

Abstract
The adenomatous polyposis coli (Apc) tumor suppressor is involved in the initiation and progression of colorectal cancer via regulation of the Wnt signaling cascade. In addition, Apc plays an important role in multiple cellular functions, including cell migration and adhesion, spindle assembly, and chromosome segregation. However, its role during adult hematopoiesis is unknown. We show that conditional inactivation of Apc in vivo dramatically increases apoptosis and enhances cell cycle entry of hematopoietic stem cells (HSCs)/ hematopoietic progenitor cells (HPCs), leading to their rapid disappearance and bone marrow failure. The defect in HSCs/HPCs caused by Apc ablation is cell autonomous. In addition, we found that loss of Apc leads to exhaustion of the myeloid progenitor pool (common myeloid progenitor, granulocyte-monocyte progenitor, and megakaryocyte-erythroid progenitor), as well as the lymphoid-primed multipotent progenitor pool. Down-regulation of the genes encoding Cdkn1a, Cdkn1b, and Mcl1 occurs after acute Apc excision in candidate HSC populations. Together, our data demonstrate that Apc is essential for HSC and HPC maintenance and survival.
Hematopoiesis is a tightly regulated process in which hematopoietic stem cells (HSCs) undergo self-renewal and give rise to more differentiated progenitor populations, such as common lymphoid progenitors (CLPs) and common myeloid progenitors (CMPs), as well as more restricted granulocyte-monocyte progenitors (GMPs) and megakaryocyte-erythroid progenitors (MEPs), which develop into all mature blood cell lineages (1, 2). Maintenance of the HSC pool, which is essential for homeostasis of the hematopoietic system, requires the integrated regulation of proliferation, differentiation, and apoptosis of HSCs (3). Recent studies have revealed that the PcG (Polycomb group) proteins, such as Bmi1, Pcgf2, and Phc1, are important in the regulation of HSC maintenance (3). In addition, transcription factors, such as Etv6 (4), Gfi1 (5, 6), HoxB4 (7–9), Myc (10), and Zfx (11) have been implicated in the regulation of HSC self-renewal. Deletion of Pten, which encodes an important signal transducer, leads to a transient expansion of HSCs and depletion of the HSC pool over time, indicating that it has a critical role in the maintenance of HSCs (12, 13). However, the Cdkn1a and Cdkn2c cell cycle inhibitors have an opposing role in regulating the self-renewal of HSCs (14). Deletion of Cdkn1a results in increased proliferation and impaired self-renewal of HSCs. In contrast, loss of function of Cdkn2c leads to enhanced HSC self-renewal. Finally, apoptosis is a pivotal mechanism for regulation of the HSC pool, and antiapoptotic proteins, such as Mcl1 and Birc5 (Survivin), are required for the survival of HSCs (15, 16).
The adenomatous polyposis coli (APC) tumor suppressor gene is involved in the initiation and progression of colorectal cancer, via regulation of β-catenin through the Wnt signaling pathway (17). APC is essential for the formation of the APC–Axin–GSK3-β destruction complex, which mediates the phosphorylation and subsequent ubiquitylation of β-catenin for proteosomal degradation (17). In addition to regulation of the Wnt pathway, APC plays a role in cell migration, cell adhesion, spindle assembly, and chromosome segregation. With respect to cell migration, APC regulates stabilization and polymerization of microtubules through association with EB1 (end binding 1) and microtubules. APC also binds IQGAP1 (GTPase activating protein 1), which mediates cell polarization and directional migration. The guanine nucleotide exchange factor Asef is stimulated by its interaction with APC and, subsequently, promotes cell migration (18). Recent reports indicate that APC may function with Asef and catenins to regulate cell–cell adhesion (19). Mutations in APC induce chromosome instability, suggesting that WT APC also has a role in the maintenance of chromosome stability during mitosis (20). Although poorly understood, the mechanism by which APC regulates spindle assembly and chromosomal segregation may involve interaction with EB1 to regulate the dynamics and organization of microtubules, as well as association of APC with Bub checkpoint proteins to ensure proper attachment of microtubules to the kinetochore (20).
In this paper, we have examined the function of Apc in the hematopoietic system in vivo by conditional inactivation of Apc in hematopoietic cells in the mouse. We found that loss of Apc in vivo resulted in rapid lethality. The Apc mutant mice displayed a dramatic reduction in HSCs and hematopoietic progenitor cells (HPCs) and failed to maintain normal hematopoiesis. The effect of Apc deficiency on HSC and HPC maintenance involves increased apoptosis, as well as increased proliferation. In addition, we found that depletion of Apc leads to dramatic apoptosis of hepatocytes in vivo, indicating that Apc plays a role in the regulation of cell survival in multiple tissues. Collectively, our studies reveal that endogenous Apc plays a critical role in regulating HSC and HPC maintenance and adult hematopoiesis.
RESULTS
Apc is expressed in hematopoietic stem/progenitor cells
To examine the relative expression levels of Apc transcripts in primitive hematopoietic cells, we used a combination of cell purification and real-time quantitative RT-PCR (qRT-PCR) techniques. In this study, LSK (Lin− Sca-1+ c-kit+) cells, an HSC-enriched population, are referred to as HSCs, whereas Lin− Sca-1− IL-7R− c-kit+ BM cells are referred to as HPCs. Apc was expressed in all subsets of stem/progenitor cells, although the amount of Apc mRNA is slightly higher in HSCs and CLPs than in CMPs, GMPs, and MEPs (Fig. 1 A).
Figure 1.

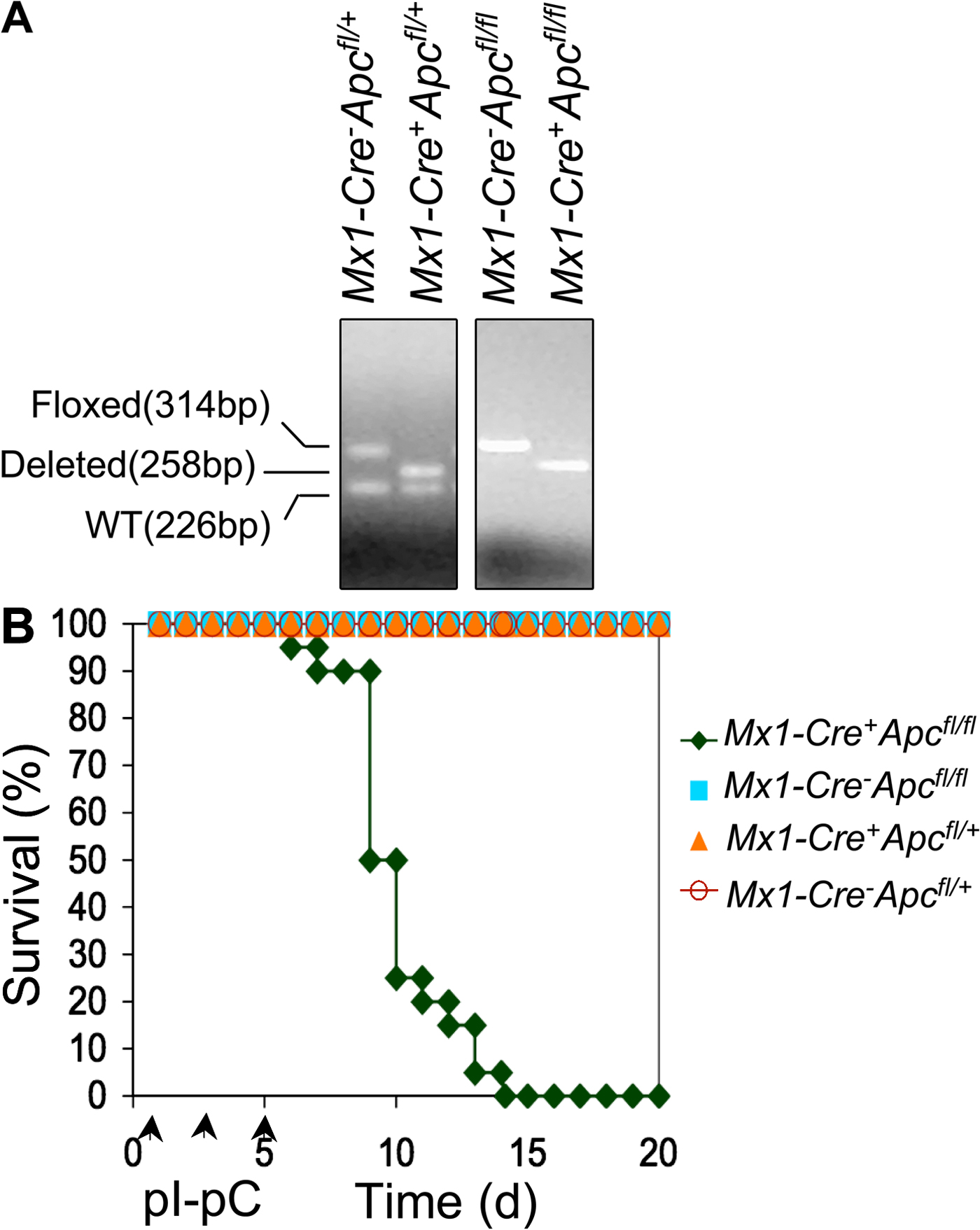
Ablation of Apc leads to rapid lethality. (A) Expression of Apc and Gata2 (control) mRNAs in progenitor populations purified from WT BM as determined by qRT-PCR (means ± SE; independent experiments were performed in triplicate). Gene expression was normalized initially to Hprt expression. Values represent the fold changes in gene expression relative to that in HSCs. (B) Comparison of engraftment of CD45.2+ Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl BM cells in WT (CD45.1+) recipient CM by flow analysis of PB (mean ± SD; n = 10). (C) Analysis of deletion of Apc in CM as determined by semiquantitative PCR of BM cells 7 d after induction. (D) Kaplan-Meier survival curve of CM recipient mice reconstituted with BM cells from Mx1-Cre+Apcfl/fl (n = 14) or control Mx1-Cre−Apcfl/fl (n = 14) mice. Arrowheads indicate pI-pC injections.
