

Abstract
Branched peptide amphiphile (PA) molecules bearing biological epitopes were designed and synthesized using orthogonal protecting group chemistry on amine groups at lysine residues. These molecules self-assemble into high-aspect-ratio cylindrical nanofibers, and their branched architecture enhances accessibility of epitopes for protein binding and also allows the presentation of more than one epitope in a single molecule. The RGDS cell adhesion epitope was used as a model bioactive signal on PA molecules for potential biomedical applications. Aggregation of the branched PA molecules into nanofibers was demonstrated by TEM and through shifts in the protonation profiles of peripheral amines. These systems also formed self-supporting gels in the presence of physiological fluids and other biologically relevant macromolecules such as synovial fluid and DNA, an important property for their potential use in medicine. Fluorescence anisotropy measurements on the PAs with tryptophan residues were performed to examine the effect of branching on packing and mobility of the peptides in the self-assembled nanofibers. The mobility of tryptophan residues was observed to be restricted upon packing of PA molecules into nanofibers. However, relative to linear analogues, branched molecules retain more mobility in the supramolecular aggregates.
Introduction
In recent years, there has been considerable interest in concept of tissue regeneration as a therapy for failed organs and trauma.1-10 These therapies require materials that can serve as scaffolds to either deliver or recruit cells. The scaffold’s function may range from mechanically supporting cells to actually signaling cells at the level of receptors to promote cell proliferation, cell survival, and cell differentiation, or even recruit the right cells to grow blood vessels that feed the cells and regenerate the organ or tissue.11
One of the chemical concepts to improve scaffolds has been to include peptide signals that can be involved in cell signaling.12-16 A favorite set of peptide epitopes have been those found in extracellular matrix proteins such as fibronectin, vitronectin, collagen, and laminin, which bind to integrin receptors important in cell survival and function. The RGDS epitope in fibronectin has been used in previous papers on the subject, and it is used here to probe molecular and supramolecular variables necessary to optimize the nature of scaffolds in regenerative medicine.14,17-19 Attaching the RGDS sequence to the surface of biomaterial scaffolds has been used to control cell adhesion, proliferation, differentiation, and organization.20-26 Biological adhesion through the RGD or RGDS sequences on synthetic substrates is known to depend on epitope density.20-26 The molecular implication is that many aspects of epitope structure and dynamics on the internal surfaces of scaffolds will be important to their recognition by receptors. There is previous work in which improved recognition of the RGDS epitope was achieved by using a cyclic structure sequence which mimics the loop presentation in natural fibronectin.16,27-29 It has also been observed that integrin mediated cell adhesion through these epitopes depends on presentation and gathering of receptors.22,24-26,30-32 Thus, different cell responses are observed with varying surface concentration of cell recognition motifs and with changes to the conformations of the presented ligands.26,32
We have previously reported on charged self-assembling peptide amphiphile (PA) molecules consisting of a β-sheet-forming segment and a bioactive peptide segment covalently grafted to a hydrophobic alkyl segment.33-38 These strong amphiphiles self-assemble into nanofibers that bond to form networks and therefore gels when their charge is eliminated by pH changes or screened by ions.37,39 The gels are known to encapsulate cells in aqueous three-dimensional networks of the nanofibers.39,40
Recently, we reported on enhanced recognition of biotinylated forms of the branched PAs containing the RGDS signal and synthesized using the lysine residues as the branch point.35 Biotin-labeled branched PAs and linear analogues were compared in terms of accessibility to fluorescently labeled avidin. It was clearly observed that biotins on the branched PAs are significantly more available to avidin than on nanofibers formed by the linear analogues. In this paper, branched PA molecules were studied by fluorescence polarization to elucidate accessibility of bioactive sequences with tryptophan, a natural chromophore, and to observe its fluorescence anisotropy in branched and linear architectures. The branched architecture provides further control over gel formation and enables presentation of multiple bioactive groups on the same molecule and decoration of the periphery of the nanofibers with functional groups. Additional branched PAs were also synthesized to present various bioactive signals on self-assembling high-aspect-ratio nanofibers.
