

Abstract
There is growing evidence that the maturation state of dendritic cells (DCs) is a critical parameter determining the balance between tolerance and immunity. We report that mouse CD4+ effector memory T (TEM) cells, but not naive or central memory T cells, constitutively expressed CD40L at levels sufficient to induce DC maturation in vitro and in vivo in the absence of antigenic stimulation. CD4+ TEM cells were excluded from resting lymph nodes but migrated in a CD62P-dependent fashion into reactive lymph nodes that were induced to express CD62P, in a transient or sustained fashion, on high endothelial venules. Trafficking of CD4+ TEM cells into chronic reactive lymph nodes maintained resident DCs in a mature state and promoted naive T cell responses and experimental autoimmune encephalomyelitis (EAE) to antigens administered in the absence of adjuvants. Antibodies to CD62P, which blocked CD4+ TEM cell migration into reactive lymph nodes, inhibited DC maturation, T cell priming, and induction of EAE. These results show that TEM cells can behave as endogenous adjuvants and suggest a mechanistic link between lymphocyte traffic in lymph nodes and induction of autoimmunity.
DCs orchestrate a repertoire of immune responses that ranges from tolerance to self-antigens to resistance to infectious pathogens (1, 2). In lymph nodes, immature DCs continually present tissue antigens to self-reactive naive T cells, leading to abortive T cell proliferation and establishment of T cell tolerance (3–5). In contrast, mature DCs present microbial antigens and typically induce robust effector and memory T cell responses. In the latter case, DC maturation is initiated by microbial products, endogenous danger signals, or signals delivered by antigen-activated T cells, primarily through CD40L (6–8). It is generally believed that full-blown T cell immune responses are dependent on a combination of stimuli derived from microbial products, endogenous danger signals, and feedback from T cells that act in a temporally and spatially ordered fashion to induce DC maturation (9, 10). However, it has been shown in some experimental systems that the triggering of CD40, by CD40L expressed on antigen-activated CD4+ T cells or by agonistic antibodies, is sufficient to license DCs for priming of naive T cells against antigens administered in the absence of adjuvant (11–14).
CD40L is a TNF family member that is rapidly up-regulated on antigen-activated T cells and released by activated platelets, and plays an important role in immune responses by regulating DC and B cell function (15). Several studies have reported that CD4+ effector and memory T cells contain preformed CD40L in intracellular stores that can be rapidly mobilized to the cell surface after TCR stimulation (16–18). Interestingly, mouse naive CD4+ T cells also constitutively express low amounts of CD40L, which appears to be sufficient to induce survival of autoreactive B cells in the absence of T cell activation (19). Whether circulating naive or memory T cells may influence the DC maturation state in peripheral tissues or lymph nodes through constitutive expression of CD40L remains to be established.
Under steady-state conditions, migration of naive T cells and central memory T (TCM) cells to peripheral lymph nodes is dependent on the expression of CD62L and CCR7, which mediate interaction with the cognate ligands peripheral node addressin (PNAd) and CCL21, respectively, expressed on high endothelial venules (HEVs) (20). Effector and effector memory T (TEM) cells, which lack CD62L and CCR7, are largely excluded from resting lymph nodes (21–23). However, we recently demonstrated that CD8+ TEM cells, as well as CCR7− NK cells, can efficiently migrate into lymph node draining sites of injection of mature DCs or adjuvants (24, 25). In reactive lymph nodes, CD8+ TEM and NK cells modulate ongoing responses by killing antigen-bearing DCs, thus limiting secondary immune responses, or by producing IFN-γ, which enhances Th1 polarization. Recruitment of CD8+ TEM and NK cells is dependent on the expression of CXCR3 on migrating cells and coincides with a transient expression of its ligand, CXCL9, on HEVs of reactive lymph nodes (24, 25).
In this paper, we report that mouse CD4+ TEM cells, but not naive T or TCM cells, constitutively expressed CD40L at levels sufficient to trigger phenotypic maturation of DCs and license them for priming of naive CD4+ T cells in vitro and in vivo. We also found that CD4+ TEM cells were largely excluded from resting lymph nodes but migrated in a CD62P-dependent fashion into reactive lymph nodes that were induced to express, in a transient (acute) or sustained (chronic) fashion, CD62P on HEVs. Strikingly, presentation of myelin oligodendrocyte glycoprotein (MOG) peptides by TEM cell–licensed DCs in chronic reactive lymph nodes was sufficient to prime self-reactive T cells, leading to experimental autoimmune encephalomyelitis (EAE).
RESULTS
CD4+ TEM cells constitutively express CD40L and trigger DC maturation
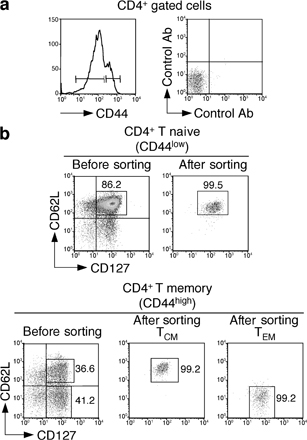
It has been recently reported that naive CD4+ T cells constitutively express CD40L in amounts that are sufficient to promote autoreactive B cell survival (19). We asked whether constitutive expression of CD40L on naive and memory T cell subsets would be sufficient to induce DC maturation. To generate memory T cells, BALB/c mice were either immunized with KLH in CFA or were adoptively transferred with OVA-specific DO11.10 TCR transgenic naive T cells and immunized with OVA-pulsed mature DCs. At least 4 wk after immunization, CD4+ naive T cells (CD44low, CD62L+, CD127+), TCM cells (CD44high, CD62L+, CD127+), and TEM cells (CD44high, CD62L−, CD127+) were isolated from spleens of BALB/c mice (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20081212/DC1) and stained with antibodies to CD40L. Naive T and TCM cells isolated from KLH-immunized mice did not express detectable levels of surface CD40L (Fig. 1 a). In contrast, CD4+ TEM cells homogeneously expressed low amounts of surface CD40L, which was significantly higher compared with naive T and TCM cells (Fig. 1 a). Upon in vitro stimulation with CD3 antibodies, CD40L expression increased in all three subsets, although in TEM cells the kinetics were more rapid and the plateau level was ∼10-fold higher than that reached by naive T or TCM cells (Fig. 1 b). CD40L was also expressed on DO11.10 TEM cells but not on DO11.10 naive or TCM cells (unpublished data). CD40L mRNA was found to be expressed in comparable amounts in TCM and TEM cells (unpublished data), suggesting that the difference in protein expression may be caused by differences in translation and/or storage. Consistent with this possibility, previous studies showed the presence of prestored CD40L in memory T cells of both human and mouse origin (16–19).
Figure 1.

CD4+ TEM cells constitutively express CD40L and promote DC maturation in vitro through CD40L–CD40 interaction. (a) Surface expression of CD40L (black lines) in CD4+ T naive (CD44lowCD62L+CD127+), TCM (CD44highCD62L+CD127+), and TEM (CD44highCD62L−CD127+) cells. Gray lines represent control staining with isotype-matched control antibodies. (b) Kinetics of CD40L surface expression on CD4+ T cell subsets sorted as in panel a and stimulated in vitro with anti-CD3 and PdBU. Data are the means ± SD of three separate experiments. Student's t test on values at time 0: TEM versus TCM, P = 0.041; TEM versus T naive, P = 0.023; TCM versus T naive, NS. Unstimulated T cells cultured for the same time had a level of CD40L expression comparable to time 0 (not depicted). (c) Expression of CD40, CD86, and MHC class II (black lines) on DCs that had been cultured for 12 h with the indicated CD4+ T cell subsets or stimulated with LPS. Gray lines represent staining of untreated immature DCs. Results are representative of four independent experiments. (d) Expression of CD86 (black lines) in wild-type DCs (top) or CD40−/− DCs (bottom) cultured for 12 h with live or fixed CD4+ TEM cells or with LPS as control. Gray lines represent staining of untreated immature DCs. Results are representative of three independent experiments.
CD4+ naive T, TCM, and TEM cells were then cultured with syngeneic immature BM-derived DCs, and the expression of DC activation markers was measured after 24 h. Although naive T and TCM cells did not induce DC maturation, TEM cells induced up-regulation of CD40, CD86, and MHC class II molecules to an extent comparable to that induced by LPS (Fig. 1 c). DC maturation was also triggered by glutaraldehyde-fixed CD4+ TEM cells (Fig. 1 d, top), indicating that the latter trigger DC maturation in the absence of antigen-dependent or -independent signaling. In addition, CD4+ TEM cells did not induce maturation of CD40−/− DCs (Fig. 1 d, bottom), indicating that the maturation signal is delivered to DCs through a CD40–CD40L interaction. Collectively, these data indicate that mouse CD4+ TEM cells, but not naive T or TCM cells, constitutively expressed CD40L at levels sufficient to trigger in vitro phenotypic maturation of DCs.