Loss of Apc results in rapid BM failure
To determine whether Apc is required for hematopoiesis in vivo, we used the Cre–loxP system to inactivate Apc in hematopoietic cells in vivo. Conditional Apc-floxed (Apcfl/fl) mice (21, 22) were bred with transgenic mice expressing the Cre recombinase under control of the type I interferon-inducible Mx1 promoter (23). We induced ablation of Apc in vivo by intraperitoneal injection of the interferon-α inducer polyinosinic-polycytidylic acid (pI-pC) into Mx1-Cre+Apcfl/fl, Mx1-Cre−Apcfl/fl, Mx1-Cre+Apc fl/+, or Mx1-Cre−Apc fl/+ mice. All Mx1-Cre+Apcfl/fl (also referred to as Apc mutant mice) and Mx1-Cre+Apc fl/+ mice treated with pI-pC had Cre-mediated deletion of exon 14 of Apc in the majority of BM cells (Fig. S1 A, available at http://www.jem.org/cgi/content/full/jem.20080578/DC1). Within 7–14 d after treatment with pI-pC (referred to here as days after induction), all Mx1-Cre+Apcfl/fl died, whereas all Mx1-Cre−Apcfl/fl (also referred to as Apc control mice), Mx1-Cre+Apc fl/+, and Mx1-Cre−Apc fl/+ mice survived (Fig. S1 B) and showed normal hematological parameters in peripheral blood (PB) 2 mo after induction (not depicted).
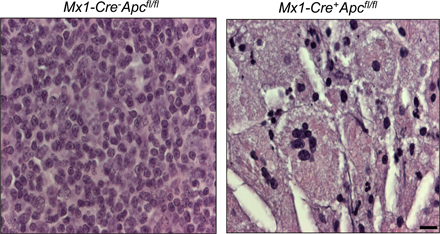
Because Mx1-Cre is expressed in multiple tissues, ablation of the targeted gene may be induced in various interferon-responsive tissues in Mx1-Cre+ mice carrying a floxed allele after treatment with pI-pC (23). Therefore, death of the Mx1-Cre+Apcfl/fl mice in a short period of time may be caused by dysfunction of multiple tissues resulting from loss of Apc. In this regard, conditional inactivation of Apc in osteoblasts (24), intestinal epithelium (25), and renal epithelium (26) leads to lethality. To distinguish the hematopoietic effects from nonhematopoietic effects of Apc deletion, we transplanted BM cells from untreated Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice (CD45.2+) into lethally irradiated WT recipient mice (CD45.1+). More than 90% of PB cells in recipient mice were reconstituted from donor-derived CD45.2+ cells (Fig. 1 B), confirming successful reconstitution. 2 mo after transplantation, which is a time when HSCs have ceased expanding, the chimeric mice (CM) were treated with pI-pC. By 7 d after induction, deletion of Apc is nearly complete in total BM cells (Fig. 1 C). The expression of Apc mRNA in Apc-deficient LSK (Lin− Sca-1+ c-kit+) cells was undetectable by qRT-PCR analysis (unpublished data). All recipient CM reconstituted with Mx1-Cre+Apcfl/fl BM cells became moribund and died within 15–20 d after the first injection of pI-pC (Fig. 1 D). Both Mx1-Cre+Apcfl/fl and control Mx1-Cre−Apcfl/fl chimeras showed normal white blood cell and differential counts, platelet counts, red blood cell counts, and hemoglobin levels before treatment (Fig. 2 A). In contrast, after treatment, all hematological parameters were decreased dramatically in Mx1-Cre+Apcfl/fl CM, and myeloid cells were almost absent in PB. However, the control Mx1-Cre−Apcfl/fl CM showed normal hematological parameters (Fig. 2 A). Furthermore, the Apc mutant CM had a markedly lower BM cellularity than did the Apc control chimeras (Fig. 2, B and C). These data indicate that Apc has a cell-intrinsic function in hematopoiesis and that loss of Apc leads to rapid hematopoietic failure.
Figure 2.

Apc is essential for adult hematopoiesis. (A) Absolute number of white blood cells (WBC), neutrophils (NE), lymphocytes (LY), monocytes (MO), eosinophils (EO), basophils (BA), red blood cells (RBC), platelets (PLT), and hemoglobin (Hb) levels in PB from Mx1-Cre+Apcfl/fl and control Mx1-Cre−Apcfl/fl CM before (mean ± SD; n = 15) and after (7–15 d after induction; mean ± SD; n = 10–12) pI-pC treatment. *, P < 0.001. (B) Histological analysis of hematoxylin and eosin–stained sternum from Mx1-Cre−Apcfl/fl CM (top) and Mx1-Cre+Apcfl/fl CM (bottom) 12 d after induction. Bar, 100 μm. (C) Numbers of BM cells in Mx1-Cre+Apcfl/fl moribund CM (red) as compared with Mx1-Cre−Apcfl/fl CM (blue; BM cells were collected from both tibias and femurs of mice 7–15 d after induction; mean ± SD; n = 9). *, P < 0.001.
