

Abstract
Since the introduction of the concepts of allostery about four decades ago, much advancement has been made in elucidating the structure-function correlation in allostery. However, there are still a number of issues that remain unresolved. In this review we employed mammalian pyruvate kinase (PK) as a model system to understand the role of protein dynamics in modulating cooperativity. PK has a TIM (α/β)8 barrel structural motif. PK is an ideal system to address basic questions regarding regulatory mechanisms about this common (α/β)8 structural motif. The simplest model accounting for all the solution thermodynamic and kinetic data on ligand-enzyme interactions involves two conformational states, inactive ET and active ER. These conformational states are represented by domain movements. Further studies provide the first evidence for a differential effect of ligand binding on the dynamics of the structural elements, not major secondary structural changes. These data are consistent with our model that allosteric regulation of PK is the consequence of perturbation of the distribution of an ensemble of states in which the inactive ET and active ER represent the two extreme end states. Sequence differences and ligands can modulate the distribution of states leading to alterations of functions. The future work includes: defining the network of functionally connected residues; elucidate the chemical principles governing the sequence differences which affect functions; and probe the nature of mutations on the stability of the secondary structural elements which in turn modulate allostery.
Keywords: allostery, thermodynamics, protein dynamics, protein fold, human genetics
One of the major aims of the post-genomic era is to establish the correlation between protein structural folds and functions. The premise is based on the assumption that protein folds are designed for specific functions, analogous to the early concept of one-gene-one-function. However, it is soon recognized that diverse functions are carried out by proteins with essentially identical folds but different sequences. Many protein structural folds have been identified e.g. the various folds involved in signal transduction [1–3]. The chemical principles behind these observations are still unclear. Thus, we focus on the investigation of the chemical principles that govern the relationship linking sequence-fold-function.
We have chosen the biological phenomenon of allosteric regulation as the focus to tackle the issue of fold-function correlation. The rationale for the choice is that allostery is a predominant regulatory mechanism and an allosteric system consists of a variety of functions that can be modulated by a change in sequence.
An ideal system that has an outstanding chance for revealing the chemical principles of allostery should consist of the following properties: 1. Structure-defined at the atomic level; 2. Function-identified linked reactions that lead to allostery; 3. Identities of natural mutants that affect allostery. Rabbit muscle pyruvate kinase (RMPK) consists of properties that satisfy these criteria.
The outstanding features that make RMPK an ideal system are: 1,The allosteric behavior can be modulated from a non-allosteric to an allosteric form; 2. Only 22 amino acid changes out of 530 residues per subunit are required to accomplish this significant functional change; 3. No significant structural changes in PK are associated with these changes in residues [4,5].
PK has a TIM (α/β)8 barrel structural motif [6–9]. Although, there is a paucity of knowledge on the chemical ground rules on how its functions are regulated, PK is an ideal system to address basic questions regarding regulatory mechanisms about this common (α/β)8 structural motif [10]. Muscle PK is a homo-tetramer [11–14], as shown in Fig. 1. Each subunit consists of 530 amino acids and multiple domains. In each subunit, there is a complete set of binding sites to bind the substrates (ADP and PEP), inhibitor (Phe) and activator (fructose-1,6-bisphosphate, FBP), as shown in Fig. 2. The active site resides between the A and B domains and at a long distance from the effectors binding sites which are distributed throughout the subunit structure. Thus, the regulatory mechanism of RMPK involves communication through distant sites. The tetrameric RMPK communicates through two intersubunit interfaces mainly between adjacent A domains and C domains. The only amino acid sequence differences between the mammalian allosteric kidney PK and the non-allosteric muscle PK isozymes are located in the C-domain and are involved in intersubunit interactions (Fig. 1 and Fig. 2). Thus, embedded in these two isoforms of PK are the rules involved in engineering the popular TIM (α/β)8 motif to modulate its allosteric properties.
Fig. 1. Structure of tetrameric muscle PK.