CD4+ TEM cell–matured DCs prime naive T cells in vitro
We next asked whether TEM cell–matured DCs would be capable of priming naive T cells. Hemagglutinin (HA) peptide–pulsed immature DCs were cultured with CFSE-labeled HA-specific 6.5 TCR transgenic CD4+ naive T cells alone or together with naive T, TCM, and TEM cells specific for a different antigen (DO11.10 TCR transgenic T cells recognizing OVA). HA-specific naive T cells did not proliferate when stimulated with HA-pulsed immature DCs (Fig. 2 a), consistent with the notion that immature DCs have poor co-stimulatory capacity and fail to prime naive T cells (26). However, these cells proliferated vigorously and differentiated to IFN-γ–producing effector T cells when stimulated with immature DCs in the presence of OVA-specific TEM cells but not naive T or TCM cells (Fig. 2 a and not depicted). Similarly, CFSE-labeled OVA-specific CD4+ naive T cells proliferated in response to OVA-pulsed immature DCs when cultured with polyclonal CD4+ TEM cells but not naive T or TCM cells (Fig. 2 b). Finally, in the presence of polyclonal CD4+ TEM cells, OVA-specific CD4+ naive T cells proliferated in response to OVA peptide presented by wild-type but not CD40−/− DCs (Fig. 2 c). The results of these in vitro experiments indicate that CD4+ TEM cells can license immature DCs for priming of naive T cells in an antigen-independent and CD40-dependent fashion, and raise the question of whether CD4+ TEM cells might induce DC maturation in vivo.
Figure 2.

CD40L+ CD4+ TEM cells license DCs for T cell priming in vitro. (a) Proliferation of CFSE-labeled HA-specific 6.5 transgenic naive CD4+ T cells cultured with 50 μM HA110-119–pulsed syngeneic immature DCs in the absence or presence of ex vivo–isolated OVA-specific DO11.10 transgenic CD4+ T naive, TCM, or TEM cells. (b) Proliferation of CFSE-labeled OVA-specific OT-II transgenic naive CD4+ T cells cultured with 0.1 μM OVA323-339–pulsed syngeneic immature DCs in the absence or presence of ex vivo–isolated polyclonal T naive, TCM, or TEM cells. (c) Proliferation of CFSE-labeled OVA-specific OT-II transgenic naive CD4+ T cells cultured with polyclonal TEM cells in the presence of OVA-pulsed wild-type or CD40−/− immature DCs. Shown is the CFSE profile of T cells measured on day 5. Results are representative of at least three independent experiments.
CD4+ TEM cells rapidly migrate into reactive lymph nodes in a CD62P-dependent, CXCR3-independent fashion
Mouse CD4+ and CD8+ TEM cells lack CCR7 and CD62L and are therefore largely excluded from lymph nodes in the steady state (21–23). However, it is increasingly recognized that lymph nodes undergoing an immune response show an increased cellularity and become capable of recruiting CD8+ TEM and NK cells (27). Recruitment of CD8+ TEM and NK cells is dependent on expression of CXCR3 on migrating cells and coincides with a transient expression of its ligand, CXCL9, on HEVs of reactive lymph nodes (24, 25). To investigate whether CD4+ TEM cells would be also recruited to reactive lymph nodes, we injected i.v. in vivo–generated OVA-specific memory T cells, comprising both TCM (CD127+CD62L+) and TEM (CD127+CD62L−) cells, into syngeneic mice and measured their migration into a reactive lymph node, which had been induced by s.c. injection of LPS-matured DCs, and into a resting lymph node as control (Fig. 3 a). As expected, TCM cells, which represented a minor fraction of the transferred cell population, were highly enriched in resting lymph nodes, accounting for up to 90% of the recruited cells. In contrast, TEM cells were virtually excluded from resting lymph nodes but efficiently migrated into reactive lymph nodes, where they accounted for up to 85% of the migrated cells, a figure similar to that found in the spleen and inflamed peritoneum (Fig. 3 a). Transferred TEM cells were detected in reactive lymph nodes as early as 20 min after i.v. injection (Fig. 3 b), consistent with a direct migration of circulating T cells through HEVs rather than transit from tissues. In addition, 16 h after transfer, TEM cells colocalized with naive T cells in the T cell areas of reactive lymph nodes (Fig. S2, available at http://www.jem.org/cgi/content/full/jem.20081212/DC1).
Figure 3.

CD4+ TEM cells rapidly migrate to reactive lymph nodes in a CD62P-dependent, CXCR3-independent manner. Adoptively transferred TCR transgenic OVA-specific naive T cells were primed by immunization with an s.c. injection of 106 OVA-pulsed syngeneic LPS-matured DCs. 3 wk later, memory CD4+ T cells were enriched from spleens and lymph nodes, and 3 × 106 T cells were injected i.v. into mice in which reactive lymph nodes had been induced 24 h before by injection of 106 syngeneic LPS-matured DCs. Some mice received also an i.p. injection of thioglycolate 48 h before T cell transfer. (a) Relative proportion of DO11.10 CD4+ TCM (CFSE−KJ1-26+CD62L+CD127+) and TEM (CFSE−KJ1-26+CD62L−CD127+) cells in the population before transfer and in the indicated organs 12 h after transfer (percentages are shown). (b) Absolute number of DO11.10 CD4+ TEM (KJ1-26+CD62L−CD127+) cells in reactive lymph nodes of mice 20 or 60 min after T cell transfer. (c) Expression of CD62P ligands and CXCR3 (black lines) on gated CD4+CD62L− TEM cells. The gray dashed lines represent background staining. (d) Percentages of TEM-like cells from wild-type and CXCR3−/− mice in reactive lymph nodes and spleen 24 h after i.v. injection. T cells were mixed at a ratio of 1:1, and 107 cells were injected in each mouse in which a reactive lymph node was produced by s.c. injection of 3 × 106 mature DCs in the footpad. (e) Absolute number of OT-II CD4+ TEM (Ly5.1+CD62L−CD127+) cells in reactive lymph nodes of wild-type C57BL/6 or CD62P/E double-deficient mice. (f) Absolute number of DO11.10 CD4+ TEM (KJ1-26+CD62L−CD127+) cells in reactive lymph nodes of mice 12 h after injection of blocking antibodies to CD62P or CD62E or of isotype-matched control antibodies. Data are the means ± SD of two or three independent experiments each performed with two mice per condition. p-values were obtained with the Student's t test.
In vivo–generated CD4+ TEM cells homogeneously expressed CXCR3, the receptor for CXCL9, a chemokine expressed on HEVs of reactive lymph nodes, and P-selectin glycoprotein ligand 1, the ligand for CD62P/CD62E expressed on endothelial cells and platelets (Fig. 3 c) (28). To investigate the requirements for CXCR3 in the process of CD4+ TEM cell migration to reactive lymph nodes, TEM-like cells (96% CD62L negative) were generated in vitro from wild-type and CXCR3-deficient mice, mixed at a 1:1 ratio, and injected i.v. into mice carrying a reactive lymph node induced by s.c. injection of LPS-matured DCs. Surprisingly, wild-type and CXCR3-deficient T cells were recovered in similar proportions in spleens and reactive lymph nodes (Fig. 3 d), indicating that CXCR3 is not required for migration of CD4+ TEM cells into reactive lymph nodes. In contrast, TEM cells generated in vivo by immunization of wild-type mice and transferred into mice lacking CD62P and CD62E selectins failed to migrate to reactive lymph nodes (Fig. 3 e). Furthermore, migration of TEM cells to reactive lymph nodes of wild-type mice was inhibited by injection of antibodies blocking CD62P but not CD62E (Fig. 3 f). Collectively, these results indicate that recruitment of CD4+ TEM cells into reactive lymph nodes is dependent on CD62P expression on endothelial cells but independent of CXCR3 expression on migrating T cells.
Transient and sustained expression of CD62P on HEVs of reactive lymph nodes
We next investigated whether and under which conditions CD62P would be expressed on HEVs of lymph nodes. Reactive lymph nodes were induced by s.c. injection of either LPS-matured DCs or adjuvants. After 2 or 30 d, mice were injected i.v. with fluorescinated antibodies to CD62P to stain the luminal side of HEVs. Reactive and resting lymph nodes were collected and examined by immunohistology after counterstaining with PNAd antibodies to identify HEVs. As shown in Fig. 4 a, CD62P was not expressed on the HEVs of resting lymph nodes. After a single injection of mature DCs (DC 1×), CD62P was readily detected on day 2, persisted for ∼6 d, and was no longer detectable on day 30 (Fig. 4 a and not depicted). CD62P was also detected on HEVs of draining lymph nodes 2 d after injection of adjuvants such as CFA, CpG, LPS, IFA (Fig. 4 b), R848, and TNF (not depicted). Remarkably, however, two consecutive injections 2 d apart of DCs (DC 2×) or a single injection of CFA, but not one or two injections of CpG or LPS, led to a sustained expression of CD62P on HEVs for >30 d (Fig. 4 a and not depicted). The different staining patterns of CD62P and PNAd on HEVs may be caused by differences in cellular localization or by technical reasons, because antibodies to CD62P were injected i.v., whereas anti-PNAd antibodies were added on tissue sections.