Experimental Section
Solid-phase peptide synthesis (SPPS) technique was used to prepare peptide amphiphile (PA) molecules. Amino acids, MBHA rink amide resin, and HBTU [2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate] were purchased from NovaBiochem. All other chemicals were purchased from Fisher or Aldrich and used as provided. Peptides were constructed by standard Fmoc (9-Fluorenylmethoxycarbonyl) solid-phase peptide synthesis. Amino acid couplings were carried out with 4 equiv of Fmoc-protected amino acid, 3.95 equiv of HBTU, and 6 equiv of DIEA (N,N-diisopropylethylamine) for 4 h. Fmoc deprotections were performed with 30% piperidine/DMF solutions for 10 min. 4-Methyl trityl (Mtt) protecting groups were deprotected with 4% TFA/dichloromethane solutions for 5 min. The first arm of 8 shown in Chart 2 was synthesized on the α-amine of the Fmoc-Lys(Mtt)-OH at the branching point. The L-aspartic acid di-tert-butyl ester hemiscucinate 41 residue was coupled to the first branch, and after completion of the first peptide branch, the Mtt on the ∈-amine of the lysine residue was removed to add the bioactive RGDS peptide sequence. Cleavage of peptides from the resin was carried out using a mixture of TFA/ TIS/H2O (95:2.5:2.5) for 3 h. Excess TFA was removed by rotary evaporation. The remaining viscous peptide solution was triturated with cold ether, and the resulting white product was dried under vacuum. Cyclic RGDS containing molecule 9 shown in Chart 3 was synthesized by coupling Fmoc-Asp-OAllyl after the branching point for further cyclization of the RGDS peptide sequence. The O—allyl group is stable under peptide coupling and Fmoc deprotection conditions. The RGDS sequence was made following the Fmoc-Asp-OAllyl residue. A glycine residue was coupled as a spacer after the arginine residue. Cyclization was accomplished by attaching the N-terminus of the glycine to the α-carboxylic acid of the aspartic acid residue. Pd(PPh3)4 was used to remove the O—allyl group.42 The resin was swelled in a CHCl3/acetic acid/N-methylmorpholine (37/2/1) mixture under nitrogen gas. Three equivalents of the Pd complex were added and mixed for 3 h under nitrogen gas followed by the Fmoc deprotection. Cyclization was performed with 3 equiv of DPTS/DIPC43 for 4 h in DMF. PAs were characterized using a PE Biosystems Voyager-DE PRO matrix-assisted laser desorption ionization time-of-flight mass spectrometer (MALDI-TOF MS) and a Thermo Finnigan LCQ Advantage electrospray ionization mass spectrometer (ESI-MS). Reverse-phase HPLC was performed on a Hewlett-Packard 1050 liquid chromatograph equipped with Agilent Zorbax 300SB-C18 4.6 × 150 mm column or Waters Atlantis C-18 4.6 × 150 mm column in elution gradient of water (0.1% TFA) and acetonitrile (0.1% TFA). Preparative HPLC was performed on a Varian chromatograph using a Waters Atlantis C-18 30 × 250mm column under similar elution gradients as previously mentioned. The purities of 1–4 shown in Chart 1 were higher than 95% after HPLC purification and 5–12 shown in Charts 2-5 were more than 80% pure by HPLC. Molecules 1–5 were purified by HPLC, and they are reported to be more than 95% pure. Fluorescence anisotropy measurements were done with these samples. Molecules 5–12 were analyzed by HPLC, and their purities were higher than 80%. We did not observe any changes in the self-assembly behavior of these molecules at this purity compared to those found to be more than 95% pure.
Chart 2.
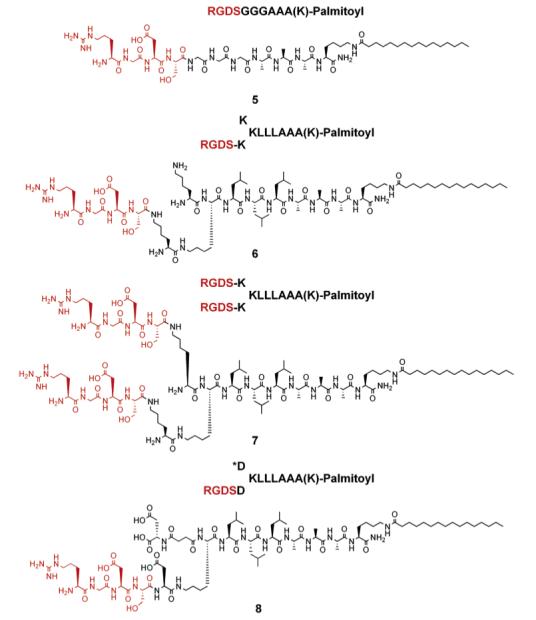
RGDS Presentation on Peptide Amphiphiles
Chart 3.
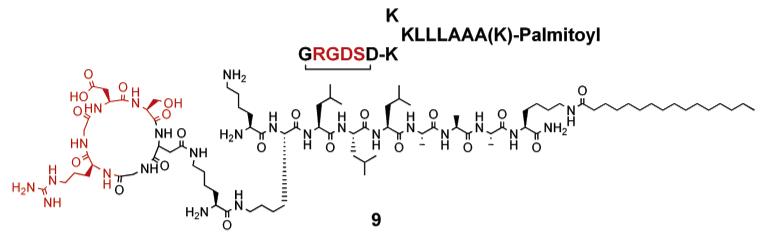
Cyclic RGDS Presentation on Branched Peptide Amphiphile
Chart 1.
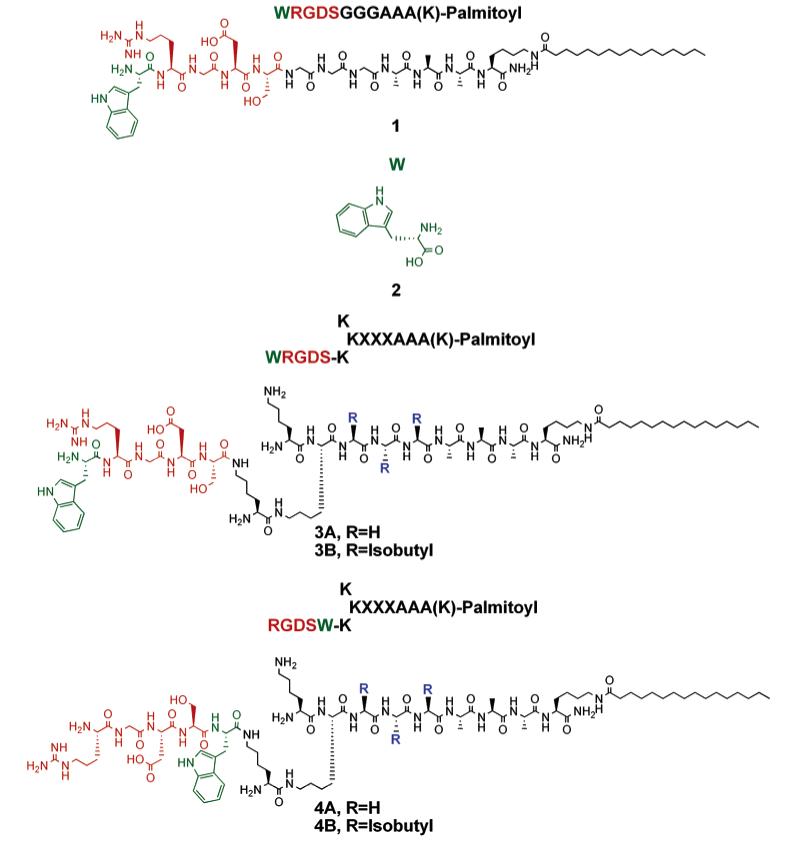
Tryptophan Containing Peptide Amphiphiles
Chart 5.