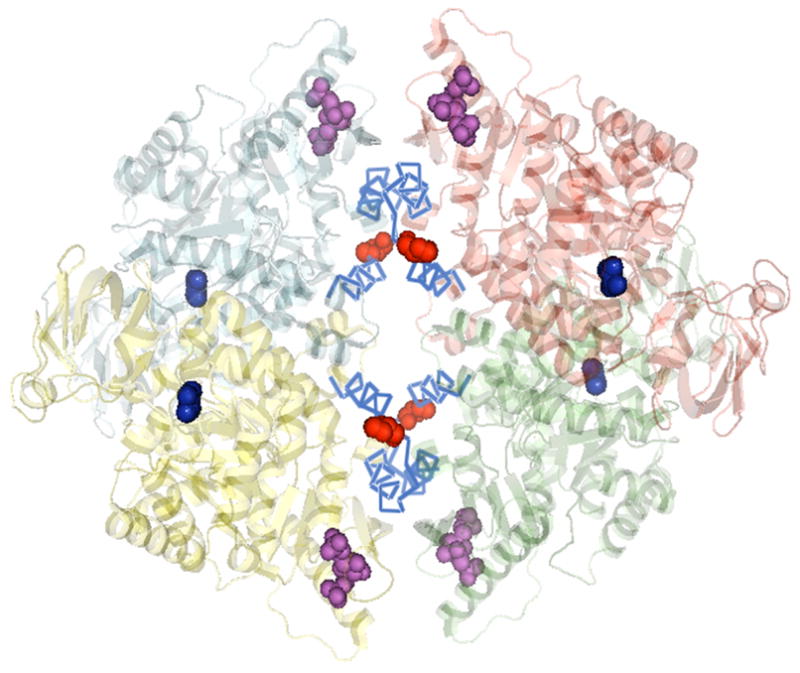
The four subunits are in different colors. The 22 amino acids in cyan; the molecules of Mn2+ and pyruvate (blue) in the active sites; the activator FBP in magenta; and the position of residue 402 are shown in red spheres (PyMol software presentation of PDB 1F3W).
Fig. 2. Momoneric PK showing the A, B and C domains.
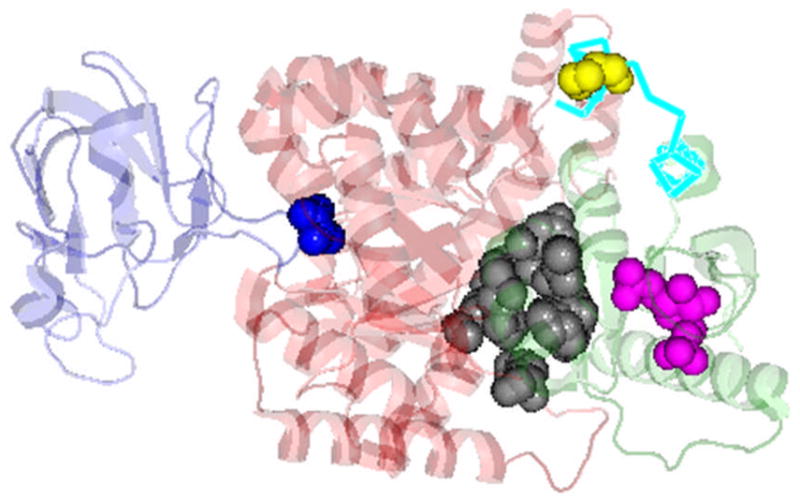
Color and domain identity are: B (blue), A (red), C (green). The ligand binding sites are: substrates (blue sphere); FBP (magenta sphere); pocket for Phe (inhibitor, black sphere) (based on PDB 2G50), location of the 22 amino acid differences between the PK isoforms in C-domain (cyan) and residue 402 (yellow sphere) (PyMol software presentation of PDB 1F3W).
Functional Linkage Scheme of Allostery for RMPK
As a result of a series of extensive steady-state kinetic, equilibrium and solution structural studies, a concerted, allosteric model was developed for quantitative interpretation of the kinetic and equilibrium binding data of RMPK [15–18]. The simplest model accounting for all the experimental data involves two conformational states, inactive ET and active ER, shown in Fig. 3. E, S, P and I are enzyme, substrate (phosphoenolpyruvate, PEP or ADP), product (pyruvate) and inhibitor (Phe), respectively.
Fig. 3. Proposed two-state model for the allosteric behavior of RMPK.
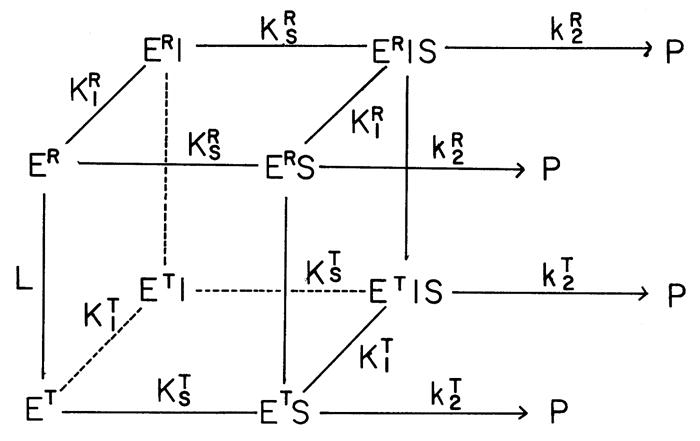
E, S, P and I are enzyme, substrate (phosphoenolpyruvate, PEP or ADP), product (pyruvate) and inhibitor (Phe), respectively. ER and ET are the two structural states. The species are interconnected with equilibrium constant, L=[ET]/[ER]; KR and KT are the ligand binding equilibrium constants associated to the ER- and ET-state, respectively, while KI and KS are the equilibrium constants associated with the binding of inhibitor Phe and substrate, respectively. kT and kR are the catalytic rates of ER and ET, respectively [16].