Figure 4.

Expression of CD62P on HEVs of reactive lymph nodes correlates with increased lymph node cellularity and selective increase of TEM cell recruitment. (a) Reactive lymph nodes were induced by one or two consecutive (2 d apart) injections of 106 LPS-matured DCs (DC 1× and DC 2×, respectively). After 2 or 30 d, mice received an i.v. injection of 100 μg FITC-labeled anti-CD62P antibody. After 30 min, lymph nodes were collected, snap frozen, and processed for immunohistochemistry. Shown is the expression of PNAd (red) and CD62P (green) in resting and reactive lymph nodes. Bar, 100 μm. (b) CD62P expression in reactive lymph nodes detected by in vivo staining 2 d after s.c. injection of the indicated adjuvants. Results are representative of three independent experiments. Bar, 100 μm. (c) Absolute cell number in resting and reactive draining lymph nodes at different time points after s.c. injection of PBS, as control or CFA, or one or two consecutive s.c. injections of mature DCs. The asterisk indicates the Student's t test on values: CFA versus PBS, P = 0.013; DC 2× vs. PBS, P = 0.001. (d) Fold increase of the indicated cell populations in day 30 reactive lymph nodes compared with resting lymph nodes (naive, CD44lo; memory, CD44hi; TCM, CD44hiCD62L+; TEM, CD44hiCD62L−; NK, CD3− NK1.1+; NKT, CD3+ NK1.1+; B, CD3− CD19+; pDC, B220+ CD11clo; dermal DC, B220− CD11clo; LC, B220− CD11chi). (e) Absolute number of endogenous TEM cells recovered in resting and reactive lymph nodes 30 d after injection of DCs or adjuvants, as indicated. Data are the means ± SD of two to three separate experiments, each performed with two or three mice per condition. p-values were obtained with the Student's t test.
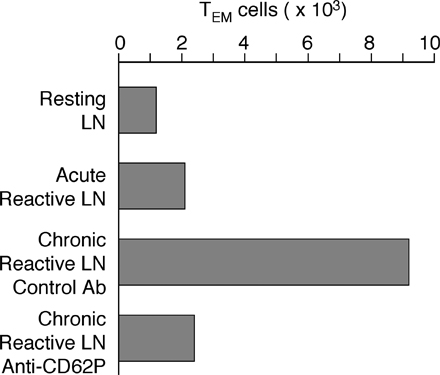
The sustained expression of CD62P on HEVs of lymph nodes stimulated by two injections of mature DCs or a single injection of CFA correlated with a significant increase of lymph node cellularity that was sustained for up to 60 d after initial stimulation (Fig. 4 c and not depicted). In spite of the marked increase in total cell number, the relative proportion of different cell subsets—including CD8+ T, NK, NKT, and B cells, as well as plasmacytoid DCs, dermal DCs, and Langerhans cells—was comparable in resting and DC-reactive lymph nodes, with the only exception being CD4+ TEM cells, which were highly enriched in reactive lymph nodes (Fig. 4 d). Lymph nodes stimulated 30 d earlier with two injections of DCs or one injection of CFA contained significantly higher numbers of endogenous CD4+ CD44highCD127+ CD62L− TEM cells compared with resting lymph nodes, or lymph nodes stimulated by one injection of DCs or one or two injections of CpG or LPS (Fig. 4 e). In addition, reactive lymph nodes induced 6 or 30 d before by one or two consecutive injections of LPS-matured DCs recruited i.v.-injected OVA-specific TEM cells, indicating that TEM cell migration into reactive lymph nodes is not dependent on the presence of a specific antigen (Fig. S3 a, available at http://www.jem.org/cgi/content/full/jem.20081212/DC1).
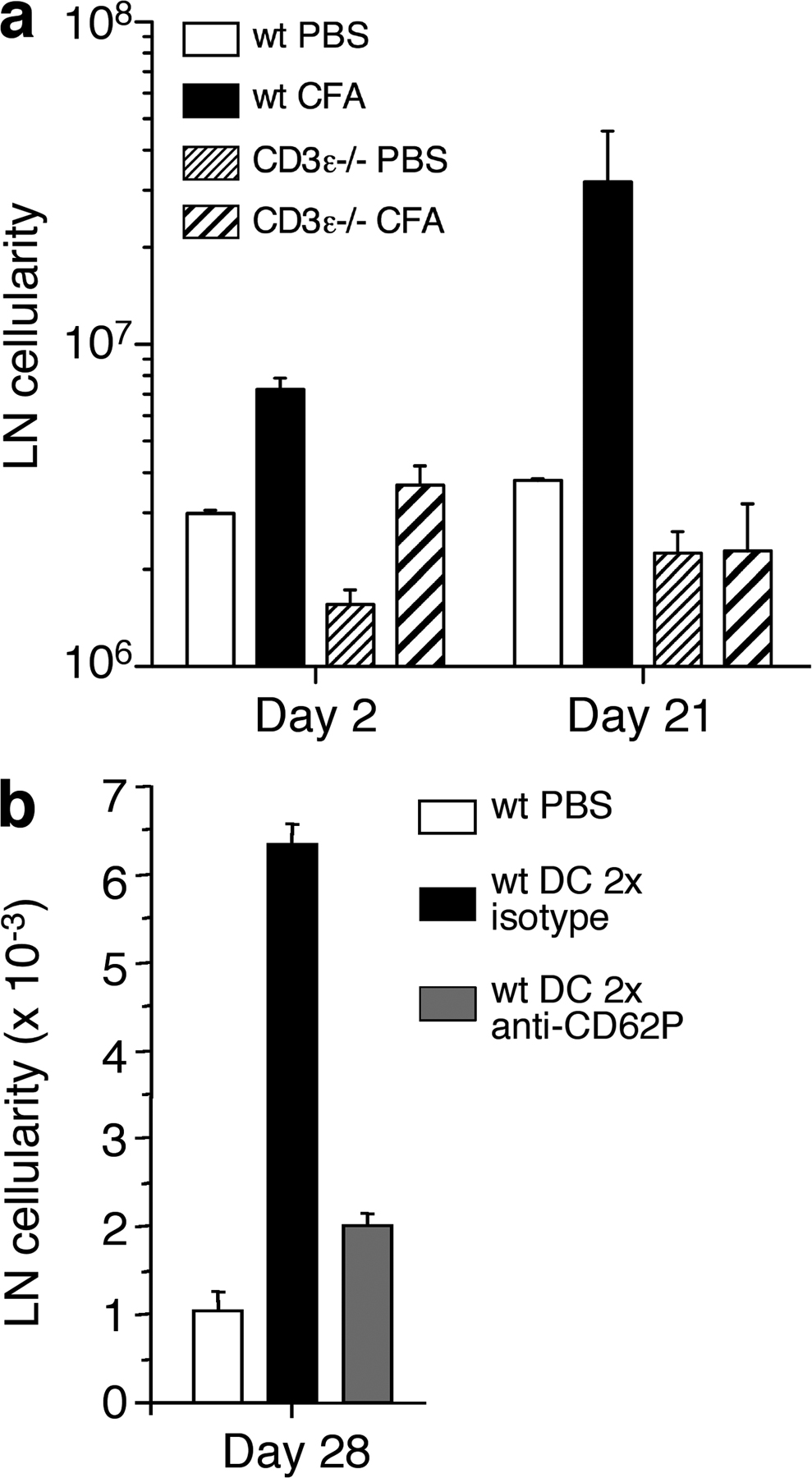
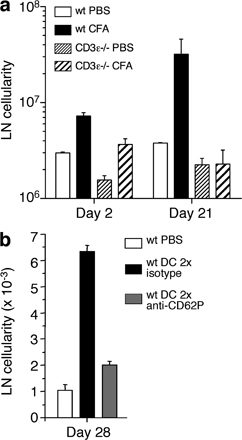
The increased number of TEM cells found in reactive lymph nodes 30 d after injection of DCs or CFA may result from an increase in resident TEM cells or may depend on a continuous recruitment of TEM cells from the blood. To distinguish between these possibilities, we injected antibodies to CD62P, which, as shown in Fig. 3 f, block TEM cell recruitment into reactive lymph nodes. Within a few hours after antibody injection, the number of TEM cells in reactive lymph nodes decreased substantially (Fig. S3 b), indicating that TEM cells continually recirculate from blood to lymph node and from there to efferent lymph. We conclude that expression of CD62P on the luminal side of HEVs of reactive lymph nodes can be induced in a transient or sustained fashion depending on the nature of the eliciting stimulus, and coincides with increased lymph node cellularity, which was caused by a uniform increase of all major cell subsets and a much higher increase of CD4+ TEM cells.
We also investigated the role of T cells in induction and maintenance of reactive lymph nodes. When CFA was injected s.c. in CD3ε−/− mice, cell numbers in draining lymph nodes increased on day 2, but this increase was not sustained at later time points (Fig. S4 a, available at http://www.jem.org/cgi/content/full/jem.20081212/DC1), suggesting that T cells may be required for the establishment and/or maintenance of chronic reactive lymph nodes. When wild-type mice were injected with antibodies to CD62P that prevent migration of TEM cells into reactive lymph nodes (Fig. 3 f), the increase in lymph node cellularity was almost entirely prevented (Fig. S4 b), consistent with the possibility that TEM cells migrating in a CD62P-dependent fashion are required for long-term maintenance of chronic reactive lymph nodes.