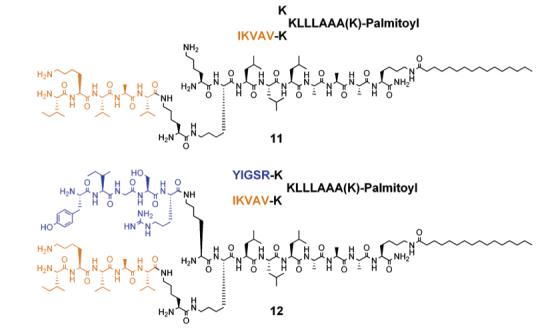
Presentation of IKVAV and YIGSR Peptide Sequences on Branched Peptide Amphiphiles
Transmission Electron Microscopy (TEM)
A Hitachi H-8100 transmission electron microscope was used for TEM imaging. The samples were prepared from 1 wt % gels of the PAs on a holey carboncoated copper TEM grid. Negative staining was carried out with 2 wt % phosphotungstic acid in water. TEM grids were prepared by casting onto the carbon side of the grid 3 μL of PA gel diluted to 0.01 wt % in water followed by wicking off of excess moisture with filter paper after 3 min. Negative staining was then performed by placing the grid carbon-side down on a droplet of filtered phosphotungstic acid solution for 1 min. The TEM samples were air-dried for at least 3 h before imaging. In all cases, electron microscopy was performed at an acceleration voltage of 200 kV.
Circular Dichroism (CD) spectroscopy
The CD spectra were obtained using a Jasco J-715 CD spectrometer at concentrations of 3.33 × 10-6 M with 1 and 2 mm cell path lengths. The CD spectra were collected from 260 to 190 nm.
Fluorescence
Both steady-state fluorescence and fluorescence polarization experiments were performed on an ISS PC1 spectrofluorimeter using the standard right-angle configuration. Sample concentrations at 20 μM were taken before every experiment using a Cary 500 spectrometer. Experiments were carried out at least in triplicate. Excitation and emission monochromator slits were set to 16 and 8 nm band-pass, respectively. A wavelength of 280 nm was used to excite the tryptophan residue. Fluorescence anisotropy measurements were performed at room temperature. The g-factor bias was minimal and not included in the calculations.
pH Titration of PA Fibers in Solution
pH titrations of PA solutions were carried out using an Accumet pH meter and electrode. Each PA was solubilized at 30 mg/mL in 0.1 M KCl. The pH probe was submerged in the PA solution, and a magnetic stir bar was added to aid in mixing. The solutions were titrated in 1-3 μL increments with 0.1 M KOH to pH ∼11, then titrated with 0.1 M HCl to pH <4. The pH meter was allowed to stabilize for several seconds after each addition to obtain a stable reading before adding more acid/base.
Synovial Fluid Isolation and Gelation
Synovial fluid was isolated from a bovine articular joint. Front hooves from 18–24 month old Holstein cattle were obtained from a local slaughterhouse within 1 h of sacrifice and maintained on ice for no more than 8 h prior to dissection. Hooves were washed thoroughly with antibiotic soap and opened using sterilized instruments to reveal the metacarpophlangeal joint. As each joint was opened, the surrounding synovial fluid was collected with a sterile syringe and needle and stored in sterile tubes at 4°C prior to use. Synovial fluid samples from six different joints were mixed 1:1 with the specified PA solutions (10 mg/mL in water), at volumes of 50 μL each. Hemispherical gels were made on glass coverslips and could support their own weight within seconds of mixing. Images of synovial fluid gels formed in a sample tube were obtained as frames of a movie, with an Olympus C4000 digital camera. Negatively charged biomolecules including calf thymus DNA, chondroitin sulfate A, chondroitin sulfate C, bovine serum albumin (fraction V), and glucose were each solubilized in 1X TNE at 50 and 10 mg/mL. Gelation of these negatively charged biomolecules with a branched PA was examined by forming a hemisphere of 50 μL of each solution with 50 μL of PA solution on a glass coverslip. The hemispherical bubbles were examined after 5 min to determine whether a gel had formed.
Subcutaneous Injection of PA Nanofibers
As an initial test of immunogenicity, 10 μL of either vehicle alone (water) or an aqueous solution (10 mg/mL) of 5 or 6 was injected subcutaneously in male C57 BL/6 mice (28-30 g). The animals were sacrificed 4 days postinjection, and the skin was biopsied for histology, with the site of injection confirmed by immunofluorescence. All animal experiments were performed in accordance with protocols approved by Northwestern’s Animal Care and Usage Committee.
