

Abstract
The current goal of diabetes therapy is to reduce time-averaged mean levels of glycemia, measured as HbA1c, to prevent diabetic complications. However, HbA1c only explains <25% of the variation in risk of developing complications. Because HbA1c does not correlate with glycemic variability when adjusted for mean blood glucose, we hypothesized that transient spikes of hyperglycemia may be an HbA1c–independent risk factor for diabetic complications. We show that transient hyperglycemia induces long-lasting activating epigenetic changes in the promoter of the nuclear factor κB (NF-κB) subunit p65 in aortic endothelial cells both in vitro and in nondiabetic mice, which cause increased p65 gene expression. Both the epigenetic changes and the gene expression changes persist for at least 6 d of subsequent normal glycemia, as do NF-κB–induced increases in monocyte chemoattractant protein 1 and vascular cell adhesion molecule 1 expression. Hyperglycemia-induced epigenetic changes and increased p65 expression are prevented by reducing mitochondrial superoxide production or superoxide-induced α-oxoaldehydes. These results highlight the dramatic and long-lasting effects that short-term hyperglycemic spikes can have on vascular cells and suggest that transient spikes of hyperglycemia may be an HbA1c–independent risk factor for diabetic complications.
Diabetes is a leading cause of blindness, end-stage renal failure, and peripheral neuropathy in most developed countries. Hyperglycemia-induced reactive oxygen species (ROS) initiate the complex series of molecular events that result in diabetic tissue damage, and transgenic expression of superoxide dismutase prevents diabetic complications in animal models (1). Consistent with this, multiple variations in SOD1 are significantly associated with persistent microalbuminuria and severe nephropathy in patients with Type 1 diabetes from the DCCT/EDIC (Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications) study (2). Diabetes is also a major independent risk factor for atherosclerotic cardiovascular disease (CVD). Patients with type 2 and type 1 diabetes both have a two- to fourfold higher risk of CVD compared with those without diabetes (3–5), and subjects with impaired glucose tolerance have a CVD risk almost as high as that seen in patients with type 2 diabetes (6).
In the DCCT/EDIC study, reduction of time-averaged mean levels of glycemia, measured as HbA1c, decreased the risk of sustained retinopathy progression, the primary DCCT outcome, by 73%. However, for the entire study group, HbA1c only explained <25% of the variation in risk of developing this complication (7). Because chronic hyperglycemia has been shown to cause increased acetylation of various histone lysine residues, a general epigenetic marker associated with increased gene transcription (8), we hypothesized that transient exposure to hyperglycemia would cause persistent increases in proatherogenic gene expression during subsequent periods of normal glycemia because of specific long-lasting epigenetic changes induced by ROS and its consequences. These transient spikes of hyperglycemia could be an HbA1c–independent risk factor for diabetic complications. The p65 subunit of the pleiotropic transcription factor NFκB was selected for study because NF-κB–driven proinflammatory gene expression appears to play a major role in the pathogenesis of atherosclerosis (9, 10), and p65 expression is significantly increased in aorta of diabetic ApoE-null mice and in circulating mononuclear cells of diabetic patients (11, 12).
RESULTS
Transient hyperglycemia promotes p65 gene transcription and NF-κB activation
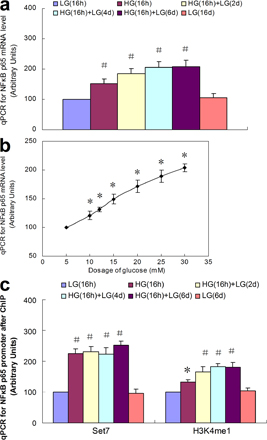
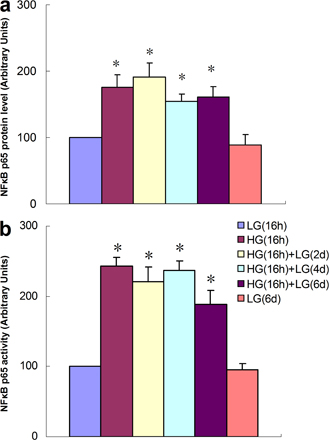
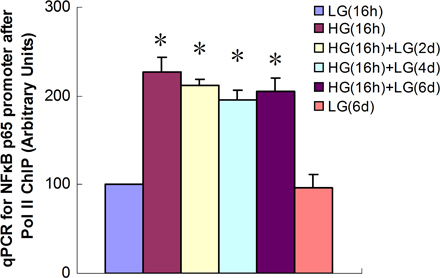
To create a model of transient hyperglycemia, we first incubated either primary bovine aortic endothelial cells (BAECs) or primary human aortic endothelial cells (HAECs) in high glucose (HG) for 16 h and then returned the media glucose concentration to physiological glucose levels for 6 d (Fig. 1 a). Transcription of p65 was increased by transient exposure to HG in both HAECs and BAECs (Fig. 1 a and Fig. S1 a, available at http://www.jem.org/cgi/content/full/jem.20081188/DC1). In contrast, transient hyperglycemia did not increase expression of the NF-κB subunits RElB, c-Rel, p50/p105, or p52/p100 (unpublished data). Remarkably, this increase in p65 transcription persisted during subsequent incubation at physiological glucose levels for the entire 6-d experimental period (Fig. 1 b and Fig. S1 a). Expression of p65 increased with glucose concentration from 5 to 30 mM (Fig. S1 b). Protein levels of p65 and NF-κB p65 activity were also increased by transient exposure to HG, and these also persisted for 6 d (Fig. S2). Addition of actinomycin D to the physiological glucose media after 16 h of HG reduced the elevated p65 expression to normal levels at 2 d, which is consistent with a primary role for transcription (unpublished data). To exclude the possibility that increased p65 expression occurs as a result of osmotic stress, vascular endothelial cells were incubated in 30 mM mannitol. In these cells, there was no change in p65 expression (unpublished data). Association of RNA polymerase II with the p65 promoter was also increased by transient exposure to HG, and this also persisted for 6 d (Fig. S3), confirming that p65 transcription was indeed increased.