The upper and lower faces of the cube contain all of those species assuming ER and ET state, respectively. The species are interconnected with equilibrium constant, L=[ET]/[ER]; KR and KT are the ligand binding equilibrium constants associated to the ER- and ET-state, respectively, while KI and KS are the equilibrium constants associated with the binding of inhibitor Phe and substrate, respectively. The conceptual significance of this model is that the two states are in a pre-existing equilibrium, L. This crucial aspect of the model was independently substantiated by the literature [19,20]. The distribution of ligands bound to these two states is defined by their respective equilibrium constants to these states. The ligands shift the state changes in PK by mass action. This model differs from models which assume a mechanism consisting of a ligand induced state change not in a pre-existing equilibrium.
The simple model shown in Fig. 3 is NOT adequate to account for the experimentally defined energy landscape of the linked equilibria that govern the enzymatic reactions. Added novel features are derived from global analysis of calorimetric and fluorescence data. These features are: 1. Coupling between the bindings of ADP and Phe; 2. Temperature dependence of the preferential binding of ADP to the ER- or ET-state of PK; 3. At high temperature, ADP actually prefers binding to the inactive ET state. The consequence is a more sigmoidal response of PK activity to fluctuation of substrate concentration (Herman and Lee, data to be published).
In summary, our thermodynamic studies have established quantitatively the intricate network of interactions among ligands and PK.
Structural Properties of the ER and ET States
The transition between ER- and ET-state of RMPK can be monitored by sedimentation velocity or gel filtration techniques [15,21]. These results imply significant changes in the quaternary structure of the enzyme. To characterize this change in global structure of RMPK, we studied the effects of ligands on the structure of RMPK by small angle neutron scattering [22]. The radius of gyration, RG, decreases by about 1 Å in the presence of substrate PEP but increases by the same magnitude in the presence of inhibitor Phe. When the scattering data were analyzed as a function of P(r) vs r, where P(r) is the frequency distribution of all the point-to-point pair distances, r, between scattering centers of the particle, the results indicate that the increase in RG is associated with a pronounced increase in the probability for interatomic distance between 80 and 110 Å, shown in Fig. 4 (upper panel). With the aid of computer modeling, these changes in interatomic distance is consistent with the rotation of the B domain relative to the A domain, leading to the closure or opening of the cleft between these domains as a consequence of binding to PEP and Phe, respectively, as shown in the lower panel of Fig. 4. These results show that one of the dynamic motions in RMPK includes change in domain-domain interaction between A and B domains.
Fig. 4. Comparison between active and inactive RMPK conformation.
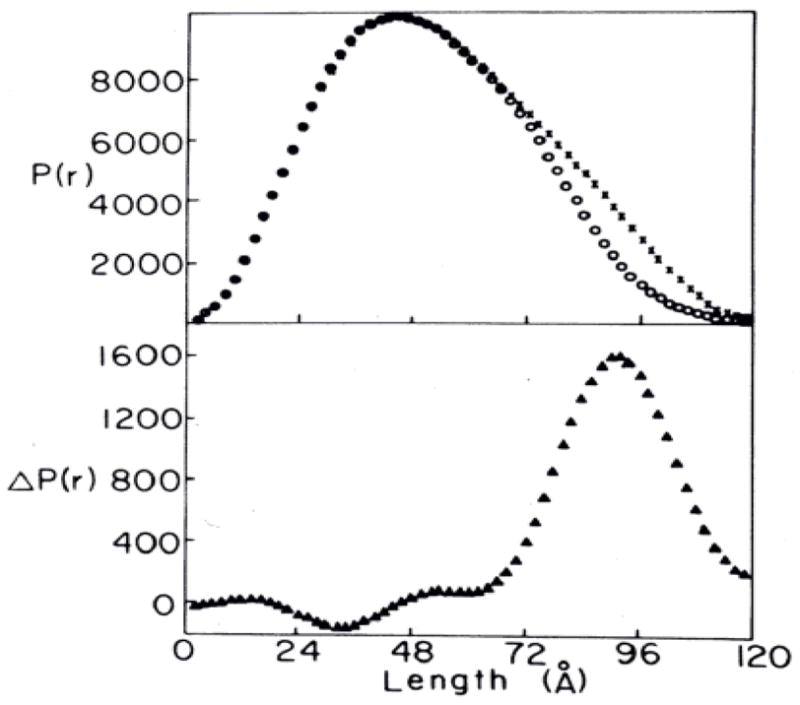
Upper panel, experimental length distribution functions; lower panel, difference length distribution function. X and O, enzyme in the presence of 15 mM Phe and 2 mM PEP, respectively [20].