Ablation of Apc leads to rapid exhaustion of the HSC pool in vivo
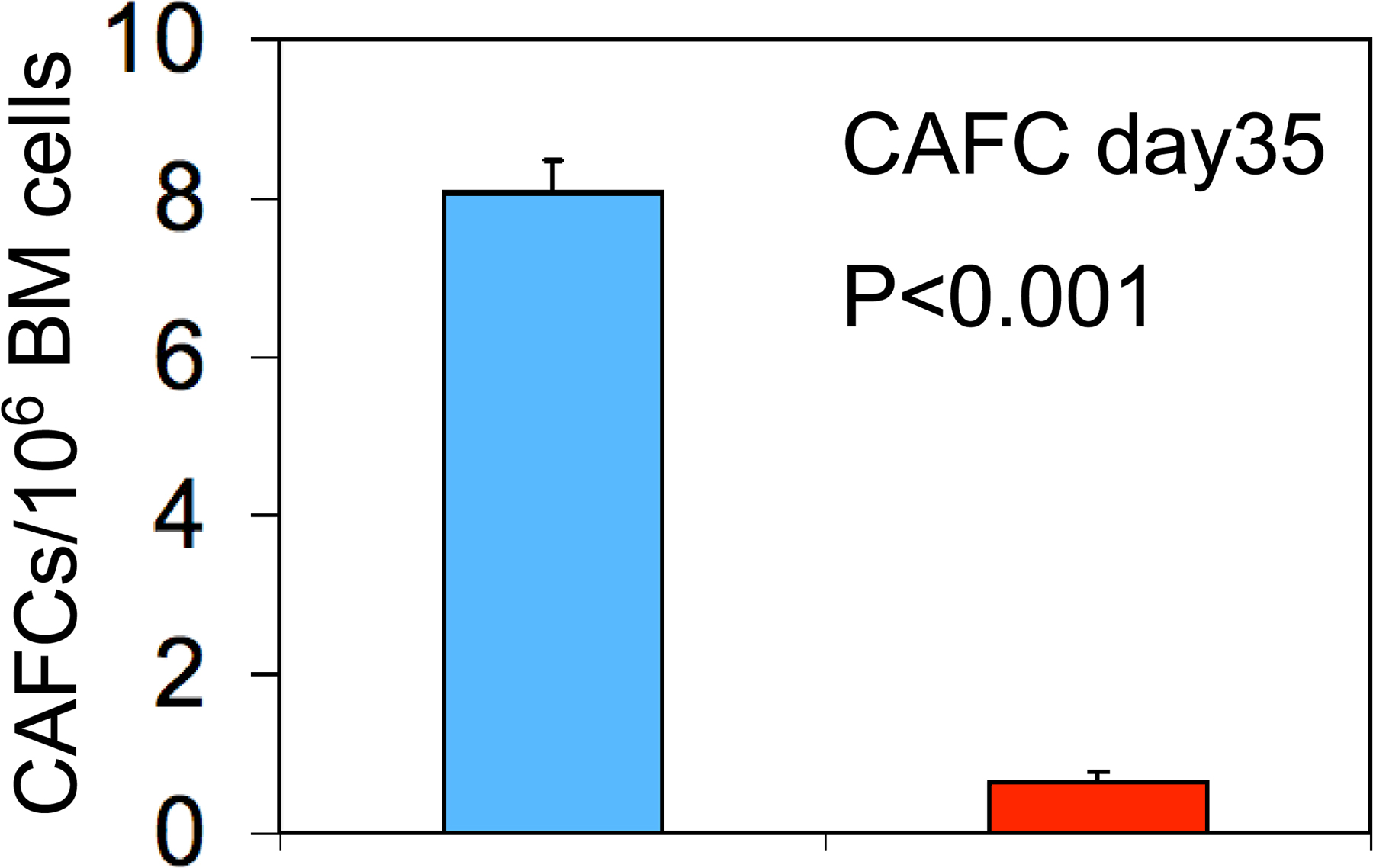
The observation of multilineage hematopoietic abnormalities and rapid lethality after ablation of Apc suggested that Apc function plays a critical role in HSCs and progenitors. To determine whether BM failure is caused by ineffective HSC function in Apc-deficient mice, we examined the HSC compartment by FACS. After induction of Apc ablation, primary Mx1-Cre+Apcfl/fl mice (4 d after induction) and CM (7 d after induction) reconstituted with Mx1-Cre+Apcfl/fl BM cells had a mean of 3- or 10-fold fewer total HSCs (LSK; Lin− Sca-1+ c-kit+ cells; Fig. 3, A and B) than did the control Mx1-Cre−Apcfl/fl mice or control Mx1-Cre−Apcfl/fl CM, respectively. To characterize the primitive hematopoietic cell compartment of Apc mutant mice further, we performed the cobblestone area–forming cell (CAFC) assay (27). At 4 d after induction (two treatments with pI-pC), the BM cells were isolated from Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl mice and cultured on monolayers of FBMD1 stromal cells. The frequency of CAFCs (day 35) in Apc mutant BM cells was 10-fold lower than in Apc-control mice, indicating that Apc mutant HSCs had reduced CAFC activity in vitro (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20080578/DC1).
Figure 3.

Characterization of Apc-deficient HSCs. (A) Comparison of the frequency of HSCs in primary Mx1-Cre+Apcfl/fl mice 4 d after induction (top) and Mx1-Cre+Apcfl/fl CM mice 7 d after induction (bottom). The percentage of LSK cells is indicated (mean ± SD of six to eight animals). **, P < 0.01. (B) Total number of HSCs (LSK) in BM from primary mice and CM. **, P < 0.01. (C and D) Hematopoietic reconstitution after ablation of Apc was assessed by the proportion of donor-derived CD45.2 cells in PB (C) and populations of B cells (B220+), myeloid cells (Gr-1+ Mac-1+), erythroid-myeloid progenitor cells (HPCs; Lin− Sca1− c-kit+ IL-7R−), and HSCs (LSKs) in BM from CM (D; mean ± SD of five animals) after Apc ablation 7 wk after transplantation. *, P < 0.05; **, P < 0.01.
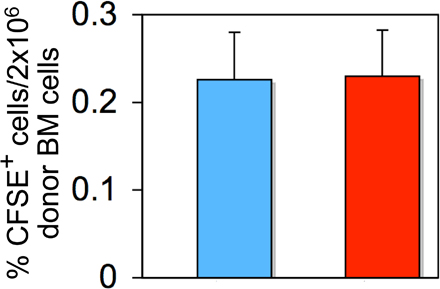
Next, we performed competitive reconstitution assays to examine the function of Apc mutant HSCs in vivo. At 4 d after induction (two injections of pI-pC), equal numbers of BM cells (CD45.2+) from induced Mx1-Cre+Apcfl/fl mice or control Mx1-Cre−Apcfl/fl mice together with competitor BM cells (CD45.1+) were transplanted into lethally irradiated WT CD45.1 recipient mice. Analysis of CM 7 wk after transplantation revealed an extremely low contribution of the donor cells (CD45.2+) in PB and BM cells (Fig. 3, C and D). In addition, Apc mutant donor HSCs gave rise to very few HSCs (LSKs) and HPCs in recipient mice, whereas control donor cells generated a normal proportion of primitive and mature hematopoietic cells, indicating that Apc mutant HSCs are defective in reconstituting hematopoiesis in irradiated recipient mice (Fig. 3 D). Moreover, we performed in vivo homing assays and determined that deletion of Apc does not affect homing of Apc-deficient BM cells to the BM in irradiated recipient mice (Fig. S3, available at http://www.jem.org/cgi/content/full/jem.20080578/DC1).
Loss of Apc results in exhaustion of the myeloid progenitor pool in vivo
Analysis of PB in Mx1-Cre+Apcfl/fl CM revealed that loss of Apc function results in rapid loss of multilineage hematopoietic cells. We analyzed myeloid and erythroid lineage cells by flow cytometric analysis of cell surface markers in CM 7–10 d after induction. The mature myeloid cells (Gr-1+ Mac-1+) were reduced dramatically in BM (4 ± 3 vs. 52 ± 10%; P < 0.01) and in spleen (0.43 ± 0.3 vs. 7.9 ± 3.5%; P < 0.01) from Apc mutant CM as compared with Apc-control CM (Fig. 4 A). Apc mutant CM had a significantly increased proportion of the proerythroblast (RI, Ter119lowCD71hi; 2.5 ± 2 vs. 0.7 ± 0.2%; P < 0.05) and basophilic erythroblast (RII, Ter119hiCD71hi; 53 ± 8.5 vs. 17 ± 6.5%; P < 0.01) populations as compared with Apc control CM; however, the frequency of late erythroblasts (RIII, Ter119hiCD71med; and RIV, Ter119hiCD71low) was unaffected (Fig. 4 B). Thus, loss of Apc function led to ineffective erythropoiesis as a result of differentiation arrest of late erythroblasts. Although the relative proportion of Ter119-positive cells was increased, the total number of Ter119-positive cells was reduced in Apc mutant CM as compared with Apc control (unpublished data).
Figure 4.

Loss of Apc results in exhaustion of myeloid progenitor cells. (A and B) Proportion of myeloid cells (Gr-1+ Mac-1+; A) in BM and spleen (SP and erythroid cells; B) in BM from chimeric Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice (mean ± SD of five to six animals). (C) In vitro analysis of HPCs. The number of BM CFU-GEMM, CFU-GM, CFU-G, CFU-M, and BFU-E was examined in Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice 4 d after induction (mean ± SD of three animals). *, P < 0.05; **, P < 0.01. (D and E) Proportion (D) and absolute number (E) of myeloid progenitor cells in BM from chimeric Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice (mean ± SD of five to six animals). *, P < 0.05; **, P < 0.02.