Figure 1.

Persistent increase in Set7-mediated histone methylation and p65 gene expression caused by transient hyperglycemia. (a) Schematic representation of experimental model. (b) NF-κB p65 subunit mRNA levels in response to transient hyperglycemia in HAECs. (c) Schematic representation of the human NF-κB p65 proximal promoter region. (d) ChIP of NF-κB p65 promoter by Set7 and H3K4me1 after transient hyperglycemia in HAECs. (e) H3K4me1 associated with the p65 promoter after transient hyperglycemia in WT and SET7 knockdown cells. (f) p65 expression after transient hyperglycemia in WT HAECs and SET7 knockdown cells. (g) Effect of p65 and SET7 knockdown on MCP-1 and VCAM-1 expression after transient hyperglycemia. *, P < 0.05 versus LG; ¶, P < 0.05 versus WT group. For B and D–F, n = 3. Error bars show SEM.
Transient hyperglycemia induces persistent mobilization of Set7 to the p65 promoter
Based on this observation, we postulated that transient exposure to HG induces persistent activation of p65 expression by inducing specific activating methylation of histones associated with the p65 promoter. Histone methylation is an important posttranslational modification involved in fundamental processes such as transcriptional regulation and genome stability (13, 14). In particular, methylation of H3K4 (lysine 4 of histone 3) favors transcriptional activation (15). In mammalian cells, H3K4 methylation is mediated by several histone methyltransferases (HMTs). The mammalian HMT Set7 has been shown to monomethylate histone H3K4 (16). In contrast, SET1a, SET1b, and four MLL-family HMTs function as tri- and dimethyltransferases (17).
To determine whether Set7 is mobilized by HG to maintain the active transcriptional state, chromatin immunoprecipitations (ChIPs) were performed with antibody to Set7, and association with the proximal p65 promoter was determined by quantitative real time PCR (Fig. 1, c and d; and Fig. S1 c) (18). Chromatin from both HAECs and BAECs exposed to transient hyperglycemia (16 h) was significantly enriched for Set7 on the p65 promoter when compared with antibody controls. Remarkably, the increased Set7 association with the p65 promoter, like the increased expression of NF-κB, persisted in a normoglycemic environment for the entire 6-d experimental period (P < 0.05). To exclude the possibility that recruitment of Set7 occurs as a result of osmotic stress, vascular endothelial cells were incubated in 30 mM mannitol. In these cells, there was no change in Set7 enzyme binding on the p65 promoter (unpublished data). To confirm that recruitment of Set7 did indeed result in increased H3K4 methylation, we analyzed methylation of histone H3K4. ChIP analysis indicated that there was a specific and persistent increase in histone H3K4 monomethylation on p65 chromatin compared with no antibody control (Fig. 1 c and Fig. S1 c).
Transient hyperglycemia increases a specific epigenetic mark, histone 3 lysine 4 monomethylation (H3K4me1), in the proximal promoter region of the p65 gene
Although neither di- nor trimethyl H3K4 was affected by transient hyperglycemia in the single amplicon we examined (unpublished data), these marks are frequently found downstream of transcription start sites (TSS) (19). We therefore performed ChIP walking for H3K4me1, H3K4 dimethylation (H3K4me2), and H3K4 trimethylation 4 (H3K4me3) from nt 1 to +1500 of the p65 gene, as well as from nt −1 to −450 of the p65 promoter in HAECs (Fig. 2 a). The only statistically significant change occurred with H3K4me1 in the proximal promoter near the TSS (Fig. 2 b). This 2.5-fold increase was present after 16 h of HG and remained at this level after 6 d of subsequent exposure to low glucose (LG). Neither the small increases of H3K4me1 at +450 and +750 nor the slight increase of H3K4me2 and H3K4me3 near the TSS were significantly different from LG (Fig. 2, c and d). To show that our optimized ChIP assays are specific for the indicated epitopes, we performed ChIPs using an antibody that recognizes unmodified histone 3 (unpublished data). Association of the trimethyl H3K4 binding partner NURF with the p65 promoter was also not increased by transient exposure to HG (unpublished data). Consistent with these data, neither MLL1 nor hSET1 association with the p65 promoter was changed at any time point (unpublished data). Because hyperacetylation of histones by p300 and other histone acetyl transferases has been associated with recruitment of chromatin remodeling proteins, we also examined levels of pan-acetylated H3, acetyl H3K9, and acetyl H3K14 associated with the p65 promoter. Transient hyperglycemia increased all three, and this increase also persisted during subsequent incubation at physiological glucose levels for 6 d (Fig. S4, a and b, respectively, available at http://www.jem.org/cgi/content/full/jem.20081188/DC1). To determine whether this increased histone acetylation is sufficient to induce the observed effects of HG, we used the histone deacetylase inhibitor trichostatin A (TSA). TSA treatment of cells incubated in LG increased levels of pan-acetylated H3, H3K9, and H3K14 acetylation to acetylation levels observed in cells incubated in HG (Fig. S4 c). However, this increase in H3 acetylation was not associated with the Set7 recruitment and increased H3K4me1, which are both induced by incubation in HG (Fig. S4 d). Similarly, this increase in acetylation was also not associated with the increased expression of p65, MCP-1, or VCAM-1 induced by incubation in HG (Fig. S4 e). Together, these data indicate that increased histone acetylation is not sufficient to induce the effects observed after exposure to HG.
Figure 2.

Partial quantitative PCR–based chromosomal walking of the p65 gene by ChIP. (a) Schematic representation of the p65 proximal promoter and downstream sequence mapped. (b) Profiles of H3K4me1 after 16-h exposure to LG, 16-h exposure to HG, and 16-h exposure to HG followed by 6 d of LG. (c) Profiles of H3K4me2 after 16-h exposure to LG, 16-h exposure to HG and 16-h exposure to HG followed by 6 d of LG. (d) Profiles of H3K4me3 after 16-h exposure to LG, 16-h exposure to HG, and 16-h exposure to HG followed by 6 d of LG. *, P < 0.05 versus LG for HG group; #, P < 0.05 versus LG for HG (16 h) + LG (6 d) group. For B–D, n = 3. Error bars show SEM.