We further studied the structural perturbations by Fourier transform infrared (FT-IR) spectroscopy [23], shown in Fig. 5. The experiments were designed to monitor the secondary structure of RMPK in the presence of saturating amounts of various ligands. Mg2+ is a divalent cation essential for activity. PEP and ADP are the two substrates while Phe is the inhibitor. Fig. 5 shows that in all experimental conditions there is no significant change in the areas encompassed by the peaks assigned to either α or β structures. That implies that there is no detectable conversion of secondary structure of PK in the presence of all these ligands. A closer examination of Fig. 5 shows that the maximum wavenumber associated with α-helix remains the same while that of the β-strand shifts as a function of ligand. In the presence of Mg2+, PEP and ADP the maximum wavenumber is less than that in buffer alone or Phe. These results imply that the local environments of the β-strands are perturbed by these ligands, although the amount of β strands has not changed. The origin of the differential perturbations by ligands in the environments of secondary structures might be the modulation of structural dynamics of RMPK. Thus, the structural dynamics of RMPK in the presence of various ligands was probed by H/D exchange monitored by FT-IR. The second derivative spectra as a function of time of H/D exchange were monitored and compared. The spectra in the presence of K+ and Mg2+ were compared to those of RMPK in buffer. They show that the basic pattern of exchange was retained. However, a larger change in intensity was observed even at the 1-min time point. These results imply that the activating metal ions induce an increase in the number of rapidly exchangeable amide protons. The presence of either PEP or ADP shows a pattern of exchange that is quite similar to each other, namely, a very rapid exchange was observed in both the helices and sheets. There is a clear indication of the presence of two different populations of helices. The change in the second derivative spectra reflecting the amide proton exchange in the presence of Phe showed that within the time frame of the experiment no exchangeable amide proton was detected in the β-sheets, an observation that differs from that of the helices. Thus, these H/D exchange experiments show that substrates (ADP or PEP) and activating metal ions (Mg2+) lock PK in a more dynamic ER structure while Phe exerts an opposite effect.
Fig. 5. FT-IR amide spectra in D20.
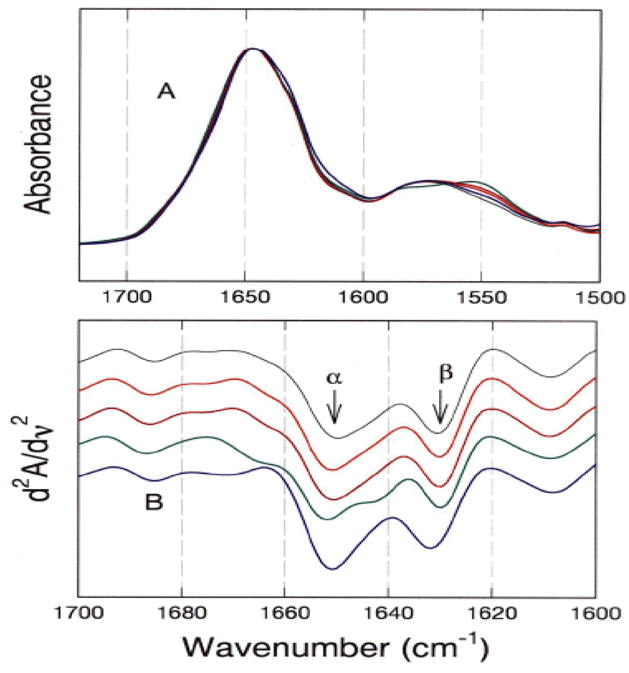
(A) Averaged absorption spectra of the amide I and II regions. (B) Second-derivative of amide I spectra. The bar represents the maximum deviations. The spectra are displaced in the Y-axis for clarity in presentation. The solution composition and color of the line are: (Black), Tris buffer; (Red), +7.2 mM Mg2+; (Dark red), +2 mM ADP; (Green), +2 mM PEP; and (Blue), +10 mM Phe [23].
These results provide the first evidence for a differential effect of ligand binding on the dynamics of the structural elements, not major conformational changes, in RMPK. These data are consistent with our model that allosteric regulation of RMPK is the consequence of perturbation of the distribution of an ensemble of states in which the observed change in RG represents the two extreme end states. Sequence differences and ligands can modulate the distribution of states leading to alterations of functions.