CD4+ TEM cells in lymph node license DCs for T cell priming
The finding that under certain conditions recruitment of TEM cells in lymph nodes can be sustained for several weeks suggests the possibility that these cells might influence the DC maturation state through the constitutive expression of CD40L. To test this possibility, mice were transferred with Thy1.1+ OVA-specific DO11.10 naive T cells and primed with one or two consecutive s.c. injections of OVA-pulsed DCs (OVA-DC 1× and OVA-DC 2×, respectively) to generate TEM cells and to induce an acute or a chronic reactive lymph node (Fig. 5 a). Some mice were injected weekly with anti-CD62P blocking antibodies or isotype-matched control antibodies to inhibit TEM cell migration and the induction of chronic reactive lymph nodes. 30 d after priming, high numbers of DO11.10 TEM cells were recovered from chronic reactive but not acute reactive lymph nodes (Fig. S5, available at http://www.jem.org/cgi/content/full/jem.20081212/DC1). Interestingly, on day 30 after priming all DCs recovered from chronic reactive lymph nodes expressed increased levels of CD40 and MHC class II molecules as compared with DCs recovered from resting or acute reactive lymph nodes (Fig. 5 b). At this late time point, there was no evidence of persisting OVA antigen in reactive lymph nodes, as shown by the lack of proliferation of i.v.-injected CFSE-labeled OT-II T cells (unpublished data). Strikingly, in mice treated with blocking CD62P antibodies, DCs did not show increased expression of CD40 and MHC class II molecules (Fig. 5 b). These results are consistent with a role for lymph node–migrating CD4+ TEM cells in the induction of DC maturation in vivo.
Figure 5.

TEM cells migrating into reactive lymph nodes maintain DCs in a mature immunostimulatory state. (a, d, and g) Experimental design. (a) Mice were transferred with Thy1.1+ DO11.10 OVA-specific T cells and either left untreated or primed with one or two s.c. injections of OVA-pulsed DCs to generate TEM cells and acute or chronic reactive lymph nodes. Some mice were injected weekly with 100 μg of control antibodies or anti-CD62P blocking antibodies. (b) Expression of CD40 and MHC class II on CD11c+ DCs purified 30 d after priming from resting (gray lines) or reactive (black lines) lymph nodes. (c) On day 30, mice were transferred with CFSE-labeled HA-specific 6.5 naive T cells and challenged with an i.v. injection of 10 μg HA in PBS. Shown are the percentages and the CFSE profiles of 6.5+ T cells in resting or reactive lymph nodes 5 d after challenge. (d) Mice received an s.c. injection of CFA to generate chronic reactive lymph nodes. Control mice received an s.c. injection of PBS. (e) Expression of CD40 and MHC class II on CD11c+ DCs purified 30 d after priming from resting or chronic reactive lymph nodes. (f) On day 30, mice were transferred with CFSE-labeled 6.5 naive T cells and challenged with an i.v. injection of 10 μg of HA protein in PBS. Shown are the percentages and the CFSE profiles of 6.5+ T cells in resting or chronic reactive lymph nodes 5 d after challenge. (g) Naive mice were adoptively transferred with CFSE-labeled 6.5 naive T cells, and a reactive lymph node was induced by s.c. injection of LPS. 5 d later, mice received an s.c. injection of HA in PBS together with an i.v. transfer of DO11.10 TEM cells that had been treated with control or CD40L-blocking antibodies. (h) Proliferative response of 6.5+ T cells in reactive lymph nodes in the absence or presence of untreated or anti-CD40L–treated TEM cells. Results are representative of two to five separate experiments.
We next asked whether TEM cell–matured DCs in chronic reactive lymph node would be capable of priming naive T cells to an antigen administered in the absence of adjuvant. Mice treated as above were adoptively transferred on day 30 with CFSE-labeled HA-specific 6.5 naive T cells and challenged by i.v. injection of HA in PBS (Fig. 5 c). As expected, no proliferative response was observed in control mice in response to HA administered in the absence of adjuvants. In addition, HA-specific T cells did not proliferate in mice in which a transient reactive lymph node was induced by a single injection of DCs (1×). In contrast, a strong proliferative response of HA-specific T cells was observed in chronic reactive lymph nodes induced by two injections of DCs (2×) and containing OVA-specific TEM cells. Strikingly, HA-specific T cells did not proliferate in mice chronically treated with CD62P antibodies, which blocked TEM cell migration into lymph nodes and DC maturation (Fig. 5 c and Fig. S5). Comparable results were obtained in mice in which a reactive lymph node was induced by injection of CFA and that did not receive DO11.10 T cell transfer (Fig. 5, d–f), suggesting that CFA-induced TEM cells emerging from the endogenous repertoire are sufficient to induce DC maturation and T cell priming.
In another experimental set up, we measured the proliferative response of naive T cells in the presence of adoptively transferred TEM cells (Fig. 5 g). Mice transferred with CFSE-labeled HA-specific 6.5 naive T cells received a s.c. injection of LPS to induce a reactive lymph node. On day 5 after LPS injection, some mice were also transferred with OVA-specific TEM cells, which had been either untreated or treated with anti-CD40L antibodies and then challenged i.v. with HA in PBS. HA-specific T cells failed to proliferate in LPS-treated mice, consistent with our preliminary observation that the adjuvant effect of LPS is rapidly lost in vivo (unpublished data). In contrast, HA-specific T cells proliferated in lymph nodes containing TEM cells but not CD40L antibody–treated TEM cells (Fig. 5 h). Of note, treatment of CD4+ TEM cells with anti-CD40L antibodies did not prevent their migration into reactive lymph nodes (unpublished data). Collectively, these results indicate that CD4+ TEM cells migrating into chronic reactive lymph nodes act as an antigen-nonspecific endogenous adjuvant by continuously triggering DC maturation in a CD40L-dependent fashion and facilitating T cell immune responses to otherwise nonimmunogenic antigens.
Induction of EAE in reactive lymph nodes in the absence of adjuvants
The findings described in the previous section raise the possibility that in chronic reactive lymph nodes, CD4+ TEM cells may license DCs for priming of autoreactive T cells. To test this possibility, we induced reactive lymph nodes by two consecutive injections of mature DCs or a single injection of CFA, and 1 mo later challenged the mice by s.c. injection of MOG peptides in PBS at sites draining into the reactive lymph node. Control mice were injected with MOG peptide in PBS or CFA. The clinical score of EAE was recorded in a blinded fashion by two researchers. In addition, brains and spinal cords were collected at the end of the experiments and analyzed for the presence of infiltrating CD4+ T cells.
Injection of MOG peptide in CFA led to rapid development of clinical symptoms of EAE and to accumulation of CD4+ T cells in the central nervous system, whereas injection of MOG peptide in PBS did not result in any clinical symptoms or T cell infiltrate (Fig. 6, a and b). Remarkably, injection of MOG peptide in PBS in mice carrying a reactive lymph node, which had been induced by injection of either CFA or mature DCs 1 mo earlier, led to the development of clinical symptoms and T cell infiltrates that were comparable in severity and extent to those induced by the administration of MOG in CFA (Fig. 6, c and d). Pretreatment of mice with anti-CD62P antibodies did not interfere with EAE induced by immunization with MOG peptide in CFA (Fig. 6 a), consistent with the fact that CD62P is not required for migration of effector T cells into the brain (29). Remarkably, however, anti-CD62P treatment prevented EAE and T cell infiltration in the central nervous system induced by injection of MOG peptide in PBS draining into reactive lymph nodes (Fig. 6, c and d). Collectively, these data are consistent with a model in which CD4+ TEM cells recruited into reactive lymph nodes provide a licensing signal to resident DCs that facilitates response to self-antigens and the development of pathogenic autoreactive T cells.
Figure 6.

EAE can be induced by presentation of MOG peptides in reactive lymph nodes and is inhibited by CD62P blockade. Groups of mice (n = 16), either untreated (a and b) or that had received 30 d earlier an s.c. injection of CFA or two consecutive s.c. injections of LPS-matured DCs to induce a chronic reactive lymph node (c and d), were challenged i.v. with 100 μg MOG35-55 peptide in CFA (a) or in PBS (b–d). For each experimental group, half of the mice received once a week an i.v. injection of 100 μg CD62P antibodies (circles) or isotype matched control antibodies (squares) for the entire duration of the experiment. Mice were graded daily for clinical manifestation of EAE. Shown are mean clinical scores of eight mice per group. Error bars represent the SE for each point. (right) CD4 expression on lymphocytes isolated from the brain and spinal cords of mice 20 d after priming with MOG peptide. Numbers are the percentage of the gated population. Results are representative of two separate experiments.