Set7 knockdown prevents glucose-induced up-regulation of p65 and the NF-κB–dependent genes MCP-1 and VCAM-1
Having demonstrated that Set7 and H3K4me1 are associated with p65 promoter, we next wished to investigate the effect of loss of Set7 on p65-mediated transcription in HAECs using lentivirus shRNA. As shown in Fig. 1 e, in Set7 knockdown HAECs (SET7 shRNA), transient hyperglycemia failed to induce increased H3K4 monomethylation. Similarly, knockdown of Set7 prevented the increase and persistence of NF-κB p65 expression induced by transient hyperglycemia (Fig. 1 f). Finally, we examined the effects of transient hyperglycemia on the expression of two NF-κB p65-activated genes relevant to hyperglycemia-induced arterial pathology, monocyte chemoattractant protein 1 (MCP-1), and vascular cell adhesion molecule 1 (VCAM-1). MCP-1 is a chemokine involved in the recruitment of plasma monocytes in the early stages of atherosclerosis, and VCAM-1 promotes monocyte adhesion to arterial endothelial cells (20). Expression of both MCP-1 and VCAM-1 was increased by transient hyperglycemia (Fig. 1 g) and remained elevated during 6 d of subsequent incubation at physiological glucose levels. Expression of three other NF-κB p65-dependent proinflammatory genes, the cytokine IL-6, inducible NOS2 (nitric oxide synthase), and the proinflammatory adhesion molecule ICAM1, also increased after exposure to transient HG and this increase persisted for 6 d of subsequent exposure to 5 mM glucose (unpublished data).
To link this increased expression to the changes in p65 expression and activity, we measured the effect of p65 knockdown on hyperglycemia-induced MCP-1 and VCAM-1 expression. Similarly, to link this increased expression of MCP-1 and VCAM-1 to Set7, we also determined the effect of SET7 knockdown on hyperglycemia-induced MCP-1 and VCAM-1 expression. Both knockdown of p65 and SET7 prevented the increase in MCP-1 and VCAM-1 expression induced by transient hyperglycemia (Fig. 1 g).
Mitochondrial ROS and GLO-1 substrate participate in glucose-induced changes in p65 gene expression and in remodeling of the p65 promoter
Because mitochondrial overproduction of superoxide has been shown to initiate a large number of hyperglycemia-induced mechanisms related to the pathogenesis of diabetic complications (21, 22), we next investigated the effect of inhibiting mitochondrial superoxide production on p65 expression. As shown in Fig. 3 a, the increase in p65 expression induced by transient hyperglycemia was completely prevented by overexpression of either uncoupling protein-1 (UCP-1) or manganese superoxide dismutase (MnSOD), both of which prevent hyperglycemia-induced superoxide accumulation (21, 22). Transient hyperglycemia had no effect on endogenous MnSOD expression (unpublished data), a finding which is consistent with our observation that the NF-κB subunit c-Rel was not induced by transient hyperglycemia (23). In addition, we found that overexpression of the α-oxoaldehyde degradation enzyme glyoxalase 1 (GLO1) also prevented the increase in p65 expression induced by transient hyperglycemia (Fig. 3 a). The major physiological substrate for GLO1, methylglyoxal, is a highly reactive dicarbonyl that accumulates in several cell types exposed to hyperglycemia as a consequence of increased mitochondrial superoxide production. This results in functionally significant covalent modifications of intracellular proteins (24). Overexpression of UCP-1, MnSOD, or GLO1 also prevented the increased association of Set7 and H3K4me1 with the p65 promoter in response to transient hyperglycemia alone (Fig. 3 b).
Figure 3.

UCP1, MnSOD, and GLO1 prevent persistent increases in Set7-mediated histone methylation and p65 gene expression. (a) p65 mRNA after transient hyperglycemia, measured by quantitative PCR. (b) ChIP analyses for association of Set7 and H3K4m1 with the proximal p65 promoter after transient hyperglycemia. (c) Chromatin remodeling after transient hyperglycemia as determined by susceptibility to Eag1 digestion. Nuclei from treated cells were isolated, digested by Eag1, and the extracted DNA amplified by quantitative PCR over the indicated p65 promoter region. *, P < 0.05 versus LG group. For each graph, n = 3. Error bars show SEM.
Transcriptional competence is typically associated with changes in chromatin structure. Therefore, we next examined the effect of transient hyperglycemia on remodeling of the p65 locus (Fig. 3 c). HAECs were infected with UCP-1, MnSOD, or GLO1 adenovirus and then treated as described previously. Nuclear extracts were digested with the restriction endonuclease Eag1 (Fig. 1 c), and a 161-bp fragment of the p65 promoter was quantified by quantitative PCR amplification. Transient hyperglycemia caused active remodeling of the p65 promoter, proximal to the TSS, with an increased susceptibility to Eag1 digestion indicating transition to an open chromatin conformation (Fig. 3 c). This remodeling of the p65 promoter also persisted for 6 d of normoglycemia and was prevented by overexpression of UCP-1, MnSOD, or GLO1.