Composite Effects in the Model of Allostery Induced by the 22 Residues that Differ between the Isozymes of PK
The differences in the various parameters of the allostery model induced by the 22 amino acids that are different between the RMPK and rabbit kidney PK (RKPK) isozymes are summarized in Table 1. It is apparent that a change of <5% of amino acids in strategically important structural elements leads to changes in allosteric parameters. Six out of nine parameters are affected and the magnitude of changes is significant, up to 1000-fold. Thus, a change in a small number of residues has major impact on the behavior of the RMPK. These changes are NOT elicited either by changes in the sequences involving the active sites or by changes in the other ligand binding sites. These changes in the binding constants of ligands are strictly elicited by changes of 22 amino acids in a localized region, as shown in Fig. 1 and Fig. 2. These amino acids are located in the C-domain of each subunit and are obviously involved in intersubunit interactions. They are far removed from the active sites and yet changes in these residues lead to the very significant changes in the allosteric properties of the enzyme including favoring the ET state and higher affinities of substrate and inhibitor to both states.
Table 1.
Summary of the equilibrium and kinetic parameters [42]
| Muscle PK | Kidney PK | Muscle PK | Kidney PK | ||
|---|---|---|---|---|---|
| L | 0.06±0.04 | 800±530 | 10000±3400 | 0.06±0.02 | |
| 60±6.8 | 64±9.3 | >50000 | 1.3±0.27 | ||
| 480±29 | 7000±3100 | 1.0±0.02 | 0.97±0.01 | ||
| 13000±5800 | 850±330 | 0.00 | 0.03 | ||
| 780±320 | 90±24 |
L=[ET]/[ER] is a unitless parameter. The K values are in μM and S, I and A are substrate (PEP), inhibitor (Phe) and activator (fructose-bis-phosphate, FBP) respectively. The k values are expressed as relative activity, 1.0 being equal Vmax.
Effects of Single Residue Mutation Derived from the 22 Amino Acids that Differ between the Isozymes
S402P
In our study to elucidate the role of each of these 22 residues in conferring allosteric properties to the RMPK, we mutated residue 402 of RMPK from S to P, in accordance to the difference in sequence between the two isozymes. Converting S402 to P changes neither the secondary, nor the tetrameric structure [24]. The S402P RMPK mutant exhibits steady-state kinetic behavior that indicates that it is more responsive to regulation by effectors, as shown in Fig. 6. The data are shown as initial enzyme velocity vs substrate PEP concentration. The sigmoidicity of the curves is a reflection of cooperativity of substrate binding. The RMPK data show almost no sigmoidicity, as expected for an enzyme exhibiting little allosteric behavior; whereas the RKPK shows pronounced sigmoidicity. The data for the S402P RMPK are intermediary to the muscle and kidney isozymes. The presence of 12 mM inhibitor Phe shifts the curve to the right, as expected, since Phe would shift the conformational state equilibrium towards ET which has a weaker affinity for PEP. The presence of 10 nM activator FBP in addition to 12 mM Phe shifts the curve to the left. In RMPK, it would require millimolar concentrations of FBP to achieve the same effect. Thus, a S402P mutation confers partial restoration of allosteric behavior to the RMPK.
Fig. 6. Kinetic characterization of S402P RMPK.
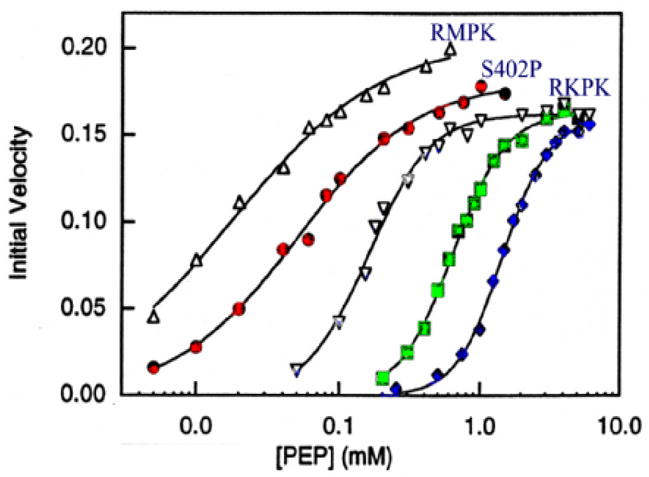
PEP was the variable substrate. The symbols and labels are RMPK, S402P and RKPK for WT RMPK, mutant RMPK and rabbit kidney PK, respectively, in the absence of any allosteric effectors. The other symbols for S402P RMPK are: (◇), in the presence of 12 mM Phe and (■) in the presence of both 12 mM Phe and 10 mM FBP [24].
We have elucidated the atomic structure of the S402P RMPK variant by X-ray crystallography [25]. Although the overall S402P PK structure is nearly identical to the wild-type (WT) structure within experimental error, significant differences in the conformation of the backbone are found at the site of mutation. We found that the ratio of B-factors of mutant/WT provides a good representation of the pattern of long range communications between distant sites [25]. The most obvious increase in B-factor in the S402P PK is around the residue 402 indicating a significant increase in dynamics in that region. There are also significant changes in the ratio of B-factor for residues 50 to 200. In addition, there is an increase in the number of heterogeneity in the angle assumed by the B-domain with respect to the A-domain i.e., increase dynamics in domain movements. Closer examination of the X-ray data shows a disruption of a salt bridge between residues 341 and 177 of an adjacent subunit. This salt bridge and residue 402 reside in different subunit interfaces. Thus, these structural data show a communication between these two different subunit interfaces. A similar conclusion was derived from the results of our study of subunit assembly [26,27]. It is evident that a mutation at residue 402 leads to increased dynamics in distant sites through long range communication without significant changes in secondary structures.