Results and Discussion
We previously studied the availability of biotin for avidin binding on the periphery of PA nanofibers using fluorescence measurements.35 In these molecules, the biotin moiety was covalently bound at the terminus of the peptide segment and found to be most accessible in PAs with branched architectures. In this work, the solvent accessibility to peptide sequences with similar architectures was studied using the amino acid tryptophan as a natural fluorophore. Fluorescence anisotropy of the tryptophan residue was measured to conceive the rotational freedom of the chromophore in the self-assembled supramolecular structure using the linear and branched PAs synthesized as described in Chart 1. A tryptophan residue was incorporated before and after the RGDS sequence by SPPS to gain insight into the molecular packing of the RGDS bioactive sequences on the periphery of the nanofibers. Peptides with sequences XXXAAA-(K)-lipophilic segment were synthesized with X = G or L. Alanine, glycine, and leucine residues were used to promote hydrogen bonding among peptide segments and to form the characteristic β-sheet structures that drive formation of cylindrical nanofibers as opposed to the spherical micelles typically observed in amphiphilic self-assembly.33,44 The lysine residue was introduced to form branching on the peptide sequence, altering the structural presentation of the bioactive peptide sequence after self-assembly. Orthogonal protecting groups on the α and ∈ amines of the lysine residue enabled synthesis of asymmetric or symmetric branches on the PA system. Fmoc, Boc, and Mtt protecting groups on the amines of the lysine residues were used to control the design of peptides, as each of these protecting groups can be manipulated independently (see Figure 1). The Fmoc-protected amines were deprotected to couple amino acids, Boc-protected amines were used to block peptide growth on the lysine branches, and Mtt-protected amines were deprotected selectively to build asymmetrical branches on a single peptide amphiphile molecule.
Figure 1.
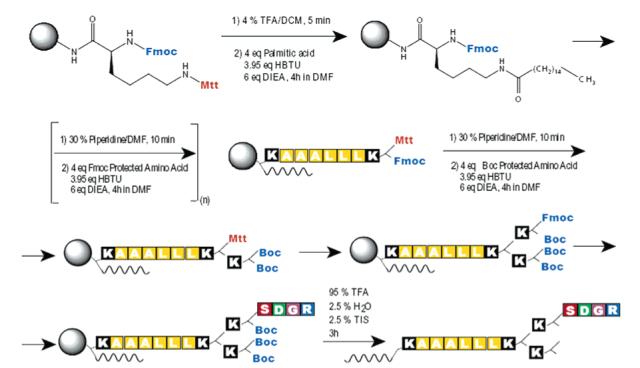
Synthesis of 6 on solid-phase peptide synthesis is shown as a representative synthesis of branched PAs. Use of Fmoc-Lys(Mtt)-OH at branching points allows for creation of the lysine dendron, as well as selective growth of the individual branches. This synthesis method results in amine termination at the peptide periphery.
The PAs shown in Chart 1 were observed to be soluble at pH 4 and formed self-supporting gels at concentrations greater than 0.5 wt % when the pH was increased above 6.5. Oscillatory rheology for the self-supporting gels obtained from 1 wt % PA gels indicated that the storage modulus for the gels was always higher than the loss modulus. Therefore, these molecules form self-supporting gels rather than solutions. Storage modulus measured at 10 rad/s was found to be in the range 200–1000 pascals. Transmission electron microscopy (TEM) of negatively stained gel samples revealed high-aspect-ratio nanofiber formation (see Figure 2 and Supporting Information). The nanofibers observed were 7 ± 1 nm in diameter and range in length from hundreds of nanometers to several microns. The height of the nanofibers were also confirmed with atomic force microscopy (AFM) (data not shown). Circular dichroism (CD) spectroscopy of the PAs revealed the presence of hydrogen-bonded structures among peptide segments (see Figure 3), including β-sheets and α-helices, as well as segments in random coil conformations. This is indicated by the observation of a broad signal between 200 and 230 nm. Molecules 3B and 4B containing leucine residues reveal stronger β-sheet peaks at 215 nm, whereas PAs with glycine residues reveal greater content of α-helix structure.
Figure 2.
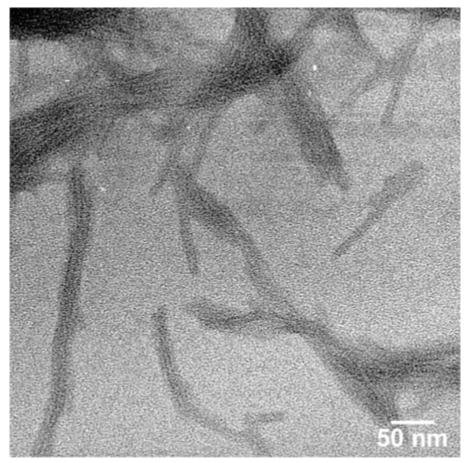
Negatively stained TEM micrograph of nanofibers formed by molecule 4B at pH 7.4.
Figure 3.

Circular dichroism spectra of molecules shown in Chart 1 at pH 7.4.
