science.6165080. [DOI] [PubMed] [Google Scholar]
- 81.Wreschner DH, McCauley JW, Skehel JJ, Kerr IM. Interferon action–sequence specificity of the ppp(A2′p)nA-dependent ribonuclease. Nature. 1981;289:414–417. doi: 10.1038/289414a0. [DOI] [PubMed] [Google Scholar]
- 82.Zhou A, et al. Interferon action and apoptosis are defective in mice devoid of 2′, 5′-oligoadenylate-dependent RNase L. EMBO J. 1997;16:6355–6363. doi: 10.1093/emboj/16.21.6355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xiang Y, et al. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J. Virol. 2002;76:5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bonnevie-Nielsen V, et al. Variation in antiviral 2′, 5′-oligoadenylate synthetase (2′5′AS) enzyme activity is controlled by a single-nucleotide polymorphism at a splice-acceptor site in the OAS1 gene. Am. J. Hum. Genet. 2005;76:623–633. doi: 10.1086/429391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yakub I, et al. Single nucleotide polymorphisms in genes for 2′-5′-oligoadenylate synthetase and RNase L inpatients hospitalized with West Nile virus infection. J. Infect. Dis. 2005;192:1741–1748. doi: 10.1086/497340. [DOI] [PubMed] [Google Scholar]
- 86.Sperling J, et al. Possible involvement of (2′5′)oligoadenylate synthetase activity in pre-mRNA splicing. Proc. Natl Acad. Sci. USA. 1991;88:10377–10381. doi: 10.1073/pnas.88.23.10377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meurs E, et al. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell. 1990;62:379–390. doi: 10.1016/0092-8674(90)90374-N. [DOI] [PubMed] [Google Scholar]
- 88.Roberts WK, Hovanessian A, Brown RE, Clemens MJ, Kerr IM. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature. 1976;264:477–480. doi: 10.1038/264477a0. [DOI] [PubMed] [Google Scholar]
- 89.Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2α by the RNA-dependent protein kinase PKR. Cell. 2005;122:887–900. doi: 10.1016/j.cell.2005.06.044. [DOI] [PubMed] [Google Scholar]
- 90.Ank N, et al. λ interferon (IFN-λ), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J. Virol. 2006;80:4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gelev V, et al. Mapping of the auto-inhibitory interactions of protein kinase R by nuclear magnetic resonance. J. Mol. Biol. 2006;364:352–363. doi: 10.1016/j.jmb.2006.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nanduri S, Rahman F, Williams BR, Qin J. A dynamically tuned double-stranded RNA binding mechanism for the activation of antiviral kinase PKR. EMBO J. 2000;19:5567–5574. doi: 10.1093/emboj/19.20.5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.George CX, Thomis DC, McCormack SJ, Svahn CM, Samuel CE. Characterization of the heparin-mediated activation of PKR, the interferon-inducible RNA-dependent protein kinase. Virology. 1996;221:180–188. doi: 10.1006/viro.1996.0364. [DOI] [PubMed] [Google Scholar]
- 94.Ruvolo PP, Gao F, Blalock WL, Deng X, May WS. Ceramide regulates protein synthesis by a novel mechanism involving the cellular PKR activator RAX. J. Biol. Chem. 2001;276:11754–11758. doi: 10.1074/jbc.M011400200. [DOI] [PubMed] [Google Scholar]
- 95.Patel RC, Sen GC. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998;17:4379–4390. doi: 10.1093/emboj/17.15.4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dey M, et al. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2α substrate recognition. Cell. 2005;122:901–913. doi: 10.1016/j.cell.2005.06.041. [DOI] [PubMed] [Google Scholar]
- 97.Romano PR, et al. Autophosphorylation in the activation loop is required for full kinase activity in vivo of human and yeast eukaryotic initiation factor 2α kinases PKR and GCN2. Mol. Cell Biol. 1998;18:2282–2297. doi: 10.1128/MCB.18.4.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Taylor DR, et al. Autophosphorylation sites participate in the activation of the double-stranded-RNA-activated protein kinase PKR. Mol. Cell Biol. 1996;16:6295–6302. doi: 10.1128/MCB.16.11.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nanduri S, Carpick BW, Yang Y, Williams BR, Qin J. Structure of the double-stranded RNA-binding domain of the protein kinase PKR reveals the molecular basis of its dsRNA-mediated activation. EMBO J. 1998;17:5458–5465. doi: 10.1093/emboj/17.18.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nallagatla SR, et al. 5′-triphosphate-dependent activation of PKR by RNAs with short stem-loops. Science. 2007;318:1455–1458. doi: 10.1126/science.1147347. [DOI] [PubMed] [Google Scholar]