To determine the frequency of myeloid progenitor cells in BM, we performed in vitro colony-forming unit assays. BM cells harvested from primary Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice 4 d after induction were plated in methylcellulose medium containing SCF, IL3, and IL6. Apc mutant BM cells gave rise to fewer colony forming units and CFU-GEMM, CFU-GM, CFU-G, CFU-M, and BFU-E colonies than did the Apc control BM cells (Fig. 4 C), indicating that Apc mutant BM cells contained a reduced number of myeloid progenitors as compared with Apc control BM cells.
Next, we characterized the myeloid progenitor compartment. Apc mutant chimeras had a mean sevenfold decrease in the frequency of myeloid progenitor cells (Lin− Sca-1− IL-7R− c-kit+) as compared with Apc control chimeras (Fig. 4 D). In addition, the CMP (Lin− Sca-1− IL-7R− c-kit+ FcγRII/IIIlow CD34hi) and GMP (Lin− Sca-1− IL-7R− c-kit+ FcγRII/IIIhi CD34hi) populations were nearly absent, whereas the proportion of MEP (Lin− Sca-1− IL-7R− c-kit+ FcγRII/IIIlow CD34low) were increased significantly in BM from Apc mutant CM as compared with Apc control chimeras (Fig. 4 D). However, the total number of CMP, MEP, and GMP are significantly reduced in BM from Apc mutant CM as compared with Apc control chimeras (Fig. 4 E), suggesting that Apc is required for survival of different myeloid progenitors.
Loss of Apc blocks B cell and T cell development
Adolfsson et al. (28) have proposed an alternative model for hematopoietic development based on the development of a population of lymphoid-primed multipotent progenitor pool (LMPP, Lin− Sca-1+ c-kit+ Flt3+) cells primarily with B cell, T cell, and monocyte potential. The frequency of LMPPs (Fig. 5 A) is increased in Apc mutant CM as compared with control CM. However, the total number of LMPPs is decreased because of a dramatic decrease in the absolute numbers of LSKs in Apc-mutant CM (Fig. 5 C, top). Phenotypic analysis demonstrated that these mice had substantially fewer pro-B cells (B220+ CD43+ IgM−), Pre-B cells (B220+ CD43− IgM−), and immature B cells (Fig. 5, B and C, bottom). The proportion of mature B cells was increased, but there was a decrease in the absolute numbers because of a significantly lower number of total BM cells in Apc mutant mice. These data suggest that loss of Apc blocks B cell development early at the HSC/LMPP stage. Examination of the thymus from primary Mx1-Cre+Apcfl/fl mice 4 d after induction revealed that the cellularity was decreased by >10-fold, which is consistent with the morphology which showed depletion of lymphoid cells (Fig. S4, available at http://www.jem.org/cgi/content/full/jem.20080578/DC1), indicating that Apc is also required for survival of T cells. With respect to the early stages of T cell development, we showed that early T cell progenitors (ETPs, Lin−c-kit+ CD25−), DN2 (Lin−c-kit+ CD25+), and DN3 (Lin−c-kit− CD25+) were nearly absent in Apc mutant mice as compared with Apc control mice (Fig. 5 D, right), indicating that Apc is required for survival of T cells at early developmental stages. Alternatively, it is possible that the putative T cell progenitors are unable to reach the thymus as a result of HSC failure. Consistent with previous findings (29), the CD4, CD8 double-positive population was dramatically decreased in Apc mutant mice (Fig. 5 D, left), as compared with control mice. Collectively, our data suggest that Apc is a key regulator of hematopoietic cell development.
Figure 5.

Loss of Apc function blocks B cell and T cell differentiation. (A–C) Analysis of B cells. Frequency (A) and total number (C, top) of LMPPs and frequency (B) and total number (C, bottom) of pro-B cells (B220+ CD43+ IgM−), preB cells (B220+ CD43− IgM−), immature B cells (B220low CD43− IgM+), and mature B cells (B220hi CD43− IgM+) in BM cells from chimeric Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice 7 d after induction (mean ± SD of three to five animals). (D) FACS analysis of c-Kit and CD25 expression in Lin− thymocytes (left) and of CD4 and CD8 expression in thymocytes (right) from primary Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice 2–4 d after induction (mean ± SD of three to five animals). *, P < 0.05; **, P < 0.01.
Induction of Apc ablation in preestablished CM
To examine the long-term function of Apc mutant HSCs in a normal microenvironment, we performed competitive repopulation assays by generating CM (WT) reconstituted with a mixture of uninduced Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl donor cells (CD45.2+) and competitor WT donor cells (half the number of Apcfl/fl cells; CD45.1+; Fig. 6 A). After 8 wk, the chimeras were injected with pI-pC to induce ablation of Apc in engrafted BM. All treated mice survived and appeared to be healthy. We found that the Apc mutant myeloid cells (Gr-1+ and Mac-1+) were depleted within 20 d after induction, and the proportion of Apc mutant B cells (B220+) and T cells (CD3+) were gradually reduced in PB after Apc ablation in recipient mice (Fig. 6 B). In addition, the proportion of Apc mutant HSCs (LSKs) and HPCs was substantially lower as compared with control HSCs in recipient mice after 3 mo (Fig. 6 C). These data are consistent with our analysis of noncompetitive reconstituted chimeras and suggest that Apc regulates the survival of HSCs (LSKs) and HPCs, independent of the hematopoietic microenvironment.
Figure 6.

Loss of Apc function abolishes HSC function in preestablished BM chimeras. (A) Schematic diagram of the experimental strategy. (B) Evaluation of the proportions of donor-derived CD45.2 cells contributing to the major lineages in PB after induction of Apc deletion (mean ± SD of 4 animals). (C) Fractions of donor-derived cells in each BM cell subset 3 mo after pI-pC injection (mean ± SD of four animals). **, P < 0.01.
Apc regulates HSC/HPC proliferation
Next, we analyzed the cell cycle profile of HSCs (LSKs) to determine whether loss of Apc function results in altered cell cycle parameters. This analysis revealed that the proportion of LSKs in the S and G2-M phases was increased by 2–3-fold in Apc mutant mice as compared with control mice (Fig. 7 A). To examine the fraction of LSKs in the G0-G1 phases, we stained LSKs with both pyronin Y and HOECHST 33342 to assess the RNA and DNA content, as previously described (12). The Apc mutant mice showed a decreased proportion of LSKs in the G0 phase and an increased proportion of LSKs in the S-G2-M phases (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20080578/DC1). We performed further analysis of the cell cycle profile of erythromyeloid progenitor cells (Lin− Sca1−c-Kit+ IL-7R−) in primary and Apc mutant and control mice CM. The proportion of S-phase HPCs 4 d after induction is slightly increased in primary Apc mutant mice as compared with control mice; however, there is a significant increase of S-phase HPCs in Apc mutant CM as compared with control CM by 7 d after induction, indicating that the proliferation of HPCs is increased after Apc ablation (Fig. S6). In addition, the genes encoding the cyclin-dependent kinase inhibitors Cdkn1a and Cdkn1b, which control cell cycling and repopulation of HSCs (30, 31), respectively, were markedly down-regulated in Apc mutant LSK cells. Notably, there was no significant alteration in expression of Ccnd1, Myc, Myb, Gata2, Gata1, Bmi1, and Hoxb4 in Apc mutant HSCs (Fig. 7 B).
Figure 7.

Analysis of the cell cycle and expression of regulatory genes in Apc-deficient LSKs. (A) Cell cycle profile of LSKs from BM 4 d after induction (mean ± SD of three animals). **, P < 0.01. (B) Analysis of gene expression in Apc mutant LSK cells by qRT-PCR analysis of RNA isolated from sorted LSK cells from primary Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl mice 4 d after induction. The relative expression is shown normalized to gene expression in control HSCs (mean ± SD of three animals). *, P < 0.05.