Fluorescence anisotropy values of the tryptophan-tagged PAs were measured in the self-assembled state as indicated in Table 1. The difference in the observed anisotropy is attributed to the nature of molecular motion of the chromophores in the various molecules. When the rotational freedom of the chromophore decreases, a higher anisotropy value is observed.45 A significantly higher anisotropy from the tryptophan residue was obtained when it was placed before the RGDS sequence in molecule 1. We propose that it is due to its localization in the sterically hindered environment of hydrogen-bonded nanofiber formed by the linear PAs. On the other hand, anisotropy values were slightly higher than tryptophan amino acid (2) alone when the tryptophan residue was placed before or after the RGDS sequence in the branched PAs, indicating that in the branched molecular architecture rotational freedom of the RGDS segment may be largely preserved in these nanostructures vs the ones formed by the linear PAs in which rotation is more hindered. Fluorescence anisotropy of the tryptophan amino acid alone in solution was measured as a reference and was lower than in any of the PA molecules, due to restricted mobility of the molecules in the self-assembled nanofibers.
Table 1.
Anisotropy Values for Tryptophan
| molecule | 1 | tryptophan (2) | 3A | 3B | 4A | 4B |
|---|---|---|---|---|---|---|
| anisotropy (<0.05) | 0.33 | 0.02 | 0.11 | 0.13 | 0.06 | 0.13 |
In light of the results discussed above, we designed additional PA architectures that presented the RGDS bioactive peptide sequence at their N-terminus. The RGDS was incorporated on linear and branched PA systems, with self-assembly into nanofibers controlled by pH change or electrolyte addition. The use of lysine residues or aspartic acid residues enabled modulation of the conditions necessary for self-assembly (see Chart 2), as the PAs rich in lysine were soluble at acidic pH (<5) and self-assembled at basic pH (>6), while the aspartic acid PAs were soluble at high pH and assembled at low pH. TEM micrographs of the PAs shown in Chart 2 also revealed nanofibers 7 ± 1 nm in diameter and micrometers in length, similar to those observed from tryptophan containing PAs (see Figure 4 and Supporting Information). CD spectroscopy revealed hydrogen bonding among peptide segments as described above (see Figure 5). β-sheets at 215 nm were observed more predominantly (in addition to contributions from α-helix and random coils) for molecules 6 and 7 compared to 5, revealing some significant differences in the internal hydrogen-bonded structure of the branched versus linear PA architecture. Aspartic acid rich branched PA, 8, was synthesized in order to change the self-assembly conditions of the system to low pH (<5). However, when Ca2+ ions are added to the solution of 8 at physiological pH, the ions also induce self-assembly of the molecule to form a self-supporting gel. TEM micrographs of Ca2+ and pH gels reveal similar nanofibers formed by molecule 8 upon self-assembly (see Supporting Information). CD spectroscopy of 8 at pH 7.4 in the presence of Ca2+ ions revealed predominantly β-sheet hydrogen bonding at 215 nm (see Figure 5).
Figure 4.
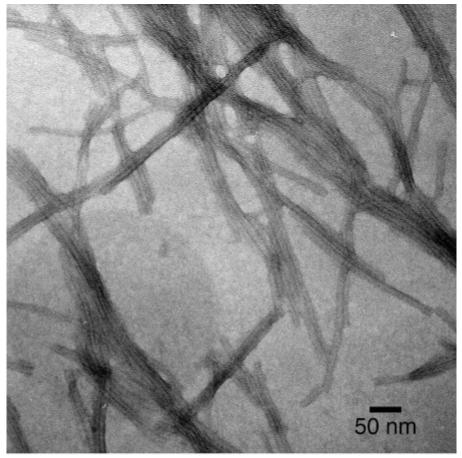
Negatively stained TEM micrograph of nanofibers formed by 6 at pH 7.4.
Figure 5.

Circular dichroism spectra of molecules shown in Chart 2 at pH 7.4.
In addition, a cyclic RGDS sequence was also incorporated into the branched PA architecture to mimic the loop conformation of RGDS found in fibronectin (see Chart 3). A TEM micrograph of nanofibers formed by 9 is shown in Figure 6. CD spectroscopy of 9 at pH 7.4 revealed predominantly β-sheet at 215 nm in addition to some α-helix hydrogen bonding (see Figure 7). To enhance the potential biological function of the PA matrixes, a synergistic peptide sequence, PHSRN, was also incorporated on the branched PA architecture by attaching it on one of the lysine residues (see Chart 4). Designing such supramolecular systems provides opportunities to present multiple functional groups in various potential orientations for biological recognition systems. CD spectroscopy of 10 at pH 7.4 revealed α-helix in addition to β-sheet hydrogen bonding (see Figure 7). The PAs shown in Chart 5 were also synthesized to demonstrate that various peptidic bioactive groups in addition to RGDS can be incorporated into the self-assembling peptide amphiphile architecture. Branched PAs containing the epitopes IKVAV and YIGSR, which are known to promote neuronal cell differentiation and neurite outgrowth,40,46 were synthesized and behaved similarly to the PAs shown above. TEM micrographs of these PAs revealed similar nanofibers at physiological pH (see Figure 8 and Supporting Information). CD spectroscopy of 11 and 12 at pH 7.4 revealed α-helix in addition to β-sheet hydrogen bonding (see Figure 7). Hence, many functional groups can be potentially presented on high-aspect-ratio nanofibers formed by self-assembling branched PAs.
Figure 6.