DISCUSSION
We have shown that CD4+ TEM cells, which migrate with low efficiency into resting lymph nodes, migrate in a CD62P-dependent fashion into reactive lymph nodes that have been induced to express, in a transient or sustained manner, CD62P on HEVs. In reactive lymph nodes, CD4+ TEM cells trigger maturation of DCs in an antigen-independent fashion via constitutively expressed CD40L and license them for T cell priming against antigens, including self-antigens, administered in the absence of exogenous adjuvants.
We previously reported that, after s.c. injection of adjuvants or mature DCs, draining lymph nodes become capable of attracting leukocytes, such as CD8+ TEM and NK cells, that are largely excluded from resting lymph nodes (24, 25). Entry of CD8+ TEM and NK cells requires expression of CXCR3 on migrating cells and coincides with expression of the cognate chemokines on HEVs of reactive lymph nodes. Although the ligand for CD62P is expressed on CD8+ effector T cells, this adhesion molecule is not required for homing of CD8+ T cells to reactive lymph nodes, as demonstrated by experiments using specific blocking antibodies (unpublished data). The present study shows that migration of CD4+ TEM cells into reactive lymph nodes occurs through a different mechanism independent of CXCR3 and dependent on CD62P. This conclusion is based on the findings that wild-type and CXCR3-deficient CD4+ TEM cells showed a comparable capacity to migrate into reactive lymph nodes, and that CD4+ TEM cell migration was not observed in mice lacking CD62P/E and could be inhibited by antibodies against CD62P but not CD62E. In addition, migration of CD4+ TEM cells in lymph nodes coincides with expression of CD62P on HEVs that was induced, in an acute or chronic fashion, by adjuvants or mature DCs. Interestingly, chronic reactive lymph nodes did not appear to efficiently recruit cytotoxic CD8+ TEM cells as acute lymph nodes do (25), indicating that entry of these cells is restricted to the early phases of an immune response.
A recent study demonstrated that antigen-primed lymph nodes contain a resident population of antigen-specific CXCR5+ICOS+ follicular helper T cells (30). In contrast to follicular helper T cells, the CD4+ TEM cells described in this paper continually recirculate through the reactive lymph nodes because they disappeared rapidly when entry was blocked by injection of CD62P antibodies. It remains to be established which lymphocyte integrin is involved in this process and whether chemokine-dependent activation of integrins is required, because the high amounts of LFA-1 and α4-integrins expressed on memory T cells may promote efficient adhesion of rolling cells even in the absence of chemokine signaling. In addition, one should consider the possibility that CD62P-expressing platelets adhering to HEVs may facilitate extravasation of P-selectin glycoprotein ligand 1+ TEM cells, as it has been suggested to allow for CD62P-dependent homing of CD62L-deficient leukocytes (31).
CD62P expression is known to be induced on the endothelial cells of inflamed peripheral tissues and to regulate recruitment of effector cells of the innate and adaptive immune system (32). Previous studies have shown that under conditions of intravital microscopy (but not in situ), CD62P can be expressed on HEVs of Peyer's patches and subiliac lymph nodes and can account for residual migration of leukocytes in the absence of CD62L or β7-integrins (33, 34). CD62P was also reported to be expressed at low levels on HEVs in the course of experimental mouse listeriosis (35). Our results show for the first time that CD62P is induced in peripheral lymph node HEVs by s.c. injection of adjuvants or mature DCs and that it can mediate migration of CD4+ TEM cells. Intriguingly, CD62P expression on HEVs can be acute or chronic, depending on the nature of the eliciting stimuli. Although soluble TLR agonists (LPS, CpG, and R848) or a single injection of mature DCs induced a transient expression of CD62P (up to day 6), a single injection of CFA or two consecutive injections, 2 d apart, of mature DCs led to an up-regulation of CD62P that was sustained for >60 d. Importantly, the persistence of CD62P on HEVs correlates with increased lymph node cellularity, recruitment of TEM cells, and increased immunostimulatory capacity of lymph node–resident DCs. Although the effect of CFA may be explained by a sustained release of microbial products from a tissue depot, it remains to be established how two injections of mature DCs, which are known to be short lived, may induce a long-lasting change in the draining lymph node in the apparent absence of residual antigen, as shown by the finding that 30 d after injection of OVA-pulsed DCs, CFSE-labeled OVA-specific T cells failed to proliferate (unpublished data).
We have provided evidence that T cells may be required for maintenance of chronic reactive lymph nodes. Indeed, although the initial increase of cell number in lymph nodes stimulated by adjuvants or mature DCs can be induced in T cell–deficient CD3ε−/− mice, the sustained increase in lymph node cellularity is dependent on the presence of T cells. The findings that antibodies to CD62P prevented migration of TEM cells and increased lymph node cellularity would be consistent with a role for these cells in the development of chronic reactive lymph nodes. However, at this point we cannot rule out the possibility that cells other than TEM cells requiring CD62P to enter lymph node might also be involved. In addition, it is possible that other changes besides sustained expression of CD62P may take place in reactive lymph nodes. For instance, innate stimuli have been shown to promote remodeling of the primary feed arteriole, leading to an increase in lymphocyte input (36). It is tempting to speculate that, once the inducing stimulus has reached a sufficient threshold, the reactive state of the lymph node may be maintained by irreversible structural changes or by continuous recruitment of TEM and/or innate immune cells.
Another major finding of this study is that CD4+ TEM cells constitutively express CD40L at levels sufficient to trigger DC maturation and to license them for T cell priming (37). Constitutive CD40L expression was significantly higher in TEM cells as compared with naive or TCM cells, and only TEM cells, but not naive or TCM cells, triggered DC maturation and promoted T cell priming in vitro in an antigen-independent and CD40-dependent fashion. Importantly, this effect was also observed in vivo. DCs in chronic reactive lymph nodes, exposed to recirculating CD4+ TEM cells, homogeneously expressed high levels of MHC and co-stimulatory molecules and were able to trigger naive T cell proliferation in response to antigens administered in the absence of adjuvants. The critical role of TEM cells was demonstrated by the fact that injection of CD62P antibodies, which blocked TEM cell migration into reactive lymph nodes, inhibited both DC maturation and T cell priming. The efficiency of CD4+ TEM cells in inducing DC maturation may rely on the anatomy of the lymph node, which places extravasating TEM cells in direct contact with DCs, and on the high numbers and motility of TEM cells, and hence, their ability to interact with multiple DCs in a serial fashion (38). Although our results indicate that DC maturation and licensing is selectively induced by TEM cells but not by naive or TCM cells, we cannot rule out the possibility that constitutive expression of CD40L on the latter may contribute to DC survival and function. Indeed, CD40L on mouse naive T cells has been shown to be sufficient to promote survival of autoreactive B cells (19).
Microbial products have been shown to be required to break tolerance to tissue antigens, leading to autoimmunity (39). In addition, LPS may activate autoreactive CD4+ cells in a bystander fashion (40). Based on our findings, we hypothesized that the DC licensing effect exerted by CD40L+ CD4+ TEM cells trafficking into reactive lymph nodes may facilitate immune responses to poorly immunogenic antigens, such as tissue antigens, in the apparent absence of microbial products. This possibility was demonstrated by the finding that EAE could be induced by injection of a MOG peptide at a site draining into the chronic reactive lymph node accessible to circulating antigen-nonspecific TEM cells. Although endogenous danger signals, such as ATP or uric acid (41), may play a synergistic role in the priming of autoreactive T cells, the finding that the mere exclusion of TEM cells form the reactive lymph nodes by CD62P antibody treatment was sufficient to prevent immunopathology points to TEM cells as the primary inducing factor in eliciting this pathogenic response. Although it is premature to envisage a role for CD40L constitutively expressed on CD4+ TEM cells in induction of autoimmune diseases, it is of note that increased levels of CD40L-expressing effector and memory T cells have been observed in autoimmune-prone mice (17) and in human memory T cells of patients with multiple sclerosis (42–44), and targeting the CD40–CD40L interaction has been proposed as a treatment for autoimmune diseases or to induce allograft tolerance (45, 46). In addition, activated CD4+ T cells have been shown to prime and expand endogenous autoreactive CD8+ T cells in an antigen-dependent or -independent fashion through the licensing of DCs via CD40L (47, 48). We found that clinical symptoms and the extent of T cell infiltration were comparable in mice immunized with MOG in CFA or with soluble MOG draining into a reactive lymph node. It remains to be established whether these distinctive priming environments generate a comparable spectrum of polarized effector T cells (49).
In conclusion, our study provides evidence for a mechanistic link between changes in cell traffic in lymph nodes and predisposition to mount effector responses to otherwise nonimmunogenic antigens. We are currently testing the possibility that induction of chronic reactive lymph nodes at sites that drain tumor antigens may facilitate priming of effector T cells against the tumor.
MATERIALS AND METHODS
Mice.