In nondiabetic mice, transient hyperglycemia induces increased H3K4me1 at the p65 promoter and increases p65 gene transcription
To validate our in vitro observations in an animal model, we examined the effect of transient hyperglycemia on H3K4me1 and p65 expression in aortic endothelial cells of nondiabetic mice (Fig. 4). Mice were exposed to hyperglycemia (20 mM glucose) for 6 h using pancreatic insulin clamps and killed immediately and after 2, 4, and 6 d of subsequent euglycemia. Aortic endothelial cells were isolated from these mice by laser capture microdissection (LCM), and the levels of H3K4me1 at the NF-κB p65 promoter were determined by carrier ChIP (cChIP; Fig. 4 a) (25). Transient hyperglycemia induced an increase in this activating H3K4 methylation, which persisted for the subsequent 6 d of exposure to normal levels of blood glucose. These epigenetic changes were associated with an increase in NF-κB p65 expression that also persisted for the subsequent 6 d of exposure to normal levels of blood glucose (Fig. 4 b). Because both of these hyperglycemia-induced effects were prevented by overexpression of UCP-2 in vitro, we also analyzed aortic endothelial cells isolated from nondiabetic UCP-2+/− mice, which produce excess intracellular ROS at normal glucose levels. In the absence of hyperglycemia, both the level of H3K4me1 at the NF-κB p65 promoter and the level of p65 expression were increased to the same extent as they were in WT mice exposed to transient hyperglycemia. Expression of MCP-1 and VCAM-1 was also increased (Fig. 4, c and d, respectively). Similarly, because these hyperglycemia-induced effects were also prevented by overexpression of glyoxalase 1 in vitro, we analyzed the same variables in aortic endothelial cells isolated from nondiabetic glyoxalase 1 knockdown mice. The effects on both H3K4m1 at the NF-κB promoter and p65 expression were qualitatively similar to those observed with both transient hyperglycemia and in UCP-2+/− mice (Fig. 4, e and f, respectively). These results demonstrate that increased intracellular ROS, which normally are generated by hyperglycemia, are sufficient to induce both increased H3K4me1 at the NF-κB promoter and p65 expression in the absence of hyperglycemia. Similarly, they show that increased glyoxalase 1 substrate, which normally occurs as a consequence of hyperglycemia (26), is sufficient to induce both increased H3K4me1 at the NF-κB promoter and p65 expression in the absence of hyperglycemia. Thus, the proximate mechanistic events mediating increased p65 expression are HG-induced ROS and subsequent methylglyoxal formation. The distal mechanistic events are chromatin remodeling, Set7 recruitment, and increased H3K4 monomethylation in the p65 promoter.
Figure 4.

Persistent increases in Set7-mediated histone methylation and p65 gene expression in nondiabetic mice. (a and b) WT mice were exposed to 20 mM glucose for 6 h using a hyperinsulinemic hyperglycemic clamp. Aortas were removed at the indicated times, and aortic endothelial cells were isolated by LCM. (a) cChIP of NF-κB p65 promoter by H3K4m1 antibody (n = 2). (b) NF-κB p65 subunit mRNA levels (n = 3). *, P < 0.05 versus LG. Error bars show SEM. (c and d) Aortic endothelial cells were isolated from UCP-2+/− mice by LCM. (c) cChIP of NF-κB p65 promoter by H3K4m1 antibody (n = 4). (d) p65, MCP-1, and VCAM-1 mRNA levels (n = 4). *, P < 0.05 versus LG. Error bars show SEM. (e and f) Aortic endothelial cells were isolated from Glo1 KD mice by LCM. (e) cChIP of NF-κB p65 promoter by H3K4m1 antibody (n = 4). (f) p65, MCP-1, and VCAM-1 mRNA levels (n = 4). *, P < 0.05 versus LG. Error bars show SEM.
DISCUSSION
In the present study, we show that transient exposure of aortic endothelial cells to hyperglycemia induces persistent epigenetic changes in the promoter of the NF-κB p65 subunit in both cultured human aortic endothelial cells and in nondiabetic mice. In the proximal promoter region of p65, increased monomethylation of histone 3 lysine 4 by the histone methyltransferase Set7 caused a sustained increase in p65 gene expression, leading to a sustained increase in expression of the NF-κB–responsive proatherogenic genes MCP-1 and VCAM-1. These epigenetic changes are caused by increased generation of methylglyoxal because of hyperglycemia-induced ROS formation by the mitochondrial electron transport chain.
Our epigenetic findings are particularly novel for two reasons. First, to our knowledge there are no data about Set7 increasing H3K4 monomethylation modification of a promoter and altering gene expression. It has been commonly assumed, based on studies in yeast, that H3K4 methyltransferases function mainly during elongation after recruitment by elongating RNA polymerase complexes (27). Although recent studies in animal cells have shown activator-dependent interactions and recruitment of other methyltransferases (28), thus indicating promoter-related functions that may complement the elongation-related functions, no studies have implicated Set7 and H3K4 monomethylation. Second, and most important clinically, our study is the first to demonstrate that transient hyperglycemia induces chromatin remodeling and vascular epigenetic changes that cause persistent increases in proatherogenic gene expression during subsequent normoglycemia.
Miao et al. (29) reported that nine candidate genes displayed increased H3K4Me2 after chronic exposure of the human monocytic cell line THP-1 to HG. Three of these showed increased gene expression, four showed decreased expression, and two showed no difference in gene expression. Whether or not these changes persisted during subsequent normoglycemia was not investigated. During exposure to HG, they found no change in H3K4 dimethyllysine in the NF-κB p65 promoter sequence, which is consistent with the data reported here (Fig. 2 c). In a separate publication, Miao et al. (8) reported that after chronic exposure of the human monocytic cell line THP-1 to HG, H3 acetylation at Lys 9 and Lys 14 was increased at the TNF-α and COX-2 promoters. However, in contrast to our findings with p65 expression (Fig. S4), the HDAC inhibitor TSA stimulated transcription of these two genes in normal glucose.
In addition to posttranslational modification of histones, DNA methylation may also play an epigenetic role in controlling gene expression in adults (30). In a recent study, Ling et al. (31) provided a compelling example of how genetic and epigenetic factors may interact to confer an age-dependent susceptibility to insulin resistance. In muscle from young and elderly identical twins, a polymorphism in the promoter of a nuclear-encoded electron transport chain protein was associated with increased DNA methylation in this promoter in the older subjects, which correlated with lower levels of gene expression and increased insulin resistance. The role of DNA methylation in gene expression changes related to metabolic memory is a fertile area for future investigation.