- 101.Hsu LC, et al. The protein kinase PKR is required for macrophage apoptosis after activation of Toll-like receptor 4. Nature. 2004;428:341–345. doi: 10.1038/nature02405. [DOI] [PubMed] [Google Scholar]
- 102.Gil J, et al. TRAF family proteins link PKR with NF-κB activation. Mol. Cell Biol. 2004;24:4502–4512. doi: 10.1128/MCB.24.10.4502-4512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gil J, Esteban M. The interferon-induced protein kinase (PKR), triggers apoptosis through FADD-mediated activation of caspase 8 in a manner independent of Fas and TNF-α receptors. Oncogene. 2000;19:3665–3674. doi: 10.1038/sj.onc.1203710. [DOI] [PubMed] [Google Scholar]
- 104.Saelens X, Kalai M, Vandenabeele P. Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-α phosphorylation. J. Biol. Chem. 2001;276:41620–41628. doi: 10.1074/jbc.M103674200. [DOI] [PubMed] [Google Scholar]
- 105.Abraham N, et al. Characterization of transgenic mice with targeted disruption of the catalytic domain of the double-stranded RNA-dependent protein kinase, PKR. J. Biol. Chem. 1999;274:5953–5962. doi: 10.1074/jbc.274.9.5953. [DOI] [PubMed] [Google Scholar]
- 106.Yang YL, et al. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14:6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Scheuner D, et al. The double-stranded RNA-activated protein kinase mediates viral-induced encephalitis. Virology. 2003;317:263–274. doi: 10.1016/j.virol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- 108.Ladiges W, et al. Expression of human PKR protein kinase in transgenic mice. J. Interferon Cytokine Res. 2002;22:329–334. doi: 10.1089/107999002753675758. [DOI] [PubMed] [Google Scholar]
- 109.Balachandran S, et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13:129–141. doi: 10.1016/S1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- 110.Stojdl DF, et al. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000;74:9580–9585. doi: 10.1128/JVI.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bergmann M, et al. Influenza virus NS1 protein counteracts PKR-mediated inhibition of replication. J. Virol. 2000;74:6203–6206. doi: 10.1128/JVI.74.13.6203-6206.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Noguchi T, et al. Effects of mutation in hepatitis C virus nonstructural protein 5A on interferon resistance mediated by inhibition of PKR kinase activity in mammalian cells. Microbiol. Immunol. 2001;45:829–840. doi: 10.1111/j.1348-0421.2001.tb01322.x. [DOI] [PubMed] [Google Scholar]
- 113.Chen CW, et al. The double-stranded RNA-activated kinase, PKR, can phosphorylate hepatitis D virus small δ antigen at functional serine and threonine residues. J. Biol. Chem. 2002;277:33058–33067. doi: 10.1074/jbc.M200613200. [DOI] [PubMed] [Google Scholar]
- 114.Samuel MA, et al. PKR and RNase L contribute to protection against lethal West Nile Virus infection by controlling early viral spread in the periphery and replication in neurons. J. Virol. 2006;80:7009–7019. doi: 10.1128/JVI.00489-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nagai K, et al. Induction of CD4 expression and human immunodeficiency virus type 1 replication by mutants of the interferon-inducible protein kinase PKR. J. Virol. 1997;71:1718–1725. doi: 10.1128/jvi.71.2.1718-1725.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gorchakov R, Frolova E, Williams BR, Rice CM, Frolov I. PKR-dependent and -independent mechanisms are involved in translational shutoff during Sindbis virus infection. J. Virol. 2004;78:8455–8467. doi: 10.1128/JVI.78.16.8455-8467.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yeung MC, Chang DL, Camantigue RE, Lau AS. Inhibitory role of the host apoptogenic gene PKR in the establishment of persistent infection by encephalomyocarditis virus in U937 cells. Proc. Natl Acad. Sci. USA. 1999;96:11860–11865. doi: 10.1073/pnas.96.21.11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chinsangaram J, Koster M, Grubman MJ. Inhibition of L-deleted foot-and-mouth disease virus replication by α/β interferon involves double-stranded RNA-dependent protein kinase. J. Virol. 2001;75:5498–5503. doi: 10.1128/JVI.75.12.5498-5503.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Al-khatib K, Williams BR, Silverman RH, Halford W, Carr DJ. The murine double-stranded RNA-dependent protein kinase PKR and the murine 2′, 5′-oligoadenylate synthetase-dependent RNase L are required for IFN-β-mediated resistance against herpes simplex virus type 1 in primary trigeminal ganglion culture. Virology. 2003;313:126–135. doi: 10.1016/S0042-6822(03)00298-8. [DOI] [PubMed] [Google Scholar]