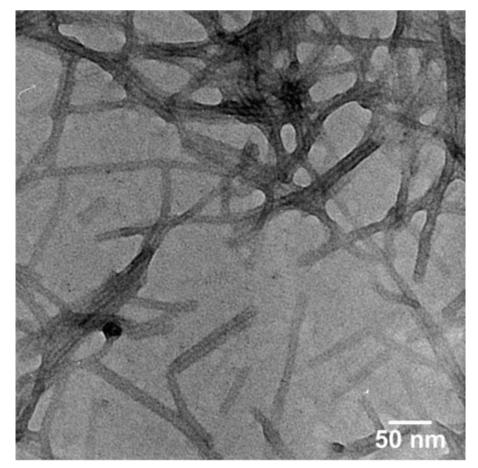
Negatively stained TEM micrograph of nanofibers formed by 9 at pH 7.4.
Figure 7.

Circular dichroism spectra of molecules shown in Charts 3, 4, and 5 at pH 7.4.
Chart 4.
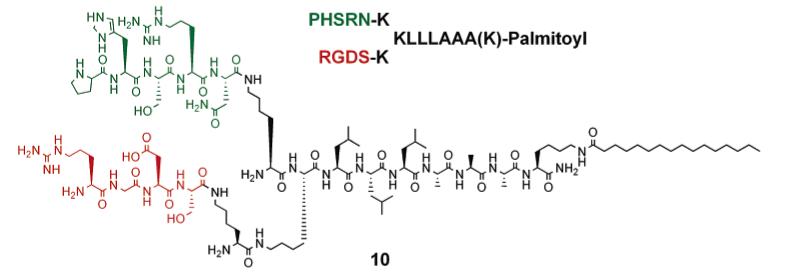
RGDS and PHSRN Presentation on Branched Peptide Amphiphile
Figure 8.
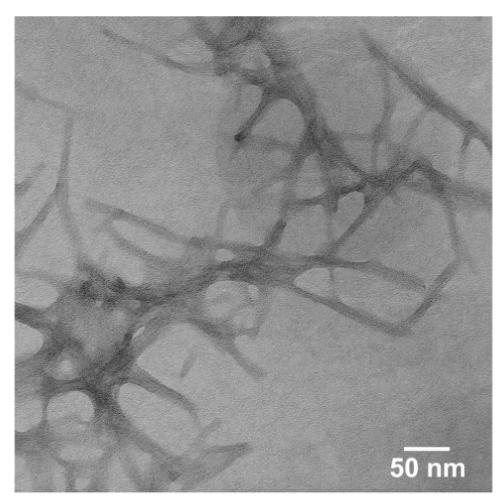
Negatively stained TEM micrograph of nanofibers formed by 12 at pH 7.4.
The supramolecular assembly of these systems was also assessed by pH titration of the PAs in 0.1 M KCl (see Figure 9). As previously shown by other researchers, the close spatial arrangement of high-generation lysine dendrimers can influence the protonation behavior of the terminal α and ∈ amines.47 While the reported pKa values for the α and ∈ amines on free lysine are 8.9 and 10.3, respectively, the observed gelation for our branched PA molecules occurs near pH ∼6.0–6.5. The inconsistency between these observations can be resolved by considering the spatial effects of close neighboring amines on protonation. As shown in Figure 9, the pKa transitions for the terminal amines on branched PA assemblies have broadened and shifted downward. This response for closely packed lysine branches in supramolecular assemblies matches the behavior seen in covalently bound systems, such as the lysine dendrimers described above and polyamidoamine (PAMAM) starburst dendrimers.47
Figure 9.

pH titration of 6 in 0.1 N KCl solution. A downward shift in protonation of the terminal amines is observed, indicating close association of the peripheral lysines.
The gels formed by the PA molecules have potential use in the tissue engineering field as a biomaterial scaffold mimicking the extracellular matrix for encapsulating cells. In addition to pH- and ionic-based gelation methods, we have tested the gelation behaviors of the PAs using biological fluids. We have previously demonstrated the gelation of other PA systems in situ using cerebrospinal fluid (CSF).40 Investigations with chondrocytes and cartilage motivated an interest in the synovial fluid which bathes tissue within an articular joint. The majority of nonfluid components of synovial fluid are the negatively charged highly branched macromolecules, glycosaminoglycans. Because of its natural abundance within joints and its relative ease of isolation, synovial fluid could make an excellent natural carrier for repairing joint tissues. PAs shown in Charts 2-4 were tested with freshly isolated bovine synovial fluid to determine if any gelation could be induced with these molecules. PAs were dissolved in water at 10 mg/mL and mixed 1:1 (v/v) with multiple samples of synovial fluid. Amine-terminated PAs or PAs with basic residues near their periphery (5, 6, 7, 9, and 10) were able to form a strong self-supporting hydrogel with synovial fluid (see Figure 10). However, the negatively charged branched PA 8 did not form a self-supporting hydrogel with synovial fluid.
Figure 10.

Images of a gel formed upon mixing 1:1 volumes of bovine synovial fluid and 1 wt % solution of 6.
In addition, 5, 6, 7, 9, and 10 were found to form gels with the addition of other biologically relevant macromolecules. These PA solutions were mixed 1:1 with solutions of calf-thymus DNA (∼8–9 MDa), chondroitin sulfate (a long, linear polysaccharide), bovine serum albumin (∼60 kDa negatively charged protein), and glucose, all of which were buffered at pH 6.5. The larger macromolecules, DNA and chondroitin sulfate, were able to gel solutions of 5, 6, 7, 9, and 10, while addition of the smaller BSA and glucose only resulted in slight viscosity increases. These findings point to the strong influence of electrostatics in the gelation of these systems, as well as significant opportunities for in situ gelation with a variety of physiological or biologically relevant fluids.