Apc plays a pivotal role in survival of HSCs/HPCs
Next, we sought to determine whether altered apoptosis of the HSCs (LSKs) as a result of loss of Apc function accounts for the rapid exhaustion of the HSC pool in Apc mutant mice. The frequency of HSCs undergoing apoptosis was significantly higher in primary Apc mutant mice 4 d after induction as compared with control mice, whereas the frequency of apoptosis in HPCs was unaltered (Fig. 8 A). By 7 d after induction, the frequency of apoptosis of both HSCs and HPCs was increased significantly in Apc mutant CM as compared with control CM (Fig. 8, B and C). In addition, the frequency of apoptosis in Lin+ BM cells representing the mature population is comparable among Apc mutant and control CM 7 d after induction (Fig. S7, available at http://www.jem.org/cgi/content/full/jem.20080578/DC1) but is significantly increased in the moribund CM 12 d after induction (Fig. 8 D), suggesting that loss of Apc function had a progressive cell-intrinsic effect on survival of HSCs and HPCs, as well as mature BM cells. Autopsy and gross examination revealed liver pathology (numerous white patches on the surface of the liver) in primary Apc mutant mice (Fig. S8 A) but not in control mice (not depicted); however, there was no obvious evidence of organ damage in the kidneys, lungs, and heart. Histological analysis of liver in primary Apc mutant mice revealed many necrotic regions, which were characterized by a significant number of apoptotic cells identified by TUNEL staining (Fig. S8 B). These results suggest that loss of Apc function leads to hepatocyte apoptosis in vivo and that the rapid mortality of the primary Apc mutant mice is caused by injury to major organs.
Figure 8.

Analysis of apoptosis and expression of genes involved in the regulation of apoptosis in Apc-deficient HSCs. (A and B) Frequency of apoptosis in gated HSCs (LSKs) and HPCs (Lin− Sca-1− c-kit+ IL-7R−) stained with Annexin V and 7-ADD from Mx1-Cre+Apcfl/fl and Mx1-Cre−Apcfl/fl mice 4 d after induction (primary mice; A) or 7 d after induction (CM; B; mean ± SD of three animals). *, P < 0.05; **, P < 0.01. (C) Representative histograms of flow cytometric analysis of the frequency of apoptotic cells in gated HSCs (LSKs) and HPCs 7 d after induction (CM). (D) Representative TUNEL staining of BM cells from Mx1-Cre−Apcfl/fl CM mice (left) and Mx1-Cre+Apcfl/fl CM mice (right), analyzed 12 d after three doses of pI-pC induction. Apoptotic cells are shown in brown (TUNEL positive). Bar, 20 μm. (E) Analysis of gene expression in Apc mutant LSK cells by qRT-PCR analysis of RNA isolated from sorted LSK cells from primary Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl mice 4 d after induction. The relative expression is shown normalized to gene expression in control HSCs (mean ± SD of three animals). *, P < 0.05.
To determine the molecular mechanism underlying increased apoptosis of Apc mutant HSCs (LSKs), we performed expression analysis of several genes involved in regulating cell death of HSCs. Loss of genes encoding survival factors, such as Mcl1 or Birc5 (survivin), leads to depletion of HSCs (15, 16), whereas constitutive expression of Bcl2 results in accumulation of HSCs in vivo and enhances the capacity of HSCs to reconstitute the hematopoietic system in irradiated recipient mice (32). The LSK cells were isolated from the primary Apc mutant and control mice 4 d after two doses of pI-pC induction. In sorted Apc mutant LSK cells, Mcl1 is down-regulated; however, the expression of other genes involved in cell death and survival was not significantly altered (Fig. 8 E).
Loss of Apc leads to an accumulation of β-catenin in hematopoietic cells in vivo
Mutant APC contributes to colorectal tumorigenesis through stabilization of β-catenin, resulting in activation of the Wnt pathway. To determine if ablation of Apc results in accumulation of β-catenin, we analyzed protein lysates from hematopoietic cells from Mx1-Cre−Apcfl/fl and Mx1-Cre+Apcfl/fl CM 4 d after induction by immunoblotting using antibodies to β-catenin. β-Catenin levels were elevated in hematopoietic cells from thymus and spleen from Mx1-Cre+Apcfl/fl CM (Fig. 9 A). The expression of β-catenin in BM cells was not detectable by immunoblotting (unpublished data), which may be because of the heterogeneity of hematopoietic cells in the BM. Thus, we assessed the intracellular level of β-catenin in primitive hematopoietic cells and subsets of mature hematopoietic cells in BM by flow cytometry. Our data revealed that ablation of Apc stabilizes intracellular β-catenin in LSKs, HPCs, and mature B cells (B220+) but not in mature (Gr-1+ and Mac-1+) myeloid cells, which is indicative of activation of the Wnt pathway in subsets of BM cells (Fig. 9 B).
Figure 9.

Ablation of Apc results in the accumulation of β-catenin in hematopoietic cells in vivo. (A) Expression of β-catenin in Thymus (T) and Spleen (SP) from CM 4 d after induction (two doses). (B) Expression of intracellular β-catenin in subsets of BM cells from Mx1-Cre−Apcfl/fl (blue profile) and Mx1-Cre+Apcfl/fl (red profile) CM 4 d after induction (two doses). Data are representative of three independent experiments.
DISCUSSION
The APC tumor suppressor gene is located on chromosome band 5q22 (17). Loss of a whole chromosome 5 or a del(5q) are recurring cytogenetic abnormalities in myeloid neoplasms and are noted in 15% of patients with a primary myelodysplastic syndrome or acute myeloid leukemia de novo and in >40% of patients with therapy-related myelodysplastic syndrome or acute myeloid leukemia (33). However, the role of APC in the function of HSCs and hematopoiesis is poorly understood. In this paper, we used a conditional knockout animal model to examine the function of Apc in the hematopoietic system in vivo and demonstrate that Apc is a key regulator of HSC and HPC maintenance, survival, and differentiation.
The maintenance of HSCs is required for hematopoietic homeostasis throughout the lifespan of an animal. In this paper, we demonstrate that endogenous Apc is an essential regulator of HSC maintenance. Ablation of Apc in both primary Mx1-Cre+Apcfl/fl mice and chimeric Mx1-Cre+Apcfl/fl mice results in rapid BM failure and lethality. Further characterization of Apc-deficient mice revealed that loss of hematopoietic homeostasis correlates with a rapid loss of the HSC and HPC compartments. As compared with other animal models for important regulators of HSC maintenance, such as Bmi1 (34, 35), Pten (12, 13), Cdkn1a (31), and Mcl1 (16), the effect of Apc ablation on HSC maintenance is more rapid and striking. In addition, Apc mutant HSCs failed to reconstitute hematopoiesis in lethally irradiated recipient mice. This intrinsic functional failure was also observed in the context of a WT microenvironment, indicating that the effect is cell autonomous.
Regulation of the cell cycle of HSCs plays an important role in the maintenance of the HSC pool. Most HSCs are maintained in a quiescent state and divide once within a 2–4-wk period in the mouse (36). Enforced cell cycle entry as a result of deletion of Cdkna1 or Pten leads to exhaustion of the HSC pool (12, 13, 31). Consistent with previous findings that overexpression of Apc blocks cell cycle progression from G0/G1 to the S phase (37), we demonstrated that deletion of Apc increased the proportion of proliferating HSCs and myeloid progenitors markedly. The proportion of HSCs that exit G0/G1 and enter the S phase is increased about threefold on average in Apc-deficient mice (Fig. 7 A). Thus, an increase in the proportion of proliferating cells caused by ablation of Apc contributes to the exhaustion of the HSC pool in Apc mutant mice. In addition, we found that both Cdkn1a and Cdkn1b are expressed at lower levels in Apc mutant LSK cells, suggesting that the role of Apc in regulating cell cycle progression may be associated with its function in negatively modulating the activity of cyclin–CDK complexes.
Regulation of apoptosis is also critical for the maintenance of the HSC pool. Loss of survival factors, such as Mcl1 and Birc5, leads to depletion of BM cells (15, 16). Consistent with previous results that conditional inactivation of Apc in the neural crest induces massive apoptosis during development (38), we found that loss of Apc significantly increased the frequency of apoptosis in HSCs and HPCs, which contributes to the loss of hematopoietic homeostasis. In addition, we demonstrated that deletion of Apc also induces apoptosis of hepatocytes in vivo. The expression of Mcl1 is down-regulated in the Apc mutant LSK population, which may contribute to induction of apoptosis in Apc-deficient HSCs. Loss of Apc is associated with chromosomal instability and missegregation (20), raising the possibility that induction of apoptosis by loss of Apc in hematopoietic cells may result, in part, from an increased frequency of chromosome missegregation.