Data from the EDIC study, which followed patients with type 1 diabetes after they completed the DCCT, show that early chronic exposure to a moderately high level of hyperglycemia has prolonged effects on diabetic complications during subsequent periods of improved glycemia, a phenomenon termed “metabolic memory.” For example, atherosclerotic changes not even present at the end of the DCCT appeared subsequently in the previously higher HbA1c group, followed by a twofold increase in myocardial infarction, strokes, and cardiovascular death. This occurred despite the fact that their HbA1c since the end of the DCCT was identical to that of the formerly intensive control group during the entire time that these arterial changes developed (32, 33). Whether persistent epigenetic changes induced by transient spikes of hyperglycemia play a role in metabolic memory remains to be determined by future investigations.
In summary, the observations reported here show that transient hyperglycemia causes persistent atherogenic effects during subsequent normoglycemia by inducing long-lasting changes in chromatin remodeling, recruitment of the histone methyltransferase Set7, and increased H3K4 monomethylation in the proximal NF-κB promoter, leading to increased expression of p65, MCP-1, and VCAM-1. Together, these results provide a molecular basis for understanding some of the variation in risk for diabetic complications that is not explained by HbA1c.
MATERIALS AND METHODS
Cells and reagents.
Primary HAECs (Cascade Biologics) were maintained in medium EGM-2MV bullet kit (Cambrex) supplemented with 10% FBS and antibiotics. BAECs were maintained in MEM (Invitrogen) with 0.5% FBS and nonessential amino acids (Invitrogen) containing either 5 mM glucose (LG) or 30 mM glucose (HG). The formulation sheets for the media used stated that no LPS was present.
We also tested for the presence of LPS using the Pyrosate test kit (Associates of Cape Cod, Inc.). Antibodies for SET7, LSD1, H3-Ac, H3K9-Ac, H3K14-Ac, and H3K4me3 were purchased from Millipore. Antibodies for H3K4me1 and H3K4me2 were purchased from Abcam. Antibody for RNA Pol II was obtained from Santa Cruz Biotechnology, Inc., and antibody for unmodified H3 was obtained from Cell Signaling Technology. Antibody to MLL1 was obtained from the laboratory of R.G. Roeder (Rockefeller University, New York, NY) and hSET1 antibody was obtained from Bethyl Laboratories, Inc. Non-targeted shRNA and shRNA for Set7 lentiviral particles were obtained from Sigma-Aldrich. Negative controls for p65 siRNA and p65 siRNA (siRNA ID: 216912) were obtained from Ambion. Adenovirus for UCP1, MnSOD, GLO1 and empty vectors were prepared by the Gene Transfer Vector Core (University of Iowa, Iowa City, IA). Viruses were infected 24 h preceding treatment with HG or LG where stated. Drosophila melanogaster SL2 cells were grown at 26°C in Schneider's medium (Invitrogen). Protein concentrations were measured by Coomassie Protein Assay kit (Thermo Fisher Scientific) using BSA as a standard.
RT reaction and real-time quantitative PCR.
Total RNA from cells was extracted using the RNeasy Mini kit or RNeasy Micro kit (QIAGEN), and the RNA was reverse transcribed with the SuperScript III First Strand Synthesis System (Invitrogen). Real-time quantitative PCR was run on a LightCycler 480 with LightCycler 480 SYBR Green I Master kit (Roche). PCR was performed by denaturing at 95°C for 7 min, followed by 45 cycles of denaturation at 95°C, annealing at 60°C, and extension at 72°C for 10 s, respectively. 1 μl of each cDNA was used to measure target genes, and the results were normalized by β-actin for HAECs and hydroxymethylbilane synthase for BAECs. Total RNA from aorta was prepared after homogenization in TRIZOL (Invitrogen) with the Ultra-Turrax (IKA).
ChIP.
The procedure of ChIP used here was that described by Metivier et al. (34) with minor modification. In brief, treated cells were washed and cross-linked using 1% formaldehyde for 20 min. After stopping cross-linking by addition of 0.1 M glycine, cell lysates were sonicated and centrifuged. 500 μg of protein was precleared by BSA/salmon sperm DNA and a slurry of Protein A Agarose beads. Immunoprecipitations were performed using the indicated antibodies or preimmune rabbit IgG in the presence of BSA/salmon sperm DNA and 50% slurry of Protein A agarose beads. Input and immunoprecipitates were washed, eluted, and then incubated for 2 h at 42°C in the presence of Proteinase K followed by 6 h at 65°C to reverse the formaldehyde cross-linking. DNA fragments were recovered by phenol/chloroform extraction and ethanol precipitation. A 162-bp fragment for the bovine NF-κB p65 promoter and a 161-bp fragment from the human NF-κB p65 promoter were amplified by real-time PCR (quantitative PCR). Quantitative PCR values were normalized to input DNA and to the values obtained with normal rabbit IgG. The mean fraction of input for 5 mM glucose was arbitrarily set as 100 U, and relative fold changes induced by various experimental conditions were calculated from that.
Preparation of NF-κB subunit p65 and Set7 knockdown HAECs.
HAECs were either transfected by negative control siRNA or siRNA for NF-κB subunit p65 using Lipofectamine reagent (Invitrogen). For each experiment, an aliquot of cells were harvested for measurement of mRNA level and used if the p65 mRNA was decreased by 80% compared with control cells. shRNA lentivirus for human Set7 (NM030648) was obtained from Sigma-Aldrich. 80% confluent HAECs (passage 3) in 96-well plates were infected by ∼200 MOI of either nontargeted shRNA or Set7 shRNA lentivirus particles in the presence of 8 μg/ml polybrene in 0.2 ml of medium. Fresh medium containing 10 μg/ml puromycin was added after 36 h to select puromycin-resistant cells. Cells were trypsinized and seeded at a density of 100 cells/10-cm dish, and single colonies were picked and characterized. Cells that showed at least an 80% decrease in Set7 mRNA levels compared with control cells were designated Set7 knockdown-positive cells and used for later experiments.
H3K4me1-3 ChIP coupled with quantitative PCR-based chromosomal walking.