Given the immunogenic potential of some peptide-based biomaterials, it will be important to assess the immune response to these self-assembled nanofibers if they are to be used in vivo for tissue engineering applications. This is particularly true given the nonnatural lysine branching used to display the RGDS peptide epitope. As an initial test of immunogenicity, we studied examples of immunological response of some PAs. No evidence of any large-scale inflammatory reaction was observed (see Supporting Information). This could indicate that the peptides themselves are too short to elicit an immune response on their own and that their self-assembly into nanofibers does not induce a significant foreign-body response, at least at the concentration and time point tested. Studies are underway to examine other time points as well as staining for specific immune cell markers. Further studies will be necessary to confirm the biocompatibility of the PA nanofibers at other tissue sites as well as in other species.
Conclusions
We have reported on formation of cylindrical nanostructures by branched peptide amphiphile molecules that present high densities of bioactive peptide sequences on their surfaces. The RGDS cell adhesion epitope was incorporated into the branched PAs for potential use as a cell matrix material for regenerative medicine. These molecules were observed to form 3D networks of high-aspect-ratio nanofibers, which result in gel formation that may be useful for in situ encapsulation of cells. The PA architecture was modified using a branching strategy for appropriate presentation of recognition elements on the surface of the nanofibers. This branched architecture provides further control over gel formation at physiological pH, in addition to enabling presentation of multiple bioactive groups on the same molecule, and decoration of the periphery of the nanofibers with functional groups. Orthogonal protecting group chemistry provided a facile approach to present RGDS in different configurations, which may prove important in mediating cell interactions. Linear and cyclic RGDS sequences were presented on the periphery of the PA nanofibers as well as the synergistic PHSRN peptide sequence, which could potentially affect the recognition of RGDS by integrins on the cell surface. These molecules were also shown to gel in a variety of physiologically relevant conditions, either through pH or ionic gradients or through exposure to large biomacromolecules.
Supplementary Material
Acknowledgment
We thank the Electron Probe Instrumentation Center, the Keck Biophysics Facility, and the Analytical Services Laboratory at Northwestern University. We are also grateful to Prof. Jon W. Lomasney and Dr. Xiaoqiang Han (Pathology Department, Northwestern University) for immunological testing, and to Randy Claussen, Bryan Rabatic, Leslie Lee, and Mukti Rao of the authors’ laboratory for helpful discussions. This work was supported by the U.S. Department of Energy under award no. DE-FG02-00ER54810 and the National Institutes of Health under award nos. R01 EB003806-01 and R01 DE15920-01A1.
Footnotes
Supporting Information Available. Additional TEM images,MALDI-MS results, and histology. This material isavailable free of charge via the Internet at http://pubs.acs.org.
References and Notes
- (1).Griffith LG, Naughton G. Science. 2002;295:1009–1014. doi: 10.1126/science.1069210. [DOI] [PubMed] [Google Scholar]
- (2).Hirano Y, Mooney DJ. Adv. Mater. 2004;16:17–25. [Google Scholar]
- (3).Langer R. Acc. Chem. Res. 2000;33:94–101. doi: 10.1021/ar9800993. [DOI] [PubMed] [Google Scholar]
- (4).Langer R, Peppas NA. AIChE J. 2003;49:2990–3006. [Google Scholar]
- (5).Langer R, Tirrell DA. Nature (London) 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- (6).Langer RS, Vacanti JP. Sci. Am. 1999;280:86–89. doi: 10.1038/scientificamerican0499-86. [DOI] [PubMed] [Google Scholar]
- (7).Lee KY, Mooney DJ. Chem. Rev. 2001;101:1869–1879. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- (8).Pratt AB, Weber FE, Schmoekel HG, Muller R, Hubbell JA. Biotechnol. Bioeng. 2004;86:27–36. doi: 10.1002/bit.10897. [DOI] [PubMed] [Google Scholar]
- (9).Vogel V, Baneyx G. Annu. Rev. Biomed. Eng. 2003;5:441–463. doi: 10.1146/annurev.bioeng.5.040202.121615. [DOI] [PubMed] [Google Scholar]
- (10).Welsh ER, Tirrell DA. Biomacromolecules. 2000;1:23–30. doi: 10.1021/bm0002914. [DOI] [PubMed] [Google Scholar]
- (11).Stupp SI. MRS Bull. 2005;30:546–553. [Google Scholar]
- (12).Dillow AK, Ochsenhirt SE, McCarthy JB, Fields GB, Tirrell M. Biomaterials. 2001;22:1493–1505. doi: 10.1016/s0142-9612(00)00304-5. [DOI] [PubMed] [Google Scholar]
- (13).Kowalczynska HM, Nowak-Wyrzykowska M, Kolos R, Dobkowski J, Kaminski J. J. Biomed. Mater. Res., Part A. 2005;72A:228–236. doi: 10.1002/jbm.a.30238. [DOI] [PubMed] [Google Scholar]