C57BL/6 (H-2b) and BALB/c (H-2d) mice were purchased from Harlan. Transgenic DO11.10-Thy1.2 (H-2d) mice were purchased from Taconic. BALB/c-Thy1.1 mice, a gift of S.L. Swain (The Trudeau Institute, Saranac Lake, NY), were bred onto DO11.10-Thy1.2 mice in the animal facility of the Institute for Research in Biomedicine. Transgenic OT-II (H-2b) (50) and HA-TCR (H2d) (51) mice were obtained from J. Kirberg (Max-Plank-Institute of Immunobiology, Freiburg, Germany). The OT-II transgenic mice were also bred onto backgrounds of different CD45 alleles. CD62P/CD62E double-deficient mice (H-2b) were a gift of R.O. Hynes (Massachusetts Institute of Technology, Boston, MA) through B. Engelhardt (Theodor Kocher Institute, Bern, Switzerland). CD40-deficient mice (H-2b) (52) were obtained from K. Schwarz (Cytos Biotechnology AG, Zurich, Switzerland). CXCR3−/− mice were provided by C. Gerard (Harvard Medical School, Boston, MA). CD3e−/− mice (53) were obtained from B. Malissen (Centre d'Immunologie Marseille-Luminy, Marseille, France). All transgenic mice used in this study were on a Rag2−/− background. Mice were treated in accordance with the Swiss Federal Veterinary Office guidelines, and approval for animal experiments was obtained by the committee of the same office.
DCs and cell transfer.
BM-derived DCs were generated as previously described (54) by culturing BM precursors from femurs of mice in RPMI 1640 medium supplemented with 10% (vol/vol) FCS, 1% (vol/vol) Glutamax, 1% (vol/vol) nonessential amino acids, 1% (vol/vol) pyruvate, 50 U/ml penicillin, 50 μg/ml streptomycin (all from Invitrogen), and 50 μg/ml 2-mercaptoethanol (Merck) in the presence of recombinant GM-CSF (R&D Systems). DC maturation was induced by overnight incubation with 0.5 μg/ml LPS (Sigma-Aldrich). Whole spleen cells were labeled with 0.3 or 2.5 μM CFSE (Invitrogen) or with 10 μM 5- and 6-(4-chloromethyl)benzoyl-amino-tetramethylrhodamine (Invitrogen). For in vivo blocking experiments, purified anti-CD62L, anti-CD62P, anti-CD62E, or isotype control antibodies (all from BD Biosciences) were injected i.v (100 μg per mouse). These experiments were performed either before or after transfer of fluorescently labeled cells and before or after injection of DCs, as indicated in the figures.
Immunization.
The following proteins and peptides were used: whole OVA protein was obtained from Sigma-Aldrich and KLH was obtained from EMD; purified HA of influenza A/IVR116 (H1N1) virus was a gift of G. Galli (Novartis, Siena, Italy); and OVA323-339 (ISQAVHAAHAEINEAGR), HA110-119 (SFERFEIFPK), and MOG35-55 (MEVGWYRSPFSRVVHLYRNGK) peptides were synthesized by the Servei de Proteòmica (Pompeu Fabra University, Barcelona, Spain). For immunization with adjuvant, 50 μg of whole protein was emulsified in CFA (DIFCO) and injected s.c. in the flank. Where indicated in the figures, immunization was performed i.v. with 10 μg OVA or HA in PBS. 3–4 wk after priming, CD4+ T cells were enriched from spleen and peripheral lymph nodes with CD4 magnetic beads (Miltenyi Biotec) and sorted using a FACSAria (BD Biosciences) according to the expression of CD44, CD62L, and CD127. Transgenic Thy1.1+ DO11.10 CD4+ T cells were purified from spleen and lymph nodes and labeled with 2.5 μM CFSE, and 106 cells were injected i.v. into syngeneic hosts. Adoptively transferred mice received one (DC 1×) or two consecutive (2 d apart; DC 2×) s.c. injections of 106 OVA-loaded, LPS-matured DCs. 3–4 wk after priming, DO11.10 CD4+ T cells were enriched from spleen and lymph nodes by cell sorting according to the expression of CD44, CD62L, and CD127 (TCM, CD44highCD62L+CD127+; TEM, CD44highCD62L−CD127+). Naive T cells (CD44lowCD62L+CD127+) were purified from spleens of Rag2−/− DO11.10 mice. CD4+ naive T, TCM, and TEM cells were adoptively transferred into syngeneic hosts or used in vitro, as indicated in the figures. Reactive lymph nodes were in some experiments induced by s.c. injection of LPS, CpG1826 oligodeoxynucleotide (5′-CCATGACGTTCCTGACGTT-3′; Microsynth), IFA (DIFCO), or CFA. Peritonitis was induced by an i.p. injection of a 3% solution (wt/vol) of thioglycolate (Sigma-Aldrich) in PBS.
In vitro experiments.
Naive antigen-specific CD4+ T cells were primed in vitro with immature or with LPS-matured DCs pulsed with the peptides indicated in the figures (T cell/DC ratio = 10:1). CD4+ naive T, TCM, or TEM cells specific for a different antigen were added to these cultures. Polyclonal and OT-II CD4+ T cells were primed in vivo, and 3 wk later TEM CD4+ T cells were sorted as before and expanded in vitro with 10 μg/ml each of anti-CD3 and anti-CD28 (both from BD Biosciences). On day 4, cultures were supplemented with 10 U/ml of human recombinant IL-2 (55), and cells were collected 7–9 d later. To measure surface expression of CD40L, CD4+ T cells were activated in vitro with soluble 10 μg/ml anti-CD3 (BD Biosciences) and 10 ng/ml phorbol-12,13 dibutyrate (PDBu; Sigma-Aldrich). At the time points after stimulation indicated in the figures, cold PBS containing 5% FCS was added to the cells. Immature BM-DCs were prepared and co-cultured in vitro with naive or in vivo–generated TCM or TEM CD4+ T cells. DCs were analyzed for expression of CD40, CD86, and MHC-II 12 h after co-culture. For fixation of T cells, 5 × 106 CD4+ naive T, TCM, and TEM cells were washed in PBS/1% FCS and fixed in 1 ml 0.05% glutaraldehyde (Merck) for 30 s at room temperature. 1 ml of 0.2 M Glycin (Sigma-Aldrich) was then added for 30 s. Cells were finally washed and co-cultured with immature DCs.
Flow cytometry and cell sorting.
The following monoclonal antibodies were used: anti-CD62L (MEL14), anti-CD62P (RB40.34), anti-CD62E (10E9.6), anti-CD44 (IM7), anti-CD4 (L3T4), anti-CD8 (Ly-2), anti-CD3 (145-2C11), anti-CD11c (HL3), anti-CD40 (3/23), anti-IA/IEd (2G9), anti-CD86 (PO3), anti–IFN-γ (XMG1.2), anti–IL-2 (JES6-5H4), anti-TNF (MP6-XT22), and anti–mouse DO11.10 clonotypic TCR (KJ1-26; all from BD Biosciences). The anti-CD45.1 (A20), anti-CD45.2 (104), anti-CD90.1 (HIS51), anti-CD127 (A7R34), and anti-CCR7 (4B12) antibodies were from eBioscience. The anti-CXCR3 (220803) antibody was from R&D Systems. The anti-CD154 (MR-1) antibody was from SouthernBiotech. The anti–mouse HA clonotypic TCR (clone 6.5) was a gift of J. Kirberg. CD62P ligands on T cells were detected by staining with a human IgG-CD62P fusion protein (BD Biosciences), followed by incubation with a biotinylated-goat F(ab′)2 anti–human IgG antibody (SouthernBiotech) and streptavidin-allophycocyanin (APC; Invitrogen). Cytokine production was measured by intracellular staining after 4 h of stimulation with 10−7 M PMA and 1 μg/ml ionomycin (both from Sigma-Aldrich), with the last 2 h of culture in the presence of 10 μg/ml brefeldin A (Sigma-Aldrich). APC-labeled antibodies to IFN-γ, IL-2, and TNF (BD Bioscience) were used after cell fixation in 4% (wt/vol) paraformaldehyde and permeabilization with 0.5% (wt/vol) saponin (Sigma-Aldrich). 6-color staining of the cell surface was done with the appropriate combinations of antibodies conjugated to FITC, PE, PerCP, PE-Cy7, APC, APC-Cy7, or biotin, and with streptavidin labeled with PE-Cy7 or APC-Cy7 (BD Biosciences). Samples were acquired on a FACSCanto (BD Biosciences) and were analyzed with FlowJo software (Tree Star, Inc.).
Immunohistochemistry and histology.
For detection of CD62P, reactive lymph nodes were induced by s.c. injection of LPS-matured DCs or adjuvant, as indicated in the figures. In some experiments, 50 μg of FITC-labeled anti-CD62P or anti-CD62E antibodies (BD Biosciences) per mouse were injected i.v. 30 min after antibody treatment. Draining reactive and control resting lymph nodes were collected and snap frozen in liquid nitrogen. Tissue sections of 6-μm thickness were prepared and directly processed for microscopic visualization without further counterstaining. For PNAd detection, tissue sections were fixed in cold (4°C) acetone for 10 min and incubated for 30 min with a 1:100 dilution of PE-labeled rat IgM MECA-79 antibody (BD Biosciences), followed by incubation with Alexa Fluor 594 donkey anti–rat IgM (Invitrogen). A microscope (Eclipse E800; Nikon) was used for immunofluorescence analysis. Separate images were collected for each fluorochrome, and images were overlaid to produce a multicolor image with Openlab 5 software (Improvision). Individual images were juxtaposed with Photoshop software (Adobe) to reconstitute the image of lymph node cross sections.