HAECs were treated with LG for 16 h, HG for 16 h, or HG for 16 h followed by LG for 6 d. Cells were fixed and cross-linked, the chromatin was sonicated to achieve 300–400-bp DNA fragments, and the chromatin was immunoprecipitated by H3K4-me1, me2, and me3 antibodies, respectively. We designed a series of PCR primers so as to produce a series of adjacent partially overlapping PCR products ranging in size from 130 to 170 bp spanning nucleotides +1 to +1500 downstream of the TSS and nucleotides −450 to −1 upstream of the TSS. Quantitative PCR and data analysis were performed as described in RT reaction and real-time quantitative PCR.
Eag1 digestion of NF-κB p65 promoter.
Treated HAECs were washed with PBS and cross-linked by 1% formaldehyde for 15 min at room temperature. Cells were washed by cold washing solution containing 150 mM sucrose, 80 mM KCl, 35 mM Hepes, pH 7.4, 5 mM K2HPO4, 5 mM MgCl2, and 0.5 mM CaCl2, and then permeabilized for 2 min at room temperature with ice-cold 0.5 mg/ml lysolecithin (Sigma-Aldrich) dissolved in the cold washing solution. Cells were then scraped with a rubber policeman and suspended and homogenized in 700 μl of nuclei buffer containing 25 mM Hepes, pH 7.8, 1.5 mM MgCl2, 10 mM KCl, 0.1% NP-40, 1 mM DTT, and protease inhibitor cocktail (Sigma-Aldrich). Nuclei were then sedimented by 15-min centrifugation at 4°C at 1,400 g. The nuclear pellet was then resuspended in 100 μl of digestion buffer (50 mM Tris-HCl, 100 mM NaCl, 10 mM MgCl2, and 1 mM DTT) containing 50 U Eag1 restriction enzyme and incubated for 30 min at 37°C. The reaction was stopped by addition of stop solution containing 50 mM Tris, pH 8, 300 mM NaCl, 25 mM EDTA, 0.2% SDS, and 0.2 mg/ml Proteinase K. After Proteinase K digestion, genomic DNA was purified by DNeasy Tissue kit (QIAGEN). 2 μl DNA samples were used for the analysis of the NF-κB p65 promoter by real-time PCR using the forward primer 5′-gtgcagcctcttcgtcctc-3′ and reverse primer 5′-gtgcactacagacgagccatt-3′.
Nondiabetic mouse protocol and sample preparation.
Adult male C57B/6 mice were catheterized for pancreatic insulin clamp studies under conscious and unrestrained conditions. A primed-continuous infusion of 3 μg/kg/min of somatostatin, ∼0.3 mU/kg/min of insulin, and 4 ng/kg/min of glucagon was administered, and a variable infusion of a 25% glucose solution was started at time 0 and periodically adjusted to clamp plasma glucose concentration at ∼5 mM (euglycemic protocol) or at ∼20 mM (hyperglycemic protocol) for 6 h. Groups of mice from each treatment group (for each time point, n = 3) were killed at days 0, 2, 4, and 6 after the clamping procedure. The aortas were isolated and snap frozen in OCT compound. 10-μm sections were cut by clean microtome and mounted on PEN membrane slides (2.0 μm; Leica). 200–2,000 aortic endothelial cells per mouse were isolated by LCM (Leica) and used for either mRNA preparation or cChIP analysis. The animal study protocol was reviewed and approved by the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine.
UCP-2+/− and Glo1 knockdown mice.
UCP-2 heterozygous KO mice (UCP-2+/−) on a C57/B6 background were kindly provided S. Collins (CIIT Centers for Health Research, Research Triangle Park, NC). Glyoxalase 1 knockdown mice on a C57/B6 background were created in the laboratory of M. Brownlee (Albert Einstein College of Medicine, Bronx, NY). In brief, short oligonucleotides with a target sequence to mouse Glo1 nt 235–255 in a hairpin sequence with restriction enzyme cohesive end sequences were ordered and cloned into the pSilencer 1.0-U6 vector (Ambion). Fragments, including the mouse U6 promoter and inserts, were subcloned into a lentiviral vector (FUGW; gift from D. Baltimore's laboratory, California Institute of Technology, Pasadena, CA). The recombined plasmids were used to generate lentiviral particles. shRNA lentivirus was injected into the perivitelline space of single-cell C57/B6 mouse embryos. After incubation of 4–6 h, embryos were implanted into pseudopregnant females and were carried to term. Mice whose genome contained a single copy of the insert were identified by Southern blotting and used to establish founder lines. Glo1 mRNA and protein levels were determined by qPCR and WB and further confirmed by measurement of glyoxalase activity. Heterozygous offspring of founder #14 had a 45–65% decrease in tissue glyoxalase 1 activity, and these mice were used in all experiments.
cChIP.
The cChIP was performed using a published method with minor modification (25). In brief, around 2 × 107 SL2 cells were pelleted, mixed with ∼1,000 aorta endothelial cells isolated from mice aorta by LCM, and then suspended in 1 ml NB buffer (15 mM Tris-HCl, pH 7.4, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 0.1 mM EGTA, 0.5 mM 2-mercaptoethanol, and 0.1 mM PMSF) supplemented with 5 mM sodium butyrate and homogenized. Nuclei were released and pelleted at 1,250 g for 15 min at 4°C, resuspended in 1 ml NB buffer plus 0.32 M sucrose, and pelleted at 1,800 g for 25 min at 4°C. Nuclei were resuspended in 1 ml of digestion buffer (50 mM Tris-HCl, pH 7.4, 0.32 M sucrose, 4 mM MgCl2, 1 mM CaCl2, and 0.1 mM PMSF). 50 U micrococcal nuclease (Sigma-Aldrich) were added and the mixture was incubated for 5 min at 28°C. The subsequent solution was used for ChIP by the indicated antibody after the standard ChIP protocol described in the ChIP section of Materials and methods. To account for potential changes in background during different stimulation conditions, we analyzed α-satellite DNA in cell culture experiments after exposure to transient hyperglycemia and found that this locus is not affected by the H3K4m1 modification. We also have performed ChIP for Set7, and found that there is no significant change in Set7 recruitment to the α-satellite DNA. We also confirmed that there is no change in the total amount of unmodified H3 associated with the p65 promoter by ChIP. Finally, we measured both H3K4m1 and unmodified histone H3 levels by immunoblotting and found that the experimental conditions did not alter levels of either.