Establishment of Functional Coupling among Residues
T340M RMPK mutant
In an effort to establish functional coupling among residues in RMPK, we incorporated into our studies the human genetic data [28–33]. Our choice of residue 340 is based on the combined results of our structural studies and human genetic data. The crystallographic structures of yeast and mammalian PK have identified two intersubunit interfaces [4,5,11–14,34], one involves the C-domain while the other comprises mainly the A domain, as shown in Fig. 1. In our modeling study we have further identified the residues whose inter-α carbon distances are within 15 Å [35] along the axis between the adjacent A-domains. These include residues 330–350. Human genetic data identified T340M as a mutant which is observed in patients suffering from Pyruvate Kinase Deficiency. The T340M RMPK and RKPK mutants are only half as active as the WT PKs. The T340M RMPK enzyme is more susceptible than RKPK to inhibition by Phe or to the activator FBP. Evidently the 22 residues in the C-domain modulate the effect of residue 340, which reside in different subunit interfaces. The resultant is a differential effect on the functional energetics of PK. This study demonstrates the linkage between distant residues in different interfacial interactions i.e., establishing the functional coupling among residues and the identities of these residues [24,26,27].
Prospectus
While we are successful in discovering that protein dynamics are intimately coupled to allostery and the impact of differences in protein sequence on the functional energy landscape of PK, there are serious limitations: 1. Identification of connectivity among residues to modulate functions is a very time consuming effort and limited to a pair at a time. It is very inefficient in trying to establish the goal of defining the network of functionally connected residues; 2. Even if the complete network has been identified, we might not have gained deeper insight on the chemical principles involved in these results which are basically simply phenomenological. It is difficult to assign significance in sequence differences to the chemical principles that govern the changes in functions i.e. failure to establish the ground rules in linking sequence-fold-function. Hence, our future direction of research on RMPK focuses on: 1. Cast both structure and function in terms of energetics, allowing us to correlate the energy landscapes. 2. Elucidate the chemical principles governing the sequence differences which affect functions. Probe the nature of mutations on the stability of the secondary structural elements e.g. N- and C-caps of helices; helices and loops. 3. Use the algorithm such as COREX/BEST [36–41] to probe the connectivity among distant residues. Probe the changes in this connectivity pattern due to mutations. 4. Integration of results from 1 to 3 will enhance our chances to identify the chemical principles which govern the correlation of sequence-fold-function.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM77551) and the Robert A. Welch Foundation
References
- 1.Pawson T, Nash P. Assembly of cell regulatory systems through protein interaction domains. Science. 2003;300:445–452. doi: 10.1126/science.1083653. [DOI] [PubMed] [Google Scholar]
- 2.Copley RR, Bork P. Homology among (βα)8 barrels: implications for the evolution of metabolic pathways. J Mol Biol. 2000;303:627–640. doi: 10.1006/jmbi.2000.4152. [DOI] [PubMed] [Google Scholar]
- 3.Heggi H, Gerstein M. The relationship between protein structure and function: a comprehensive survey with application to the yeast genome. J Mol Biol. 1999;288:147–164. doi: 10.1006/jmbi.1999.2661. [DOI] [PubMed] [Google Scholar]
- 4.Williams R, Holyoak T, McDonald G, Gui C, Fenton AW. Differentiating a ligand’s chemical requirements for allosteric interactions from those for protein binding. Phenylalanine inhibition of pyruvate kinase. Biochemistry. 2006;45:5421–5429. doi: 10.1021/bi0524262. [DOI] [PubMed] [Google Scholar]
- 5.Dombrauckas JD, Santarsiero BD, Mesecar AD. Structural basis for tumor pyruvate kinase M2 allosteric regulation and catalysis. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 6.Gerlt JA, Babbitt PC. Divergent evolution of enzymatic function: mechanistically diverse superfamilies and functionally distinct superfamilies. Annu Rev Biochem. 2001;70:209–246. doi: 10.1146/annurev.biochem.70.1.209. [DOI] [PubMed] [Google Scholar]