EAE.
Groups of 6-wk-old C57BL/6 mice were immunized s.c. with CFA in PBS (50:50, vol/vol). 30 d later mice were challenged i.v. with 100 μm MOG35-55 peptide in PBS containing 200 ng pertussis toxin (PTX; Sigma-Aldrich). A second injection of PTX was given 2 d later. Control 8–10-wk-old C57BL/6 female mice were immunized s.c. on day 0 with 100 μg MOG35-55 peptide emulsified in CFA. PTX was injected i.v. twice in 100 μl saline, on days 0 and 2. Groups of mice received 100 μm anti-CD62P antibodies or isotype-matched irrelevant antibodies as controls once a week. Control mice received IFA alone. Mice were graded for clinical manifestation of EAE on a 0–5 scale in a blinded manner. A 5-point disease severity scale was used, as follows: 0, no disease; 1, tail weakness; 2, paraparesis; 3, paraplegia; 4, paraplegia with forelimb weakness or paralysis; and 5, moribund or dead animal. For the isolation of lymphocytes infiltrating the central nervous system, brains and spinal cords were excised and dissociated for 1 h at 37°C in 1 mg/ml collagenase D (Roche) in RPMI 1640 medium. Cells were passed through a nylon mesh, pelleted, and resuspended in RPMI 1640 medium, and layered onto a Lympholyte-M density gradient (Cedarlane). Tubes were centrifuged at room temperature for 20 min at 800 g. Viable lymphocytes were removed from the interface.
Statistical analysis.
Data, presented as means ± SD, were analyzed with a paired Student's t test using the SPSS software (v16). P < 0.05 was considered significant.
Online supplemental material.
Fig. S1 shows the sorting strategy for T naive, TCM, and TEM cells. Fig. S2 shows that adoptively transferred naive T and TEM cells localized in the same area in reactive lymph nodes. Fig. S3 shows that TEM cell migration into the reactive lymph node is not dependent on the presence of specific antigen, and that TEM cells do not represent a resident population in reactive lymph nodes. Fig. S4 shows that T cells may be required for induction of chronic reactive lymph nodes. Fig. S5 shows the absolute number of DO11.10 TEM cells in chronic reactive lymph nodes in the experiment in Fig. 5.
Supplementary Material
Acknowledgments
We thank J. Kirberg, S.L. Swain, R.O. Hynes, K. Schwarz, B. Engelhardt, and C. Gerard for transgenic and knockout mice; J. Stein for critical reading and discussions; D. Jarrossay, E. Mira-Catò, and L. Perlini for technical help; and K. Kuscher for help in manuscript editing.
Supported by the Swiss National Science Foundation (grants 31-63885 and 31-109832 to F. Sallusto and A. Martín-Fontecha, respectively), the European Commission FP6 “Network of Excellence” initiative (grants LSHG-CT-2003-502935 MAIN, LSHB-CT-2004-512074 DC-THERA, and LSHG-CT-2005-005203 MUGEN), and the Helmut Horten Foundation (to the Institute for Research in Biomedicine). D. Baumjohann is the recipient of a Boehringer Ingelheim Fonds PhD Scholarship. A. Reboldi is supported by the European Union–funded International Graduate Program in Molecular Medicine.
The authors declare no competing financial interests.
Abbreviations used: EAE, experimental autoimmune encephalomyelitis; HA, hemagglutinin; HEV, high endothelial venule; MOG, myelin oligodendrocyte glycoprotein; PNAd, peripheral node addressin; TCM, central memory T; TEM, effector memory T.
A. Martín-Fontecha's present address is Dept. of Nephrology and Transplantation, King's College London and Guy's and St Thomas' Hospital, SE1 9RT London, England, UK.
G. Guarda's present address is Dept. of Biochemistry, University of Lausanne, 1066 Epalinges, Switzerland.
M. Hons's present address is Theodor Kocher Institute, University of Bern, 3012 Bern, Switzerland.
References
- 1.Steinman, R.M., and J. Banchereau. 2007. Taking dendritic cells into medicine. Nature. 449:419–426. [DOI] [PubMed] [Google Scholar]
- 2.Steinman, R.M., D. Hawiger, and M.C. Nussenzweig. 2003. Tolerogenic dendritic cells. Annu. Rev. Immunol. 21:685–711. [DOI] [PubMed] [Google Scholar]
- 3.Kurts, C., H. Kosaka, F.R. Carbone, J.F. Miller, and W.R. Heath. 1997. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8+ T cells. J. Exp. Med. 186:239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adler, A.J., D.W. Marsh, G.S. Yochum, J.L. Guzzo, A. Nigam, W.G. Nelson, and D.M. Pardoll. 1998. CD4+ T cell tolerance to parenchymal self-antigens requires presentation by bone marrow–derived antigen-presenting cells. J. Exp. Med. 187:1555–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hernandez, J., S. Aung, W.L. Redmond, and L.A. Sherman. 2001. Phenotypic and functional analysis of CD8+ T cells undergoing peripheral deletion in response to cross-presentation of self-antigen. J. Exp. Med. 194:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janeway, C.A., Jr., and R. Medzhitov. 2002. Innate immune recognition. Annu. Rev. Immunol. 20:197–216. [DOI] [PubMed] [Google Scholar]
- 7.Matzinger, P. 2002. The danger model: a renewed sense of self. Science. 296:301–305. [DOI] [PubMed] [Google Scholar]
- 8.Reis e Sousa, C. 2006. Dendritic cells in a mature age. Nat. Rev. Immunol. 6:476–483. [DOI] [PubMed] [Google Scholar]
- 9.Schulz, O., A.D. Edwards, M. Schito, J. Aliberti, S. Manickasingham, A. Sher, and C. Reis e Sousa. 2000. CD40 triggering of heterodimeric IL-12 p70 production by dendritic cells in vivo requires a microbial priming signal. Immunity. 13:453–462. [DOI] [PubMed] [Google Scholar]
- 10.Napolitani, G., A. Rinaldi, F. Bertoni, F. Sallusto, and A. Lanzavecchia. 2005. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat. Immunol. 6:769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ridge, J.P., F. Di Rosa, and P. Matzinger. 1998. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 393:474–478. [DOI] [PubMed] [Google Scholar]
- 12.Schoenberger, S.P., R.E. Toes, E.I. van der Voort, R. Offringa, and C.J. Melief. 1998. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 393:480–483. [DOI] [PubMed] [Google Scholar]
- 13.Bennett, S.R., F.R. Carbone, F. Karamalis, R.A. Flavell, J.F. Miller, and W.R. Heath. 1998. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 393:478–480. [DOI] [PubMed] [Google Scholar]
- 14.Bonifaz, L., D. Bonnyay, K. Mahnke, M. Rivera, M.C. Nussenzweig, and R.M. Steinman. 2002. Efficient targeting of protein antigen to the dendritic cell receptor DEC-205 in the steady state leads to antigen presentation on major histocompatibility complex class I products and peripheral CD8+ T cell tolerance. J. Exp. Med. 196:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grewal, I.S., and R.A. Flavell. 1998. CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 16:111–135. [DOI] [PubMed] [Google Scholar]
- 16.Casamayor-Palleja, M., M. Khan, and I.C. MacLennan. 1995. A subset of CD4+ memory T cells contains preformed CD40 ligand that is rapidly but transiently expressed on their surface after activation through the T cell receptor complex. J. Exp. Med. 181:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lettesjo, H., G.P. Burd, and R.A. Mageed. 2000. CD4+ T lymphocytes with constitutive CD40 ligand in preautoimmune (NZB × NZW)F1 lupus-prone mice: phenotype and possible role in autoreactivity. J. Immunol. 165:4095–4104. [DOI] [PubMed] [Google Scholar]
- 18.Koguchi, Y., T.J. Thauland, M.K. Slifka, and D.C. Parker. 2007. Preformed CD40 ligand exists in secretory lysosomes in effector and memory CD4+ T cells and is quickly expressed on the cell surface in an antigen-specific manner. Blood. 110:2520–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lesley, R., L.M. Kelly, Y. Xu, and J.G. Cyster. 2006. Naive CD4 T cells constitutively express CD40L and augment autoreactive B cell survival. Proc. Natl. Acad. Sci. USA. 103:10717–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Andrian, U.H., and T.R. Mempel. 2003. Homing and cellular traffic in lymph nodes. Nat. Rev. Immunol. 3:867–878. [DOI] [PubMed] [Google Scholar]
- 21.Reinhardt, R.L., A. Khoruts, R. Merica, T. Zell, and M.K. Jenkins. 2001. Visualizing the generation of memory CD4 T cells in the whole body. Nature. 410:101–105. [DOI] [PubMed] [Google Scholar]