Statistical analysis.
Results are given as mean ± SEM. All experiments were performed at least in triplicate, unless otherwise mentioned. Data distribution was analyzed, and statistical differences for different treatments were evaluated by the analysis of variance and Tukey-Kramer statistical tests using PHStat2 (Pearson Prentice Hall). A P value of <0.05 was considered significant.
Online supplemental material.
Fig. S1 shows the effects of transient HG on p65 expression, Set 7 recruitment, and H3K4me1 in BAECs. Fig. S2 displays the effect of transient HG on total p65 protein level and nuclear p65 activity in HAECs. Fig. S3 shows the effect of transient HG on Po II association with the p65 promoter. Fig. S4 shows the effect of transient HG alone on total H3, H3K9, and H3K14 acetylation in the proximal p65 and in the presence of TSA. The effects of TSA on recruitment of Set 7, H3K4, and expression of p65, MCP-1, and VCAM-1 at physiological glucose concentration are also shown in Fig. S4. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20081188/DC1.
Supplementary Material
Acknowledgments
We thank Dr. Qida Ju for making the glyoxalase 1 knockdown mice while working in the laboratory of M. Brownlee. We are indebted to Dr. Shiela Collins (Senior Investigator, Division of Biological Sciences, Director of the Endocrine Biology Program at CIIT Centers for Health Research, Research Triangle, NC) for providing us with UCP-2 KO mice.
This study was funded by a Center Grant from the Juvenile Diabetes Research Foundation (M. Brownlee, D. Yao, and M.E. Cooper), a JDRF Scholar Award (M. Brownlee), NIDDK grant DK060764 (R.G. Roeder), and grants from the National Health and Medical Research Council of Australia, National Heart Foundation of Australia, and Juvenile Diabetes Research Foundation (A. El-Osta and M.E. Cooper).
The authors declare no competing financial interests.
Abbreviations used: BAEC, bovine aortic endothelial cell; ChIP, chromatin immunoprecipitation; cChIP, carrier ChIP; CVD, cardiovascular disease; GLO1, glyoxalase 1; H3k4me1, histone 3 lysine 4 monomethylation; H3K4me2, H3K4 dimethylation; H3K4me3, H3K4 trimethylation 4; HAEC, human aortic endothelial cell; HG, high glucose; HMT, histone methyltransferases; LCM, laser capture microdissection; LG, low glucose; MCP-1 monocyte chemoattractant protein 1; MnSOD, manganese superoxide dismutase; ROS, reactive oxygen species; TSA, trichostatin A; TSS, transcription start site; UCP-1, uncoupling protein 1; VCAM-1, vascular cell adhesion molecule 1.
D. Brasacchio and D. Yao contributed equally to this paper.
References
- 1.Brownlee, M., L.P. Aiello, M.E. Cooper, A.I. Vinik, R. Nesto, and A.J.M. Bolton. 2008. Diabetic Complications. In Williams Textbook of Endocrinology, 11th ed. Larsen, P.R., Kronenberg, H., Melmed, S., and Polonsky, K., editors. W.B. Saunders, Philadelphia. 1417–1501.
- 2.Al-Kateb, H., A.P. Boright, L. Mirea, X. Xie, R. Sutradhar, A. Mowjoodi, B. Bharaj, M. Liu, J.M. Bucksa, V.L. Arends, et al. 2008. Multiple superoxide dismutase 1/splicing factor serine alanine 15 variants are associated with the development and progression of diabetic nephropathy: the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Genetics study. Diabetes. 57:218–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu, F.B., M.J. Stampfer, C.G. Solomon, S. Liu, W.C. Willett, F.E. Speizer, D.M. Nathan, and J.E. Manson. 2001. The impact of diabetes mellitus on mortality from all causes and coronary heart disease in women: 20 years of follow-up. Arch. Intern. Med. 161:1717–1723. [DOI] [PubMed] [Google Scholar]
- 4.Fox, C.S., S. Coady, P.D. Sorlie, D. Levy. B. Meigs, R.B. D'Agostino Sr., P.W. Wilson, and P.J. Savage. 2004. Trends in cardiovascular complications of diabetes. JAMA. 292:2495–2499. [DOI] [PubMed] [Google Scholar]
- 5.Krolewski, A.S., E.J. Kosinski, J.H. Warram, O.S. Leland, E.J. Busick, A.C. Asmal, L.I. Rand, A.R. Christlieb, R.F. Bradley, and C.R. Kahn. 1987. Magnitude and determinants of coronary artery disease in juvenile-onset, insulin-dependent diabetes mellitus. Am. J. Cardiol. 59:750–755. [DOI] [PubMed] [Google Scholar]
- 6.Qiao, Q., K. Pyorala, M. Pyorala, A. Nissinen, J. Lindstrom, R. Tilvis, and J. Tuomilehto. 2002. Two-hour glucose is a better risk predictor for incident coronary heart disease and cardiovascular mortality than fasting glucose. Eur. Heart J. 23:1267–1275. [DOI] [PubMed] [Google Scholar]
- 7.1995. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes. 44:968–983. [PubMed] [Google Scholar]
- 8.Miao, F., I.G. Gonzalo, L. Lanting, and R. Natarajan. 2004. In vivo chromatin remodeling events leading to inflammatory gene transcription under diabetic conditions. J. Biol. Chem. 279:18091–18097. [DOI] [PubMed] [Google Scholar]
- 9.Thurberg, B.L., and T. Collins. 1998. The nuclear factor-kappa B/inhibitor of kappa B autoregulatory system and atherosclerosis. Curr. Opin. Lipidol. 9:387–396. [DOI] [PubMed] [Google Scholar]
- 10.Glass, C.K., and J.L. Witztum. 2001. Atherosclerosis. the road ahead. Cell. 104:503–516. [DOI] [PubMed] [Google Scholar]