- 7.Wierenga RK. The TIM-barrel fold: a versatile framework for efficient enzymes. FEBS Lett. 2001;492:193–198. doi: 10.1016/s0014-5793(01)02236-0. [DOI] [PubMed] [Google Scholar]
- 8.Farber GK, Petsko GA. The evolution of α/β barrel enzymes. Trends Biochem Sci. 1990;15:228–234. doi: 10.1016/0968-0004(90)90035-a. [DOI] [PubMed] [Google Scholar]
- 9.Branden C, Tooze J. Introduction to Protein Structure. 2. Garland Publishing, Inc; New York, NY: 1999. [Google Scholar]
- 10.Mattevi A, Bolognesi M, Valentini G. The allosteric regulation of pyruvate kinase. FEBS Lett. 1996;289:15–19. doi: 10.1016/0014-5793(96)00462-0. [DOI] [PubMed] [Google Scholar]
- 11.Muirhead H, Clayden DA, Barford D, Lorimer CG, Fothergill-Gilmore LA, Schiltz E, Schmitt W. The structure of cat muscle pyruvate kinase. EMBO J. 1986;5:475–481. doi: 10.1002/j.1460-2075.1986.tb04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larsen TM, Laughlin T, Holden HM, Rayment I, Reed GH. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry. 1994;33:6301–6309. doi: 10.1021/bi00186a033. [DOI] [PubMed] [Google Scholar]
- 13.Larsen TM, Benning MM, Wesenberg GE, Rayment I, Reed GH. Ligand-induced domain movement in pyruvate kinase: structure of the enzyme from rabbit muscle with Mg2+, K+, and L-phospholactate at 2.7 Å resolution. Arch Biochem Biophys. 1997;345:199–206. doi: 10.1006/abbi.1997.0257. [DOI] [PubMed] [Google Scholar]
- 14.Larsen TM, Benning MM, Rayment I, Reed GH. Structure of the bis (Mg2+)-ATP-oxalate complex of the rabbit muscle pyruvate kinase at 2.1Å resolution: ATP binding over a barrel. Biochemistry. 1998;37:6247–6255. doi: 10.1021/bi980243s. [DOI] [PubMed] [Google Scholar]
- 15.Oberfelder RW, Lee LL, Lee JC. Thermodynamic linkages in rabbit muscle pyruvate kinase: kinetic, equilibrium, and structural studies. Biochemistry. 1984;23:3813–3821. doi: 10.1021/bi00312a004. [DOI] [PubMed] [Google Scholar]
- 16.Oberfelder RW, Barisas BG, Lee JC. Thermodynamic linkages in rabbit muscle pyruvate kinase: Analysis of experimental data by a two-state model. Biochemistry. 1984;23:3822–3826. doi: 10.1021/bi00312a005. [DOI] [PubMed] [Google Scholar]
- 17.Consler TG, Jennewein MJ, Cai GZ, Lee JC. Synergistic effects of proton and phenylalanine on the regulation of muscle pyruvate kinase. Biochemistry. 1990;29:10765–10771. doi: 10.1021/bi00500a007. [DOI] [PubMed] [Google Scholar]
- 18.Consler TG, Jennewein MJ, Cai GZ, Lee JC. Energetics of allosteric regulation in muscle pyruvate kinase. Biochemistry. 1992;31:7870–7878. doi: 10.1021/bi00149a018. [DOI] [PubMed] [Google Scholar]
- 19.Lonhienne TG, Reilly PE, Winzor DJ. Further evidence for the reliance of catalysis by rabbit muscle pyruvate kinase upon isomerization of the ternary complex between enzyme and products. Biophys Chem. 2003;104:189–98. doi: 10.1016/s0301-4622(02)00366-6. [DOI] [PubMed] [Google Scholar]
- 20.Lonhienne TG, Winzor DJ. Calorimetric demonstration of the potential of molecular crowding to emulate the effect of an allosteric activator on pyruvate kinase kinetics. Biochemistry. 2002;41:6897–6901. doi: 10.1021/bi020064h. [DOI] [PubMed] [Google Scholar]
- 21.Heyduk E, Heyduk T, Lee JC. Global conformational changes in allosteric proteins. A study of Escherichia coli cAMP receptor protein and muscle pyruvate kinase. J Biol Chem. 1992;267:3200–3204. [PubMed] [Google Scholar]
- 22.Consler TG, Uberbacher EC, Bunick GJ, Liebman MN, Lee JC. Domain-interaction in rabbit muscle pyruvate kinase, II: Small-angle neutron scattering and computer simulation. J Biol Chem. 1988;263:2794–2801. [PubMed] [Google Scholar]
- 23.Yu S, Lee L, Lee JC. Effects of metabolites on the structural dynamics of rabbit muscle pyruvate kinase. Biophys Chem. 2003;103:1–11. doi: 10.1016/s0301-4622(02)00146-1. [DOI] [PubMed] [Google Scholar]
- 24.Friesen RH, Castellani RJ, Lee JC, Braun W. Allostery in rabbit pyruvate kinase: development of a strategy to elucidate the mechanism. Biochemistry. 1998;37:15266–15276. doi: 10.1021/bi981273y. [DOI] [PubMed] [Google Scholar]