Our data also suggests that inactivation of Apc in vivo leads to a block in multilineage differentiation. Differentiation of the erythroid lineage was blocked at early erythroblast stages, whereas development of B cells was blocked at the LMPP stage. Gounari et al. (29) demonstrated that loss of Apc disrupts thymic development. We have extended these observations and determined that Apc ablation results in an absence of ETP, DN2, and DN3 T cells and a delay in the DN4 and DP transitions during T cell development. Collectively, our data indicate that Apc is a key regulator of hematopoiesis.
In line with the role of Apc as an antagonist of the Wnt pathway, loss of Apc leads to the accumulation of intracellular β-catenin in hematopoietic cells from thymus and spleen, HSCs, HPCs, and subsets of mature BM cells, indicating that loss of Apc may activate the Wnt pathway by stabilizing β-catenin in HSCs, HPCs, and other hematopoietic cells. The involvement of Wnt signaling in regulating HSC activity is controversial (39–43). Activation of Wnt signaling by overexpression of stabilized β-catenin enhances the self-renewal of HSCs (39). Conversely, enhanced expression of Axin, a negative regulator of the Wnt pathway, resulted in inhibition of HSC proliferation in vitro and decreased reconstitution capacity of HSCs (39). In addition, stimulation of the Wnt pathway with purified Wnt protein in vitro (40), or a small-molecule inhibitor of GSK3β in vivo (41), promotes the self-renewal of HSCs. Nonetheless, in vivo studies have yielded unexpected results. Conditional inactivation of β-catenin did not affect the function of HSCs and hematopoiesis in vivo (44). However, Kirstetter et al. (42) and Scheller et al. (43) showed that conditional expression of stabilized β-catenin in hematopoietic cells in vivo results in disrupted function of HSCs and lethality in mice. A caveat is that it is not yet known whether the magnitude of Wnt signaling resulting from expression of stabilized β-catenin reflects physiological conditions in hematopoietic cells. Our data showed that Apc-deficient mice share some common phenotypic features, such as enhanced cell cycle entry of HSC, as well as multilineage defects, with mice expressing stabilized β-catenin in hematopoietic cells, suggesting that the function of Apc in the hematopoietic system may, in part, be mediated by the regulation of β-catenin stabilization. In addition, tight regulation of the magnitude of signaling through the Wnt pathway by Apc may be required for the maintenance and differentiation of HSCs and HPCs. However, loss of Apc results in more severe defects in HSCs as compared with overexpression of β-catenin, suggesting that additional signaling pathways may be involved in mediating the effect. Recently, down-regulation of Apc has been implicated in activation of the mTOR pathway independent of β-catenin–dependent transcription (45). Whether activation of the mTOR signaling pathway resulting from loss of Apc enhances protein synthesis, leading to a synergistic growth-stimulatory effect with stabilization of β-catenin in Apc mutant HSCs, remains to be determined.
Although there are some common features, our data indicate that acute physiological loss of Apc in HSCs has some consequences that are distinct from those of aberrant stabilization and constitutive activation of β-catenin. For example, Apc ablation resulted in extremely rapid depletion of HSCs, which is in contrast to the expansion of HSCs observed after expression of constitutively active β-catenin in vitro (39) and in vivo (42, 43). In addition, Apc ablation increased the frequency of apoptosis of HSCs and HPCs and led to a marked reduction in the pool of myeloid progenitors, whereas expression of constitutively active β-catenin did not affect apoptosis in HSCs or the size of the myeloid progenitor pool (42, 43). Thus, the mechanism by which loss of Apc increases the frequency of apoptosis in HSCs may be independent of Wnt/β-catenin signaling. Apc is a multidomain protein that interacts with several other proteins, and has been implicated in multiple cellular functions in addition to regulation of the Wnt pathway (17, 18). In this regard, the Drosophila melanogaster homologue of Apc plays a role in the orientation of asymmetrical cell division of male germline stem cells, suggesting that Apc may contribute to maintaining the balance between self-renewal and differentiation of stem cells (46). Whether Apc regulates the self-renewal of HSCs by controlling asymmetrical cell division is unknown. In addition, Apc regulates thymocyte development through regulation of Wnt/β-catenin signaling and control of chromosome segregation (29). Collectively, the available data suggest that other cellular functions of Apc may also contribute to its critical role in maintaining the integrity of the HSC and progenitor compartments. The rapid lethality of Apc mutant mice may be the result of synergistic effects resulting from defects in multiple cellular functions of Apc.
In the gastrointestinal system, Apc is expressed in colonic epithelial cells, where its expression is lower at the bottom and higher in the upper portions of the crypts, suggesting that there is an increase in the level of Apc expression with maturation of the epithelium (47, 48). Loss of Apc function is believed to lead to expansion of the intestinal stem cell compartment (17). Collectively, these data suggest that Apc has a differential effect in HSCs and intestinal stem cells, i.e., exhaustion of the HSC pool versus expansion of the intestinal stem cell pool, and that the function of Apc in regulating self-renewal of adult stem cells is cell context dependent.
MATERIALS AND METHODS
Apc mutant mice.
Apcfl/fl mice carrying a conditionally targeted allele of Apc (APC580s; reference 21) were generated from embryonic stem cells obtained from T. Noda (The Cancer Institute, Tokyo, Japan; reference 22). Apcfl/+ mice carrying the Mx1-Cre transgene (23) were crossed with Apcfl/fl mice to generate Mx1-Cre+Apcfl/fl, Mx1-Cre−Apcfl/fl, Mx1-Cre+Apcfl/+, and Mx1-Cre−Apcfl/+ mice. To induce excision of exon 14 of Apc, primary mice or transplanted recipient mice received 6–10 μg pI-pC (GE Healthcare) per gram of body weight per injection i.p. every second day for two or three doses. The deletion status of Apc was monitored by PCR analysis. All animal research was approved by the University of Chicago Institutional Animal Care and Use Committee.
Flow cytometric analysis.
Single cell suspensions from BM, spleen, PB, and thymus were stained with the indicated fluorochrome-conjugated antibodies (BD Biosciences and eBioscience). Flow cytometric analysis of HSCs and subsets of HPCs have been described previously (1, 2). For lineage analysis, whole BM cells were stained with various combinations of antibodies for different cell populations: Percp-B220, PE-CD43, and APC-IgM for B cells; PE-Gr-1 and APC-Mac-1 for myeloid cells; APC-Ter119 and PE-CD71 for erythroid cells; CD4 and CD8 for mature T cells; and CD4, CD8, TCRβ, TCRγδ, CD11c, NK1.1, Mac-1, B220, and Ter119 with APC-c-kit and PE-CD25 for early T cells. APC-Cy7-CD45.2 or FITC-CD45.2 and PE-CD45.1 were used to identify the donor cells in recipient mice. For sorting of HSCs and HPCs, BM cells were stained with biotinylated rat antibodies specific for lineage markers (CD3, B220, IgM, Ter119, Mac-1, Gr-1, and CD19). The Lin+ cells were depleted with sheep anti–rat IgG-conjugated magnetic beads (Dynabeads M-450; Invitrogen), and the remaining cells were resuspended and stained with APC-c-kit, PE-Sca-1, PE-Cy5-Flk2 (Flt3), and Streptavidin-FITC for HSCs and LMPPs, stained with APC-c-kit, PE-Sca-1, PE-Cy5-IL-7R, and Streptavidin-FITC for CLPs, or stained with APC-c-kit, PE-Sca-1, FITC-CD34, PE-Cy7-CD16/32 (Fcr), and Streptavidin-PE-Cy5 for myeloid progenitors. Dead cells were excluded by DAPI or propidium iodide staining. Flow cytometry was performed using FACSCanto or LSRII flow cytometers. Cell cycle analysis with pyronin Y and HOECHST 33342 staining was performed as described previously (12). For the detection of apoptosis, BM cells were stained with antibody conjugates, Annexin V, and 7-ADD (BD Biosciences). All data were analyzed by FlowJo software (Tree Star, Inc.).
Colony-forming unit and CAFC assays.