- 22.Masopust, D., V. Vezys, A.L. Marzo, and L. Lefrancois. 2001. Preferential localization of effector memory cells in nonlymphoid tissue. Science. 291:2413–2417. [DOI] [PubMed] [Google Scholar]
- 23.Iezzi, G., D. Scheidegger, and A. Lanzavecchia. 2001. Migration and function of antigen-primed nonpolarized T lymphocytes in vivo. J. Exp. Med. 193:987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martin-Fontecha, A., L.L. Thomsen, S. Brett, C. Gerard, M. Lipp, A. Lanzavecchia, and F. Sallusto. 2004. Induced recruitment of NK cells to lymph nodes provides IFN-gamma for T(H)1 priming. Nat. Immunol. 5:1260–1265. [DOI] [PubMed] [Google Scholar]
- 25.Guarda, G., M. Hons, S.F. Soriano, A.Y. Huang, R. Polley, A. Martin-Fontecha, J.V. Stein, R.N. Germain, A. Lanzavecchia, and F. Sallusto. 2007. L-selectin-negative CCR7− effector and memory CD8+ T cells enter reactive lymph nodes and kill dendritic cells. Nat. Immunol. 8:743–752. [DOI] [PubMed] [Google Scholar]
- 26.Hawiger, D., K. Inaba, Y. Dorsett, M. Guo, K. Mahnke, M. Rivera, J.V. Ravetch, R.M. Steinman, and M.C. Nussenzweig. 2001. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J. Exp. Med. 194:769–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin-Fontecha, A., G. Guarda, A. Lanzavecchia, and F. Sallusto. 2008. Activated lymph nodes recruit blood borne NK cells and effector T cells: implication for adaptive T cell responses. Curr. Immunol. Rev. 4:20–27. [Google Scholar]
- 28.McEver, R.P., and R.D. Cummings. 1997. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J. Clin. Invest. 100:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelhardt, B., D. Vestweber, R. Hallmann, and M. Schulz. 1997. E- and P-selectin are not involved in the recruitment of inflammatory cells across the blood-brain barrier in experimental autoimmune encephalomyelitis. Blood. 90:4459–4472. [PubMed] [Google Scholar]
- 30.Fazilleau, N., M.D. Eisenbraun, L. Malherbe, J.N. Ebright, R.R. Pogue-Caley, L.J. McHeyzer-Williams, and M.G. McHeyzer-Williams. 2007. Lymphoid reservoirs of antigen-specific memory T helper cells. Nat. Immunol. 8:753–761. [DOI] [PubMed] [Google Scholar]
- 31.Diacovo, T.G., K.D. Puri, R.A. Warnock, T.A. Springer, and U.H. von Andrian. 1996. Platelet-mediated lymphocyte delivery to high endothelial venules. Science. 273:252–255. [DOI] [PubMed] [Google Scholar]
- 32.Ley, K., C. Laudanna, M.I. Cybulsky, and S. Nourshargh. 2007. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7:678–689. [DOI] [PubMed] [Google Scholar]
- 33.Kunkel, E.J., C.L. Ramos, D.A. Steeber, W. Muller, N. Wagner, T.F. Tedder, and K. Ley. 1998. The roles of L-selectin, beta 7 integrins, and P-selectin in leukocyte rolling and adhesion in high endothelial venules of Peyer's patches. J. Immunol. 161:2449–2456. [PubMed] [Google Scholar]
- 34.Harakawa, N., A. Shigeta, M. Wato, G. Merrill-Skoloff, B.C. Furie, B. Furie, T. Okazaki, N. Domae, M. Miyasaka, and T. Hirata. 2007. P-selectin glycoprotein ligand-1 mediates L-selectin-independent leukocyte rolling in high endothelial venules of peripheral lymph nodes. Int. Immunol. 19:321–329. [DOI] [PubMed] [Google Scholar]
- 35.Lopez, S., N. Prats, and A.J. Marco. 1999. Expression of E-selectin, P-selectin, and intercellular adhesion molecule-1 during experimental murine listeriosis. Am. J. Pathol. 155:1391–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Soderberg, K.A., G.W. Payne, A. Sato, R. Medzhitov, S.S. Segal, and A. Iwasaki. 2005. Innate control of adaptive immunity via remodeling of lymph node feed arteriole. Proc. Natl. Acad. Sci. USA. 102:16315–16320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinman, R.M., and M.C. Nussenzweig. 2002. Avoiding horror autotoxicus: the importance of dendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA. 99:351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bajenoff, M., J.G. Egen, H. Qi, A.Y. Huang, F. Castellino, and R.N. Germain. 2007. Highways, byways and breadcrumbs: directing lymphocyte traffic in the lymph node. Trends Immunol. 28:346–352. [DOI] [PubMed] [Google Scholar]
- 39.Waldner, H., M. Collins, and V.K. Kuchroo. 2004. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J. Clin. Invest. 113:990–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nogai, A., V. Siffrin, K. Bonhagen, C.F. Pfueller, T. Hohnstein, R. Volkmer-Engert, W. Bruck, C. Stadelmann, and T. Kamradt. 2005. Lipopolysaccharide injection induces relapses of experimental autoimmune encephalomyelitis in nontransgenic mice via bystander activation of autoreactive CD4+ cells. J. Immunol. 175:959–966. [DOI] [PubMed] [Google Scholar]
- 41.Petrilli, V., C. Dostert, D.A. Muruve, and J. Tschopp. 2007. The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 19:615–622. [DOI] [PubMed] [Google Scholar]
- 42.Balashov, K.E., D.R. Smith, S.J. Khoury, D.A. Hafler, and H.L. Weiner. 1997. Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc. Natl. Acad. Sci. USA. 94:599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Teleshova, N., W. Bao, P. Kivisakk, V. Ozenci, M. Mustafa, and H. Link. 2000. Elevated CD40 ligand expressing blood T-cell levels in multiple sclerosis are reversed by interferon-beta treatment. Scand. J. Immunol. 51:312–320. [DOI] [PubMed] [Google Scholar]
- 44.Jensen, J., M. Krakauer, and F. Sellebjerg. 2001. Increased T cell expression of CD154 (CD40-ligand) in multiple sclerosis. Eur. J. Neurol. 8:321–328. [DOI] [PubMed] [Google Scholar]
- 45.Howard, L.M., and S.D. Miller. 2004. Immunotherapy targeting the CD40/CD154 costimulatory pathway for treatment of autoimmune disease. Autoimmunity. 37:411–418. [DOI] [PubMed] [Google Scholar]
- 46.Quezada, S.A., L.Z. Jarvinen, E.F. Lind, and R.J. Noelle. 2004. CD40/CD154 interactions at the interface of tolerance and immunity. Annu. Rev. Immunol. 22:307–328. [DOI] [PubMed] [Google Scholar]
- 47.Calzascia, T., M. Pellegrini, A. Lin, K.M. Garza, A.R. Elford, A. Shahinian, P.S. Ohashi, and T.W. Mak. 2008. CD4 T cells, lymphopenia, and IL-7 in a multistep pathway to autoimmunity. Proc. Natl. Acad. Sci. USA. 105:2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Behrens, G.M., M. Li, G.M. Davey, J. Allison, R.A. Flavell, F.R. Carbone, and W.R. Heath. 2004. Helper requirements for generation of effector CTL to islet beta cell antigens. J. Immunol. 172:5420–5426. [DOI] [PubMed] [Google Scholar]
- 49.Bettelli, E., T. Korn, and V.K. Kuchroo. 2007. Th17: the third member of the effector T cell trilogy. Curr. Opin. Immunol. 19:652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barnden, M.J., J. Allison, W.R. Heath, and F.R. Carbone. 1998. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol. Cell Biol. 76:34–40. [DOI] [PubMed] [Google Scholar]
- 51.Kirberg, J., A. Baron, S. Jakob, A. Rolink, K. Karjalainen, and H. von Boehmer. 1994. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex–restricted receptor. J. Exp. Med. 180:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawabe, T., T. Naka, K. Yoshida, T. Tanaka, H. Fujiwara, S. Suematsu, N. Yoshida, T. Kishimoto, and H. Kikutani. 1994. The immune responses in CD40-deficient mice: impaired immunoglobulin class switching and germinal center formation. Immunity. 1:167–178. [DOI] [PubMed] [Google Scholar]
- 53.Malissen, M., A. Gillet, L. Ardouin, G. Bouvier, J. Trucy, P. Ferrier, E. Vivier, and B. Malissen. 1995. Altered T cell development in mice with a targeted mutation of the CD3-epsilon gene. EMBO J. 14:4641–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin-Fontecha, A., S. Sebastiani, U.E. Hopken, M. Uguccioni, M. Lipp, A. Lanzavecchia, and F. Sallusto. 2003. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J. Exp. Med. 198:615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Traunecker, A., F. Oliveri, and K. Karjalainen. 1991. Myeloma based expression system for production of large mammalian proteins. Trends Biotechnol. 9:109–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}