- 25.Wooll JO, Friesen RH, White MA, Watowich SJ, Fox RO, Lee JC, Czerwinski EW. Structural and functional linkages between subunit interfaces in mammalian pyruvate kinase. J Mol Biol. 2001;312:525–540. doi: 10.1006/jmbi.2001.4978. [DOI] [PubMed] [Google Scholar]
- 26.Friesen RH, Chin AJ, Ledman DW, Lee JC. Interfacial communications in recombinant rabbit kidney pyruvate kinase. Biochemistry. 1998;37:2949–2960. doi: 10.1021/bi971990c. [DOI] [PubMed] [Google Scholar]
- 27.Friesen RH, Lee JC. The negative dominant effects of T340M mutation on mammalian pyruvate kinase. J Biol Chem. 1998;273:14772–14779. doi: 10.1074/jbc.273.24.14772. [DOI] [PubMed] [Google Scholar]
- 28.Zanella A, Fermo E, Bianchi P, Chiarelli LR, Valentini G. Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 2007;21:217–231. doi: 10.1016/j.blre.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Miwa S, Kanno H, Fujii H. Concise review: pyruvate kinase deficiency: historical perspective and recent progress of molecular genetics. Am J Hematol. 1993;42:31–35. doi: 10.1002/ajh.2830420108. [DOI] [PubMed] [Google Scholar]
- 30.Neubauer B, Lakomek M, Winkler H, Parke M, Hofferbert S, Schröter W. Point mutations in the L-type pyruvate kinase gene of two children with hemolytic anemia caused by pyruvate kinase deficiency. Blood. 1991;77:1871–1875. [PubMed] [Google Scholar]
- 31.Valentine WN, Tanaka KR, Paglia DE. Pyruvate kinase and other enzyme deficiency disorders of the erythrocyte. In: Seriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic Basis of Inherited Disease. Vol. 6. New York: McGrans-Hill; 1989. [Google Scholar]
- 32.Kanno H, Fujii H, Hirono A, Miwa S. cDNA cloning of human R-type pyruvate kinase and identification of a single amino acid substitution (Thr384 → Met) affecting enzymatic stability in a pyruvate kinase variant (PK Tokyo) associated with hereditary hemolytic anemia. Proc Natl Acad Sci USA. 1991;88:8218–8221. doi: 10.1073/pnas.88.18.8218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baronciani L, Beutler E. Molecular study of pyruvate kinase deficient patients with hereditary nonspherocytic hemolytic anemia. J Clin Invest. 1995;95:1702–1709. doi: 10.1172/JCI117846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jurica MS, Mesecar A, Heath PJ, Shi W, Nowak T, Stoddard BL. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure. 1998;6:195–210. doi: 10.1016/s0969-2126(98)00021-5. [DOI] [PubMed] [Google Scholar]
- 35.Consler TG, Liebman MN, Lee JC. Structural elements involved in the allosteric switch in mammalian pyruvate kinase. In: Liebman MN, Kumosinski T, editors. Molecular Modeling. 1994. pp. 466–485. ACS Symposium Series 576, Chapter 25. [Google Scholar]
- 36.Pan H, Lee JC, Hilser VJ. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc Natl Acad Sci USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hilser VJ, Freire E. Structure-based calculation of the equilibrium folding pathway of proteins. Correlation with hydrogen exchange protection factors. J Mol Biol. 1996;262:756–772. doi: 10.1006/jmbi.1996.0550. [DOI] [PubMed] [Google Scholar]
- 38.Hilser VJ, Freire E. Predicting the equilibrium protein folding pathway: structure-based analysis of staphylococcal nuclease. Proteins. 1997;27:171–183. doi: 10.1002/(sici)1097-0134(199702)27:2<171::aid-prot3>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 39.Hilser VJ, Townsend BD, Freire E. Structure-based statistical thermodynamic analysis of T4 lysozyme mutants: structural mapping of cooperative interactions. Biophys Chem. 1997;64:69–79. doi: 10.1016/s0301-4622(96)02220-x. [DOI] [PubMed] [Google Scholar]
- 40.Hilser VJ, Dowdy D, Oas TG, Freire E. The structural distribution of cooperative interactions in proteins: analysis of the native state ensemble. Proc Natl Acad Sci USA. 1998;95:9903–9908. doi: 10.1073/pnas.95.17.9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friere E. The propagation of binding interactions to remote sites in proteins: analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc Natl Acad Sci US. 1999;96:10118–10122. doi: 10.1073/pnas.96.18.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consler TG, Woodard SH, Lee JC. Effects of primary sequence differences on the global structure and function of an enzyme: a study of pyruvate kinase isozymes. Biochemistry. 1989;28:8756–8764. doi: 10.1021/bi00448a012. [DOI] [PubMed] [Google Scholar]