- 120.Knapp S, et al. Polymorphisms in interferon-induced genes and the outcome of hepatitis C virus infection: roles of MxA, OAS-1 and PKR. Genes Immun. 2003;4:411–419. doi: 10.1038/sj.gene.6363984. [DOI] [PubMed] [Google Scholar]
- 121.Carritt B, et al. A gene from human chromosome region 3p21 with reduced expression in small cell lung cancer. Cancer Res. 1992;52:1536–1541. [PubMed] [Google Scholar]
- 122.Kok K, et al. A gene in the chromosomal region 3p21 with greatly reduced expression in lung cancer is similar to the gene for ubiquitin-activating enzyme. Proc. Natl Acad. Sci. USA. 1993;90:6071–6075. doi: 10.1073/pnas.90.13.6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carpten J, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nature Genet. 2002;30:181–184. doi: 10.1038/ng823. [DOI] [PubMed] [Google Scholar]
- 124.Urisman A, et al. Identification of a novel γ-retrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 125.Silverman RH, et al. Skin allograft rejection is suppressed in mice lacking the antiviral enzyme, 2′, 5′-oligoadenylate-dependent RNase L. Viral Immunol. 2002;15:477–483. doi: 10.1089/088282402317340242. [DOI] [PubMed] [Google Scholar]
- 126.Field LL, et al. OAS1 splice site polymorphism controlling antiviral enzyme activity influences susceptibility to type 1 diabetes. Diabetes. 2005;54:1588–1591. doi: 10.2337/diabetes.54.5.1588. [DOI] [PubMed] [Google Scholar]
- 127.Bonnevie-Nielsen V, Larsen ML, Frifelt JJ, Michelsen B, Lernmark A. Association of IDDM and attenuated response of 2′, 5′-oligoadenylate synthetase to yellow fever vaccine. Diabetes. 1989;38:1636–1642. doi: 10.2337/diab.38.12.1636. [DOI] [PubMed] [Google Scholar]
- 128.Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-κB by phosphorylating IκB. Proc. Natl Acad. Sci. USA. 1994;91:6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Uetani K, et al. Central role of double-stranded RNA-activated protein kinase in microbial induction of nitric oxide synthase. J. Immunol. 2000;165:988–996. doi: 10.4049/jimmunol.165.2.988. [DOI] [PubMed] [Google Scholar]
- 130.Lee JH, et al. Double-stranded RNA-activated protein kinase is required for the LPS-induced activation of STAT1 inflammatory signaling in rat brain glial cells. Glia. 2005;50:66–79. doi: 10.1002/glia.20156. [DOI] [PubMed] [Google Scholar]
- 131.Karehed K, Dimberg A, Dahl S, Nilsson K, Oberg F. IFN-γ-induced upregulation of Fcγ-receptor-I during activation of monocytic cells requires the PKR and NF-κB pathways. Mol. Immunol. 2007;44:615–624. doi: 10.1016/j.molimm.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 132.Lee SB, Esteban M. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology. 1994;199:491–496. doi: 10.1006/viro.1994.1151. [DOI] [PubMed] [Google Scholar]
- 133.Kadereit S, et al. Negative regulation of CD8+ T cell function by the IFN-induced and double-stranded RNA-activated kinase PKR. J. Immunol. 2000;165:6896–6901. doi: 10.4049/jimmunol.165.12.6896. [DOI] [PubMed] [Google Scholar]
- 134.Hardenberg G, et al. Specific TLR ligands regulate APRIL secretion by dendritic cells in a PKR-dependent manner. Eur. J. Immunol. 2007;37:2900–2911. doi: 10.1002/eji.200737210. [DOI] [PubMed] [Google Scholar]
- 135.Rager KJ, et al. Activation of antiviral protein kinase leads to immunoglobulin E class switching in human B cells. J. Virol. 1998;72:1171–1176. doi: 10.1128/jvi.72.2.1171-1176.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bonnet MC, Weil R, Dam E, Hovanessian AG, Meurs EF. PKR stimulates NF-κB irrespective of its kinase function by interacting with the IκB kinase complex. Mol. Cell Biol. 2000;20:4532–4542. doi: 10.1128/MCB.20.13.4532-4542.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Haller O, Kochs G, Weber F. Interferon, Mx, and viral countermeasures. Cytokine Growth Factor Rev. 2007;18:425–433. doi: 10.1016/j.cytogfr.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Langland JO, Cameron JM, Heck MC, Jancovich JK, Jacobs BL. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006;119:100–110. doi: 10.1016/j.virusres.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 139.Leib DA, Machalek MA, Williams BR, Silverman RH, Virgin HW. Specific phenotypic restoration of an attenuated virus by knockout of a host resistance gene. Proc. Natl Acad. Sci. USA. 2000;97:6097–6101. doi: 10.1073/pnas.100415697. [DOI] [PMC free article] [PubMed] [Google Scholar]