- 11.Lewis, P., N. Stefanovic, J. Pete, A.C. Calkin, S. Giunti, V. Thallas-Bonke, K.A. Jandeleit-Dahm, T.J. Allen, I. Kola, M.E. Cooper, et al. 2007. Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation. 115:2178–2187. [DOI] [PubMed] [Google Scholar]
- 12.Bierhaus, A., S. Schiekofer, M. Schwaninger, M. Andrassy, P.M. Humpert, J. Chen, M. Hong, T. Luther, T. Henle, I. Kloting, et al. 2001. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 50:2792–2808. [DOI] [PubMed] [Google Scholar]
- 13.Martin, C., and Y. Zhang. 2005. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 6:838–849. [DOI] [PubMed] [Google Scholar]
- 14.Kondo, Y., L. Shen, and J.P. Issa. 2003. Critical role of histone methylation in tumor suppressor gene silencing in colorectal cancer. Mol. Cell. Biol. 23:206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lachner, M., and T. Jenuwein. 2002. The many faces of histone lysine methylation. Curr. Opin. Cell Biol. 14:286–298. [DOI] [PubMed] [Google Scholar]
- 16.Cheng, X., R.E. Collins, and X. Zhang. 2005. Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 34:267–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou, Y., T.A. Milne, A.J. Ruthenburg, S. Lee, J.W. Lee, G.L. Verdine, C.D. Allis, and R.G. Roeder. 2006. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 13:713–719. [DOI] [PubMed] [Google Scholar]
- 18.Harikrishnan, K.N., M.Z. Chow, E.K. Baker, S. Pal, S. Bassal, D. Brasacchio, L. Wang, J.M. Craig, P.L. Jones, S. Sif, et al. 2005. Brahma links the SWI/SNF chromatin-remodeling complex with MeCP2-dependent transcriptional silencing. Nat. Genet. 37:254–264. [DOI] [PubMed] [Google Scholar]
- 19.Barski, A., S. Cuddapah, K. Cui, T.Y. Roh, D.E. Schones, Z. Wang, G. Wei, I. Chepelev, and K. Zhao. 2007. High-resolution profiling of histone methylations in the human genome. Cell. 129:823–837. [DOI] [PubMed] [Google Scholar]
- 20.Denk, A., M. Goebeler, S. Schmid, I. Berberich, O. Ritz, D. Lindemann, S. Ludwig, and T. Wirth. 2001. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J. Biol. Chem. 276:28451–28458. [DOI] [PubMed] [Google Scholar]
- 21.Brownlee, M. 2001. Biochemistry and molecular cell biology of diabetic complications. Nature. 414:813–820. [DOI] [PubMed] [Google Scholar]
- 22.Brownlee, M. 2005. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 54:1615–1625. [DOI] [PubMed] [Google Scholar]
- 23.Pizzi, M., I. Sarnico, F. Boroni, M. Benarese, N. Steimberg, G. Mazzoleni, G.P. Dietz, M. Bahr, H.C. Liou, and P.F. Spano. 2005. NF-kappaB factor c-Rel mediates neuroprotection elicited by mGlu5 receptor agonists against amyloid beta-peptide toxicity. Cell Death Differ. 12:761–772. [DOI] [PubMed] [Google Scholar]
- 24.Shinohara, M., P.J. Thornalley, I. Giardino, P. Beisswenger, S.R. Thorpe, J. Onorato, and M. Brownlee. 1998. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Invest. 101:1142–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O'Neill, L.P., M.D. VerMilyea, and B.M. Turner. 2006. Epigenetic characterization of the early embryo with a chromatin immunoprecipitation protocol applicable to small cell populations. Nat. Genet. 38:835–841. [DOI] [PubMed] [Google Scholar]
- 26.Nishikawa, T., D. Edelstein, X.L. Du, S. Yamagishi, T. Matsumura, Y. Kaneda, M.A. Yorek, D. Beebe, P.J. Oates, H.P. Hammes, et al. 2000. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 404:787–790. [DOI] [PubMed] [Google Scholar]
- 27.Shilatifard, A. 2006. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu. Rev. Biochem. 75:243–269. [DOI] [PubMed] [Google Scholar]
- 28.Kouzarides, T. 2007. Chromatin modifications and their function. Cell. 128:693–705. [DOI] [PubMed] [Google Scholar]
- 29.Miao, F., X. Wu, L. Zhang, Y.C. Yuan, A.D. Riggs, and R. Natarajan. 2007. Genome-wide analysis of histone lysine methylation variations caused by diabetic conditions in human monocytes. J. Biol. Chem. 282:13854–13863. [DOI] [PubMed] [Google Scholar]
- 30.Klose, R.J., and A.P. Bird. 2006. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 31:89–97. [DOI] [PubMed] [Google Scholar]
- 31.Ling, C., P. Poulsen, S. Simonsson, T. Ronn, J. Holmkvist, P. Almgren, P. Hagert, E. Nilsson, A.G. Mabey, P. Nilsson, et al. 2007. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J. Clin. Invest. 117:3427–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nathan, D.M., J. Lachin, P. Cleary, T. Orchard, D.J. Brillon, J.Y. Backlund, D.H. O'Leary, and S. Genuth. 2003. Intensive diabetes therapy and carotid intima-media thickness in type 1 diabetes mellitus. N. Engl. J. Med. 348:2294–2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nathan, D.M., P.A. Cleary, J.Y. Backlund, S.M. Genuth, J.M. Lachin, T.J. Orchard, P. Raskin, and B. Zinman. 2005. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 353:2643–2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metivier, R., G. Penot, M.R. Hubner, G. Reid, H. Brand, M. Kos, and F. Gannon. 2003. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 115:751–763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}