For colony-forming unit assays, total BM cells, isolated from Mx1-Cre+Apcfl/fl and Mx1-Cre−Apcfl/fl mice 4 d after induction, were plated in duplicate in methylcellulose medium (MethoCult; StemCell Technologies, Inc.) supplemented with IL3, IL6, and SCF, and scored after 10 d. The CAFC assay was performed as previously described (27). The FDMB1 stromal cell line was provided by G. Van Zant (University of Kentucky, Lexington, KY).
Transplantation assays.
BM transplants were performed as previously described (49). For competitive repopulation assays, 0.5 × 106 BM cells (CD45.2) from Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl (control) mice 4 d after two injections of pI-pC were mixed 1:1 with the competitor BM cells (CD45.1) from B6.SJL mice and transplanted into lethally irradiated B6.SJL mice by retroorbital injection. For generating preestablished BM chimeras, 106 BM cells from untreated Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl mice were mixed 2:1 with the competitor BM cells from B6.SJL and injected into lethally irradiated B6.SJL mice.
Quantitative real-time PCR.
To evaluate the expression of Apc, subsets of hematopoietic stem and progenitor cells were sorted from pooled BM cells from three to four C57/BL6 mice 2–3 m of age. To examine the expression of candidate genes in Apc mutant mice, LSK cells were sorted from Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl primary mice (n = 3) 4 d after the last pI-pC injection (two doses) into Trizol Reagent (Invitrogen). Total RNA was isolated and amplified using the full spectrum complete transcriptome RNA amplification kit (System Biosciences). The amounts of RNA were determined by real-time RT-PCR using SYBR Green incorporation. The gene expression levels were normalized to that of Hprt1 and expressed relative to the indicated reference sample. The primer sequences and the mean threshold value (Ct) for each gene for both Mx1-Cre+Apcfl/fl or Mx1-Cre−Apcfl/fl LSKs are listed in Tables S1 and S2 (available at http://www.jem.org/cgi/content/full/jem.20080578/DC1), respectively.
Statistical analysis.
Statistical significance was calculated using the two-tailed Student's t test.
Online supplemental material.
Fig. S1 shows that deletion of Apc leads to lethality. Fig. S2 shows CAFC frequency in BM from primary Mx1-Cre−Apcfl/fl and Mx1-Cre+Apcfl/fl mice after induction of Apc deletion. Fig. S3 shows analysis of homing ability of Mx1-Cre−Apcfl/fl and Mx1-Cre+Apcfl/fl BM cells in lethally irradiated recipient mice. Fig. S4 shows histological analysis of thymus from Mx1-Cre−Apcfl/fl and Mx1-Cre+Apcfl/fl primary mice 4 d after three doses of pI-pC. Fig. S5 shows the proportion of LSK cells in the G0, G1, or S+G2/M populations from Apc mutant and Apc control mice. Fig. S6 shows cell cycle profile of erythromyeloid progenitors from primary Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl mice 4 d after induction and Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl CM mice 7 d after induction. Fig. S7 shows representative histograms of flow cytometric analysis of frequency of apoptotic cells in gated CD45.2 +Lin+ bone marrow cells from Mx1-Cre+Apcfl/fl or control Mx1-Cre−Apcfl/fl CM mice 7 d after induction. Fig. S8 shows increased apoptosis of hepatocytes after induction of Apc deletion in vivo. Table S1 shows primers used in qRT-PCR analysis. Table S2 shows the mean threshold cycle value for each gene in qRT-PCR. Supplemental Materials and methods are also available. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20080578/DC1.
Supplementary Material
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health (CA40046). We thank Dr. Jiwang Zhang (Loyola University) and members of the Le Beau laboratory for helpful discussions and Dr. John Anastasi for histological analysis.
The authors have no conflicting financial interests.
Abbreviations used: APC, adenomatous polyposis coli; CAFC, cobblestone area–forming cell; CLP, common lymphoid progenitor; CM, chimeric mice; CMP, common myeloid progenitor; GMP, granulocyte-monocyte progenitor; HPC, hematopoietic progenitor cell; HSC, hematopoietic stem cell; LMPP, lymphoid-primed multipotent progenitor pool; MEP, megakaryocyte-erythroid progenitor; PB, peripheral blood; pI-pC, polyinosinic-polycytidylic acid; qRT-PCR, quantitative RT-PCR.
References
- 1.Kondo, M., I.L. Weissman, and K. Akashi. 1997. Identification of clonogenic common lymphoid progenitors in mouse bone marrow. Cell. 91:661–672. [DOI] [PubMed] [Google Scholar]
- 2.Akashi, K., D. Traver, T. Miyamoto, and I.L. Weissman. 2000. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 404:193–197. [DOI] [PubMed] [Google Scholar]
- 3.Akala, O.O., and M.F. Clarke. 2006. Hematopoietic stem cell self-renewal. Curr. Opin. Genet. Dev. 16:496–501. [DOI] [PubMed] [Google Scholar]
- 4.Hock, H., E. Meade, S. Medeiros, J.W. Schindler, P.J. Valk, Y. Fujiwara, and S.H. Orkin. 2004. Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 18:2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hock, H., M.J. Hamblen, H.M. Rooke, J.W. Schindler, S. Saleque, Y. Fujiwara, and S.H. Orkin. 2004. Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature. 431:1002–1007. [DOI] [PubMed] [Google Scholar]
- 6.Zeng, H., R. Yucel, C. Kosan, L. Klein-Hitpass, and T. Moroy. 2004. Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J. 23:4116–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonchuk, J., G. Sauvageau, and R.K. Humphries. 2002. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 109:39–45. [DOI] [PubMed] [Google Scholar]
- 8.Sauvageau, G., U. Thorsteinsdottir, C.J. Eaves, H.J. Lawrence, C. Largman, P.M. Lansdorp, and R.K. Humphries. 1995. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 9:1753–1765. [DOI] [PubMed] [Google Scholar]
- 9.Krosl, J., P. Austin, N. Beslu, E. Kroon, R.K. Humphries, and G. Sauvageau. 2003. In vitro expansion of hematopoietic stem cells by recombinant TAT-HOXB4 protein. Nat. Med. 9:1428–1432. [DOI] [PubMed] [Google Scholar]
- 10.Wilson, A., M.J. Murphy, T. Oskarsson, K. Kaloulis, M.D. Bettess, G.M. Oser, A.C. Pasche, C. Knabenhans, H.R. Macdonald, and A. Trumpp. 2004. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18:2747–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galan-Caridad, J.M., S. Harel, T.L. Arenzana, Z.E. Hou, F.K. Doetsch, L.A. Mirny, and B. Reizis. 2007. Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell. 129:345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang, J., J.C. Grindley, T. Yin, S. Jayasinghe, X.C. He, J.T. Ross, J.S. Haug, D. Rupp, K.S. Porter-Westpfahl, L.M. Wiedemann, et al. 2006. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 441:518–522. [DOI] [PubMed] [Google Scholar]
- 13.Yilmaz, O.H., R. Valdez, B.K. Theisen, W. Guo, D.O. Ferguson, H. Wu, and S.J. Morrison. 2006. Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature. 441:475–482. [DOI] [PubMed] [Google Scholar]
- 14.Cheng, T. 2004. Cell cycle inhibitors in normal and tumor stem cells. Oncogene. 23:7256–7266. [DOI] [PubMed] [Google Scholar]
- 15.Leung, C.G., Y. Xu, B. Mularski, H. Liu, S. Gurbuxani, and J.D. Crispino. 2007. Requirements for survivin in terminal differentiation of erythroid cells and maintenance of hematopoietic stem and progenitor cells. J. Exp. Med. 204:1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Opferman, J.T., H. Iwasaki, C.C. Ong, H. Suh, S. Mizuno, K. Akashi, and S.J. Korsmeyer. 2005. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science. 307:1101–1104. [DOI] [PubMed] [Google Scholar]