- (14).Nikolovski J, Mooney DJ. Biomaterials. 2000;21:2025–2032. doi: 10.1016/s0142-9612(00)00079-x. [DOI] [PubMed] [Google Scholar]
- (15).Rajagopalan P, Marganski WA, Brown XQ, Wong JY. Biophys. J. 2004;87:2818–2827. doi: 10.1529/biophysj.103.037218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xiao Y, Truskey GA. Biophys. J. 1996;71:2869–2884. doi: 10.1016/S0006-3495(96)79484-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pierschbacher MD, Ruoslahti E. Proc. Natl. Acad. Sci. U.S.A. 1984;81:5985–5988. doi: 10.1073/pnas.81.19.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pierschbacher MD, Ruoslahti E. Nature (London) 1984;309:30–33. doi: 10.1038/309030a0. [DOI] [PubMed] [Google Scholar]
- (19).Swan EEL, Popat KC, Desai TA. Biomaterials. 2005;26:1969–1976. doi: 10.1016/j.biomaterials.2004.07.001. [DOI] [PubMed] [Google Scholar]
- (20).Houseman BT, Mrksich M. Biomaterials. 2001;22:943–955. doi: 10.1016/s0142-9612(00)00259-3. [DOI] [PubMed] [Google Scholar]
- (21).Irvine DJ, Ruzette AVG, Mayes AM, Griffith LG. Biomacromolecules. 2001;2:545–556. doi: 10.1021/bm015510f. [DOI] [PubMed] [Google Scholar]
- (22).Irvine DJ, Mayes AM, Griffith LG. Biomacromolecules. 2001;2:85–94. doi: 10.1021/bm005584b. [DOI] [PubMed] [Google Scholar]
- (23).Jensen TW, Hu BH, Delatore SM, Garcia AS, Messersmith PB, Miller WM. J. Am. Chem. Soc. 2004;126:15223–15230. doi: 10.1021/ja048684o. [DOI] [PubMed] [Google Scholar]
- (24).Massia SP, Hubbell JA. J. Cell Biol. 1991;114:1089–1100. doi: 10.1083/jcb.114.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Maheshwari G, Brown G, Lauffenburger DA, Wells A, Griffith LG. J. Cell Sci. 2000;113:1677–1686. doi: 10.1242/jcs.113.10.1677. [DOI] [PubMed] [Google Scholar]
- (26).Rowley JA, Mooney DJ. J. Biomed. Mater. Res. 2002;60:217–223. doi: 10.1002/jbm.1287. [DOI] [PubMed] [Google Scholar]
- (27).Auernheimer J, Hersel U, Dahmen C, Kantlehner M, Jeschke B, Enderle A, Nies B, Goodman SL, Kessler H. Biopolymers. 2003;71:402–402. [Google Scholar]
- (28).Nowlin DM, Gorcsan F, Moscinski M, Chiang SL, Lobl TJ, Cardarelli PM. J. Biol. Chem. 1993;268:20352–20359. [PubMed] [Google Scholar]
- (29).Xiong JP, Stehle T, Zhang RG, Joachimiak A, Frech M, Goodman SL, Aranout MA. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- (30).Herbert CB, McLernon TL, Hypolite CL, Adams DN, Pikus L, Huang CC, Fields GB, Letourneau PC, Distefano MD, Hu WS. Chem. Biol. 1997;4:731–737. doi: 10.1016/s1074-5521(97)90311-2. [DOI] [PubMed] [Google Scholar]
- (31).Ho MH, Wang DM, Hsieh HJ, Liu HC, Hsien TY, Lai JY, Hou LT. Biomaterials. 2005;26:3197–3206. doi: 10.1016/j.biomaterials.2004.08.032. [DOI] [PubMed] [Google Scholar]
- (32).Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT, Horwitz AR. Science. 2003;302:1704–1709. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- (33).Behanna HA, Donners J, Gordon AC, Stupp SI. J. Am. Chem. Soc. 2005;127:1193–1200. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- (34).Guler MO, Pokorski JK, Appella DH, Stupp SI. Bioconjugate Chem. 2005;16:501–503. doi: 10.1021/bc050053b. [DOI] [PubMed] [Google Scholar]
- (35).Guler MO, Soukasene S, Hulvat JF, Stupp SI. Nano Lett. 2005;5:249–252. doi: 10.1021/nl048238z. [DOI] [PubMed] [Google Scholar]
- (36).Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- (37).Hartgerink JD, Beniash E, Stupp SI. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Niece KL, Hartgerink JD, Donners J, Stupp SI. J. Am. Chem. Soc. 2003;125:7146–7147. doi: 10.1021/ja028215r. [DOI] [PubMed] [Google Scholar]
- (39).Beniash E, Hartgerink JD, Storrie H, Stendahl JC, Stupp SI. Acta Biomater. 2005;1:387–397. doi: 10.1016/j.actbio.2005.04.002. [DOI] [PubMed] [Google Scholar]
- (40).Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Science. 2004;303:1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- (41).Claussen RC, Rabatic BM, Stupp SI. J. Am. Chem. Soc. 2003;125:12680–12681. doi: 10.1021/ja035882r. [DOI] [PubMed] [Google Scholar]
- (42).Davies JS. J. Peptide Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- (43).Moore JS, Stupp SI. Macromolecules. 1990;23:65–70. [Google Scholar]
- (44).Guler MO, Claussen RC, Stupp SI. J. Mater. Chem. 2005;15:4507–4512. [Google Scholar]
- (45).Lakowicz JR, Kierdaszuk B, Callis P, Malak H, Gryczynski I. Biophys. Chem. 1995;56:263–271. doi: 10.1016/0301-4622(95)00040-5. [DOI] [PubMed] [Google Scholar]
- (46).Tashiro K, Sephel GC, Weeks B, Sasaki M, Martin GR, Kleinman HK, Yamada Y. J. Biol. Chem. 1989;264:16174–16182. [PubMed] [Google Scholar]
- (47).Ohsaki M, Okuda T, Wada A, Hirayama T, Niidome T, Aoyagi H. Bioconjugate Chem. 2002;13:510–517. doi: 10.1021/bc015525a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.