- 17.Fodde, R., R. Smits, and H. Clevers. 2001. APC, signal transduction and genetic instability in colorectal cancer. Nat. Rev. Cancer. 1:55–67. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama, T., and Y. Kawasaki. 2006. Wnt signalling and the actin cytoskeleton. Oncogene. 25:7538–7544. [DOI] [PubMed] [Google Scholar]
- 19.Bienz, M., and F. Hamada. 2004. Adenomatous polyposis coli proteins and cell adhesion. Curr. Opin. Cell Biol. 16:528–535. [DOI] [PubMed] [Google Scholar]
- 20.Hanson, C.A., and J.R. Miller. 2005. Non-traditional roles for the adenomatous polyposis coli (APC) tumor suppressor protein. Gene. 361:1–12. [DOI] [PubMed] [Google Scholar]
- 21.Shibata, H., K. Toyama, H. Shioya, M. Ito, M. Hirota, S. Hasegawa, H. Matsumoto, H. Takano, T. Akiyama, K. Toyoshima, et al. 1997. Rapid colorectal adenoma formation initiated by conditional targeting of the Apc gene. Science. 278:120–123. [DOI] [PubMed] [Google Scholar]
- 22.Qian, C.N., J. Knol, P. Igarashi, F. Lin, U. Zylstra, B.T. Teh, and B.O. Williams. 2005. Cystic renal neoplasia following conditional inactivation of apc in mouse renal tubular epithelium. J. Biol. Chem. 280:3938–3945. [DOI] [PubMed] [Google Scholar]
- 23.Kuhn, R., F. Schwenk, M. Aguet, and K. Rajewsky. 1995. Inducible gene targeting in mice. Science. 269:1427–1429. [DOI] [PubMed] [Google Scholar]
- 24.Holmen, S.L., C.R. Zylstra, A. Mukherjee, R.E. Sigler, M.C. Faugere, M.L. Bouxsein, L. Deng, T.L. Clemens, and B.O. Williams. 2005. Essential role of beta-catenin in postnatal bone acquisition. J. Biol. Chem. 280:21162–21168. [DOI] [PubMed] [Google Scholar]
- 25.Sansom, O.J., K.R. Reed, A.J. Hayes, H. Ireland, H. Brinkmann, I.P. Newton, E. Batlle, P. Simon-Assmann, H. Clevers, I.S. Nathke, et al. 2004. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 18:1385–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sansom, O.J., D.F. Griffiths, K.R. Reed, D.J. Winton, and A.R. Clarke. 2005. Apc deficiency predisposes to renal carcinoma in the mouse. Oncogene. 24:8205–8210. [DOI] [PubMed] [Google Scholar]
- 27.de Haan, G., W. Nijhof, and G. Van Zant. 1997. Mouse strain-dependent changes in frequency and proliferation of hematopoietic stem cells during aging: correlation between lifespan and cycling activity. Blood. 89:1543–1550. [PubMed] [Google Scholar]
- 28.Adolfsson, J., R. Mansson, N. Buza-Vidas, A. Hultquist, K. Liuba, C.T. Jensen, D. Bryder, L. Yang, O.J. Borge, L.A. Thoren, et al. 2005. Identification of Flt3+ lympho-myeloid stem cells lacking erythro-megakaryocytic potential a revised road map for adult blood lineage commitment. Cell. 121:295–306. [DOI] [PubMed] [Google Scholar]
- 29.Gounari, F., R. Chang, J. Cowan, Z. Guo, M. Dose, E. Gounaris, and K. Khazaie. 2005. Loss of adenomatous polyposis coli gene function disrupts thymic development. Nat. Immunol. 6:800–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng, T., N. Rodrigues, D. Dombkowski, S. Stier, and D.T. Scadden. 2000. Stem cell repopulation efficiency but not pool size is governed by p27(kip1). Nat. Med. 6:1235–1240. [DOI] [PubMed] [Google Scholar]
- 31.Cheng, T., N. Rodrigues, H. Shen, Y. Yang, D. Dombkowski, M. Sykes, and D.T. Scadden. 2000. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 287:1804–1808. [DOI] [PubMed] [Google Scholar]
- 32.Domen, J., S.H. Cheshier, and I.L. Weissman. 2000. The role of apoptosis in the regulation of hematopoietic stem cells: overexpression of BCL-2 increases both their number and repopulation potential. J. Exp. Med. 191:253–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thirman, M.J., and R.A. Larson. 1996. Therapy-related myeloid leukemia. Hematol. Oncol. Clin. North Am. 10:293–320. [DOI] [PubMed] [Google Scholar]
- 34.Park, I.K., D. Qian, M. Kiel, M.W. Becker, M. Pihalja, I.L. Weissman, S.J. Morrison, and M.F. Clarke. 2003. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 423:302–305. [DOI] [PubMed] [Google Scholar]
- 35.Lessard, J., and G. Sauvageau. 2003. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 423:255–260. [DOI] [PubMed] [Google Scholar]
- 36.Bradford, G.B., B. Williams, R. Rossi, and I. Bertoncello. 1997. Quiescence, cycling, and turnover in the primitive hematopoietic stem cell compartment. Exp. Hematol. 25:445–453. [PubMed] [Google Scholar]
- 37.Baeg, G.H., A. Matsumine, T. Kuroda, R.N. Bhattacharjee, I. Miyashiro, K. Toyoshima, and T. Akiyama. 1995. The tumour suppressor gene product APC blocks cell cycle progression from G0/G1 to S phase. EMBO J. 14:5618–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasegawa, S., T. Sato, H. Akazawa, H. Okada, A. Maeno, M. Ito, Y. Sugitani, H. Shibata, J. Miyazaki Ji, M. Katsuki, et al. 2002. Apoptosis in neural crest cells by functional loss of APC tumor suppressor gene. Proc. Natl. Acad. Sci. USA. 99:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reya, T., A.W. Duncan, L. Ailles, J. Domen, D.C. Scherer, K. Willert, L. Hintz, R. Nusse, and I.L. Weissman. 2003. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 423:409–414. [DOI] [PubMed] [Google Scholar]
- 40.Willert, K., J.D. Brown, E. Danenberg, A.W. Duncan, I.L. Weissman, T. Reya, J.R. Yates III, and R. Nusse. 2003. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 423:448–452. [DOI] [PubMed] [Google Scholar]
- 41.Trowbridge, J.J., A. Xenocostas, R.T. Moon, and M. Bhatia. 2006. Glycogen synthase kinase-3 is an in vivo regulator of hematopoietic stem cell repopulation. Nat. Med. 12:89–98. [DOI] [PubMed] [Google Scholar]
- 42.Kirstetter, P., K. Anderson, B.T. Porse, S.E. Jacobsen, and C. Nerlov. 2006. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat. Immunol. 7:1048–1056. [DOI] [PubMed] [Google Scholar]
- 43.Scheller, M., J. Huelsken, F. Rosenbauer, M.M. Taketo, W. Birchmeier, D.G. Tenen, and A. Leutz. 2006. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat. Immunol. 7:1037–1047. [DOI] [PubMed] [Google Scholar]
- 44.Cobas, M., A. Wilson, B. Ernst, S.J. Mancini, H.R. MacDonald, R. Kemler, and F. Radtke. 2004. β-Catenin is dispensable for hematopoiesis and lymphopoiesis. J. Exp. Med. 199:221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Inoki, K., H. Ouyang, T. Zhu, C. Lindvall, Y. Wang, X. Zhang, Q. Yang, C. Bennett, Y. Harada, K. Stankunas, et al. 2006. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 126:955–968. [DOI] [PubMed] [Google Scholar]
- 46.Yamashita, Y.M., D.L. Jones, and M.T. Fuller. 2003. Orientation of asymmetric stem cell division by the APC tumor suppressor and centrosome. Science. 301:1547–1550. [DOI] [PubMed] [Google Scholar]
- 47.Smith, K.J., K.A. Johnson, T.M. Bryan, D.E. Hill, S. Markowitz, J.K. Willson, C. Paraskeva, G.M. Petersen, S.R. Hamilton, B. Vogelstein, et al. 1993. The APC gene product in normal and tumor cells. Proc. Natl. Acad. Sci. USA. 90:2846–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Midgley, C.A., S. White, R. Howitt, V. Save, M.G. Dunlop, P.A. Hall, D.P. Lane, A.H. Wyllie, and V.J. Bubb. 1997. APC expression in normal human tissues. J. Pathol. 181:426–433. [DOI] [PubMed] [Google Scholar]
- 49.Wu, W.S., S. Heinrichs, D. Xu, S.P. Garrison, G.P. Zambetti, J.M. Adams, and A.T. Look. 2005. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 123:641–653. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}